
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational studies on substituted ε-caprolactams by X-ray crystallography and NMR spectroscopy†‡
Tobias
Gruber§
*a,
Amber L.
Thompson
a,
Barbara
Odell
a,
Petra
Bombicz
b and
Christopher J.
Schofield
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 275625
bInstitute of Organic Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar Tudósok körútja 2., H-1117 Budapest, Hungary
First published on 25th September 2014
Abstract
The synthesis and conformational analysis of ε-caprolactams containing a –COOMe group at the C-6 position is described. The influence of different C-2, C-6 and N substituents on ring conformation was studied using X-ray crystallography and NMR spectroscopy. The results provide evidence that all the analysed caprolactams adopt a chair type conformation with a planar lactam. In the 6-substituted caprolactam, the –COOMe residue prefers to reside in an equatorial position, but can be induced to occupy an axial orientation by the introduction of a bulky tert-butyloxycarbonyl (BOC) group on the lactam nitrogen or by C-2/C-3 ring desaturation. The BOC protected caprolactam was found to undergo exchange between two chair forms as detected by solution NMR, one with the C-6 ester equatorial (30%) and the other with it in the axial position (70%); the latter was observed by X-ray crystallography. For the C-2 dithiocarbamate substituted C-6 methyl ester seven-membered rings, a single chair form is observed for cis-isomers with both substituents equatorial. The analogous trans-isomers, however, exist as two chair forms in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equilibrium ratio of 1,NC4 and 4C1,N conformers, where either substituent can occupy axial or equatorial positions.
:1 equilibrium ratio of 1,NC4 and 4C1,N conformers, where either substituent can occupy axial or equatorial positions.
Introduction
ε-Caprolactam, or hexahydro-2-azepinone, is an important starting material in polymer chemistry; it is produced from cyclohexanone by Beckmann rearrangement.1 ε-Caprolactam is used in nylon preparation,2 and as such is the basis for the manufacturing of many useful products. Derivatives of ε-caprolactam are of interest for the production of modified nylons3 and nanogels.4 Azepinones and their unsaturated and saturated analogues play an important role in medicinal chemistry,5 including in drugs (e.g. Benazepril,6 Ivabradine,7 Telcagepant8), antibiotic research (e.g. capuramycin9), and as simple models of cyclic peptides.10 Despite the importance of caprolactams, reports on the influence of substituents on the conformations of caprolactam rings and the mutual influence of different substituents are rare11 including with respect to the solid state behavior of single component caprolactams and respective co-crystals.12Although the influence of various substituents on the conformation of cyclohexanes has been studied in detail, analogous reports on seven-membered rings are much less comprehensive. Based on studies of the conformation at the cycloheptene ring,13 caprolactams are predicted to exist in (pseudo) ‘chair’, ‘boat’ or a transition ‘twisted boat’ or ‘twisted chair’ conformations. In the case of the ‘chair’ form, two energetically favoured chair conformations can be identified (4C1,N and 1,NC4), assuming the amide C–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N–C segment is planar14 (Scheme 1). Furthermore it is known, that axial substituents on caprolactams are higher in energy than the equatorial substituents as shown for methyl and tert-butyl substituents at C-2 and C-6.15
O)–N–C segment is planar14 (Scheme 1). Furthermore it is known, that axial substituents on caprolactams are higher in energy than the equatorial substituents as shown for methyl and tert-butyl substituents at C-2 and C-6.15
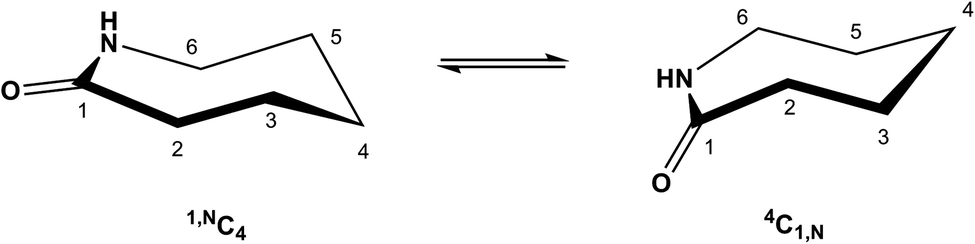 | ||
| Scheme 1 The two energetically favoured (pseudo) chair conformations of ε-caprolactam: 1,NC4 and 4C1,N. (For consistency we apply here the reported numbering system.14) | ||
Arising from studies on β-lactam antibiotic biosynthesis and mode of action,16 we were interested in a caprolactam functionalized at C-6 with a –COOMe group (1). For subsequent reactions concerning the caprolactam core we were keen to alter the conformation of the ring and position the methyl ester in an axial position. For this purpose, the effects of a C-2 substituent (adjacent to the rigid amide) and at the amide nitrogen on the conformations of the respective caprolactam derivatives were tested. Here we report solution NMR and crystallographic structural studies on the effects of substituents on caprolactam conformation.
Results and discussion
Synthesis
For the preparation of methyl ester 1 we investigated several different reactions. In contrast to the efficient γ- and δ-lactam preparation, analogous cyclization of the respective racemic aminopimelic acid with subsequent methylation had little success when using activating agents such as 3,4,5-trifluorobenzene boronic acid17 (no reaction) or SiO218 (10% yield). However, preparation of the dimethyl ester of aminopimelic acid followed by heating in refluxing p-cymene19,20 gave 1 in reasonable yield (57%, unoptimised). The free caprolactam acid 2 was obtained after saponification of 1 (LiOH, aq. THF) (70%).In order to introduce a sterically hindered tert-butyloxycarbonyl group at the amide nitrogen, 1 was reacted with (tBuOCO)2O under basic conditions in toluene to give 3. For C-2 substitution, we first carried out bromination using molecular bromine21 to yield an inseparable mixture of isomers (4). Subsequent treatment with two different dithiocarbamate salts yielded compounds 5 and 6, which could be separated into cis and trans isomers, with a significantly higher amount of the cis isomer being formed. The dithiocarbamates were chosen as, in general, they allow versatile consecutive reactions.22 After a Tschugaev-like pyrolysis,23 we obtained the C-2/C-3 unsaturated caprolactam (7), together with the rearranged C3/C4 isomer (8) in a 1:1 ratio24 (Scheme 2).
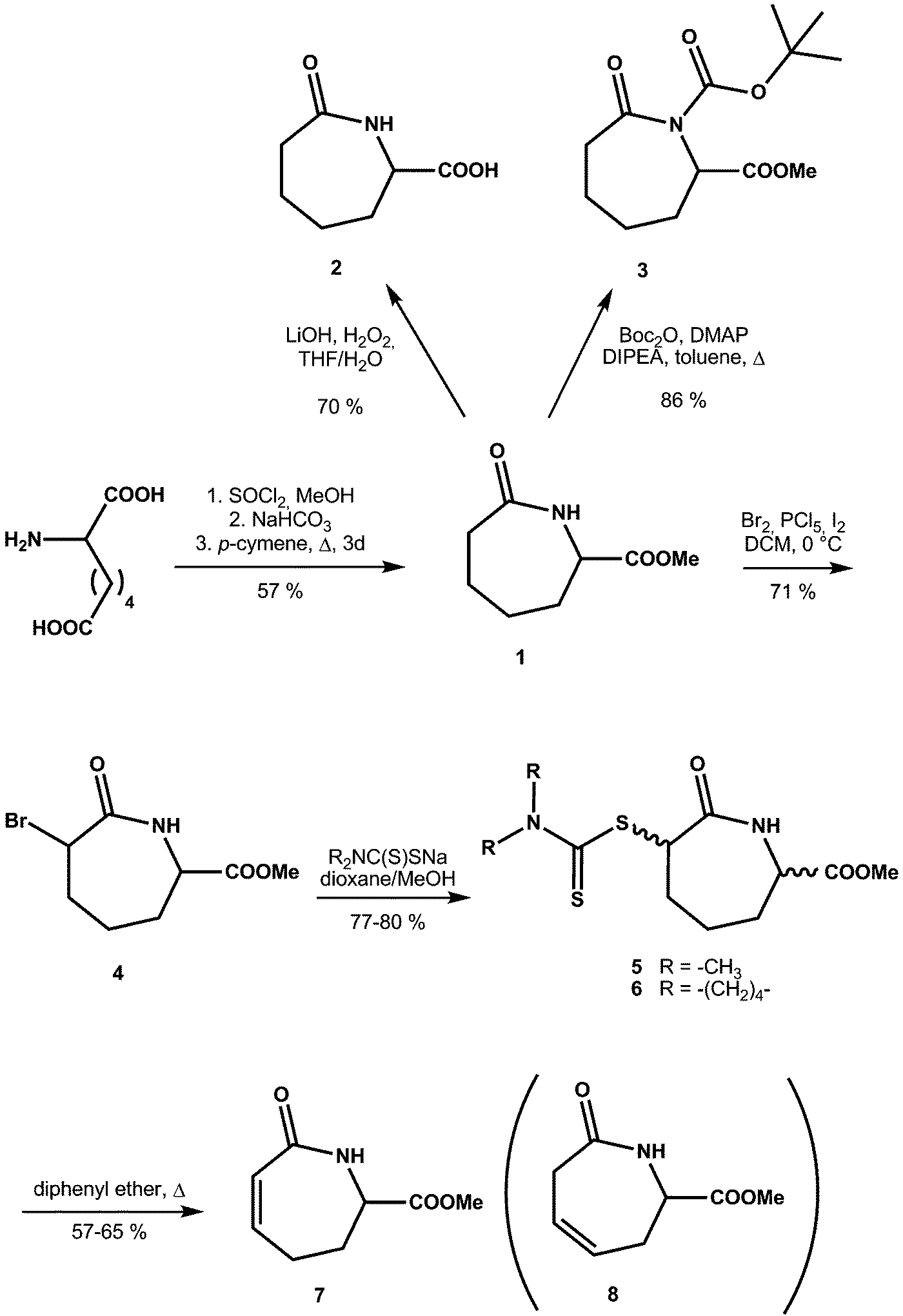 | ||
| Scheme 2 Synthesis of caprolactam derivatives 1–8. | ||
Conformational analyses of monosubstituted caprolactams
Caprolactam derivatives 1, 2, 3, 5cis, 5trans and 6cis and 7 were characterized by single-crystal X-ray diffraction. For selected crystallographic and structural refinement parameters, molecular torsion angles, and information regarding hydrogen bond geometries see Tables 1 and 2 and Table S1 (ESI‡). The molecular overlay calculation data, rmsd (= root mean square of the atomic distances) and maxd (the maximal atomic distance difference of corresponding atoms) for the caprolactam moieties with respect to ε-caprolactam are listed in Table 3. Crystal structures of ε-caprolactam have been reported;25,26 the most recently reported structure of caprolactam (published by Winkler and Dunitz in 1975), was used for comparison as it has the best reported agreement factors (Cambridge Structural Database REF CODE: CAPLAC).| Compound | 1 | 2 | 3 | 5 cis | 5 trans | 6 cis | 7 |
|---|---|---|---|---|---|---|---|
| Empirical formula | C8H13NO3 | C7H11N1O3 | C13H21NO5 | C11H18N2O3S2 | C11H18N2O3S2 | C13H20N2O3S2 | C8H11NO3 |
| Formula weight (g mol−1) | 171.19 | 157.17 | 271.31 | 290.40 | 290.40 | 316.44 | 169.18 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n | P21/c |
P |
P21/n |
P |
| a (Å) | 5.2735(2) | 10.5713(8) | 6.27304(11) | 7.54965(8) | 7.9432(6) | 7.71325(7) | 6.2079(4) |
| b (Å) | 8.6164(4) | 10.6265(5) | 8.13334(11) | 7.8511(1) | 8.4258(6) | 8.16076(7) | 7.7139(7) |
| c (Å) | 9.3766(4) | 11.0705(7) | 27.2810(5) | 23.3724(3) | 11.2990(8) | 23.7540(2) | 8.6278(8) |
| α (°) | 97.6974(18) | 72.339(5) | 90 | 90 | 89.703(6) | 90 | 82.686(8) |
| β (°) | 93.5064(18) | 78.005(6) | 91.0045(16) | 95.6507(11) | 71.855(6) | 94.6857(8) | 88.718(7) |
| γ (°) | 95.8677(19) | 80.552(5) | 90 | 90 | 73.959(6) | 90 | 89.279(6) |
| V (Å3) | 418.80(3) | 1152.29(13) | 1391.68(4) | 1378.62(3) | 687.92(9) | 1490.22(2) | 409.68(6) |
| Z | 2 | 6 | 4 | 4 | 2 | 4 | 2 |
| D c (Mg m−3) | 1.358 | 1.359 | 1.295 | 1.399 | 1.402 | 1.410 | 1.371 |
| μ (mm−1) | 0.104 | 0.896 | 0.826 | 3.539 | 0.389 | 3.323 | 0.884 |
| Data collection | |||||||
| Temperature (K) | 150 | 150 | 150 | 150 | 150 | 150 | 150 |
| Wavelength (Å) | 0.71073 | 1.54184 | 1.54184 | 1.54184 | 0.71073 | 1.54180 | 1.54184 |
| No. of collected reflections | 7659 | 10066 |
13198 |
11617 |
6132 | 19616 |
3072 |
| θ max (°) | 27.408 | 76.407 | 76.124 | 76.330 | 30.707 | 76.356 | 75.764 |
| Completeness to θmax (%) | 98.6 | 98.1 | 95.6 | 99.0 | 83.0 | 99.3 | 97.3 |
| No. of unique reflections | 1882 | 4752 | 2770 | 2866 | 3550 | 3107 | 1671 |
| R(int) | 0.028 | 0.023 | 0.028 | 0.025 | 0.037 | 0.021 | 0.011 |
| No. of refined parameters | 109 | 298 | 172 | 163 | 163 | 181 | 109 |
| No. reflections [I > 2σ(I)] | 918 | 3278 | 2364 | 2866 | 2530 | 2987 | 1592 |
| Final R-indices | |||||||
| R 1 [I > 2σ(I)] (%) | 4.24 | 5.83 | 3.49 | 2.77 | 5.65 | 2.57 | 3.27 |
| wR [I > 2σ(I)] (%) | 10.79 | 15.18 | 8.54 | 7.18 | 9.53 | 10.49 | 8.83 |
| S (= Goodness of fit on F2) | 0.9467 | 0.9925 | 0.9877 | 0.9588 | 1.0097 | 0.9997 | 1.0124 |
| Final Δρmax/Δρmin (e Å−3) | 0.28, −0.28 | 1.03, −0.32 | 0.33, −0.23 | 0.30, −0.21 | 0.66, −0.67 | 0.29, −0.21 | 0.23, −0.18 |
| Atoms | 1 | 2(1) | 2(2) | 2(3) | 3 | 5 cis | 5 trans | 6 cis | 7 |
|---|---|---|---|---|---|---|---|---|---|
a General numbering scheme for compound 3:
|
|||||||||
| C1–C2–C3–C4 | 82.8(3) | −80.8(3) | −79.3(3) | 81.7(3) | 84.34(17) | 83.88(12) | −84.3(3) | 84.96(10) | −2.1(2) |
| C2–C3–C4–C5 | −61.4(3) | 64.7(4) | 59.4(3) | −64.1(4) | −60.79(17) | −67.42(14) | 67.6(3) | −67.75(12) | 1.29(18) |
| C3–C4–C5–C6 | 59.3(3) | −62.0(4) | −61.6(3) | 61.9(3) | 60.03(17) | 63.95(15) | −60.0(3) | 63.48(12) | −44.53(15) |
| C4–C5–C6–N1 | −77.3(3) | 75.9(3) | 81.4(2) | −77.2(3) | −79.65(16) | −75.49(14) | 69.1(3) | −75.44(12) | 80.94(14) |
| C1–N1–C6–C5 | 66.5(3) | −66.1(3) | −65.1(3) | 66.2(4) | 65.01(15) | 62.76(16) | −63.2(4) | 62.63(14) | −54.82(15) |
| C6–N1–C1–C2 | −0.4(3) | 5.0(4) | −4.8(3) | −2.5(4) | −0.49(16) | 0.93(18) | 5.2(4) | 1.65(15) | −2.02(15) |
| N1–C1–C2–C3 | −68.0(3) | 61.5(3) | 70.3(3) | −63.9(4) | −67.95(16) | −66.95(14) | 62.5(3) | −67.75(11) | 25.22(18) |
| N1–C6–C7–O2 | −7.3(3) | 5.9(3) | 0.3(3) | 4.4(4) | 14.50(16) | −11.24(14) | −9.6(4) | −10.46(14) | 15.92(15) |
| Compared structures | rmsD | maxD | |
|---|---|---|---|
| a Structure is inverted related to the received one or related to the other molecule present in the asymmetric unit. | |||
| CAPLAC | 1 | 0.0391 | 0.0668 |
| 1 | 2(1)a | 0.0856 | 0.1817 |
| 1 | 2(2) | 0.0925 | 0.1907 |
| 1 | 2(3)a | 0.0626 | 0.1150 |
| 1 | 3 | 0.0429 | 0.0786 |
| 1 | 5 cis | 0.0302 | 0.0489 |
| 1 | 6 cis | 0.0290 | 0.0437 |
| 1 | 7 | 0.2273 | 0.4423 |
| 2(1)a | 2(2) | 0.0734 | 0.1227 |
| 2(1)a | 2(3)a | 0.1223 | 0.2196 |
| 2(2) | 2(3)a | 0.0894 | 0.2149 |
| 5 cis | 6 cis | 0.0084 | 0.0136 |
Generally in monosubstituted caprolactams, axial substituents are anticipated to be higher in energy than the equatorial ones, a conclusion supported by molecular mechanics calculations carried out on the C6 methyl ester (1). The 4C1,N form with the carboxylate in the axial position is calculated to be 8 kcal mol−1 higher in energy than the 1,NC4 with the methyl ester in the equatorial position (Fig. 1a). Indeed, the predicted lower energy form is observed in our crystal structure of 1 (Fig. 1b) which reveals the –COOMe moiety in an equatorial position and with the ester carbonyl group (N1–C6–C7–O2) being almost coplanar with respect to the amide (C6–N1–C1–C2) (Table 2). In the packing of 1, two molecules form hydrogen-bonded dimers via N–H⋯O hydrogen bonds [d(N⋯O) = 3.056(3) Å] (Fig. 1c). The graph set descriptor is R22(8), which is also seen in the ε-caprolactam (CAPLAC)26 and the C-2/C-6 dimethyl27 and C-2 phosphinoxide derivatives.28
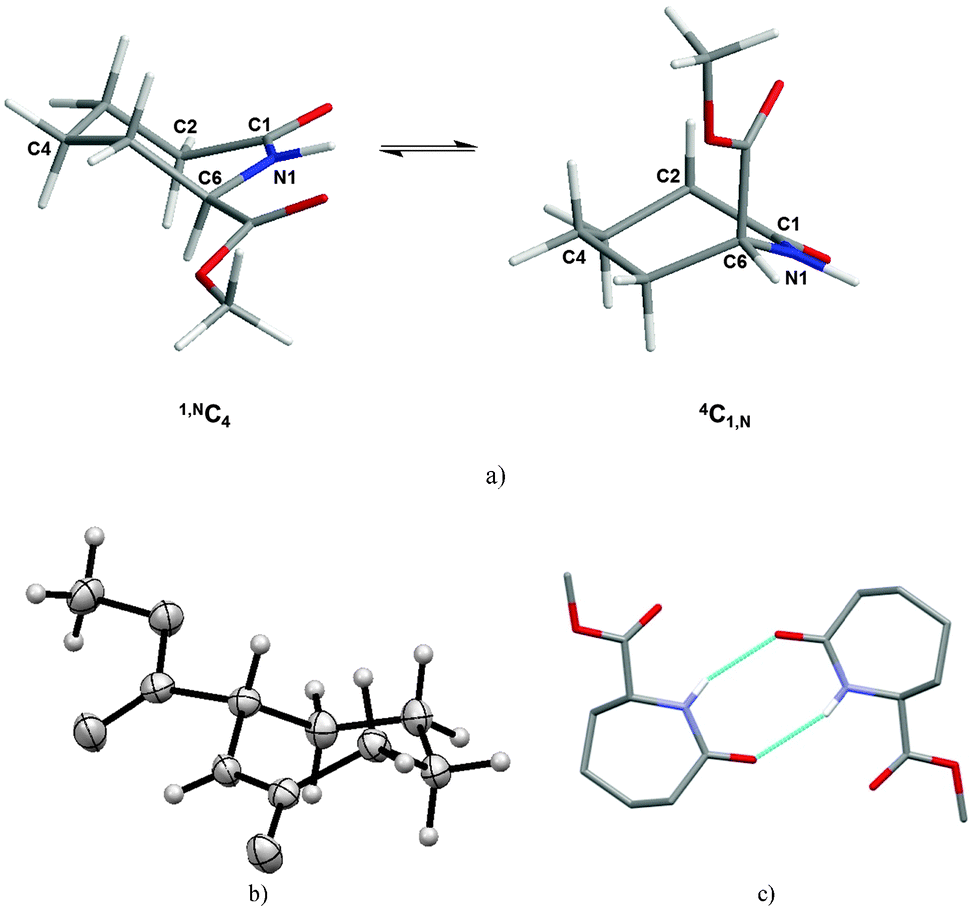 | ||
| Fig. 1 (a) Caprolactam 1 in its two chair forms. 4C1,N is 8 kcal mol−1 higher in energy than 1,NC4 as calculated by molecular modelling using MacroModel. (b) Displacement ellipsoid plot of 1 from single crystal diffraction data and (c) dimer formation in 1 showing the R22(8) hydrogen bonding motif. | ||
In contrast to 1, in the solid state, caprolactam acid 2 has three crystallographically independent molecules in the asymmetric unit; these form a trimer arranged around an approximate non-crystallographic three fold axis. The molecules are connected via three sets of strong NH⋯O and OH⋯O contacts [d(N⋯O) = 2.970(4)–3.074(4) Å; d(O⋯O) = 2.543(4)–2.555(4) Å] from the R22(8)-type (Table S1 (ESI‡), Fig. 2a). Careful examination of the three crystallographically unique molecules in 2 shows they have two different configurations at the chiral C6. Although this gives a ratio of 2:1 within the asymmetric unit, as the space group is centrosymmetric, the overall crystal is racemic,29i.e. the final ratio of the two (S) and (R) forms on C6 of the molecules is 1:1. Converting the three crystallographically independent molecules to the same configuration (i.e. inverting one) demonstrates that their molecular geometries are very similar. Hence, the loss of the methyl ester from 1 to 2, does not substantially influence the conformation (Table 2). The carboxylic acid function of 2 is in the equatorial position as observed for the –COOMe ester in 1 (Fig. 2b).
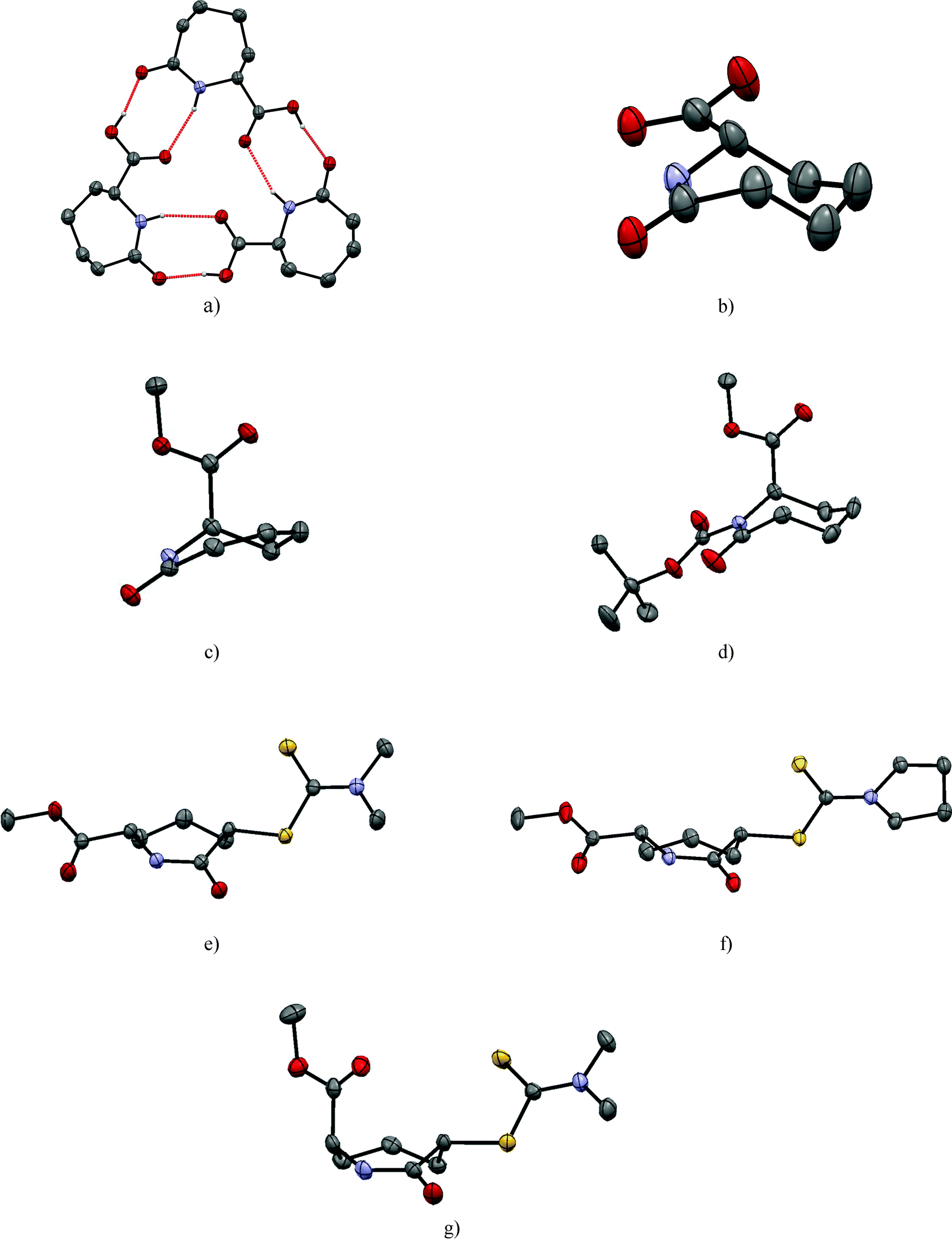 | ||
| Fig. 2 The pseudo threefold symmetry of the three crystallographic independent molecules in the asymmetric unit of 2 (a) and the structures of caprolactam acid 2 (only one molecule of the asymmetric unit is displayed for clarity) (b), the desaturated lactam 7 (c), the N-functionalized lactam 3 (d) and the dithiocarbamates 5cis (e), 6cis (f) and 5trans (g) from single crystal diffraction studies. Displacement ellipsoids are drawn at the 50% probability level and hydrogen atoms (except those illustrating hydrogen bonding interactions) are omitted for clarity. | ||
The introduction of a double bond between C2 and C3 of caprolactam ester 1 as in 7 “flattens” the seven membered ring chair conformation (Table 2 and Fig. 2c). However, the carbonyl group and the double bond are not fully coplanar, with a dihedral angle of 22.49(7)°. Notably, the placement of the –COOMe substituent on C6 has a different influence on the saturated and unsaturated compounds respectively i.e. it switches from an equatorial to an axial position, however, the dimer formation via R22(8) hydrogen bonds [d(N⋯O) = 2.8618(16) Å] of 7 is observed again.
Conformational analyses of disubstituted caprolactams
The disubstituted lactams 5 and 6 were obtained as pairs of cis/trans-diastereomers. We were able to grow single crystals from the cis isomers of both 5 and 6 and the trans isomer of 5. Atoms C-2 and C-6 of the cis-dithiocarbamates (5, 6) have the same configuration and crystallise in centrosymmetric space groups (making them racemic). In each case, both substituents adopt equatorial positions and neither the presence of the more flexible dimethyl group nor the preorganized pyrroldine dithiocarbamate group substantially influence the caprolactam ring conformation (Table 2 and Fig. 2e and f). The C2 substituent of 6 does not have a significant effect on the ring conformation as shown by the low cell similarity index (π)30 of 0.03324 for 5 and 6. Superimposing all 18 heavy atoms of 5 with the corresponding atoms of 6, the root mean square of the atomic distances rmsd = 0.008, the largest atomic distance difference of corresponding atoms maxd = 0.014, which is lowest for all structures compared herein (Table 3).
In contrast to the literature structure of caprolactam,25,26 and the structures of 1 and 7 herein, no stereotypical N–H⋯O amide dimers are seen for the cis diastereoisomers of the dithiocarbamate derivatives 5 and 6 in the crystalline state. This is interesting because the N–H of the amide of 5 and 6 do not form any hydrogen bonds at all. The strong N–H⋯O hydrogen bonds are apparently replaced by weaker C–H⋯O interactions to the amide oxygen O1 and short contacts between the ester oxygen and the dithiocarbamate. It is possible that, C–H⋯S,31 N–H⋯S32 and S⋯S33 contacts are relevant (Fig. S1, ESI‡).
In the trans-diastereomer of 5 the chair is again the favoured conformation of the caprolactam core with one substituent adopting an axial orientation (C6) whilst the other (C-2) adopting an equatorial position (Fig. 2g). Likely for steric reasons, the C-2 dithiocarbamate residue is in the equatorial position forcing the C-6 methyl ester into the axial position. Consequently, this leads to a rather short intermolecular distance of 2.424(3) Å between the carbonyl of the methyl ester and the facing methylene hydrogen. In the packing of 5trans, amide dimers featuring the R22(8) motif are the most striking feature (Table 4).
| C(6) substituent | 2nd substituent | H bonding pattern34 | |||
|---|---|---|---|---|---|
| CAPLAC | — | — | — | — | R 22(8) |
| 1 | –COOMe | Equatorial | — | — | R 22(8) |
| 2 | –COOMe | Equatorial | — | — | R 22(8) |
| 3 | –COOMe | Axial | N-BOC | Lactam and N-BOC carbonyls coplanar | — |
| 5 cis | –COOMe | Equatorial | C2–S(S)C–N(Me)2 | Equatorial | — |
| 5 trans | –COOMe | Axial | C2–S(S)C–N(Me)2 | Equatorial | R 22(8) |
| 6 cis | –COOMe | Equatorial | C2–S(S)C–N(CH2)4 | Equatorial | — |
| 7 | –COOMe | Axial | — | — | R 22(8) |
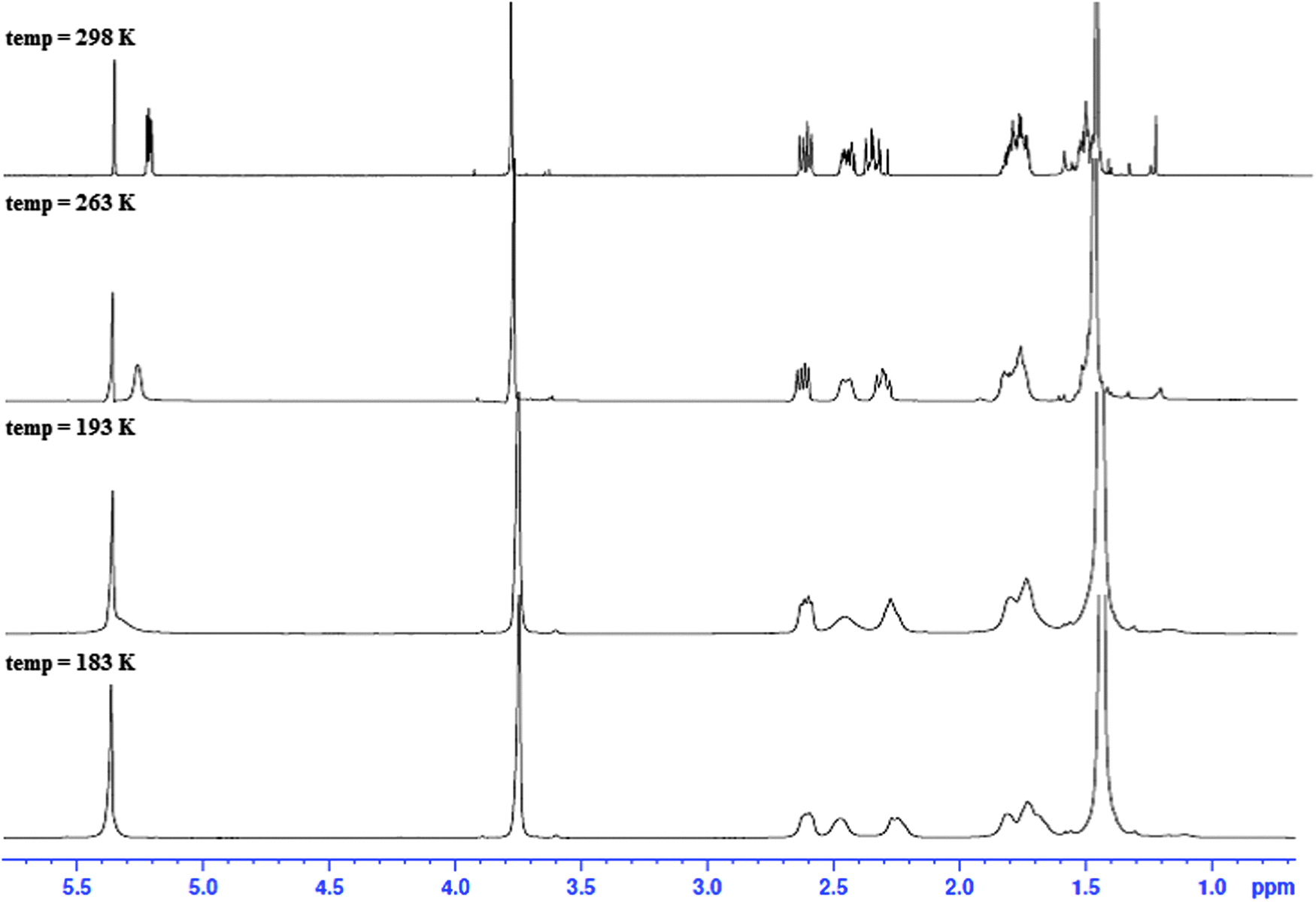 | ||
| Fig. 3 1H NMR spectra of the N-Boc protected C-6 substituted methyl ester (3) at different temperatures (183–298 K) in CD2Cl2. | ||
NOE experiments on 3 at room temperature, however, were consistent with both 1,NC4 and 4C1,N forms being present in solution due to the presence of a medium strength NOE between the methyl ester and H2′ in 4C1,N and strong-medium NOEs between H6 and H2 and H6 and H4 in 1,NC4. These two groups of mutually exclusive NOEs can only be observed if the two chair forms 1,NC4 and 4C1,N are present in fast exchange (Fig. 4a) at room temperature.
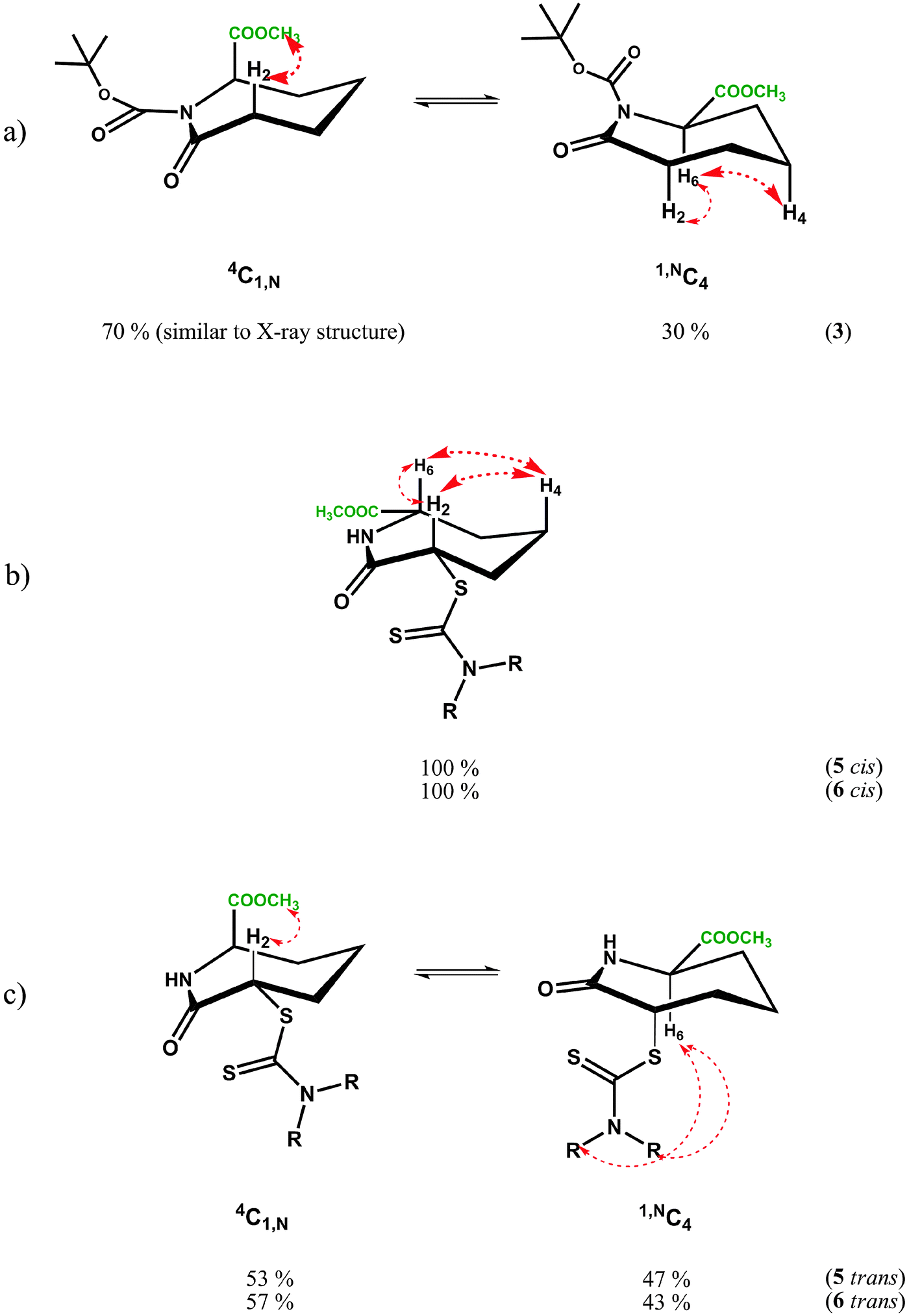 | ||
| Fig. 4 Conformational isomers of the caprolactams 3 (a), 5cis, 6cis (b), 5trans and 6trans (c). Within caprolactam 3 and trans dithiocarbamates 5 and 6, 1,NC4 and 4C1,N are in fast conformational exchange at room temperature in either CD2Cl2 or C6D6. Observed NOEs, indicated by dotted red arrows, are consistent with both forms being present in solution (spectral overlap for H4′ negated its irradiation in either CD2Cl2 or C6D6). | ||
Conformational averaging is also reflected in the vicinal J-couplings at room temperature, for example 3J(H6,H5) = 6.3 Hz, 3J(H6,H5′) = 3.0 Hz (values for 3J(Hax,Hax) = ∼11.0 Hz, 3J(Hax,Heq) = 2–5 Hz and 3J(HeqHeq) = 2–4 Hz35 in conformationally ‘frozen’ forms; see later section on cis-isomer of dithiocarbamates 5 and 6). Assuming for 3 the existence of the two chair conformers in dynamic exchange at room temperature, it is possible to calculate the conformer populations of 1,NC4 and 4C1,N from the averaged vicinal coupling constants36,37 and the literature values of 3J couplings in conformers with distinct conformations in similar systems.38 For the BOC protected methylester 3, a ratio of 70:30 was calculated with the higher populated chair form having the ester substituent in an axial position, thus minimising the steric bulk of the BOC group; the latter is as observed in the X-ray structure, see Fig. 2d. The NMR observations are consistent with the molecular dynamics calculations, where simulations commencing with the equatorial methyl ester 1,NC4, proceed to yield twisted chairs, boat and finally the more stable axial methyl ester 4C1,N conformation (by energy difference ∼8 kcal mol−1 in favour of 4C1,N). The less populated conformer of 3 is less favoured energetically due to steric clash between the equatorial ester function being in close proximity to the BOC substituent.
The NMR results for cis isomers of dithiocarbamates 5 and 6 are apparently unambiguous and irrespective of solvent (e.g. CD2Cl2 or C6D6). The proton resonances are sharp indicating a predominant conformer with both J-couplings and the NOEs consistent with a chair 4C1,N species containing both 6- and 2-substituents in equatorial positions, see Fig. 4b below. The solution analyses are thus consistent with both X-ray structures. An unusual feature of the spectra for 5 and 6 (both cis) is the low coupling constant for 3J(H2,H3) which approaches zero (1.0–1.8 Hz), see Table 5, suggesting a close to 90° dihedral angle for H2ax–C2–C3–H3eq this angle is 79° in the X-ray structure and the energy minimised structure predicts an angle of 81°, also consistent with the observed vicinal coupling constant. The other vicinal coupling constant for the corresponding protons on the other side of the seven-membered ring, 3J(H6,H5) is ‘normal’ for 3J(Hax,Heq) of 4.5–5.2 Hz corresponding to a smaller dihedral angle; in this case the X-ray structure reveals this angle is 68.5° as does the calculated value. An explanation for the unusual small coupling constant for 3J(H2ax,H3eq) and 80° dihedral angle may reside in the bulky partially delocalised dithiocarbamate group, sterically forcing the dihedral angle containing the equatorial proton H3, to twist in order to accommodate it.
| Coupling atoms | CD2Cl2 | C6D6 |
|---|---|---|
| 298 K | 298 K | |
| a Signals broadened by dynamic conformational averaging. b Sharp signal, fast conformational exchange. c J-coupling indiscernible due to line broadening from conformational exchange or signal overlap. | ||
| 3 | ||
| H2–H3 | 7.0 | — |
| H2–H3′ | 2.7 | — |
| H6–H5 | 6.3 | — |
| H6–H5′ | 3.2 | — |
| 5 (cis) | ||
| H2–H3′ | 11.6 | 11.4 |
| H2–H3 | <1.0 | 1.7 |
| H6–H5 | 10.8 | 10.8 |
| H6–H5′ | 5.2 | 4.9 |
| 6 (cis) | ||
| H2–H3′ | 11.4 | 11.2 |
| H2–H3 | 1.8 | 1.8 |
| H6–H5′ | 10.9 | 11.0 |
| H6–H5 | 4.5 | 5.2 |
The flipping of the ring of the 4C1,N to the 1,NC4 chair of 5cis and 6cis would mean both C-2 and C-6 substituents are in the axial position, which is not favoured energetically. Molecular dynamics simulations suggest the energy difference between the favoured di-equatorial 4C1,N species and the di-axial 1,NC4 conformation is ∼20 kcal mol−1, which is presumably why the later form is not observed in solution by NMR.
In the case of the trans dithiocarbamates caprolactams trans-5 and trans-6, the NMR studies reveal that at room temperature there is fast conformational exchange between the two chair forms, 1,NC4 and 4C1,N, irrespective of the type of dithiocarbamate substituent. The 3J(H2,H3), 3J(H2,H3′), 3J(H6,H5) and 3J(H6,H5′) values (Table 4) represent conformationally averaged ensembles of the two forms, although the conformer distribution may be more biased towards 4C1,N in C6D6 since 3J(H2,H3′) tends towards values for axial H2 compared to CD2Cl2 solutions (compare 3J(H2,H3′) of 9.8 to 10 Hz in C6D6versus 8.0 to 8.3 Hz in CD2Cl2). The observed NOEs are also consistent with both forms being present from the NOEs between H2 and the Me ester (4C1,N) and H6 and the dithiocarbamate NMe or NCH2 substituent (1,NC4). In this situation, where either substituent can occupy an axial orientation, the steric clash between them is minimised. The energy differences between 1,NC4 and 4C1,N conformers is predicted to be low 5–8 kcal mol−1 in favour of 4C1,N, unlike the cis isomer, where only one species is observed in solution.
For trans5 and 6, at 198 K–229 K in CD2Cl2, both 4C1,N and 1,NC4 forms are clearly observed in the proton spectra, in a ratio of 1:0.9, a slight excess of 4C1,N. The NOESY spectrum of 5trans (Fig. 5), reveals conformational exchange is evident even at 198 K, as shown from the exchange cross peaks which are of opposite phase to those of the NOEs. The NOEs, however, were also consistent with both 4C1,N and 1,NC4 forms present in dynamic exchange.
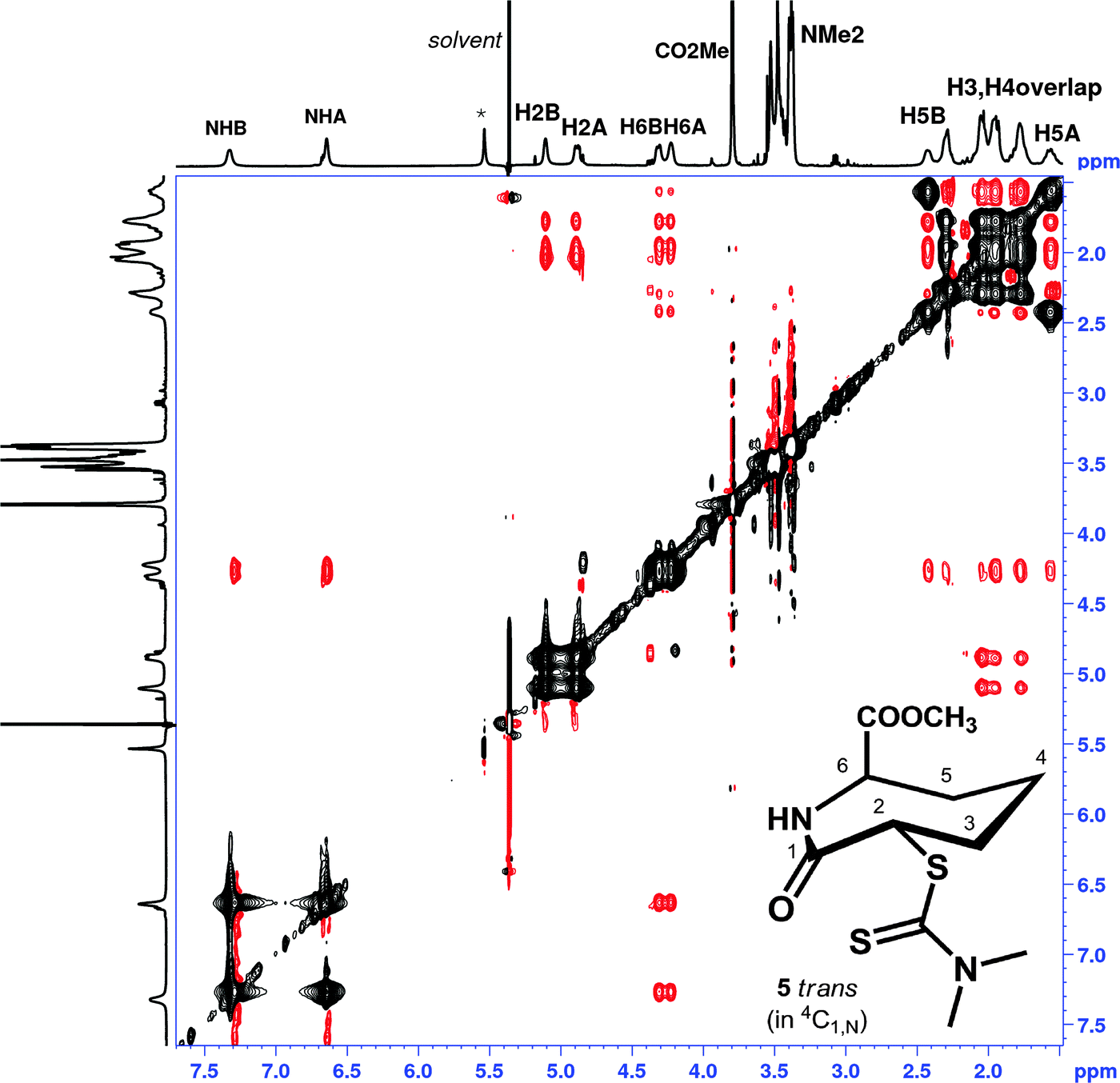 | ||
| Fig. 5 1H NMR NOESY spectrum of 5trans (CD2Cl2, 500 MHz, mixing time 800 ms, 224 K); resonance labelling follows Scheme 1. Black cross peaks indicate dynamic exchange peaks between conformers 4C1,N and 1,NC4, red cross peaks indicated inter-proton NOEs. Note the NOE between H6 and the NMe2 is not visible in the figure shown, but is present at higher contour levels, modelling suggests inter-proton distances >3.2 Å. | ||
Conclusions
Overall, the conformational analyses reveal that the investigated caprolactams prefer to adopt a chair conformation featuring a planar arrangement of the lactam group (C2–C1–N1–C6). The positions of the attached substituents relative to the ring are summarized in Table 5. Bond lengths and torsion angles of the caprolactam ring differ only slightly with C2-, C6- or N-substitution as shown by the X-ray data. Formation of amide ‘dimers’ were observed crystallographically in four of the six possible cases. The introduction of unsaturation, i.e. a C-2/C-3 alkene, has a considerable effect resulting in the seven membered ring chair conformation being partially flat along the –C1–C2C3–C4– portion of the molecule. There is no residual solvent accessible void in any of the structures of the presented ε-caprolactams, which is promoted by weak C–H⋯O contacts. Interestingly, in almost all structures, the slightly acidic proton at C-6 may be involved in these interactions. For the caprolactams 1, 5 (cis) and 6 (cis), the –COOMe residue occupies an equatorial position, but is forced into the axial position by the introduction of a BOC group at a neighbouring atom (3) or by ring desaturation (7).
The NMR studies are consistent with the chair conformations dominating in solution. Caprolactam 3 was found to undergo dynamic exchange between two chair forms, one with the ester equatorial (30%) and the other with the ester axial (70%), the latter observed in the X-ray structure. For the C2-substituted seven-membered rings 5 and 6, we observed one chair form for both cis isomers with both ester and dithiocarbamate groups occupying equatorial positions. The respective trans isomers exist in equilibrium between two chair forms in an 1:1 ratio where either substituent can occupy axial or equatorial positions. In all cases, solvent e.g. benzene or chloroform had only negligible influence on the conformational properties of the caprolactams.
Experimental section
Materials and methods
All reactions involving moisture sensitive reagents were carried out under a nitrogen atmosphere. Cooling was performed in ice-water baths (0 °C) or dry ice–acetone baths (−78 °C). Anhydrous solvents were obtained from solvent stills and were activated by passing over a short column of activated alumina. Reagents were obtained from Acros or Aldrich fine chemical suppliers and used as supplied. Thin layer chromatography (TLC) was performed on Merck DC-Kieselgel 60 F 254 0.2 mm precoated plates with fluorescence indicator. Visualization of spots was achieved using UV light (254 nm) and by developing in a basic solution of KMnO4 followed by heating. Chromatography was performed using a Biotage SP4 chromatography system, using prepacked Biotage KP-SIL SNAP columns.Proton nuclear magnetic resonance spectra (1H NMR) were recorded using a Bruker AV400 (400 MHz) and AVII 500 (500 MHz) spectrometers. Proton decoupled carbon nuclear magnetic resonance spectra (13C NMR) were recorded on a Bruker AV400 (100 MHz) and AVII 500 (125.6 MHz) with sample temperatures regulated at 298 K, unless otherwise stated. Spectra were assigned using COSY, DEPT-135, HMQC, edited-HSQC, and HMBC. All chemical shifts are quoted on the scale in ppm and referenced to residual solvent peaks; CD2Cl2 at 5.32 ppm and C6D6 at 7.2 ppm and calculated internally by the spectrometer. Resonances are described as s (singlet), d (doublet), t (triplet), m (multiplet), and br s (broad singlet). 1-D NOE experiments were performed using DPFGSE-NOE pulse sequence employing two 180 degree pulses and a mixing time of 800 ms.39
Single crystals suitable for single crystal X-ray diffraction studies were obtained from cyclohexane for compound 1 and from a mixture of dichloromethane/methanol (1:1) for 5, 6 and 7. Compound 3 was recovered as a colourless oil, that produced suitable crystals after some weeks. Single crystal X-ray diffraction data were collected using a Nonius Kappa CCD diffractometer (1) or Oxford Diffraction (Agilent) SuperNova diffractometer (2–7) fitted with an Oxford Cryosystems Cryostream open-flow nitrogen cooling device. Data collection and reduction were carried out using HKL COLLECT/DENZO-SCALEPACK40 or CrysAlisPro as appropriate. Structures were solved using SIR9241 within the CRYSTALS suite42 and optimized by full-matrix least squares on F2. Hydrogen atoms were not generally provided by the initial solution, however they were usually clearly visible in the difference Fourier map. Hydrogen atoms were positioned at geometrically sensible positions and refined using soft restraints prior to inclusion in the final refinement using a riding model.43 The rmsD and maxD data were calculated with the program Mercury44 and torsion angles were calculated using PLATON.45 CCDC numbers: 1018875–1018881.
IR spectra were recorded using a Bruker Tensor 27 ATR-FT-IR spectrophotometer. Selected absorption maxima (νmax) are given in wavenumbers (cm−1) and are uncorrected. Mass spectra were recorded on a Waters LCT premier. Melting points were recorded on a Leica VMTG heated-stage microscope melting point apparatus.
All molecular modelling work was carried out using the Schrödinger's Maestro modelling package employing MacroModel with the Schrödinger's implementation of the OPLS_2005 molecular mechanics force field.46 Conformational searches were performed as follows. Initial molecular models for each compound were energy minimised (convergence criterion: RMS energy gradient < 0.001 kcal mol−1 Å−1) using constant dielectric constant with chloroform as the solvent and used as starting points for molecular dynamics simulations at an effective temperature of 1000 K. A total of 50 samples were extracted from each dynamics trajectory with a time interval of 1 ps between samples. The sampled structures were then energy minimised, and structural duplicates removed (match criterion: heavy atom RMSD < 0.2 Å). The final set of unique minimum-energy conformers was then sorted according to calculated energy.
Syntheses
13C NMR (100 MHz, CDCl3): δ = 176.2 (COOCH3), 171.9 (CONH) 55.7, 52.9 (COOCH3, NHCH), 37.0 (CH2), 33.7 (CH2), 29.5 (CH2), 23.0 (CH2). 1H NMR (400 MHz, CDCl3): δ = 6.45 (br s, 1H, NH), 4.07–4.03 (m, 1H, NHCH), 3.74 (s, 3H, COOCH3), 2.47–2.33 (m, 2H, CH2), 2.19–2.15 (m, 1H, CH2), 2.03–1.96 (m, 1H, CH2), 1.86–1.77 (m, 1H, CH2), 1.65–1.51 (m, 3H, CH2). IR: 3270, 2949, 2919, 2865, 1740, 1645, 1557, 1513, 1468, 1437, 1404, 1343, 1312, 1298, 1261, 1241, 1214, 1196, 1183, 1136, 1107, 1089, 1062, 1012, 965, 933, 873, 850, 801, 733. m/z = 172.11 [M + H+], calc. 172.09.
13C NMR (100 MHz, CD3OD): δ = 178.9 (COOH), 173.8 (CONH), 55.9 (NHCH), 36.6 (CH2), 33.4 (CH2), 29.2 (CH2), 23.1 (CH2). 1H NMR (400 MHz, CD3OD): δ = 4.18–4.13 (m, 1H, NHCH), 2.59–2.40 (m, 1H, CH2), 2.50–2.48 (m, 1H, CH2), 2.25–2.17 (m, 1H, CH2), 2.03–1.96 (m, 1H, CH2), 1.89–1.81 (m, 1H, CH2), 1.78–1.67 (m, 1H, CH2). IR: 3234, 2922, 2850, 1703, 1612, 1443, 1413, 1358, 1343, 1327, 1265, 1250, 1229, 1220, 1203, 1191, 1152, 1086, 1026, 942, 878, 853, 834, 807, 755, 728. m/z = 156.07 [M − H+], calc. 156.17.
:1). 86% (481 mg, 1.77 mmol) of a colourless oil, which slowly crystallizes. Mp. 48–49 °C. Rf = 0.80 (SiO2; n-hexane/ethyl acetate = 1:1).
13C NMR (100 MHz, CDCl3): δ = 175.3 (COOCH3), 170.9 (CONH), 153.3 (NCOOtBu), 83.2 (C(CH3)3), 56.5 (NHCH), 52.4 (COOCH3), 39.5 (CH2), 29.8 (CH2), 27.9 (C(CH3)3), 25.6 (CH2), 22.6 (CH2). 1H NMR (400 MHz, CDCl3): δ = 5.16–5.12 (m, 1H), 3.71 (s, 3H), 2.63–2.58 (m, 1H, CH2), 2.46–2.40 (m, 1H, CH2), 2.36–2.29 (m, 1H, CH2), 1.79–1.73 (m, 3H, CH2), 1.51–1.46 (m, 2H, CH2), 1.43 (s, 9H). IR: 2983, 2961, 2933, 2869, 1715, 1701, 1454, 1434, 1380, 1368, 1295, 1285, 1251, 1235, 1204, 1144, 1127, 1083, 1050, 1022, 986, 956, 929, 912, 875, 842, 819, 806, 779, 745, 727, 704. m/z = 272.17 [M + H+], calc. 272.14.
13C NMR (100 MHz, CDCl3): δ = 171.7, 170.2, 54.5 (COOCH3), 53.2 (CHCOO), 51.1 (CHBr), 33.5 (CH2), 30.0 (CH2), 24.3 (CH2). 1H NMR (400 MHz, CDCl3): δ = 6.67 (br s, 1H, NH), 4.63 (m, 1H, CHBr), 4.55 (dd, 3J = 11.0 Hz, 2J = 3.5 Hz, 1H, NHCH), 3.79 (s, 3H, COOCH3), 2.38–2.31 (m, 1H), 2.24–2.12 (m, 1H, CH2), 2.09–1.91 (m, 3H, CH2), 1.57–1.46 (m, 1H, CH2). IR: 3347, 2952, 2933, 1734, 1672, 1434, 1379, 1360, 1337, 1303, 1269, 1240, 1223, 1177, 1147, 1107, 1076, 1061, 1011, 939. m/z = 250.02, 251.98 [M + H+], calc. 250.00, 252.00.
:1 → ethyl acetate) to yield in both cases two separable diastereomers.
Methyl 6-[(dimethylcarbamothioyl)thio]-7-oxoazepane-2-carboxylate (5). Yield 80% (202 mg, 0.70 mmol) as a mixture of 178 mg (88%) cis and 24 mg (12%) trans isomer.
cis-Methyl 6-[(dimethylcarbamothioyl)thio]-7-oxoazepane-2-carboxylate (5cis). Mp. 136–137 °C. Rf = 0.70 (SiO2; ethyl acetate). 13C NMR (100 MHz, DMSO/CD3OD): δ = 195.5, 172.5, 171.2, 57.6, 55.5, 51.1, 45.3, 41.7, 33.6 (CH2), 32.3 (CH2), 24.3 (CH2). 1H NMR (400 MHz, CDCl3): δ = 6.60 (br s, 1H, NH), 4.96 (dd, 1H, CHSCS), 4.35 (dd, 1H, NHCH), 3.80 (s, 3H, COOCH3), 3.51, 3.39 (s, NCH3, 6H), 2.30–2.20 (m, 1H), 2.12–2.03 (m, 2H, CH2), 1.96–1.84 (m, 2H, CH2), 1.59–1.49 (m, 1H, CH2). IR: 3369, 3308, 2950, 2859, 1738, 1654, 1490, 1433, 1395, 1367, 1353, 1329, 1305, 1216, 1135, 1081, 1034, 1007, 983, 943, 873, 854, 817, 786, 742, 718. m/z = 291.11 [M + H+], calc. 291.08.
trans-Methyl 6-[(dimethylcarbamothioyl)thio]-7-oxoazepane-2-carboxylate (5trans). Mp. 111–113 °C. Rf = 0.65 (SiO2; ethyl acetate). 1H NMR (400 MHz, dichloromethane): δ = 6.54 (br s, 1H, NH), 5.07 (d, 1H, 3J = 8.0 Hz, CHSCS), 4.59 (dd, 3J = 10 Hz, 2J = 4 Hz, 1H, NHCH), 3.89 (t, 3J = 10 Hz, NCH2CH2), 3.83 (s, 3H, COOCH3), 3.71 (t, 3J = 10 Hz, NCH2CH2), 2.45–2.37 (m, 1H), 2.33–2.21 (m, 1H, CH2), 2.14–2.07, 2.02–1.97 (m, NCH2CH2, 4H), 1.93–1.84 (m, 3H, CH2), 1.61–1.54 (m, 1H, CH2). 13C NMR (100 MHz, DMSO/CD3OD): δ = 195.1, 172.8, 171.3, 56.7, 54.7, 53.1, 45.7, 41.6, 33.0 (CH2), 32.5 (CH2), 25.2 (CH2). IR: 3364, 3245, 3089, 3038, 2984, 2946, 2937, 1747, 1708, 1655, 1478, 1455, 1440, 1408, 1376, 1363, 1348, 1325, 1307, 1288, 1274, 1250, 1205, 1177, 1157, 1126, 1105, 1084, 1030, 1012, 984, 945, 896, 879, 869, 841, 800, 774. m/z = 291.11 [M + H+], calc. 291.08.
Methyl 6-[(pyrrolidine-1-carbonothioyl)thio]-7-oxoazepane-2-carboxylate (6). Yield 77% (296 mg, 0.94 mmol) of a mixture consisting of 213 mg (72%) cis and 83 mg (28%) trans isomer.
cis-Methyl 6-[(pyrrolidine-1-carbonothioyl)thio]-7-oxoazepane-2-carboxylate (6cis). Mp. 129–131 °C. Rf = 0.70 (SiO2; ethyl acetate). 1H NMR (400 MHz, dichloromethane): δ = 6.54 (br s, 1H, NH), 5.07 (d, 1H, 3J = 8.0 Hz, CHSCS), 4.59 (dd, 1H, NHCH), 3.89 (t, 3J = 10 Hz, NCH2CH2), 3.82 (s, 3H, COOCH3), 3.71 (t, 3J = 10 Hz, NCH2CH2), 2.45–2.37 (m, 1H), 2.33–2.21 (m, 1H, CH2), 2.14–2.07, 2.02–1.97 (m, NCH2CH2, 4H), 1.93–1.84 (m, 3H, CH2), 1.61–1.54 (m, 1H, CH2). 13C NMR (100 MHz, CDCl3): δ = 191.0, 172.5, 171.6, 56.5, 55.2, 54.5, 53.2, 51.1, 32.8 (CH2), 32.7 (CH2), 28.9 (CH2), 26.7 (CH2), 24.3 (CH2). IR: 3369, 3303, 2950, 1736, 1657, 1431, 1396, 1353, 1307, 1221, 1161, 1083, 1066, 1035, 1008, 946, 873, 855, 821, 787, 739, 716. m/z = 317.08 [M + H+], calc. 317.09.
trans-Methyl 6-[(pyrrolidine-1-carbonothioyl)thio]-7-oxoazepane-2-carboxylate (6trans). Mp. 119–121 °C. Rf = 0.59 (SiO2; ethyl acetate). 13C NMR (100 MHz, CDCl3): δ = 189.8, 173.4, 171.4, 55.4, 55.3, 54.2, 53.1, 50.7, 33.9 (CH2), 32.5 (CH2), 26.1 (CH2), 25.2 (CH2), 24.2 (CH2). 1H NMR (400 MHz, dichloromethane): δ = 6.60 (br s, 1H, NH), 5.15 (d, 1H, CHSCS), 4.24 (m, 1H, NHCH), 3.88 (t, 3J = 10 Hz, NCH2CH2), 3.83, 3.82 (s, 3H, COOCH3), 3.68 (t, 3J = 10 Hz, NCH2CH2), 2.48–2.38 (m, 1H), 2.31–2.23 (m, 1H, CH2), 2.13–1.99 (m, NCH2CH2, 4H), 1.93–1.84 (m, 3H, CH2), 1.63–1.55 (m, 1H, CH2). IR: 3304, 3213, 3096, 2954, 2872, 1727, 1655, 1432, 1363, 1332, 1316, 1287, 1253, 1215, 1185, 1164, 1124, 1106, 1081, 1031, 1002, 957, 945, 889, 868, 846, 822, 794, 774. m/z = 317.08 [M + H+], calc. 317.09.
:1). In both cases, the pyrolysis of the respective dithiocarbamate yielded a mixture of two different desaturation products (7 and 8). Pyrolysis of 5 yielded 32% (66 mg, 0.39 mmol) of 7 and 33% (67 mg 0.40 mmol) of 8. Pyrolysis of 6 yielded 30% (10 mg, 0.059 mmol) of 7 and 27% (9 mg, 0.053 mmol) of 8.
Methyl 7-oxo-2,3,4,7-tetrahydro-1H-azepine-2-carboxylate (Δ2) (7). Mp. 101–102 °C. Rf = 0.20 (SiO2; ethyl acetate).
13C NMR (100 MHz, CDCl3): δ = 171.5 (COOCH3), 168.4 (CONH), 140.2 (COCHC), 126.0 (COCHC), 53.8, 53.0 (COOCH3, NHCH), 32.5 (CH2), 28.7 (CH2). 1H NMR (400 MHz, CDCl3): δ = 6.67 (br s, 1H, NH), 6.37–6.28 (m, 1H, COCHCH), 5.94 (dd, 3J = 12.5 Hz, 2J = 2 Hz, 1H, COCHC), 4.51–3.98 (m, 1H), 3.81 (s, 3H), 2.58–2.47 (m, 2H, CH2), 2.37–2.43 (m, 1H, CH2), 2.04–2.14 (m, 1H, CH2). IR: 3277, 3238, 3182, 3132, 3031, 2956, 2923, 1720, 1667, 1619, 1487, 1435, 1390, 1368, 1351, 1331, 1279, 1243, 1216, 1197, 1183, 1146, 1085, 1044, 1009, 989, 932, 913, 874, 862, 836, 816, 784, 731. m/z = 170.11 [M + H+], calc. 170.07.
Methyl 7-oxo-2,3,6,7-tetrahydro-1H-azepine-2-carboxylate (Δ3) (8). Mp. 155–156 °C. Rf = 0.38 (SiO2; ethyl acetate).
13C NMR (100 MHz, CDCl3): δ = 173.2 (COOCH3), 171.1 (CONH), 127.2, 120.5 (COCHC, COCHC), 53.1, 52.8 (COOCH3, NHCH), 35.4 (CH2), 33.8 (CH2). 1H NMR (400 MHz, CDCl3): δ = 6.52 (br s, 1H), 5.60–5.49 (m, 2H), 4.61–4.54 (m, 1H), 3.80 (s, 3H), 3.52–3.36 (m, 1H, CH2), 2.91–2.80 (m, 1H, CH2), 2.71–2.59 (m, 1H, CH2), 2.49–2.35 (m, 1H, CH2). IR: 3284, 3028, 2953, 1735, 1647, 1550, 1436, 1365, 1296, 1266, 1225, 1204, 1158, 1064, 1030, 998, 966, 931, 904, 860, 808, 743, 669, 610. m/z = 170.11 [M + H+], calc. 170.07.
Acknowledgements
T. G. thanks Deutsche Forschungsgemeinschaft (DFG), Germany, for generous funding (GR 3693/1-1:1). P. B. acknowledges the support from the National Scientific Research Foundation (OTKA K-100801). C. J. S. thanks the Medical Research Council for support.
References
- J. Ritz, H. Fuchs, H. Kieczka and W. C. Moran, “Caprolactam”, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- M. M. Green and H. A. Wittcoff, Organic Chemistry Principles and Industrial Practice, Wiley-VCH, Weinheim, 2003 Search PubMed.
- (a) D. A. Wicks, PCT Int. Appl., WO 2010011949 A2 20100128, 2010 Search PubMed; (b) D. A. Wicks, PCT Int. Appl., WO 2010011924 A2 20100128, 2010 Search PubMed; (c) E. Tarkin-Tas and L. J. Mathias, Macromolecules, 2010, 43, 968–974 CrossRef CAS.
- J. Ramos, A. Imaz and J. Forcada, Polym. Chem., 2012, 3, 852–856 RSC.
- J. Jampilek and K. Brychtova, Med. Res. Rev., 2012, 32, 907–947 CrossRef CAS PubMed.
- F. Hou, X. Zhang, G. Zhang, D. Xie, P. Chen, W. Zhang, J. Jiang, M. Liang, G. Wang, Z. Liu and R. Geng, N. Engl. J. Med., 2006, 354, 131–140 CrossRef CAS PubMed.
- P. Lancellotti, Rev. Med. Liege, 2008, 63, 220–224 CAS.
- E. A. MacGregor, Clin. Med. Insights: Ther., 2011, 3, 301–314 CrossRef CAS.
- (a) H. Yamaguchi, S. Sato, S. Yoshida, K. Takada, M. Itoh, H. Seto and N. Otake, J. Antibiot., 1986, 39, 1047–1053 CrossRef CAS; (b) T. Dubuisson, E. Bogatcheva, M. Y. Krishnan, M. T. Collins, L. Einck, C. A. Nacy and V. M. Reddy, J. Antimicrob. Chemother., 2010, 65, 2590–2597 CrossRef CAS PubMed; (c) E. Bogatcheva, T. Dubuisson, M. Protopopova, L. Einck, C. A. Nacy and V. M. Reddy, J. Antimicrob. Chemother., 2011, 66, 578–587 CrossRef CAS PubMed.
- B. Schneider, D. Doskočilová, P. Schmidt, J. Štokr and P. Čefelín, J. Mol. Struct., 1976, 35, 161–174 CrossRef CAS.
- (a) F. Groenewald and J. Dillen, Struct. Chem., 2012, 23, 723–732 CrossRef CAS; (b) F. H. Allen, J. A. K. Howard, N. A. Pitchford and J. G. Vinter, Acta Crystallogr., Sect. B: Struct. Sci., 1994, 50, 382–395 CrossRef; (c) R. Huisgen, H. Brade, H. Walz and I. Glogger, Chem. Ber., 1957, 90, 1437–1447 CrossRef CAS.
- (a) I. Martins, M. Martins, A. Fernandes, V. André and M. T. Duarte, CrystEngComm, 2013, 15, 8173–8179 RSC; (b) C. Guo, H. Zhang, X. Wanga, J. Xu, Y. Liu, X. Liu, H. Huang and J. Sun, J. Mol. Struct., 2013, 1048, 267–273 CrossRef CAS PubMed; (c) D.-C. Wang, D.-M. Fan, H.-Q. Liu and P.-K. Ou-yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o438 CAS; (d) D. K. Winter, A. Drouin, J. Lessard and C. Spino, J. Org. Chem., 2010, 75, 2610–2618 CrossRef CAS PubMed; (e) X.-K. Xia, Y.-G. Zhang, X. Liu, W.-P. Yuan, M.-S. Zhang, X.-J. Wang, X.-M. Meng and C.-H. Liu, Z. Kristallogr. - New Cryst. Struct., 2010, 225, 387–388 CAS; (f) H. Q. Liu, D.-M. Fan, D.-C. Wang and P.-K. Ou-Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2383 CAS; (g) J. Ondráček, J. Novotný, K. Kefurt and J. Havlíček, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 929–930 CrossRef.
- E. S. Glazer, R. Knorr, C. Ganter and J. D. Roberts, J. Am. Chem. Soc., 1972, 94, 6026–6032 CrossRef CAS.
- M. Hamerníková, S. Pakhomova, J. Havlíček, H. Votavová and K. Kefurt, Carbohydr. Res., 2000, 325, 56–67 CrossRef.
- A. Matallana, A. W. Kruger and C. A. Kingsbury, J. Org. Chem., 1994, 59, 3020–3025 CrossRef CAS.
- R. B. Hamed, J. R. Gomez-Castellanos, A. Thalhammer, D. Harding, C. Ducho, T. D. W. Claridge and C. J. Schofield, Nat. Chem., 2011, 3, 365–371 CrossRef CAS PubMed.
- K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS.
- A. Bladé-Font, Tetrahedron Lett., 1980, 21, 2443–2446 CrossRef.
- D. W. Hansen, E. A. Hallinan, T. J. Hagen, S. W. Kramer, S. Metz, K. B. Peterson, D. P. Spangler, M. V. Toth, K. F. Fok, A. A. Bergmanis, R. K. Webber, M. Trivedi, F. Tjoeng and B. S. Pitzele, US Pat., PCT/US96/05315, 1996 Search PubMed.
- T. Gruber, A. L. Thompson and C. J. Schofield, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o593–o594 CAS.
- E. Fanghänel, L. van Hinh, G. Schukat and J. Patzsch, J. Prakt. Chem./Chem.-Ztg., 1989, 331, 479–485 CrossRef.
- (a) M. Yanagawa, O. Moriya, Y. Watanabe, Y. Ueno and T. Endo, Bull. Chem. Soc. Jpn., 1998, 61, 2203–2204 CrossRef; (b) R. S. Grainger and E. J. Welsh, Angew. Chem., Int. Ed., 2007, 46, 5377–5380 CrossRef CAS PubMed; (c) B. Poladura, A. Martinez-Castaneda, H. Rodriguez-Solla, C. Concellon and V. del Amo, Tetrahedron, 2012, 68, 6438–6446 CrossRef CAS PubMed.
- S. Ahmed, L. A. Baker, R. S. Grainger, P. Innocenti and C. E. Quevedo, J. Org. Chem., 2008, 73, 8116–8119 CrossRef CAS PubMed.
- S. M. Kroner, M. P. DeMatteo, C. M. Hadad and B. K. Carpenter, J. Am. Chem. Soc., 2005, 127, 7466–7473 CrossRef CAS PubMed.
- K. P. Oya and R. M. Myasnikova, J. Struct. Chem., 1974, 15, 578–583 CrossRef.
- F. K. Winkler and J. D. Dunitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 268–269 CrossRef.
- D. K. Winter, A. Drouin, J. Lessard and C. Spino, J. Org. Chem., 2010, 75, 2610–2618 CrossRef CAS PubMed.
- V. V. Tkachev and L. O. Atovmyan, J. Struct. Chem., 1991, 32, 176–178 CAS.
- A. L. Thompson and D. J. Watkin, Tetrahedron: Asymmetry, 2009, 20, 712–717 CrossRef CAS PubMed.
- (a) A. Kálmán, L. Párkányi and G. Argay, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1039–1049 CrossRef; (b) A. Kálmán and L. Párkányi, in Isostructurality of Organic Crystals in Advances in Molecular Structure Research, ed. M. Hargittai and I. Hargittai, JAI Press Inc., 1997, vol. 3, p. 189 Search PubMed.
- (a) F. Katzsch and E. Weber, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o2354–o2355 CAS; (b) I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. – Eur. J., 2010, 16, 2442–2452 CrossRef CAS PubMed; (c) A. Saad, O. Jeannin and M. Fourmigue, CrystEngComm, 2010, 12, 3866–3874 RSC; (d) W. Lu, Z.-M. Yan, J. Dai, Y. Zhang, Q.-Y. Zhu, D.-X. Jia and W. J. Guo, Inorg. Chem., 2005, 2339–2345 CrossRef CAS; (e) J. J. Novoa, M. C. Rovira, C. Rovira, J. Veciana and J. Tarrés, Adv. Mater., 1995, 7, 233–237 CrossRef CAS.
- (a) P. M. Zorkii, A. E. Obodovskaya, R. Y. Muidinov and R. Yu, Kristallografiya, 1999, 44, 581–588 CAS; (b) T. Ishida, M. Inoue, K. Kitamura, A. Wakahara, K. Tomita and M. Shinoda, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 660–663 CrossRef.
- (a) E. W. Reinheimer, M. Fourmigué and K. R. Dunbar, J. Chem. Crystallogr., 2009, 39, 723–729 CrossRef CAS; (b) T. Kanagasabapathy, P. Krishnaswamy and J. Ramasubbu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, o579–o582 CAS; (c) L. Beer, J. F. Britten, J. L. Brusso, A. W. Cordes, R. C. Haddon, M. E. Itkis, D. S. MacGregor, R. T. Oakley, R. W. Reed and C. M. Robertson, J. Am. Chem. Soc., 2003, 125, 14394–14403 CrossRef CAS PubMed; (d) T. Ozturk, D. C. Povey and J. D. Wallis, Tetrahedron, 1994, 50, 11205–11212 CrossRef CAS.
- J. Grell, J. Bernstein and G. Tinhofer, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 1030–1043 CrossRef PubMed.
- E. Breitmaier, Structure Elucidation by NMR in Organic Chemistry: A Practical Guide, John Wiley & Sons, 3rd revised edn, 2002 Search PubMed.
- R. J. Abraham, P. Leonard, T. A. D. Smith and W. A. Thomas, Magn. Reson. Chem., 1996, 34, 71–77 CrossRef CAS.
- F. A. Bovey, P. A. Mirau and H. S. Gutowsky, Nuclear Magnetic Resonance Spectroscopy, Academic Press, 1988 Search PubMed.
- M. Kaewpet, B. Odell, M. A. King, B. Banerji, C. J. Schofield and T. D. W. Claridge, Org. Biomol. Chem., 2008, 6, 3476–3485 CAS.
- K. Stott, J. Keeler, Q. N. Van and A. J. Shaka, J. Magn. Reson., 1997, 125, 302–324 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Processing of X-ray Diffraction Data Collected in Oscillation Mode, Methods Enzymol, ed. C. W. Carter and R. M. Sweet, Academic Press., 1997, p. 276 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100–1107 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- PLATON, A Multipurpose Crystallographic Tool, ed. A. L. Spek, Utrecht, The Netherlands, 1998 CrossRef CAS; A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- E. Perrotti, N. Palladino, M. Greco and M. De Malde, Ann. Chim., 1966, 56, 1358–1372 CAS.
Footnotes |
| † Dedicated to Dr Margit Gruner, an expert on NMR studies of complex natural products and supramolecular systems, on the occasion of her 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1018875–1018881. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01339e |
| § Present address: TU Bergakademie Freiberg, Institut für Organische Chemie, Leipziger Str. 29, 09596 Freiberg/Sachsen, Germany. E-mail: tobias.gruber@chemie.tu-freiberg.de; Tel: +49 3731 392390 |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |