Syntheses and luminescence of complete solid solutions Gd1−xEux[B6O9(OH)3] and α-Gd1−xEuxB5O9†
Xuefang
Yi
,
Rihong
Cong
*,
Zhengyang
Zhou
,
Pengfei
Jiang
,
Wenliang
Gao
and
Tao
Yang
*
College of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, P. R. China. E-mail: congrihong@cqu.edu.cn; taoyang@cqu.edu.cn; Fax: +86-23-65105065; Tel: +86-23-65105065
First published on 30th September 2013
Abstract
There are very few luminescence studies for rare earth borates with hydroxyl or crystalline water molecules, which were believed to have a low luminescence efficiency because the vibrations of –OH or H2O may lead to quenching of the emission. We were motivated to study the luminescence properties of Gd1−xEux[B6O9(OH)3] (x = 0.10–1) and their dehydrated products, α-Gd1−xEuxB5O9. Efficient energy transfer from Gd3+ to Eu3+ was found in all of the studied polyborates. By TG-DSC and powder XRD experiments, we observed the dehydration of Eu[B6O9(OH)3], the re-crystallization to α-EuB5O9, and further decomposition to α-EuB3O6. During those processes, the Eu3+ luminescence spectra show interesting variations, meaning it is a good medium to understand the coordination environment evolution of Eu3+. It is observed that the symmetry of the Eu3+ coordination environment is the lowest in the amorphous state. Interestingly, this amorphous phase possesses a high efficiency of f–f transitions and a large R/O value (4.0), which implies its potential as a good red-emitting UV-LED phosphor. Anhydrous α-EuB5O9 shows the highest luminescence efficiency when excited by Eu3+ CT transition. For the first time, complete solid solutions α-Gd1−xEuxB5O9 were synthesized directly by the sol–gel method, and their luminescence properties were also studied.
Introduction
Rare earth borates are good hosts for luminescent materials due to their high transparency, good thermal stability, exceptional optical damage threshold and high luminescence efficiency.1–3 During the past decades, numerous hydrated rare earth borates were prepared by the boric acid flux and hydrothermal methods; their luminescence properties, however, were usually overlooked.4–12 A possible reason is that the vibrations of OH− or H2O may lead to emission quenching through non-radioactive pathways. On the contrary, the rare earth borates prepared under mild conditions usually have complex and large polyborate anions,6–12 which can sufficiently separate the rare earth cations to prevent the concentration quench effect.6,7,12 As a typical example, hydrothermally prepared Gd2B6O10(OH)4·H2O:Eu3+ exhibits very highly efficient Eu3+ f–f transitions and obvious energy transfer from Gd3+ to Eu3+ ions, which indicates that this series of borates may have applications in VUV- and UV-LED phosphors.12 There is no clear evidence indicating any effective energy transfer from Eu3+ to H2O molecules or hydroxyl groups.12In addition, the calcination of hydrated polyborates at appropriate temperatures can give rise to new anhydrous borates.6,7 For example, α-LnB5O9 (Ln = Pr-Eu) was first prepared by a long-time calcination of H3LnB6O12 at 650–700 °C for 5 days.6 A preliminary study of the luminescence properties of a partially solid solution of α-Gd1−xEuxB5O9 (x ≤ 0.16) was reported by Lin et al.,6 which focused on the luminescence under VUV excitation (125 nm). The appearance of strong Gd3+ absorptions implied effective energy transfer from Gd3+ to Eu3+ in this series of phosphors. However, the luminescence properties of α-GdB5O9:Eu3+ under UV excitation have not been reported.
In this work, complete solid solutions of α-Gd1−xEuxB5O9 have been prepared, not only by thermal decomposition from Gd1−xEux[B6O9(OH)3] (x = 0–1), but also directly by the sol–gel method, using Ln2O3 and H3BO3 as the starting materials. The UV-excited luminescence properties of Gd1−xEux[B6O9(OH)3] and α-Gd1−xEuxB5O9 (prepared by both methods) were systematically investigated. We also studied the structural evolution by the luminescence characteristics of Eu3+ during the dehydration of Eu[B6O9(OH)3] and the re-crystallization process to α-EuB5O9. There exists an intermediate amorphous state, the structure of which can not be studied by regular powder X-ray diffraction (XRD). Meanwhile, by analysing the Eu3+ luminescence, we noticed that the symmetry of the Eu3+ coordination environment is the lowest in the amorphous state.
Experimental section
Syntheses
The syntheses of Gd1−xEux[B6O9(OH)3] (x = 0–1) were carried out in closed Teflon autoclaves using the boric acid flux method. Typically, 0.22 mmol of Gd2O3 and 20 mmol of H3BO3 (Gd/B = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45) were first mixed and placed into a 25 mL Teflon autoclave. Then 1.0 mL of deionized water was added. The autoclave was sealed and heated at 240 °C in an oven for 7 days. The products were washed with water (25 °C) until the excess boric acid was completely removed, and then dried at 80 °C for further characterization. By the same procedure, a series of Eu-doped samples, Gd1−xEux[B6O9(OH)3] (x = 0.10–1), were prepared. Block single crystals were obtained, and there was a gradual color change from white (for Gd[B6O9(OH)3]) to light pink (for Eu[B6O9(OH)3]). Eu[B6O9(OH)3] is isostructural with Gd[B6O9(OH)3],6 so complete solid solutions of Gd1−xEux[B6O9(OH)3] (x = 0–1) can be easily obtained by the boric acid flux method. The substitution contents of the final products may not be exactly the same as the starting ratios, so elemental analyses were performed on the as-synthesized products, and the real doping contents of Eu3+ (the values of x) were determined accordingly. The x values obtained from the inductively coupled plasma-atomic emission spectrometry (ICP-AES) method on a Leeman Profile-Spec were close to the starting ratios of Eu/(Gd + Eu), and therefore were used in the following sections.
:45) were first mixed and placed into a 25 mL Teflon autoclave. Then 1.0 mL of deionized water was added. The autoclave was sealed and heated at 240 °C in an oven for 7 days. The products were washed with water (25 °C) until the excess boric acid was completely removed, and then dried at 80 °C for further characterization. By the same procedure, a series of Eu-doped samples, Gd1−xEux[B6O9(OH)3] (x = 0.10–1), were prepared. Block single crystals were obtained, and there was a gradual color change from white (for Gd[B6O9(OH)3]) to light pink (for Eu[B6O9(OH)3]). Eu[B6O9(OH)3] is isostructural with Gd[B6O9(OH)3],6 so complete solid solutions of Gd1−xEux[B6O9(OH)3] (x = 0–1) can be easily obtained by the boric acid flux method. The substitution contents of the final products may not be exactly the same as the starting ratios, so elemental analyses were performed on the as-synthesized products, and the real doping contents of Eu3+ (the values of x) were determined accordingly. The x values obtained from the inductively coupled plasma-atomic emission spectrometry (ICP-AES) method on a Leeman Profile-Spec were close to the starting ratios of Eu/(Gd + Eu), and therefore were used in the following sections.
α-Gd1−xEuxB5O9 (x = 0–1) were prepared by heating the as-synthesized Gd1−xEux[B6O9(OH)3] (x = 0–1) compounds at 670 °C for 10 hours. The powder products of α-Gd1−xEuxB5O9 (x = 0–1) also showed a gradient color change from white to light pink.
The sol–gel method was employed to synthesize α-Gd1−xEuxB5O9 (x = 0–1) directly. Stoichiometric Ln2O3 (Ln = Gd/Eu) was first dissolved in warm concentrated nitric acid. Then, H3BO3 (with a 10 mol% excess, in order to compensate for the volatilization of boron oxide at high temperature), an appropriate amount of citric acid, and deionized water were charged into the solution. After stirring and heating, a clear and homogeneous sol was formed, which was further loaded into an oven and kept at 80 °C, 120 °C and 180 °C for 5 hours, respectively. A light brown precursor was obtained, indicating the existence of carbon. The precursor was then heated in a muffle furnace at 250 °C and 550 °C for 15 hours respectively to decompose the nitrates, and it changed to a dark brown amorphous powder. Finally, the target material α-Gd1−xEuxB5O9 was obtained by grinding and re-calcination at 670 °C for 10 hours. The colour of the obtained sample turned to white. Moreover, the product was washed by deionized water in order to remove the possible excess of B2O3. All of the abovementioned samples were checked to be pure by powder XRD and ready for further characterization.
Characterization
Powder XRD data were collected at room temperature on a PANalytical Empyrean instrument equipped with a PIXcel 3D detector (Cu Kα radiation). The operation voltage and current were 40 kV and 40 mA, respectively. Le Bail refinements were performed to obtain the cell parameters using the TOPAS software package.13 Combined thermogravimetric analyses (TG) and differential scanning calorimeter (DSC) analyses for Gd1−xEux[B6O9(OH)3] (x = 0, 0.31, 0.72, 1.00) were performed on a Mettler-Toledo TGA/DSC1 instrument under a N2 flow. Photoluminescence spectra were measured on a Hitachi F4600 fluorescence spectrometer. The voltage of the Xe lamp was fixed at 700 V. The emission intensities were calculated from the integrals of the corresponding peaks.Results and discussion
Synthesis of Gd1−xEux[B6O9(OH)3] (x = 0–1)
For either Gd3+ or Eu3+, three structures, Ln[B6O9(OH)3], Ln[B9O13(OH)4]·H2O and Ln2B6O10(OH)4·H2O, can be synthesized by using the boric acid flux or hydrothermal methods.6,7,12 Differences in the synthesis conditions should be clarified first. In the literature,6,7 the only difference for the formation of Ln[B6O9(OH)3] and Ln[B9O13(OH)4]·H2O is the Ln/B ratio of the starting materials, which is Ln/B = 1:30 and 1:15, respectively. In order to improve the crystallinity of the sample in our work, a small amount of water was added; therefore the resultant Ln/B ratios were set to 1:45 and 1:30 with additional 1 mL and 0.25 mL quantities of water, respectively. The final products of Ln[B6O9(OH)3] are indeed well-qualified single crystals. More boric acid or water was used for the synthesis of Ln[B6O9(OH)3] than Ln[B9O13(OH)4]·H2O. The possible reason is that the formation of Ln[B6O9(OH)3] may require a higher inner pressure, which can be achieved by adding more boric acid or water in a closed system. Ln2B6O10(OH)4·H2O was synthesized under hydrothermal conditions, which means a larger amount of water was necessary.12
XRD study for Gd1−xEux[B6O9(OH)3] and α-Gd1−xEuxB5O9
10 single-crystal samples of Gd1−xEux[B6O9(OH)3] (x = 0–1) were finely ground and characterized by powder XRD without any observable impurity peaks (see Fig. 1a and Fig. S1, ESI†), indicating the Eu3+ is successfully doped into the structure. The XRD patterns do not show any obvious peak shifting with the increase of x, which is understandable due to the similar cationic radii for Gd3+ and Eu3+. The difference in the cell lattice parameters, including a, c and V (summarized in Table 1), can be only verified by Le Bail refinement of the whole XRD patterns. The resultant cell volumes expand slightly and linearly (see Fig. 1b).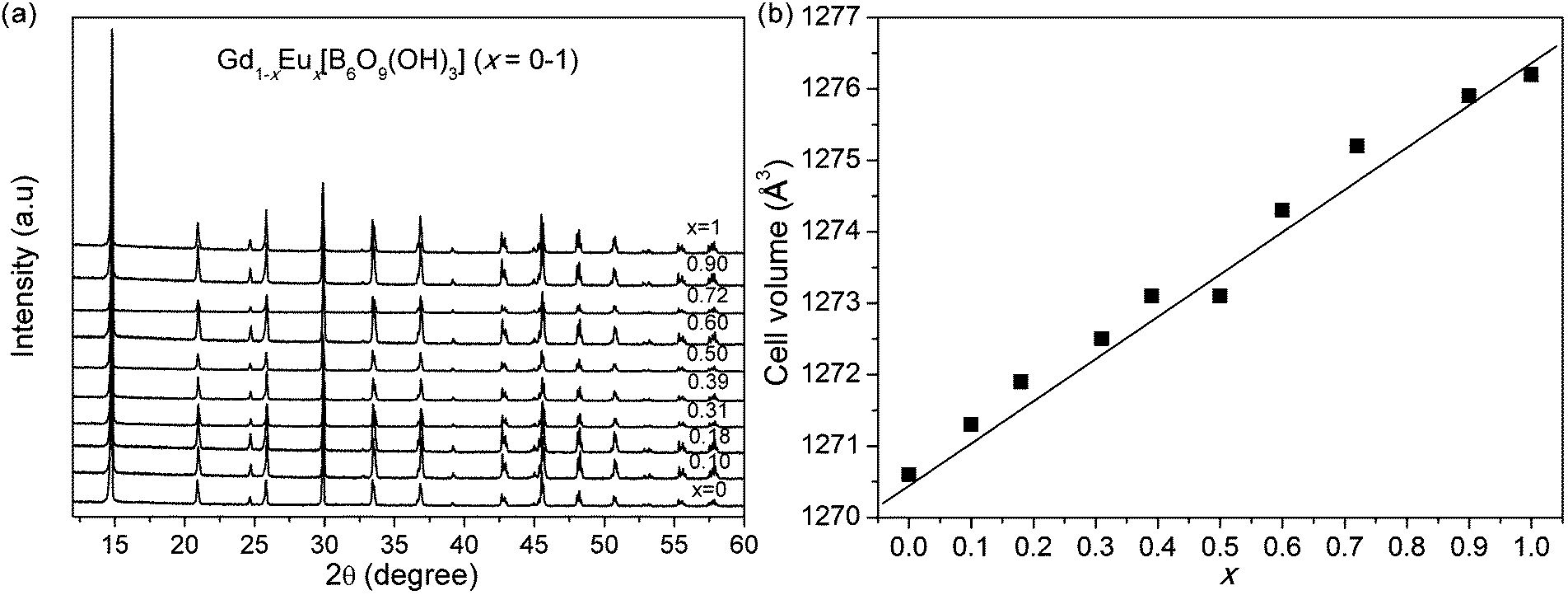 | ||
| Fig. 1 (a) X-ray diffraction patterns of Gd1−xEux[B6O9(OH)3] (x = 0–1). (b) Lattice parameters from the Le Bail fitting. | ||
| x | a (Å) | c (Å) | V (Å3) |
|---|---|---|---|
| 0 | 8.409 | 20.749 | 1270.6 |
| 0.10 | 8.411 | 20.753 | 1271.3 |
| 0.18 | 8.412 | 20.755 | 1271.9 |
| 0.31 | 8.413 | 20.759 | 1272.5 |
| 0.39 | 8.414 | 20.763 | 1273.1 |
| 0.50 | 8.415 | 20.761 | 1273.1 |
| 0.60 | 8.417 | 20.770 | 1274.3 |
| 0.72 | 8.419 | 20.773 | 1275.2 |
| 0.90 | 8.421 | 20.778 | 1275.9 |
| 1 | 8.421 | 20.781 | 1276.2 |
The powder XRD patterns of the anhydrous pentaborates α-Gd1−xEuxB5O9, prepared by both the thermal decomposition and sol–gel methods, are shown in Fig. 2. The unit cell parameters of these compounds are also obtained by Le Bail fitting using TOPAS,13 and the refined results are listed in Table 2. Fig. 3 shows the unit cell volumes against the substitution level (x) for α-Gd1−xEuxB5O9. There is a clear expanding tendency with the increasing substitution. Moreover, the cell parameters of the samples prepared by the different processes are generally consistent with each other.
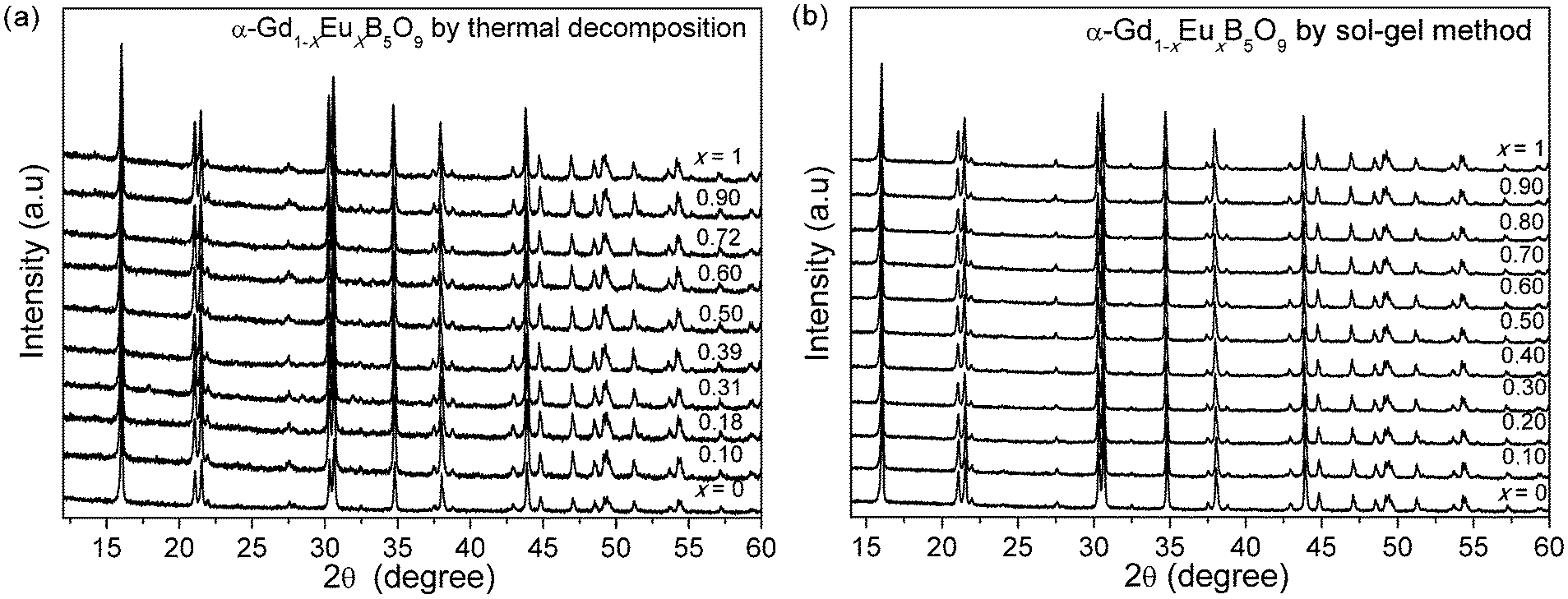 | ||
| Fig. 2 X-ray diffraction patterns of α-Gd1−xEuxB5O9 synthesized by the (a) thermal decomposition and (b) sol–gel methods. | ||
| x | a (Å) | c (Å) | V (Å3) |
|---|---|---|---|
| (a) | |||
| 0 | 8.238 | 33.645 | 2283.1 |
| 0.10 | 8.239 | 33.652 | 2284.1 |
| 0.18 | 8.241 | 33.658 | 2285.7 |
| 0.31 | 8.244 | 33.651 | 2287.0 |
| 0.39 | 8.247 | 33.654 | 2288.9 |
| 0.50 | 8.248 | 33.658 | 2289.9 |
| 0.60 | 8.248 | 33.663 | 2290.1 |
| 0.72 | 8.253 | 33.666 | 2293.2 |
| 0.90 | 8.256 | 33.672 | 2295.1 |
| 1 | 8.259 | 33.675 | 2297.0 |
| (b) | |||
| 0.0 | 8.235 | 33.657 | 2282.2 |
| 0.10 | 8.239 | 33.662 | 2285.0 |
| 0.20 | 8.241 | 33.666 | 2286.6 |
| 0.30 | 8.243 | 33.668 | 2287.7 |
| 0.40 | 8.245 | 33.675 | 2289.2 |
| 0.50 | 8.246 | 33.670 | 2289.2 |
| 0.60 | 8.248 | 33.669 | 2290.5 |
| 0.70 | 8.250 | 33.673 | 2291.6 |
| 0.80 | 8.253 | 33.678 | 2294.0 |
| 0.90 | 8.255 | 33.683 | 2295.3 |
| 1 | 8.256 | 33.678 | 2295.6 |
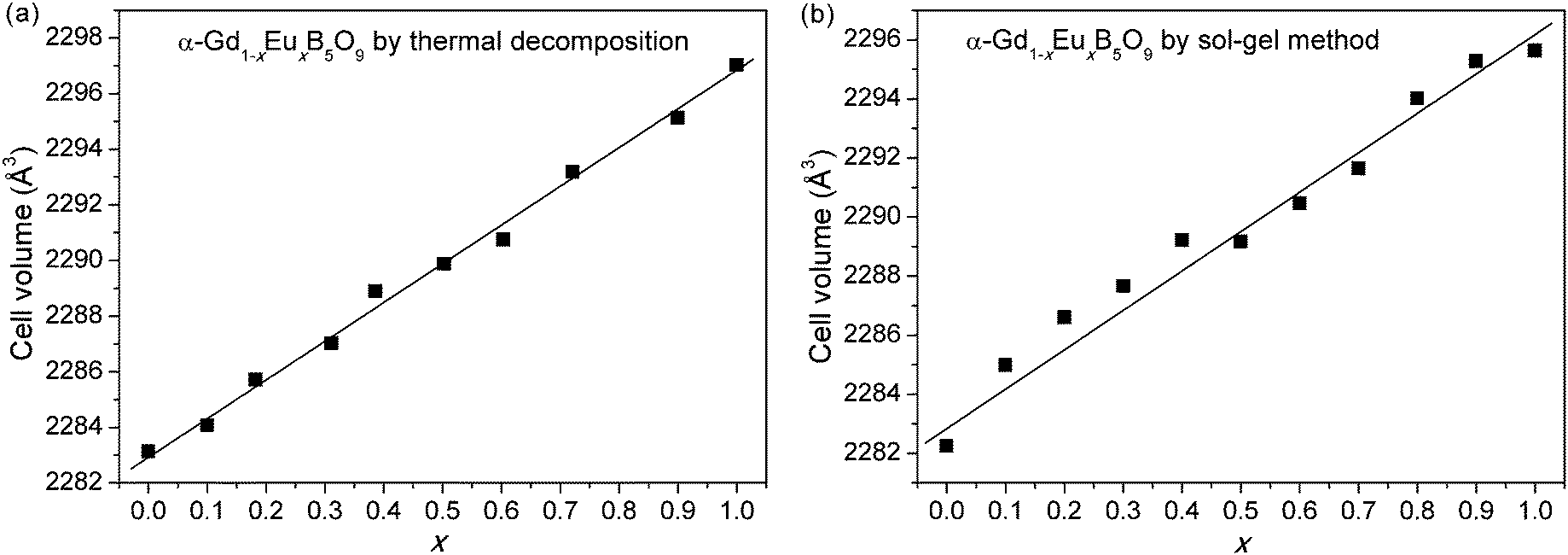 | ||
| Fig. 3 Changes in the unit cell volume for α-Gd1−xEuxB5O9 along with the doping content (x). | ||
Thermal behaviour of Gd1−xEux[B6O9(OH)3] (x = 0, 0.31, 0.72, 1)
TG-DSC analyses for Gd1−xEux[B6O9(OH)3] (x = 0, 0.31, 0.72, 1) were performed (see Fig. 4a). The thermal behavior of the as-synthesized Gd1−xEux[B6O9(OH)3] are quite similar, all of which show one sharp weight loss of dehydration from about 500 to 700 °C. There is a small weight loss (<1 wt%) from room temperature to 500 °C, which may relate to the loss of absorbed water molecules on the sample surface. From the DSC curve (Fig. 4b), this large weight loss actually comprises two endothermic processes, corresponding to two peaks at ∼578 and 630 °C for the Eu compound (see Fig. 4b). The total observed weight losses from Gd1−xEux[B6O9(OH)3] to α-Gd1−xEuxB5O9 are 6.5–6.7 wt%, which agree well with the calculated value of ∼6.6 wt%. Moreover, a small and broad exothermic band and a sharp exothermic peak are observed at about ∼733 °C and ∼797 °C, respectively. To better understand these endothermic and exothermic processes, the products obtained by heating the as-synthesized Eu[B6O9(OH)3] at different temperatures in a muffle furnace (10 hours for each step) were characterized by powder XRD (see Fig. 5). | ||
| Fig. 4 TG-DSC curves of the as-synthesized Gd1−xEux[B6O9(OH)3] (x = 0, 0.31, 0.72, 1). | ||
 | ||
| Fig. 5 X-ray diffraction patterns of Eu[B6O9(OH)3] and its calcined products at different temperatures in the furnace. | ||
The structure of Eu[B6O9(OH)3] is retained until 500 °C. At 600 °C, it is completely converted to the amorphous phase by the removal of the hydroxyl groups. Therefore, the two endothermic peaks at ∼578 and 630 °C correspond to two continuous dehydration processes. It should be noted that the structure of the parent compound apparently possesses very strong hydrogen bonds, which can only be dehydrated at a very high temperature (>550 °C). At a temperature of about 670 °C, the amorphous phase undergoes a re-crystallization to α-EuB5O9, which corresponds to the first broad exothermic band at about 733 °C on the DSC curve. The temperature difference between the DSC and heating experiments can not be neglected. We speculate that the difference comes from the different heating periods.
In the literature, the pentaborate α-LnB5O9 (Ln = Sm-Er) was first obtained by a long annealing period (650–700 °C, 5 days) using H3LnB6O12 as the precursor.6 In our case, the crystallization process of α-Gd1−xEuxB5O9 using Gd1−xEux[B6O9(OH)3] (x = 0–1) as the precursors was completed within 10 hours. Additionally, the α-Gd1−xEuxB5O9 samples remain stable below ∼780 °C and then decompose into amorphous B2O3 and α-LnB3O6, which relates to the sharp exothermic peak at 797 °C.
Luminescence properties of hydrated Gd1−xEux[B6O9(OH)3] (x = 0.10–1)
The excitation spectra of Gd1−xEux[B6O9(OH)3] (Fig. 6a) were obtained with the strongest emission at 596 nm (both of the input and output slits were set to be 2.5 nm). The broad absorption band at 220–250 nm is typical for charge transfer (CT) of O2− → Eu3+.12 In most Eu3+-doped phosphors, the CT absorption is much stronger than the f–f transitions, as the latter are parity-forbidden.14,15 However, this is the opposite in Gd1−xEux[B6O9(OH)3] (x = 0.10–1). It is known that the position of the CT band usually shifts along with the change of the local structure of Eu3+, which tends to lower the wavelength with the increasing coordination number.16,17 The positions of the CT band in the excitation spectra of the Gd1−xEux[B6O9(OH)3] (x = 0.10–1) samples are all located at relatively low wavelengths (the peak wavelength is around 225 nm, while the common CT band for other Eu3+-phosphors usually appears at ∼250 nm18,19), which is consistent with the short Gd–O distances (2.39–2.47 Å) and the high coordination number (9-coordinated).6 Moreover, the exact peak positions of the CT band show a slight red shift with the increase in Eu3+ content (as shown in Fig. 6a), indicating that the average Ln–O distance is slightly elongated. | ||
| Fig. 6 (a) Excitation and (b) emission spectra of the Gd1−xEux[B6O9(OH)3] (x = 0.10–1) samples. (c) Dependence of the emission intensity, and (d) R/O values excited at 396 nm with the doping concentration x. | ||
The sharp peaks at about 273 and 311 nm are from the 8S7/2 → 6IJ and 8S7/2 → 6P7/2 transitions of Gd3+, respectively.20,21 As a consequence, the appearance of the Gd3+ absorption indicates an energy transfer from Gd3+ to Eu3+.21 With an increase of the Eu3+ concentration, the intensities of the f–f excitation peaks of Eu3+ increase accordingly. Meanwhile, the absorptions of Gd3+ become weaker, and the intensity of the CT band does not change significantly. Gd3+ usually exhibits strong absorption in the VUV region,6,22 so it is expected that Gd1−xEux[B6O9(OH)3] samples are potential VUV-excited red phosphors (not reported in the present work).
The emission spectra of Gd1−xEux[B6O9(OH)3] (x = 0.10–1) are typical for Eu3+ (as shown in Fig. 6b). The emission spectra of the whole series of compounds with different Eu3+ concentrations generally show similar characteristic. The five emissions, at 582, 585–605, 610–640, 650–680 and 680–720 nm, are attributed to the 5D0 → 7FJ (J = 0–4) transitions.23 The 5D0 → 7F0 transition (at 582 nm) is exhibited as a single peak, because the Eu3+ ions are only present in one crystallographic site in the structure.65D0 → 7F1 (magnetic dipole–dipole transition) is stronger than 5D0 → 7F2 (electric dipole–dipole transition). The intensity ratio of R/O = I(5D0 → 7F2)/I(5D0 → 7F1) is a probe of the site symmetry of Eu3+. Generally, a lower symmetry of the crystal field around Eu3+ leads to a larger R/O.23 This result agrees with the coordination environment of Gd3+, which is close to centrosymmetric (as shown in Fig. S2, ESI,† all of the Gd–O distances are almost the same).6
When excited at 396 nm (f–f transition of Eu3+), the intensities of the 5D0 → 7F2 and 5D0 → 7F1 transitions and the total emissions increase with x, and no quenching or saturation effects are observed (Fig. 6c). This is reasonable because the Eu3+ ions are well separated and therefore diluted by borate groups (the nearest Eu–Eu distance is 6.0 Å),6 which hampers cross-relaxation or energy transfer between excited and non-excited Eu3+ ions. The values of R/O with different doping contents (x) are shown in Fig. 6d, which shows a monotonously increasing tendency from 0.27 to 0.34. Since the ionic radii of Gd3+ (1.11 Å, 9-coordinated) and Eu3+ (1.12 Å, 9-coordinated) are very similar, the two end members, Eu[B6O9(OH)3] and Gd[B6O9(OH)3], have almost the same local coordination environment for Eu3+ or Gd3+, and as a consequence, R/O shows only a small change in the Gd1−xEux[B6O9(OH)3] series.
Luminescence properties of anhydrous α-Gd1−xEuxB5O9 (x = 0.10–1)
We first discuss the luminescence properties of α-Gd1−xEuxB5O9 (x = 0.10–1) prepared by thermal decomposition. The input and output slits were selected to be 1.0 nm. As shown in Fig. 7a and b, the CT absorption is predominant. The appearance of Gd3+ absorptions at 273, 305 and 311 nm indicates that there is also energy transfer in α-Gd1−xEuxB5O9. When x = 0.60, the Gd3+ absorptions start to diminish. As shown in Fig. 7c, the 5D0–7F0 (579 nm) transition is a single peak, the 5D0–7F1 transitions (582–605 nm) are three separate sharp peaks, the 5D0–7F2 transitions (610–630 nm) also contain three peaks, with the total intensity comparable with that of the 5D0–7F1 transitions, and the 5D0–7F4 transitions (685–710 nm) contain four main peaks.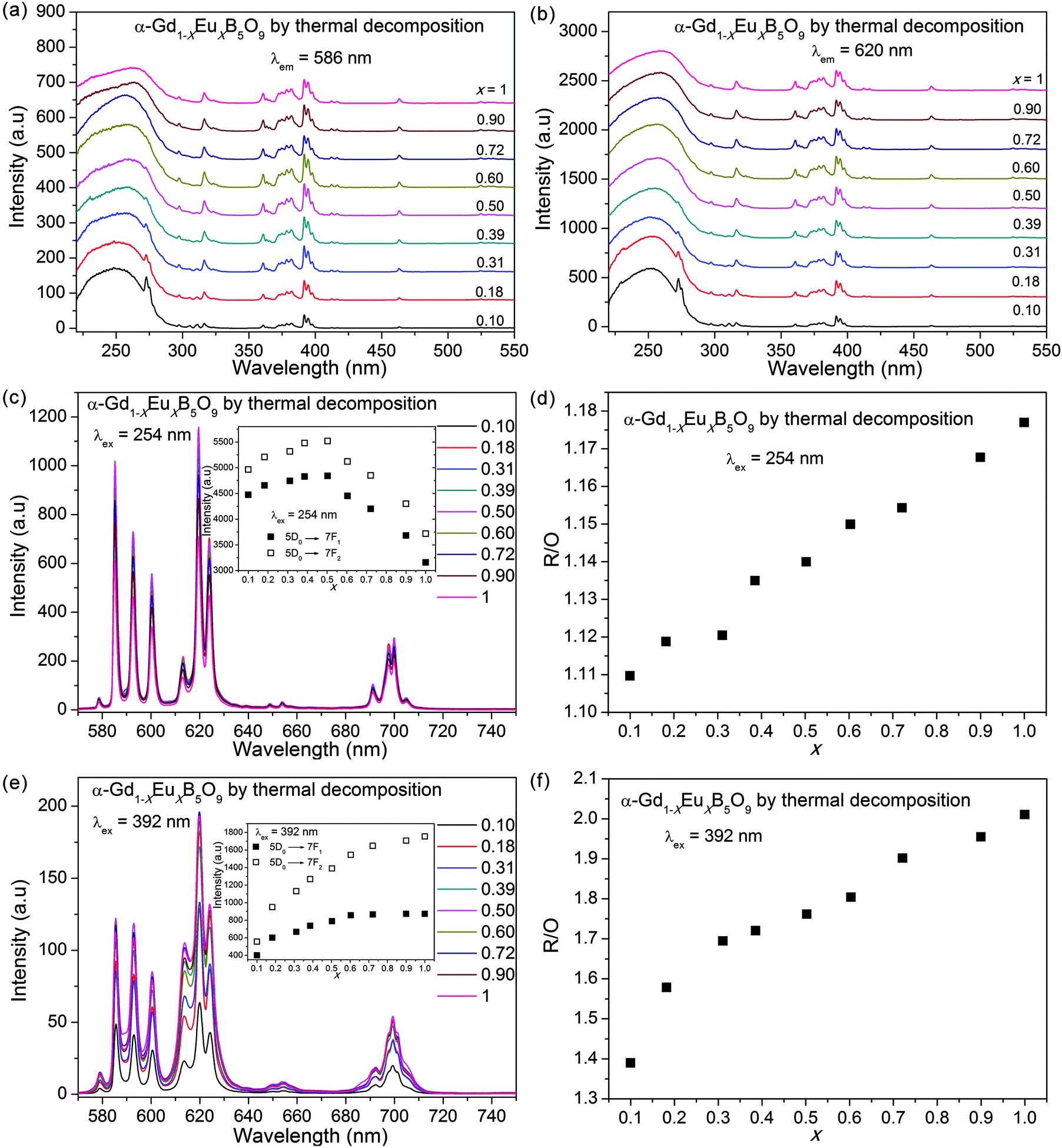 | ||
| Fig. 7 Excitation spectra of α-Gd1−xEuxB5O9, obtained by the thermal decomposition method, by monitoring at (a) 586 nm and (b) 620 nm. Emission spectra under (c) 254 nm and (e) 392 nm excitations. The insets are the calculated emission intensities. The R/O ratios are shown in (d) and (f). | ||
The influence of the Eu3+ concentration on the integrated intensity of the emissions of 5D0–7F1, 5D0–7F2 excited by 254 nm (CT of O2− → Eu3+) is shown in the inset of Fig. 7c. It is obvious that the emission first increases slowly in the range of x = 0.10–0.50, and then decreases in the subsequent range of x = 0.50–1. Usually, the saturation or quenching effect by increasing the Eu3+ concentration is mainly due to the interaction of the activated centers by the cross-relaxation or energy transfer between excited and unexcited Eu3+ ions. In α-Ln1−xEuxB5O9, the concentration quenching effect does not appear to be strong, because the Eu3+ ions are efficiently separated by large borate groups. We speculate that the decrease in the emission is due to the decreasing energy transfer from Gd3+ and Eu3+ when x increases. The values of R/O for α-Gd1−xEuxB5O9 under 254 nm excitation (see Fig. 7d) show a monotonously increasing tendency from 1.11 to 1.18.
When excited at 393 nm, the emission patterns are generally the same, as shown in Fig. 7e. The emission intensities of 5D0–7F1 and 5D0–7F2 reach maximum values, which are approximately maintained in the range of x ≥ 0.72 (inset of Fig. 7e). The calculated R/O values show a monotonously increasing tendency with x. Our study is a good comparison to the previous research of α-Gd1−xEuxB5O9 under VUV excitation, which showed a saturation at x = 0.10.6
Different synthesis methods have an impact on the luminescence intensities due to the differences in crystallinity.24–26 The excitation and emission spectra of α-Gd1−xEuxB5O9 (x = 0.10–1) prepared by the sol–gel method are shown in Fig. 8. The influence of the Eu3+ concentration on the integrated intensities of the emissions of the 5D0–7F1 and 5D0–7F2 transitions, excited at 254 nm and 392 nm, are shown in the insets of Fig. 8c and d, respectively. The CT band is much stronger and the maximum emission occurs at x = 0.40 when excited by 254 nm; meanwhile, no saturation was observed under 392 nm excitation. The emission intensities under 254 nm excitation for the samples synthesized by the sol–gel method are stronger than those prepared by thermal decomposition.
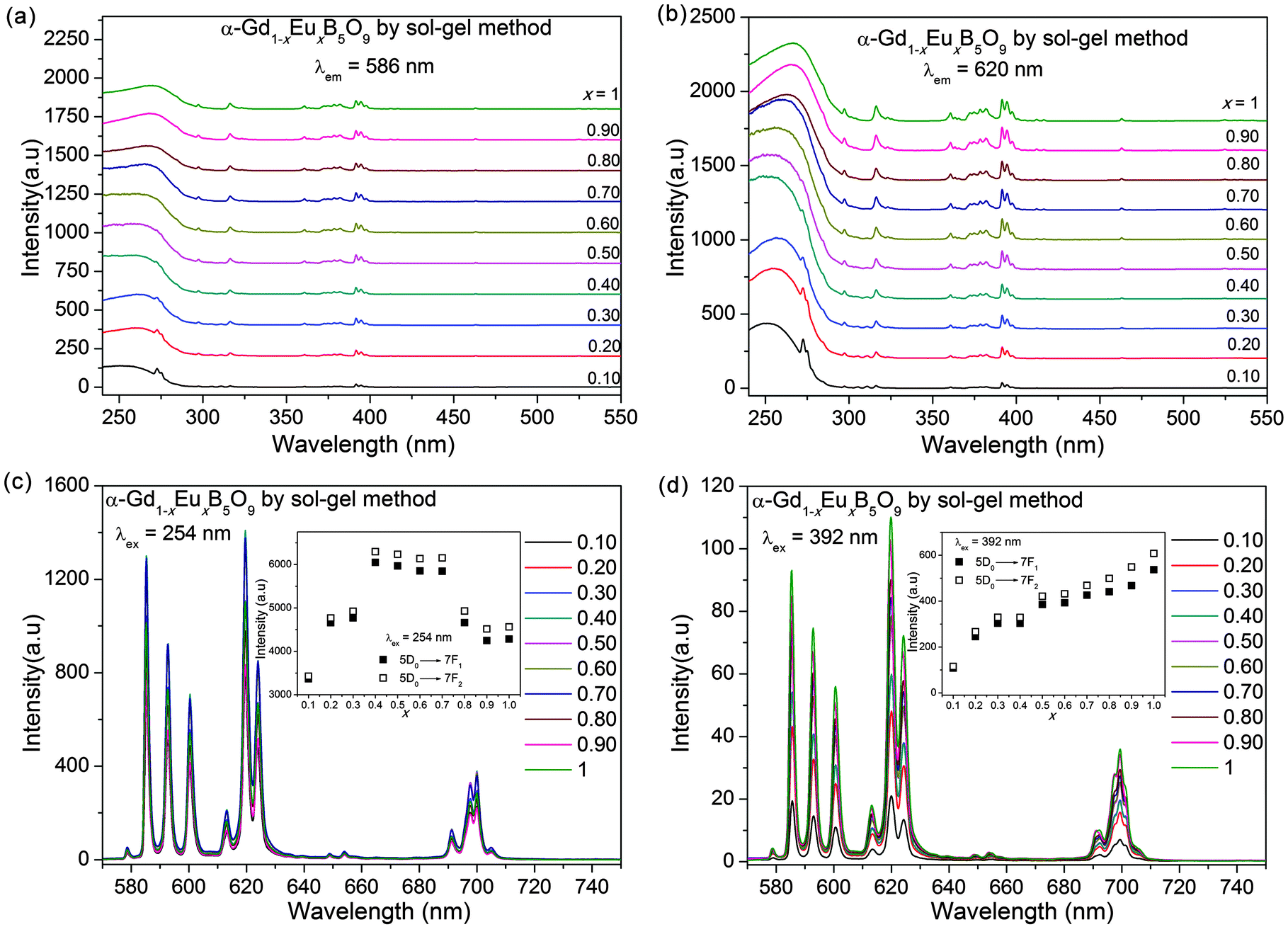 | ||
| Fig. 8 Excitation spectra of α-Gd1−xEuxB5O9 (x = 0.10–1), obtained by the sol–gel method, by monitoring at (a) 586 nm and (b) 620 nm. Emission spectra under (c) 254 nm or (d) 392 nm excitations. The insets are the calculated emission intensities. | ||
Structural evolution detected by the luminescence of Eu3+
Hydrous borates usually show rich phase transitions, including dehydration and amorphization, re-crystallization and further decomposition, as in the case of Ln2B6O10(OH)4·H2O.12 During the phase transitions, the luminescence of Eu3+ is a good medium to gain more structural information about the coordination environment of rare-earth cations, even for the intermediate amorphous phase, which can not be studied by regular XRD. Careful investigation of the thermal behavior of Eu[B6O9(OH)3] by TG-DSC and powder XRD experiments suggest that there is a dehydration/amorphization step which starts at ∼500 °C, and the re-crystallization of α-EuB5O9 at 670 °C in the furnace, which further decomposes into α-EuB3O6 at 820 °C. Therefore, it is of interest to study the luminescence properties of Eu[B6O9(OH)3] and its annealed products during these phase transitions (see Fig. 9). The similar excitation and emission spectra of Eu[B6O9(OH)3] (calcined at 300 °C) and the as-synthesized Eu[B6O9(OH)3] (25 °C) indicate that the coordination environment of Eu3+ remains unchanged. The R/O value also does not change, as shown in Fig. 9e and f, confirming that the local structure around Eu3+ is maintained. | ||
| Fig. 9 For the as-synthesized Eu[B6O9(OH)3] and its annealed products at different temperatures: (a) excitation spectra (λem ∼ 590 nm); (b) excitation spectra (λem ∼ 620 nm); (c) emission spectra (λex ∼ 254 nm); (d) emission spectra (λex ∼ 396 nm); (e) and (f) are the corresponding R/O values deduced from (c) and (d). | ||
At 500 °C, the stepwise dehydration of Eu[B6O9(OH)3] starts. As shown in Fig. 9a and b, the excitation spectra monitoring the strongest emissions of the 5D0–7F1 and 5D0–7F2 transitions are slightly different. When monitoring at 596 nm, the excitation spectrum is similar to that of the sample calcined at 300 °C, however, if monitoring at 621 nm, the intensity of the CT band is significantly enhanced by the calcination at 500 °C. As a result, the emission spectra of the sample calcined at 500 °C, which excited by CT and at 393 nm are therefore different, as shown in Fig. 9c and d. When excited by a CT transition, the emission of the 5D0–7F2 transition becomes stronger than that of the 5D0–7F1 transition, and moreover, the R/O value increases at the same time. If excited at 396 nm, the emission spectrum remains unchanged, and the R/O value is also maintained. According to its interesting and mixed luminescence behavior, we speculate that the sample calcined at 500 °C partially decomposes and contains a substantial amorphous component, which can not be clearly detected by XRD.
At 600 °C, the crystal structure collapses completely and an amorphous phase is obtained. Interestingly, the emission intensity was enhanced after amorphization when excited by CT or at 393 nm (Fig. 9c and d), which is also confirmed by the increased intensities of both CT and Eu3+ f–f transitions in the excitation spectrum after amorphization. Moreover, the emission spectrum shows an intense red emission at 616 nm and the R/O reaches a maximum value, indicating the lowest symmetry of the local structure around the Eu3+ ions. The high efficiency of the f–f transitions and the large R/O value (3.7 under 254 nm and 4.0 under 393 nm) implies that this amorphous phase may be a good red-emitting UV-LED phosphor.
At 670 °C, the amorphous phase undergoes re-crystallization to anhydrous α-EuB5O9. The inset of Fig. 9c shows the CT-excited emission spectra of the amorphous phase, α-EuB5O9 and α-EuB3O6. A significant increase in the emission intensity and a red-shift of the strongest emission peak are found during the re-crystallization. However, when excited by the Eu3+ f–f transition, the luminescence intensity of α-EuB5O9 is close to the amorphous phase. These results are consistent with the significantly increased intensity of the CT absorption and the almost unchanged Eu3+ f–f transitions in the excitation spectrum. The R/O value decreases after re-crystallization. In addition, α-EuB5O9 has a low temperature stable phase, which begins to decompose at about 780 °C and completely decomposes to α-EuB3O6 at 820 °C. Thus, we also collected the emission spectrum for α-EuB3O6 calcined at 820 °C, and found that the luminescence intensity decreases (Fig. 9c and d). Moreover, the R/O values are even smaller than that of α-EuB5O9. All of these results confirm the abundant structural evolutions during the thermal annealing.
Conclusions
In conclusion, complete solid solutions of Gd1−xEux[B6O9(OH)3] were prepared by the boric acid flux method. Careful investigations of the thermal behavior of Eu[B6O9(OH)3] by TG-DSC and powder XRD experiments reveal that there is a dehydration/amorphization step which starts at ∼500 °C, re-crystallization of α-EuB5O9 at 670 °C in the furnace, which further decomposes into α-EuB3O6 at 820 °C. Accordingly, complete solid solutions of anhydrous α-Gd1−xEuxB5O9 were prepared by thermal decomposition. In addition, the sol–gel method was also applied to obtain pure powder samples of α-Gd1−xEuxB5O9 (x = 0–1). The sol–gel method is favored for enhancing the luminescence intensity. Efficient energy transfer from Gd3+ to Eu3+ was observed for all of the compounds, as expected. Interestingly, by studying the luminescence of Eu3+, we found that the coordination symmetry of Eu3+ is the lowest in the amorphous state during the annealing treatments of Gd1−xEux[B6O9(OH)3], as indicated by the highest R/O value of 4.0. The high efficiency of the f–f excitations and the large R/O value imply that this amorphous phase may be a good red-emitting UV-LED phosphor. Our study of the luminescence properties of the hydrated borates and dehydrated products complements the traditional research of anhydrous borates. In most cases, the hydrate borates have large borate groups, which separate the luminescent activator very well and therefore avoid the concentration quenching. Many hydrated polyborates with crystalline water molecules remain unexplored, and our study shows their potential as good phosphors.Acknowledgements
This work was supported by the Nature Science Foundation of China (NSFC 21101175, 21171178, 91222106). Financial support from the Natural Science Foundation Project of Chongqing (CSTC 2012jjA0438) is also acknowledged.Notes and references
- B. Saubat, C. Fouassier, P. Hagenmuller and J. C. Bourcet, Mater. Res. Bull., 1981, 16, 193–198 CrossRef CAS.
- K. Machida, G. Adachi and J. Shiokawa, J. Lumin., 1979, 21, 101–110 CrossRef CAS.
- W. Z. Pei, Q. Su and J. Y. Zhang, J. Alloys Compd., 1993, 198, 51–53 CrossRef.
- G. K. Abdyllayev, G. G. Dzhafarov and K. S. Mamedov, Kristallografiya, 1984, 29, 1084–1088 Search PubMed.
- E. L. Belokoneva, S. Y. Stefanovich, O. V. Dimitrova and A. G. Ivanova, Russ. J. Inorg. Chem., 2002, 47, 317–323 Search PubMed.
- L. Y. Li, P. C. Lu, Y. Y. Wang, X. L. Jin, G. B. Li, Y. X. Wang, L. P. You and J. H. Lin, Chem. Mater., 2002, 14, 4963–4968 CrossRef CAS.
- L. Y. Li, X. L. Jin, G. B. Li, Y. X. Wang, F. H. Liao, G. Q. Yao and J. H. Lin, Chem. Mater., 2003, 15, 2253–2260 CrossRef CAS.
- A. G. Ivanova, E. L. Belokoneva and O. V. Dimitrova, Russ. J. Inorg. Chem., 2004, 49, 816–822 Search PubMed.
- E. L. Belokoneva, A. G. Ivanova and O. V. Dimitrova, Russ. J. Inorg. Chem., 2006, 51, 869–877 CrossRef.
- A. G. Ivanova, E. L. Belokoneva, O. V. Dimitrova and N. N. Mochenova, Russ. J. Inorg. Chem., 2006, 51, 584–588 CAS.
- A. G. Ivanova, E. L. Belokoneva, O. V. Dimitrova and N. N. Mochenova, Russ. J. Inorg. Chem., 2006, 51, 862–868 CrossRef.
- R. H. Cong, T. Yang, Z. M. Wang, J. L. Sun, F. H. Liao, Y. X. Wang and J. H. Lin, Inorg. Chem., 2011, 50, 1767–1774 CrossRef CAS PubMed.
- TOPAS, V4.1-beta, Bruker AXS, Karlsruhe, Germany, 2004 Search PubMed.
- J. H. Lin, S. Zhou, L. Q. Yang, G. Q. Yao, M. Z. Su and L. P. You, J. Solid State Chem., 1997, 134, 158–163 CrossRef CAS.
- J. Thakur, D. P. Dutta, H. Bagla and A. K. Tyagi, J. Am. Ceram. Soc., 2012, 95, 696–704 CrossRef CAS.
- G. Blasse, J. Solid State Chem., 1972, 4, 52–54 CrossRef CAS.
- H. E. Hoefdraad, J. Solid State Chem., 1975, 15, 175–177 CrossRef CAS.
- F. W. Tian, C. Fouassier and P. Hagenmuller, Mater. Res. Bull., 1987, 22, 899–909 CrossRef.
- X. X. Li, Y. H. Wang and Z. Chen, J. Alloys Compd., 2008, 453, 392–394 CrossRef CAS PubMed.
- J. G. Wang, X. P. Jing, C. H. Yan and J. H. Lin, J. Electrochem. Soc., 2005, 152, G186–G188 CrossRef CAS PubMed.
- F. T. You, Z. Yang, P. C. Lu, Y. X. Wang, J. H. Lin and T. Yang, High Energy Phys. Nucl. Phys., 2001, 25, 70–74 Search PubMed.
- R. T. Wegh, H. Donker, A. Meijerink, R. J. Lamminmäki and J. Hölsä, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13841–13848 CrossRef CAS.
- Z. Yang, J. H. Lin, M. Z. Su and L. P. You, Mater. Res. Bull., 2000, 35, 2173–2182 CrossRef CAS.
- A. E. Henkes and R. E. Schaak, J. Solid State Chem., 2008, 181, 3264–3268 CrossRef CAS PubMed.
- Y. P. Li, J. H. Zhang, X. Zhang, Y. S. Luo, S. Z. Lu, X. G. Ren, W. J. Wang, L. D. Sun and C. H. Yan, Chem. Mater., 2009, 21, 468–475 CrossRef CAS.
- X. C. Jiang, C. H. Yan, L. D. Sun, Z. G. Wei and C. S. Liao, J. Solid State Chem., 2003, 175, 245–251 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Le Bail fitting of the powder XRD patterns for Gd[B6O9(OH)3] and α-GdB5O9, crystal structure view of Gd[B6O9(OH)3]. See DOI: 10.1039/c3nj00925d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |