The amide bridge in donor–acceptor systems: delocalization depends on push–pull stress†
Mauricio
Maldonado-Domínguez
a,
Rafael
Arcos-Ramos
a,
Margarita
Romero
a,
Blas
Flores-Pérez
a,
Norberto
Farfán
*a,
Rosa
Santillan
b,
Pascal G.
Lacroix
c and
Isabelle
Malfant
c
aFacultad de Química, Departamento de Química Orgánica, Universidad Nacional Autónoma de México, 04510 México, D.F., México. E-mail: norberto.farfan@gmail.com; Fax: +52 55 56223722; Tel: +52 55 56223899 ext. 44443
bDepartamento de Química, Centro de Investigación y de Estudios Avanzados del IPN, México, D.F. Apdo. Postal 14-740 07000, México
cLaboratoire de Chimie de Coordination du CNRS, 205 Route de Narbonne, F-31077, Toulouse, France
First published on 21st October 2013
Abstract
Transmission of electronic information through amide bonds may be, under appropriate conditions, effectively achieved. In this work, a family of explicitly designed donor–(amide bridge)–acceptor architectures was synthesized. NMR studies and UV-vis absorption solvatochromism support that cross-conjugation leads to measurable polarization across push–pull, amide-bridged molecules. Computational analysis of structural parameters and frontier molecular orbitals shows the contribution of an additional, dienoid amide canonical structure to intramolecular electron delocalization, as the electron donor–acceptor strength of the substituents increases. Within the context of nonlinear optics and molecular materials, computational comparison between amide-bridged molecules and those containing typical linkers shows that there is a compromise between nonlinear optical response, ease of synthesis and chemical inertness, making the systems studied herein interesting alternatives for such applications.
Introduction
Electron delocalization is a ubiquitous phenomenon, present in virtually every physical system we can interact with. It plays a central role in processes as complex and diverse as photosynthesis,1 electrical conductivity of materials2 and dissociation of acids,3 among many others. Chemically, it is a driving force for reactivity to be present or absent;4 physically, it has a profound influence on the properties of a system and its interaction with the surrounding medium.Intramolecular electron delocalization (IED) is intimately involved in photophysical (light absorption and emission,5 nonlinear optical (NLO) response6) and photochemical (light-driven isomerizations,7 photorelease of small molecules,8 electron transfer reactions9) phenomena of academic, technological and commercial interest. Although π electron delocalization is the most broadly studied and exploited form of IED,10 electrons are also known to delocalize over σ bonds.11 Today, it is recognized and widely accepted that IED occurs through a synergistic coupling of phenomena, such as resonance, hyperconjugation,12 superexchange (SE)13 and cross-conjugation mechanisms,14 the hydrogen-bond-assisted electron delocalization being a very interesting manifestation of SE.15
In materials science, the study, design and synthesis of molecules exhibiting electron delocalization is an area of great interest, especially in the fields of optoelectronics,16 nonlinear optics, light harvesting for solar cells,17 along with more traditional applications, the dye industry being the most representative of them. A subclass of delocalized systems of particular interest is that involving a donor and an acceptor asymmetrically located in a bridging molecule, usually a conjugated one, the so-called “push–pull” systems;18 these architectures, ushered by important charge transfer (CT) processes and potential NLO capabilities, present efficient IED because π electrons facilitate overlap between the donor and acceptor functions leading to a predetermined, spatially-biased delocalization. Among the features which may be desirable within a molecular push–pull device are rigidity (unless conformational switching properties are sought for), unidirectionality, effective electronic communication between both ends of the molecule and, of course, ease of synthesis. Although many effects (superexchange, hyperconjugation) and non-π-delocalized molecular backbones (oligopeptides,19 saturated bridges,20 spirolinked polyenes21) have proven to be useful for electron delocalization to occur, the study of alternative, readily accessible bridges for IED is a seldom studied topic, classically conjugated compounds being the focus of most of the research done within this field.
Synthetically, when a molecule with push–pull electron delocalization and large dipole moment is targeted, the first line of strategies for its design and synthesis are arene,7 alkene,22 alkyne,23 azo24 and imine25 bridges even though, sometimes, they imply long and tedious synthetic routes26 and purification procedures. A molecular motif with interesting potential is the amide function. Its low chemical reactivity and restricted rotation account for a robust structural unit, while its electronic properties (C–N bond order greater than 1) render it an interesting channel for the transmission of electronic information via cross-conjugation. The amide group is also capable of forming strong intramolecular H-bonds,27 known to behave as active electron-reorganizing bridges.28 The amide moiety is usually invoked when conformational freedom29 or chemical reactivity are to be reduced; it is also relevant for the synthesis of polymers,30 dendrimers31 and peptides.32 Up to now, no studies have been carried out for the application of the electronic features of the amide group to molecular materials. Herein we propose a model for the study of the capabilities of the amide function to act as a bridge for electron delocalization in donor–acceptor substituted molecules and its potential application in molecular materials.
Results and discussion
Synthesis of the amide-bridged push–pull systems
Molecules were designed based on the push–pull stilbene scaffold, a well-known and thoroughly studied system constituted by a trans-1,2-diphenylethylene unit functionalized with electron-donating and electron-withdrawing groups (EDG and EWG, respectively) at opposite ends of the molecule.33 Substitution of the alkene bridge by the amide motif affords the N-phenylbenzamide backbone. Optimization of this structure to further exploit the amide moiety via intramolecular H-bond,34 while keeping the compounds easily accessible, led us to develop the N-phenylcoumarin-3-carboxamide as the benchmark structure for our studies (Scheme 1). | ||
| Scheme 1 Design of the N-phenylcoumarin-3-carboxamide scaffold. | ||
The amide-bridged push–pull systems 3a–i were synthesized from different substituted anilines 2a–i and 7-diethylaminocoumarin-3-carboxylic acid351 as illustrated in Scheme 2. Compound 1 was obtained by modifying a known method.36 The amide derivatives were purified by crystallization; their structures were confirmed by 1H and 13C NMR spectroscopy, High-Resolution Mass Spectrometry (ESI-TOF), reflectance IR and elemental analysis.
 | ||
| Scheme 2 Synthesis of the amide-bridged push–pull systems. | ||
The common intermediate 1 was synthesized from the reaction between Meldrum's acid and 4-(diethylamino)-salicylaldehyde with good yields (ca. 95%). The amide-bridged push–pull systems were obtained from the reaction between 1 and the respective substituted aniline 2a–i using EEDQ (N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline) in CH2Cl2 with moderate yields (51–74% see ESI† for the general procedure). The FTIR-ATR spectroscopy of all compounds 3a–i showed the characteristic bands of the amide group, N–H (3350–3100 cm−1 and ∼1540 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (∼1660 cm−1). HRMS (High Resolution Mass Spectrometry) using the Electrospray-Time of Flight technique confirmed the molecular ions. The signals in the 1H-NMR spectra at ca. 8.7, 7.4, 7.1, 6.6 and 6.5 ppm corroborate the presence of the coumarin core for all derivatives. Likewise, the 13C-NMR spectra showed the characteristic signals for the same system. The appearance of the signals between 10.8–11.1 ppm and 161.2–161.5 ppm in the 1H-NMR and 13C-NMR spectra, respectively, confirmed the presence of the amide bridge for all push–pull systems. Subtle changes are to be expected since delocalization through cross-conjugated systems is known to be relatively weak.
O (∼1660 cm−1). HRMS (High Resolution Mass Spectrometry) using the Electrospray-Time of Flight technique confirmed the molecular ions. The signals in the 1H-NMR spectra at ca. 8.7, 7.4, 7.1, 6.6 and 6.5 ppm corroborate the presence of the coumarin core for all derivatives. Likewise, the 13C-NMR spectra showed the characteristic signals for the same system. The appearance of the signals between 10.8–11.1 ppm and 161.2–161.5 ppm in the 1H-NMR and 13C-NMR spectra, respectively, confirmed the presence of the amide bridge for all push–pull systems. Subtle changes are to be expected since delocalization through cross-conjugated systems is known to be relatively weak.
X-Ray single crystal studies
Single crystals suitable for X-ray diffraction of compounds 3b, 3f, 3h and 3i were grown by slow evaporation from methylene chloride solutions; ORTEP diagrams are represented in Fig. 1. See Table S1 (ESI†) for their crystallographic data. The first collection of data for compounds 3b and 3f was performed at 298 K but, since the diethylamino group presented a severe disorder in both cases, we tried to model the disorder restricting the atomic ellipsoids using the SIMU and DELU commands unsuccessfully, contrary to that observed for compounds 3h and 3i. A second collection at lower temperature afforded the structures for 3b and 3f without disorder. | ||
| Fig. 1 ORTEP diagram of compounds (a) 3b, (b) 3f, (c) 3h and (d) 3i; thermal ellipsoids drawn at 50% probability level for all atoms other than H. | ||
All the compounds crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules per unit cell. The amide bond has the trans conformation due to the existence of one intramolecular H-bond (N–H⋯O, see Table S2, ESI†); this interaction provides certain planarity to the system (torsion angles between the averaged mean planes of the aniline ring-amide bridge-coumarin ring: 3.92° for 3b, 1.44° for 3f, 0.74° for 3h and 2.76° for 3i and interplanar angles between the same rings: 10.29° for 3b, 0.05° for 3f, 3.07° for 3h and 12.75° for 3i (see Fig. 2)), which is favourable for the effective communication between the donor and acceptor.
with two molecules per unit cell. The amide bond has the trans conformation due to the existence of one intramolecular H-bond (N–H⋯O, see Table S2, ESI†); this interaction provides certain planarity to the system (torsion angles between the averaged mean planes of the aniline ring-amide bridge-coumarin ring: 3.92° for 3b, 1.44° for 3f, 0.74° for 3h and 2.76° for 3i and interplanar angles between the same rings: 10.29° for 3b, 0.05° for 3f, 3.07° for 3h and 12.75° for 3i (see Fig. 2)), which is favourable for the effective communication between the donor and acceptor.
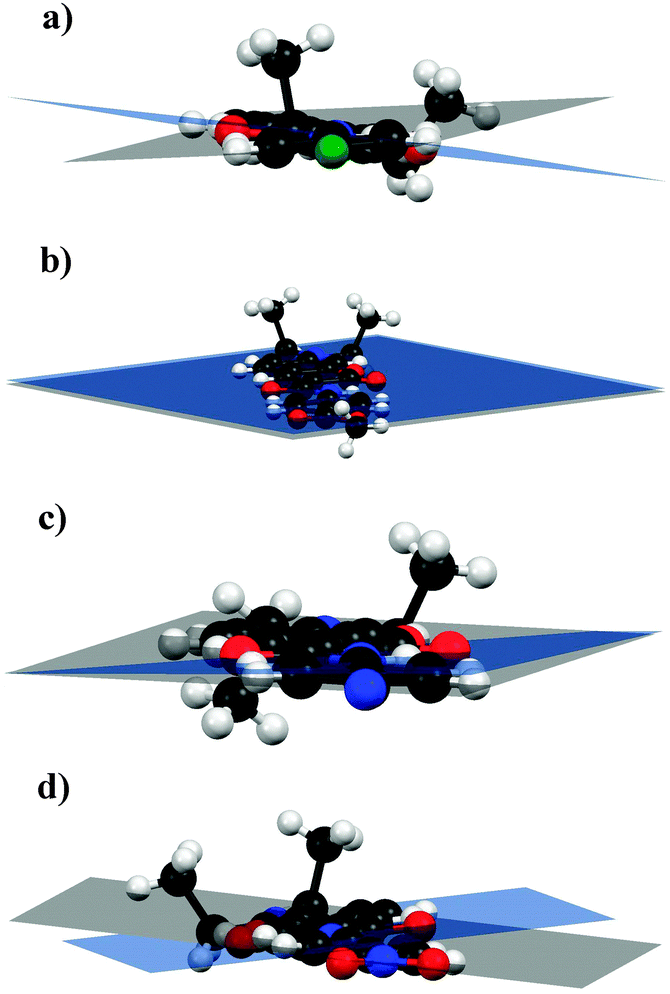 | ||
| Fig. 2 Interplanar angles for (a) 3b, (b) 3f, (c) 3h and (d) 3i. The observed quasiplanarity allows effective communication between the donor and the acceptor. | ||
Compound 3b shows intermolecular interactions including π–π stacking (face to face) of the coumarin core and the EWG ring (average interplanar distance 3.88 Å), intermolecular hydrogen bond between the EWG ring and the amide carbonyl (C–H⋯O, 2.663 Å), and intermolecular hydrogen bond between the aromatic fused rings of the coumarin core (C–H⋯O, 2.715 Å); these interactions produce 1D channels with the diethylamino groups oriented at opposite ends. Compound 3f shows two bifurcated intermolecular hydrogen bonds, one of them between the coumarin core and amide carbonyl (C–H⋯O, 2.695 Å), and the other one between the aniline ring and the coumarinic carbonyl (C–H⋯O, 2.713 Å). An unusual hydrogen bond ring R22 (10) between two adjacent methyl esters produce 1D layers with the diethylamino groups embraced at the other end (see Fig. S2, ESI†).
Compound 3h displays bifurcated hydrogen bonds between coumarins (C–H⋯O coumarinic carbonyl, 2.543 and 2.697 Å) and hydrogen bonds between the amide group and the aniline ring (C–H⋯O, 2.636 Å). It is interesting that the different orientation of the ethyl groups produces a bi-layer structural motif; the diethylamino methyls join the layers. Similarly to 3f, compound 3i shows intermolecular π–π stacking (face to face, interplanar average 3.639 Å), two hydrogen bonds between coumarins (C–H⋯O coumarinic carbonyl, 2.632 and 2.658 Å) and a hydrogen bond between the amide group and the aniline ring (C–H⋯O, 2.676 Å); it is worth noting that the nitro group does not participate in any interaction (Fig. 3).
 | ||
| Fig. 3 Representation of crystal packing motifs for (a) 3b, (b) 3f, (c) 3h and (d) 3i. | ||
These results evidence that, in the solid state, the quasiplanarity envisioned for the explored compounds is present, supporting the hypothesis that both ends of the push–pull amides may establish electronic communication.
NMR study of intramolecular electron delocalization
Since changes in electron density distribution within a set of molecules are being studied, NMR was chosen as a facile yet indirect measure of those fluctuations. Chemical shift (see Table 1) gives valuable information about the electronic environment of the nucleus under study; correlation of this experimental parameter with the presence of a given push–pull system would give us insight to conclude if there is actual communication between both ends of the molecule. Under that train of thought, the diethylamino moiety served our purpose by keeping a constant EDG structural motif among all the studied molecules while, being actively involved in electron donation, it acted as an internal probe (dependent variable) of electronic changes introduced by the different EWG explored (independent variable).| Compound | Amide group | Diethylamino group | ||
|---|---|---|---|---|
| 1H-NMR (NH) | 13C-NMR (CO) | 1H-NMR (CH2) | 13C-NMR (CH2) | |
| 3a | 10.86 | 161.09 | 3.46 | 45.14 |
| 3b | 10.90 | 161.19 | 3.46 | 45.17 |
| 3c | 10.97 | 161.16 | 3.46 | 45.17 |
| 3d | 10.90 | 161.14 | 3.46 | 45.16 |
| 3e | 10.90 | 161.15 | 3.46 | 45.17 |
| 3f | 11.10 | 161.39 | 3.46 | 45.19 |
| 3g | 11.09 | 161.50 | 3.47 | 45.20 |
| 3h | 11.18 | 161.62 | 3.48 | 45.24 |
| 3i | 11.32 | 161.71 | 3.49 | 45.26 |
As expected, the chemical shift of carbon and hydrogen atoms within the amide group correlated with the strength of the EWG under analysis. As an important result, the study showed also a good linear correlation between the 13C chemical shifts of the methylene units within the diethylamino group and the Hammett constant of the involved EWG (Fig. 4). It is noteworthy that the effect is only evident in the methylene 13C chemical shift, being highly diluted in 1H NMR and practically lost in both nuclei at the methyl termini, most likely because those groups, in addition to being less involved in inductive effects, possess the largest conformational freedom and are far more affected by the surrounding medium. These results evidence the effective communication between both ends of a push–pull system bridged by an amide spacer.
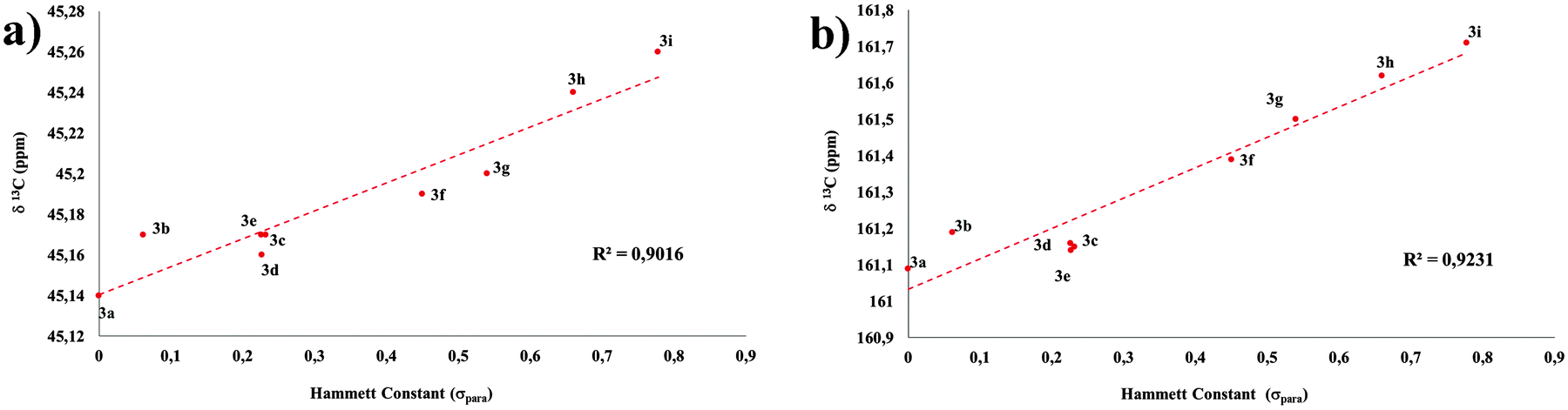 | ||
| Fig. 4 Correlation between the Hammett constant (σpara) and 13C-NMR chemical shifts for the (a) 7-diethylamino group (methylene) and (b) amide carbonylic carbon. | ||
Solvatochromic study
As an experimental means of studying the extended delocalization through the amide-bridged compounds, UV-vis absorption spectroscopy was employed in this work. Since push–pull molecules are usually accompanied by changes in dipole moment upon photoexcitation,37 a solvatochromic study was carried out.The absorption spectra of the family of compounds 3a–i display clear solvent polarity dependence (Table 2). An increase in the electron affinity of the EWG leads to an important bathochromic effect, with a maximum red shift of 31 nm, when comparing compound 3a in cyclohexane to molecule 3i in DMSO. These results suggest that charge separation is present and it is more important in the excited state compared to the polarization in the ground state; this implies an overall, solvent-dependent, narrowing of the π → π* excitation energy gap. Such an effect accounts for the observed red shift in absorption.
| Compound | R | λ max c-hexane | λ max AcOEt | λ max DMSO |
|---|---|---|---|---|
| 3a | H | 414 | 422 | 433 |
| 3b | F | 415 | 422 | 433 |
| 3c | Cl | 417 | 424 | 434 |
| 3d | Br | 418 | 424 | 433 |
| 3e | C2H | 419 | 426 | 437 |
| 3f | CO2Me | 419 | 427 | 438 |
| 3g | CF3 | 419 | 427 | 436 |
| 3h | CN | 421 | 429 | 440 |
| 3i | NO2 | 422 | 433 | 445 |
This dependence is reflected in the linear correlation between λmax and the normalized polarity index (PI) of the studied solvents (Fig. 5).38 Also, the EWG force linearly correlates with λmax red shift (Fig. 6), demonstrating that lower-energy absorption occurs due to push–pull-induced delocalization of the system, i.e., longer wavelength of the associated unidimensional potential-well, leading to a correspondingly higher degree of charge separation in the excited state as EWG force increases.
 | ||
| Fig. 5 Correlation between absorption λmax and solvent polarity index. | ||
 | ||
| Fig. 6 Correlation between absorption λmax and Hammett constant σpara. | ||
Computational studies
The double-zeta valence 6-31+G* was chosen as the basis set throughout this work. IR vibrational frequencies were computed for all the molecules at the same level of theory. All the frequencies obtained were positive and real, confirming that the stationary points correspond to stable molecular geometries. The analysis of structural parameters was made using Mercury.42
According to Linus Pauling's theory of resonance, amide bonds possess typically three canonical forms (Scheme 3A–C). In a push–pull amide, involvement of this bridge in IED would imply electron reorganization. Describing this with resonant structures leads to the appearance of an additional, dienoid form (Scheme 3D).
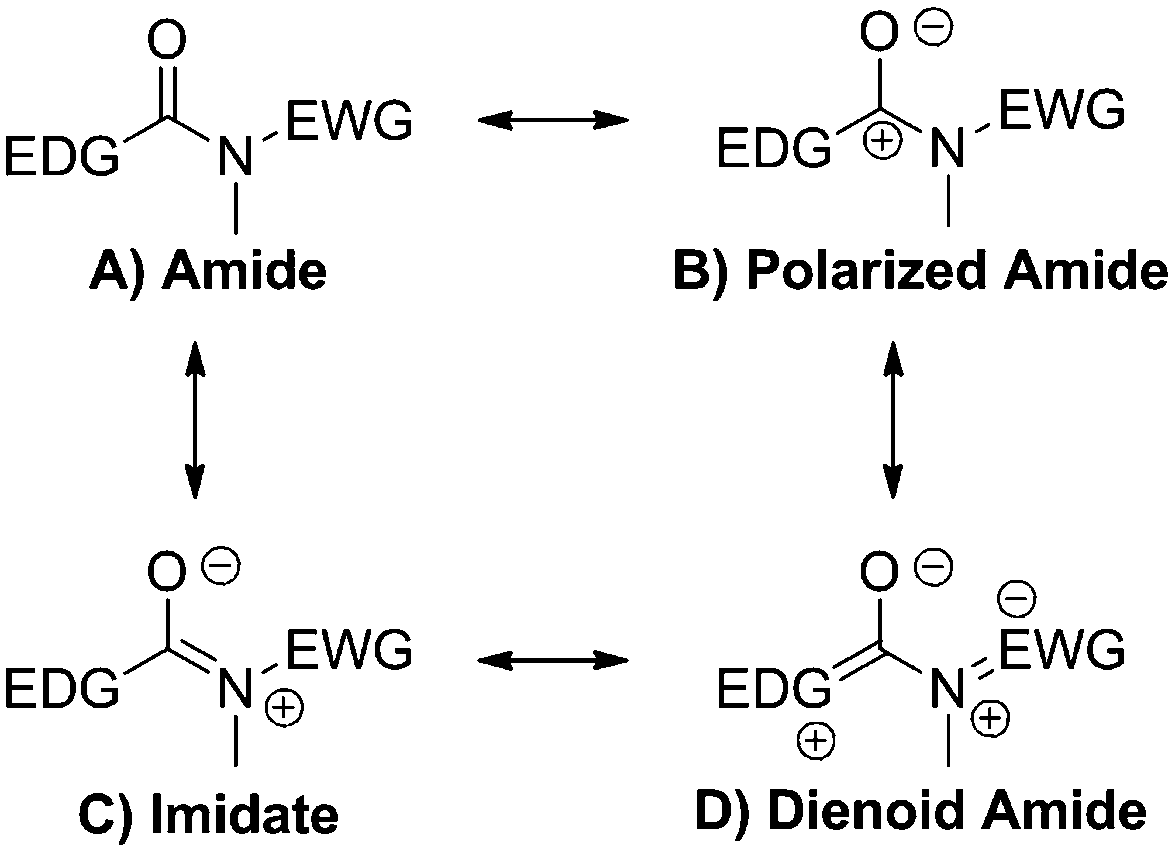 | ||
| Scheme 3 Resonance structures of the amide group under the influence of push–pull effects. | ||
Structurally, a shortening of the EDG-C and N-EWG bonds should be appreciated, with the concomitant lengthening of the central C–N bond. To confirm this hypothesis, our first studies focused on the unperturbed N-phenylcoumarin-3-carboxamide molecule, introducing the push–pull system in a stepwise fashion (Scheme 4). As will be discussed, it is evident that the dienoid form becomes more important as the push–pull effect increases.
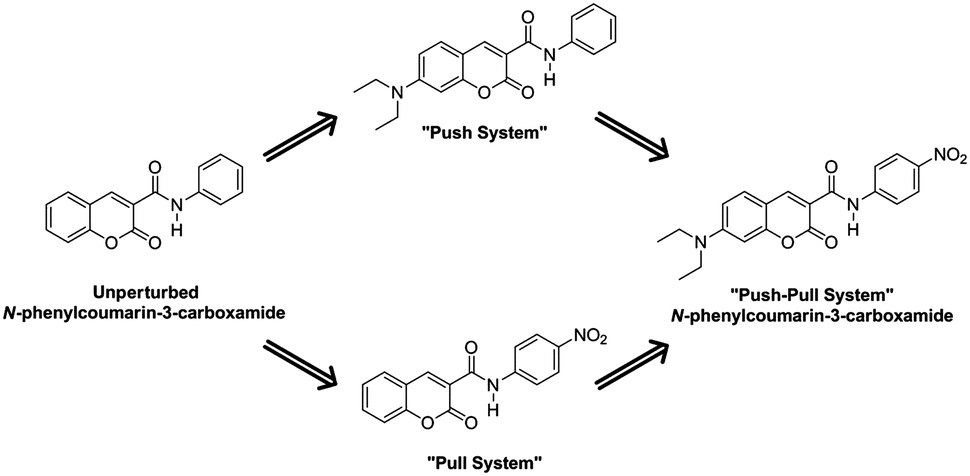 | ||
| Scheme 4 Stepwise introduction of push–pull effects. | ||
After the addition of a nitro group, the “pull system” is formed. Subtle deviations in bond lengths evidence the contribution of the dienoid canonical form; a similar effect may also be observed with the introduction of the diethylamino group in the “push system”. Communication between both units is clear in the push–pull molecule, where the net influence of the EWG and the EDG is greater than the sum of individual changes produced by each group (Table 3).
| Bond 1 | Bond 2 | Bond 3 | Bond 4 | Bond 5 | |
|---|---|---|---|---|---|
| Unpertubed | 1.513 | 1.357 | 1.408 | 1.017 | 1.875 |
| Pull system | 1.509 | 1.364 | 1.398 | 1.019 | 1.858 |
| Push system | 1.507 | 1.360 | 1.406 | 1.018 | 1.874 |
| Push–pull | 1.502 | 1.368 | 1.395 | 1.019 | 1.855 |
To study the IED in detail, the effect of different EWG keeping the EDG constant was evaluated. The optimized structures for our family of compounds exhibit the behaviour condensed in Table S3 (ESI†). For bond assignment, see Scheme 5. Furthermore, the variation in bond lengths linearly correlates with the Hammett constant, σpara, of the corresponding EWG (see Tables S3 and S4, ESI†).43,44
 | ||
| Scheme 5 Bond assignment for the study of transmission of push–pull effects through amide bridges. | ||
Assuming that the dienoid resonant form contributes to the net structure, it would imply an important degree of charge separation; thus, the optimized molecules were further refined, imposing a polar medium of a given dielectric constant. The Self-Consistent Reaction Field method (SCRF) with the Polarizable Continuum Model (PCM) was employed as implemented in Gaussian 09, selecting MeCN as a solvent due to its large dielectric constant (ε = 37.5).45 The results condensed in Fig. 7 contrast in vacuo values vs. structural parameters in MeCN. It is noteworthy that a highly polar medium does, in fact, favour charge separation leading to a more pronounced dienoid character of the amide bridge. It is also interesting that a linear correlation is, in general, higher in the polar medium, especially for bond 1 (R2 = 0.8949 in vacuo vs. 0.9469 in MeCN). This observation is attributed, as it will be later discussed, to a LUMO/LUMO + 1 stabilization and redistribution as solvent polarity increases.
 | ||
| Fig. 7 Correlation graphics between computed structural parameters (in vacuo and in MeCN) and the Hammett Constant of EWG for (a) bond length 1, (b) bond length 2, (c) bond length 3 and (d) bond length 5. | ||
The simultaneous electron delocalization through H-bond becomes evident when analyzing the N–H⋯O bond shortening with increasing EWG strength (Fig. 7d). Electron reorganization via the SE mechanism through this fragment linearly correlates with the stress exerted by the push–pull system, especially in polar media.
 | ||
| Scheme 6 Frontier molecular orbitals of molecules 3a, 3f, 3i (vacuum, MeCN). Arrows represent the transitions involved in the first excited state, according to TDDFT computations. HOMO – LUMO gaps are also depicted. | ||
It is interesting to observe the frontier molecular orbitals of compound 3f. The ester group has a well-defined electron accepting character (σpara = 0.45), but even under such push–pull stress, the LUMO does not display the substituted anilide fragment as an important acceptor. Further investigation of higher-energy MOs shows that LUMO + 1 comprises the expected topology. The anilide moiety is, again, part of the principal donor, residing in both HOMO and HOMO − 1. Electron redistribution towards EWG occurs only between HOMO − 1 and LUMO + 1, being a transition of the type n → π*. In this system, that electron promotion involves only the anilide portion of the molecule. Up to this point, no net electron donation from the Et2N group to EWG is evident. The amide bond behaves mostly as an isolating bridge for useful electron transmission.
For molecule 3h where LUMO and LUMO + 1 are relatively low in energy, the envisioned D → (amide bond) → A transmission is finally observable, though rather weak, the IED towards the amidic carbonyl group being more pronounced (see Fig. S21, ESI†).
The most marked push–pull structure studied here, compound 3i, becomes very interesting in this context. The 4-nitroanilide portion clearly increases its acceptor character to the extent of populating the LUMO. The HOMO involves, principally, the electron-rich coumarin as expected. Furthermore the HOMO, being more localized in polar media, increases its energy thus narrowing even more the HOMO – LUMO gap (261 meV in vacuo vs. 245 meV in MeCN).
This analysis shows three interesting results: (1) the dienoid canonical form, although evidently involved in the a priori resonance description, is practically nonexistent until the push–pull stress is large enough, i.e., compound 3i in vacuum; (2) under dissociating conditions, i.e., highly polar medium, the dienoid structure appears as the LUMO + 1 for the full set of molecules, but is energetic enough to be excluded from the principal transition in most cases, except for 3h and particularly 3i where its population effectively migrates to the LUMO and (3) the appearance of such delocalizing, dienoid LUMO + 1 imprints a weak but nonzero EWG-dependent attractive character to the anilide fragment, explaining the higher correlations observed in MeCN vs. vacuum.
Comparison with typical conjugated bridges
Due to the inherent asymmetry in the electronic distribution of donor–acceptor systems and their properties (electronic excitations commonly associated with important changes in dipole moment leading to strong interaction with the electric field of incident light), they are most often employed in the study, design and synthesis of materials with nonlinear optical response.Since the most common linkers in such molecules are alkene, alkyne, imine and azo bridges, the first hyperpolarizability, β, of compounds analogous to 3i (Scheme 7, molecules 4a–d) was computed under the DFT/wB97X-D framework. β, a third-rank tensor, is related to second-order nonlinear phenomena such as the Kerr effect, parametric up and down conversion, among others. These values allow a direct, numerical comparison between 3i and push–pull molecules bridged by typical conjugated moieties within the context of one of their main applications in materials science.
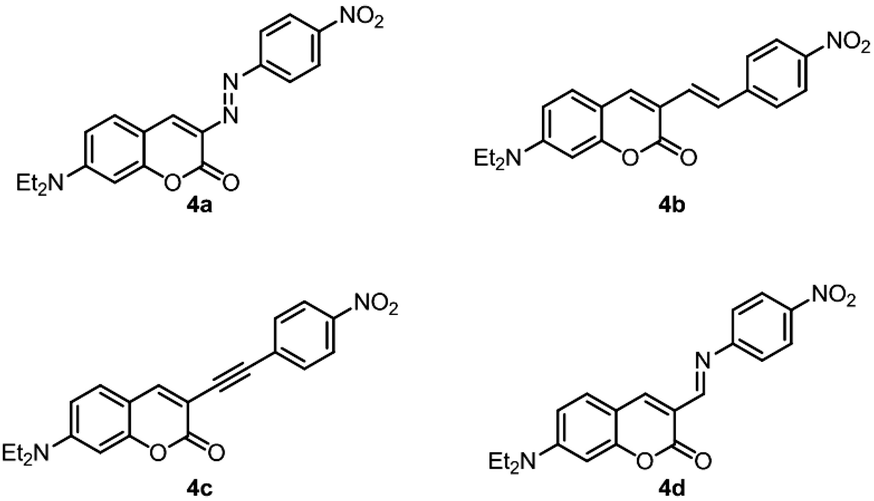 | ||
| Scheme 7 Push–pull systems analogous to 3i, bridged by common linkers. | ||
A comparison of the values of βHRS (ref. 46) obtained for the five compounds under study (Table 4) shows that the response of 3i is expected to be roughly half of that displayed by molecules bridged by double (alkene, imine, azo) or triple (alkyne) bonds. This represents an interesting result since, in some chemical contexts, it is synthetically simpler to introduce an amide group than another linker, where heavy metal-mediated synthesis (compounds 4b and 4c, for example)47 or handling of unstable intermediates (4a) is needed.
| Compound | β HRS |
|---|---|
| 3i | 27.3 |
| 4a | 63.1 |
| 4b | 48.3 |
| 4c | 51.0 |
| 4d | 42.1 |
Its low chemical reactivity makes an amide, also, a safe alternative to bridges such as the imine double bond, highly susceptible to hydrolysis. Considering this, the amide bridge offers an interesting alternative to common bridges, with the compromise between smaller, yet comparable (within the same order of magnitude), nonlinear optical response, synthetic feasibility and chemical stability. In large-scale processes or long-term applications, this compromise may tilt towards ease of synthesis and inertness.
Conclusions
A set of 9 new substituted N-phenylcoumarin-3-carboxamide derivatives was synthesized to target the capability of the amide fragment to act as an efficient, cross-conjugated bridge for the transmission of electronic effects in push–pull molecular units. NMR analysis confirms communication between the donor and acceptor substituent, comprising a synergistic through-amide (resonant) and H-bond-assisted (superexchange) delocalization.Computational studies also show the involvement of both structural motifs. Linear correlation with the Hammett constants of the studied EWG supports these results, while frontier orbital analysis shows the contribution of an additional, dienoid, canonical form of the amide-bridged systems as push–pull stress increases. UV-vis positive solvatochromism and DFT computations of polarity-dependent structural parameters suggest that a photoinduced charge-separation process may occur through amide bonds under appropriate conditions.
In the context of the potential application of amide bridges in molecular materials, there is a compromise between nonlinear optical response (smaller, yet significant, for amide bridges) and ease of synthesis and inertness, the amide group being an interesting alternative when considering these aspects.
Acknowledgements
The authors are grateful to UNAM (PAPIIT IN214010) and CONACYT for financial support and granting scholarships to M. Maldonado-Domínguez and R. Arcos-Ramos; they also thank the LIA (Laboratorio Internacional Asociado Franco-Mexicano) and the ECOS-ANUIES program for abroad mobility grants. Thanks to Rosa I. del Villar for NMR measurements.References
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802 CrossRef CAS PubMed.
- (a) Y. Che, A. Datar, X. Yang, T. Naddo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2007, 129, 6354 CrossRef CAS PubMed; (b) V. Skakálová, A. B. Kaiser, U. Dettlaff-Weglikowska, K. Hrnčariková and S. Roth, J. Phys. Chem. B, 2005, 109, 7174 CrossRef PubMed.
- P. C. Hiberty and C. P. Byrman, J. Am. Chem. Soc., 1995, 117, 9875 CrossRef CAS.
- F. Feixas, E. Matito, M. Solà and J. Poater, J. Phys. Chem. A, 2008, 112, 13231 CrossRef CAS PubMed.
- G. J. Zhao, R. K. Chen, M. T. Sun, G. Y. Li, J. Y. Liu, Y. L. Gao, K. L. Han, X. Yang and L. Sun, Chem.–Eur. J., 2008, 14, 6935 CrossRef CAS PubMed.
- (a) J. L. Oudar, J. Chem. Phys., 1977, 67, 446 CrossRef CAS; (b) D. J. Williams, Angew. Chem., Int. Ed. Engl., 1984, 23, 690 CrossRef; (c) P. J. Mendes, T. J. L. Silva, M. H. Garcia, J. P. Prates-Ramalho and A. J. Palace-Carvalho, J. Chem. Inf. Model., 2012, 52, 1970 CrossRef CAS PubMed; (d) H. S. Nalwa and S. Miyama, Nonlinear Optics of Organic Molecules and Polymers, CRC Press, Boca Raton, Florida, USA 1997 Search PubMed.
- (a) J. Akl, C. Billot, P. G. Lacroix, I. Sasaki, S. Mallet-Ladeira, I. Malfant, R. Arcos-Ramos, M. Romero and N. Farfán, New J. Chem., 2013, 37, 3518 RSC; (b) G. J. Yakatan, R. J. Juneau and S. G. Schulman, Anal. Chem., 1972, 44, 1044 CrossRef CAS.
- (a) D. M. Wagnerová and K. Lang, Coord. Chem. Rev., 2011, 255, 2904 CrossRef PubMed; (b) N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289 CrossRef CAS PubMed; (c) M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731 RSC.
- K. S. Schanze and K. Sauer, J. Am. Chem. Soc., 1988, 110, 1180 CrossRef CAS.
- (a) D. Southgate and D. Hall, J. Appl. Phys., 1972, 43, 2765 CrossRef; (b) J. Poater, M. Duran, M. Solà and B. Silvi, Chem. Rev., 2005, 105, 3911 CrossRef CAS PubMed.
- T. M. Krygowski and B. T. Stpień, Chem. Rev., 2005, 105, 3482 CrossRef CAS PubMed.
- (a) F. Feixas, E. Matito, J. Poater and M. Solà, J. Phys. Chem. A, 2011, 115, 13104 CrossRef CAS PubMed; (b) G. Frenking and I. Fernandez, Chem.–Eur. J., 2006, 12, 3617 CrossRef.
- (a) R. Hoffmann, A. Imamura and W. Hehre, J. Am. Chem. Soc., 1968, 90, 1499 CrossRef CAS; (b) N. Turro and J. Barton, JBIC, J. Biol. Inorg. Chem., 1998, 3, 201 CrossRef CAS.
- (a) Y. Zhao, S. C. Ciulei and R. R. Tykwinski, Tetrahedron Lett., 2001, 42, 7721 CrossRef CAS; (b) M. Orchin and N. F. Phelan, J. Chem. Educ., 1968, 45, 633 CrossRef.
- (a) Y. Wang, Z. Yu, J. Wu and C. Liu, J. Phys. Chem. A, 2009, 113, 10521 CrossRef CAS PubMed; (b) L. Sobczyk, S. J. Grabowski and T. M. Krygowski, Chem. Rev., 2005, 105, 3513 CrossRef CAS PubMed.
- P. B. Glenn and G. C. Bazan, Acc. Chem. Res., 2001, 34, 30 CrossRef PubMed.
- S. Günes, H. Neugebauer, N. Serdar and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- (a) M. Fanti, G. Orlandi and F. Zerbetto, J. Phys. Chem. A, 1997, 101, 3015 CrossRef CAS; (b) R. S. Butler, P. Cohn, P. Tenzel, K. A. Abboud and R. K. Castellano, J. Am. Chem. Soc., 2009, 131, 623 CrossRef CAS PubMed.
- (a) S. Isied and A. Vassilian, J. Am. Chem. Soc., 1984, 106, 1732 CrossRef CAS; (b) L. Serrano-Andrés, J. Phys. Chem. B, 2001, 105, 9323 CrossRef; (c) A. S. Serron, W. S. Aldridge III, C. N. Fleming, R. M. Danell, M. H. Baik, M. Sikora, D. M. Dattelbaum and T. J. Meyer, J. Am. Chem. Soc., 2004, 126, 14506 CrossRef PubMed; (d) D. R. Striplin, S. Y. Reece, D. G. McCafferty, C. G. Wall, D. A. Friesen, B. W. Erickson and T. J. Meyer, J. Am. Chem. Soc., 2004, 126, 5282 CrossRef CAS PubMed.
- M. N. Paddon-Row, Acc. Chem. Res., 1994, 27, 18 CrossRef CAS.
- J. Abe, Y. Shirai, N. Nemoto and Y. Nagase, J. Phys. Chem. A, 1997, 101, 1 CrossRef CAS.
- (a) M. Blanchard-Desce, I. Ledoux, J. M. Lehn, J. Malthete and J. Zyss, Chem. Commun., 1988, 737 RSC; (b) K. D. Singer, J. E. Sohn, L. A. King, H. M. Gordon, H. E. Katz and C. W. Dirk, J. Opt. Soc. Am. B, 1989, 6, 1339 CrossRef CAS.
- M. Jain and J. Chandrasekhar, J. Phys. Chem., 1993, 97, 4044 CrossRef CAS.
- (a) E. Kleinpeter, U. Bölke and J. Kreicberga, Tetrahedron, 2010, 66, 4503 CrossRef CAS PubMed; (b) O. A. Blackburn and B. J. Coe, Organometallics, 2011, 30, 2212 CrossRef CAS.
- J. M. Rivera, H. Reyes, A. Cortés, R. Santillan, P. G. Lacroix, C. Lepetit, K. Nakatani and N. Farfán, Chem. Mater., 2006, 18, 1174 CrossRef CAS.
- C. Zhang, L. R. Dalton, M. C. Oh, H. Zhang and W. H. Steier, Chem. Mater., 2001, 13, 3043 CrossRef CAS.
- N. Y. Gorobets, S. A. Yermolayev, T. Gurley, A. A. Gurinov, P. M. Tolstoy, I. G. Shenderovich and N. E. Leadbeater, J. Phys. Org. Chem., 2012, 25, 287 CrossRef CAS.
- (a) C. Turro, C. K. Chang, G. E. Leroi, R. I. Cukier and D. G. Nocera, J. Am. Chem. Soc., 1992, 114, 4013 CrossRef CAS; (b) P. J. F. De Rege, S. A. Williams and M. J. Therien, Science, 1995, 269, 1409 CrossRef CAS.
- (a) J. Lamba and J. Tour, J. Am. Chem. Soc., 1994, 116, 11723 CrossRef CAS; (b) R. L. Letsinger and T. Wu, J. Am. Chem. Soc., 1995, 117, 7323 CrossRef CAS.
- (a) M. A. A. El-Ghaffar, D. E. El-Nashar and E. A. M. Youssef, Polym. Degrad. Stab., 2003, 82, 47 CrossRef; (b) G. F. Nieckarz, J. L. Litty and D. R. Tyler, J. Organomet. Chem., 1998, 554, 19 CrossRef CAS.
- (a) M. Okazaki, I. Washio, Y. Shibasaki and M. Ueda, J. Am. Chem. Soc., 2003, 125, 8120 CrossRef CAS PubMed; (b) J. Ishida, M. Jikei and M. Kakimoto, Macromolecules, 2000, 33, 3202 CrossRef.
- (a) M. Bodanszky, Int. J. Protein Res., 1985, 25, 449 CrossRef CAS; (b) S. Plaue, S. Muller, J. P. Briand and M. H. Van Regenmortel, Biologicals, 1990, 18, 147 CrossRef CAS.
- (a) L. Antonov, K. Kamada, K. Ohta and F. Kamounah, Phys. Chem. Chem. Phys., 2003, 5, 1193 RSC; (b) P. N. Day, K. A. Nguyen and R. Pachter, J. Chem. Phys., 2006, 125, 94103 CrossRef PubMed.
- Y. Nagasawa, T. Arkadiy, P. Yartsev, K. Tominaga, P. B. Bisht, A. E. Johnson and K. Yoshihara, J. Am. Chem. Soc., 1993, 115, 7922 CrossRef CAS.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef; (b) S. Miertuš and J. Tomasi, Chem. Phys., 1982, 65, 239 CrossRef; (c) J. L. Pascual-Ahuir, E. Silla and I. Tuñón, J. Comput. Chem., 1994, 15, 1127 CrossRef CAS; (d) E. J. Jun, J. A. Kim, K. M. K. Swamy, S. Park and J. Yoon, Tetrahedron Lett., 2006, 47, 1051 CrossRef CAS PubMed; (e) C. Tablet, I. Matei, E. Pincu, V. Meltzer and M. Hillebrand, J. Mol. Liq., 2012, 168, 47 CrossRef CAS PubMed; (f) O. S. Detistov, V. D. Orlov and I. O. Zhuravel, J. Heterocycl. Chem., 2012, 49, 883 CrossRef CAS; (g) M. Frizler, M. D. Mertens and M. Gütschow, Bioorg. Med. Chem. Lett., 2012, 22, 7715 CrossRef CAS PubMed; (h) J. Y. Li, X. Q. Zhou, Y. Fang and C. Yao, Spectrochim. Acta, Part A, 2013, 102, 66 CrossRef CAS PubMed.
- G. Bardajee, F. Jafarpour and H. Afsari, Cent. Eur. J. Chem., 2010, 8, 370 CrossRef CAS.
- D. Kosenkov and L. V. Slipchenko, J. Phys. Chem. A, 2011, 115, 392 CrossRef CAS PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- M. Hanwell, D. Curtis, D. Lonie, T. Vandermeersch, E. Zurek and G. Hutchison, J. Cheminf., 2012, 4 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- C. Macrae, I. Bruno, J. Chisholm, P. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond, Cornwell University Press, 3rd edn, 1960, p. 183 Search PubMed.
- (a) L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96 CrossRef CAS; (b) D. H. McDaniel and H. C. Brown, J. Org. Chem., 1957, 23, 420 CrossRef; (c) C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207 CrossRef CAS; (d) V. L. Balke, F. A. Walker and J. T. West, J. Am. Chem. Soc., 1985, 107, 1226 CrossRef CAS; (e) D. Traschel, Helv. Chim. Acta, 2003, 86, 2754 CrossRef.
- G. Bardajee, F. Jafarpour and H. Afsari, Cent. Eur. J. Chem., 2010, 8, 370 CrossRef CAS.
- V. Rodríguez, J. Grondin, F. Adamietz and Y. Danten, J. Phys. Chem. B, 2010, 114, 15057 CrossRef PubMed.
- N. Myung, S. Connelly, B. Kim, S. J. Park, I. A. Wilson, J. W. Kelly and S. Choi, Chem. Commun., 2013, 49, 9188 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: DFT studies for computed structures 3a–i including frontier molecular orbitals, 1H and 13C NMR spectra for compounds 3a–i, crystal data for compounds 3b, 3f, 3h and 3i. CCDC 957820–957823. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj01176c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |