Luminescence and photocatalytic studies of Sm3+ ion doped SnO2 nanoparticles†
L. P.
Singh
a,
M. Niraj
Luwang
*b and
S. K.
Srivastava
*a
aDepartment of Chemistry, Manipur University, Imphal-795003, India. E-mail: sksrivastava01@yahoo.co.in; Fax: +91 385 2435831; Tel: +91 385 2435093
bChemical Engineering and Process Development Division, National Chemical Laboratory, Pune-411008, India. E-mail: mn.luwang@ncl.res.in; Fax: +91 20 25902621; Tel: +91 20 25902950
First published on 25th September 2013
Abstract
Sm3+ ion doped tetragonal SnO2 nanoparticles have been synthesized by the polyol method and its concentration and annealing effects on photoluminescence properties are studied. The XRD results show the changes of lattice parameters with the incorporation of the Sm3+ ion into the SnO2 host lattices. Fourier transform infrared (FTIR) data indicate the formation of a Sn–O bond and capping of the nanoparticles by ethylene glycol. The broad emission peaks observed around 350 to 550 nm are attributed to the emission from defects or traps present in the host SnO2. The typical emission peaks of the Sm3+ ion are observed and there is efficient energy transfer between the host, SnO2 and the dopant, Sm3+ ion. Further, the prepared samples are experimented towards the photocatalytic behavior on selected triphenylmethane dyes, Ethyl Violet (EV) and Brilliant Green (BG). The degradation of dyes was found to be enhancing in the presence of Sm3+ ions as it suppress the e−CB and h+VB recombination.
Introduction
SnO2 is an n-type semiconductor with band gap of Eg = 3.6 eV at 300 K and is used in various fields like gas-sensors, solar cells, catalysis, lithium batteries, optoelectronic devices and magnetic property applications, etc.1–6 The shape and size of the nanoparticles can be controlled by using a capping agent. Lanthanide doped semiconductor nanomaterials have a great deal of interest due to their unique optical properties and potential application in fields like fluorescent lamps, optical communication and flat panel displays.7–9 Amongst the lanthanides, samarium is a very active member having interesting properties. Due to its fluorescence property, it has applications in high-density optical storage, under sea communication and color displays and its emitting 4G5/2 level exhibits relatively high quantum efficiency.10 Because of its multiple energy-level structure and high fluorescence efficiency this lanthanide usually acts as a great emitting center as well as a red-emission center.7,11There are various synthetic methods for lanthanide doped and undoped SnO2 nanoparticles such as hydrothermal,12 precipitation,13 sol–gel,14 vapour transport method,15 microwave method16 and microemulsion.17
Nowadays, the textile industry of dyes and pigments is expanding worldwide, mainly in developing countries. Dyes and pigments are used in industry for coloring of cloth, cotton, paper, leather, wool, silk and nylon. Dyes are normally in a group of complex organic materials based fundamentally on the chromospheres structure. Wastewater containing the dyes is usually toxic, resistant to biodegradation, persistent in the environment, and difficult to be treated by general methods. As a consequence, there is a continuous increase in environmental contamination and global concerns of discharge of dyestuff effluents into the aquatic system.18 The dyeing wastewater discharge not only affects the environment but also reduces light penetration through the water surface needed for photosynthesis activity of aquatic organisms. In order to minimise such accidents in the environment, semiconductor photocatalysis has become more and more attractive and important.19 In the photocatalytic process, the semiconducting material absorbs UV light energy more than or equal to the energy gap, consequently generating the holes and electrons, which further give rise to oxidation and reduction of organic dyes.20 In the photocatalysis process, recombination of electron and hole plays a very important role. In order to perform photocatalysis successfully we have to reduce the recombination of electron and hole, to enhance the photocatalysis performance. Some techniques like rare-earth (or metal ion) doping of semiconductors or deposition (or coupling) of metal oxides to semiconductors can reduce (delay) the recombination of charges (electron and hole).21–23 But not a lot of work has been reported using lanthanide doped SnO2 nanoparticles. In this work, we have studied the photoluminescence properties of Sm3+ ion doped SnO2 nanoparticles and their application towards the degradation (photocatalysis) of the dyes, Ethyl Violet (EV) and Brilliant Green (BG). The detailed mechanism of degradation of dyes by pure and doped SnO2 nanoparticles has also been elaborated.
Experimental section
Materials
Tin metal (99.9%), samarium(III) nitrate hydrate, Sm(NO3)3·xH2O (99.9%) from Sigma-Aldrich, ethylene glycol, dyes (EV and BG) from Merck, ammonia, methanol and hydrochloric acid were used as received without further purification. Double distilled water was used throughout the experiment.Synthesis
Pure SnO2 and SmxSn1−xO2 (x = 0.02, 0.05, 0.07 and 0.10) nanoparticles were synthesized by the ethylene glycol route. In a typical synthesis procedure, stoichiometric amounts of the precursor of Sm3+ ions and tin metal were dissolved in a minimum quantity of conc. HCl to get a clear solution. The solution was evaporated five times by alternating addition of distilled water so that the excess HCl gets evaporated out. The resulting transparent solution was allowed to mix with 50 ml of ethylene glycol and heated in a round bottom flask at 40 °C for 30 minutes. Ammonia solution (stoichiometric amount) was added to the above transparent solution and was allowed to reflux at 120 °C for 2 h. The resulting white precipitate was collected by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm after washing with methanol. The precipitate was annealed at different temperatures for 2 h respectively.
000 rpm after washing with methanol. The precipitate was annealed at different temperatures for 2 h respectively.
Characterization
A Philips X-Ray diffractometer (PW 1071) with Cukα (1.5405 Å) radiation having a Ni filter was used for X-ray diffraction (XRD) study. All patterns were recorded over the angular range 10 ≤ 2θ/deg ≤ 90 with a step size of Δ2θ = 0.02. The powder samples were ground and dispersed in methanol on a glass slide and allowed to dry. The average crystallite size (t) was calculated using the Debye–Scherrer relationship, t = 0.9λ/β cosθ, where λ is the wavelength of the X-ray and β is the full width at half maximum (FWHM). The lattice parameters were calculated from the least square fitting to the diffraction peaks.
Fourier transform infrared (FTIR) spectra were recorded using an FTIR-8400S (206-72400) Shimadzu Spectrometer. Powder samples were studied by making thin pellets with KBr. A JEOL 2000 FX transmission electron microscope was used for recording TEM images. For the TEM measurements, the powder samples were ground and dispersed in methanol. A drop of the dispersed particles was put over the carbon coated copper grid and evaporated to dryness at room temperature. All the UV-visible measurements were done in a Shimadzu 2450 UV-Visible Spectrophotometer. The BET surface area measurement was performed at 77 K on a Quantachrome Instrument (V 5.02). All the luminescence spectra were recorded using a Perkin Elmer LS-55 spectrophotometer. Powder samples were dispersed in methanol and spread over the quartz slide and dried at room temperature.
Photocatalysis experiment
In the photocatalysis process (degradation of dyes), 0.233 g of the catalyst (pure SnO2 or Sm3+ ion doped SnO2 nanoparticles) was dispersed in 10 ml of deionized water and sonicated for 1 hour. Different concentrations of the dispersed nanomaterials were mixed with 1 ml of the dye solution (EV or BG) having a concentration of 2.5 × 10−4 M, as summarized in Table S1 (see ESI†). The solution mixture of the catalyst and the dye were kept in the dark prior to the excitation in order to achieve maximum adsorption of the dye on the surface of the catalyst. During the illumination time no volatility of the solution was observed.The solution mixture was irradiated with 260 nm of UV light inside a closed chamber and kept for 10 minutes. After irradiating, the catalyst was removed by centrifugation and the absorbance of the filtrate was measured. The changes in the concentration of dyes were monitored from their characteristic absorption (λmax) at 593 (EV) and 625 nm (BG) using a UV-Visible spectrophotometer. The absorbance at their respective λmax of the dyes represents the aromatic part of the dyes and its decrease of absorbance indicates the degradation of dye.
The UV illumination is obtained by using a self-deozonating 150 W Xe lamp along with a stigmatic concave diffraction grating monochromator to get the desired wavelength. The slit width was kept constant at 2.5 for all the illumination processes.
Results and discussion
XRD patterns of the pure powder and 5 at% Sm3+ ion doped SnO2 nanoparticles (as-prepared, 500 and 900 °C) are shown in Fig. 1(a and b). All the XRD patterns of the annealed samples could be indexed on the basis of a tetragonal system reported for SnO2 (JCPDS Card No. 41-1445) with lattice parameters a = 4.73 Å and c = 3.18 Å. The broad peaks of the as-prepared sample indicate the low crystallinity of the sample and the crystallinity of the sample increases with the increase in annealing temperature. The lattice parameters for undoped pure SnO2 prepared at 500 °C are a = 4.70 Å and c = 3.19 Å. In the case of Sm3+ doped SnO2 nanoparticles annealed at 500 °C, the lattice parameters are a = 4.71 Å and c = 3.18 Å and those for the 900 °C annealed sample are a = 4.69 Å and c = 3.19 Å indicating that Sm3+ ions had incorporated into the SnO2 lattice. As ionic radius of the Sm (RSm3+ = 0.1079 nm) is greater than the ionic radius of the Sn (RSn4+ = 0.076 nm),24,25 it is expected that the lattice undergoes significant change with Sm3+ incorporation. In previous studies,26,27 only small amount of lanthanides could be incorporated into the SnO2 lattice. The crystalline size calculated using the Debye–Scherer equation from the plane (110) are found to be 20 and 34 nm for pure SnO2 annealed at 500 and 900 °C and 20 and 35 nm for doped SnO2 nanoparticles annealed at 500 and 900 °C.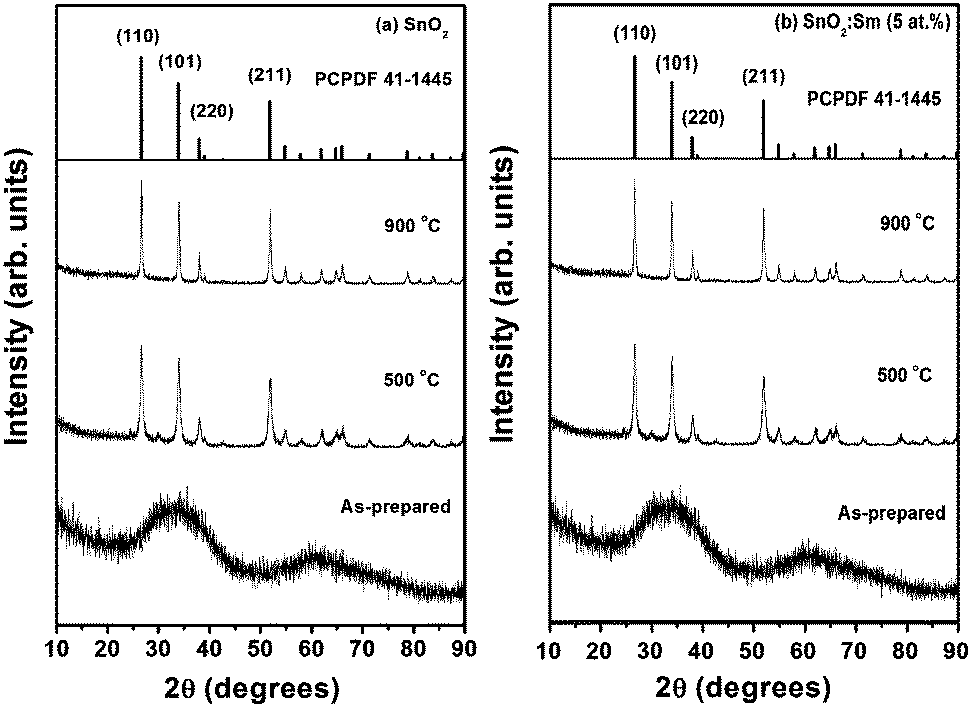 | ||
| Fig. 1 XRD patterns of (a) pure SnO2 and (b) 5 at% Sm3+ doped SnO2 nanoparticles at different annealing temperatures (as-prepared, 500 and 900 °C). | ||
Fig. 2 shows the FTIR spectra of Sm3+ (5 at%) doped SnO2 nanoparticles at different annealing temperatures (as prepared, 500 and 900 °C). Distinctly, a broadband in the range 400–750 cm−1 (580 cm−1) can be observed for all the samples under study. This peak is due to the vibration of the Sn–O bond.28 With the increase in the annealing temperature, the intensity of the Sn–O band increases indicating the crystallisation of the SnO2 particles which is also confirmed by the XRD study. There are also OH− peaks at 1635 cm−1 (bending vibration) and a broad peak at 3453 cm−1 (stretching vibration) which could be observed in the samples, vibration corresponding to C–H are observed in the region of 2700–2900 cm−1 (ref. 29) indicating the prepared samples were well capped by the ethylene glycol used during the synthesis of the nanoparticles, which is absent at the higher annealing temperature, which indicates the evaporation of the capping agent.
 | ||
| Fig. 2 FTIR spectra of SnO2:Sm3+ (5 at%) at different annealing temperatures (as-prepared, 500 and 900 °C). | ||
Fig. 3 shows the microscopy structure of the as-prepared sample of Sm3+ (5 at%) doped SnO2. The particles are spherical in shape. The sizes are 20 nm in range which is well in agreement with the calculation from XRD. The SAED shows the low crystallinity of the sample as also indicated from the XRD data. The as prepared samples have a high content of water molecules. Highly crystalline SnO2 nanomaterials are observed on annealing at higher temperatures because of the evaporation of water and organic moiety.
 | ||
| Fig. 3 (a) TEM images and (b) SAED of annealed samples of SnO2:Sm3+ (5 at%). | ||
Fig. 4a shows the emission spectrum of the pure SnO2 nanoparticles for different annealing temperatures excited at 300 nm (absorption peak of SnO2).30 A broad peak ranging from 350–550 nm is observed, which is related to the emission from defects (oxygen vacancy) or traps present in the host SnO2.24,31 The peak observed around 420 nm is related with the emission from defects or traps present in the host responsible for the luminescence. The intensity of the broad peak increases with the increase in annealing temperature, indicating the loss of the luminescence quencher hydroxyl ion (OH−) present in the sample.
 | ||
| Fig. 4 Emission spectra of (a) pure SnO2 and (b) SnO2:Sm3+ (5 at%) nanoparticles. | ||
In the excitation spectrum of Sm3+ ion doped SnO2 nanoparticles (Fig. S1, see ESI†), monitored at an emission wavelength of 4G5/2 → 6H7/2 (600 nm) of the Sm3+ ion, the whole spectrum can be divided into two regions. The peaks in the region 220–300 nm relate to the host absorption.32 The peak at 260 nm is related to the O−2 to Sm3+ charge transfer transition. The other regions from 300–450 nm is related to the f–f transition of Sm3+ ions. The peaks at 365, 406 and 425 nm can be assigned to the 6H5/2 → 4D15/2, 6H5/2 → 4K11/2 and 6H5/2 → 4M19/2 transitions of the Sm3+ ion.32
The emission spectrum of Sm3+ doped SnO2 nanoparticles (Fig. 4b) show the typical emission of the Sm3+ ion. The three sharp peaks observed at 562, 600 and 644 nm can be assigned to the 6G5/2 → 6H5/2, 6G5/2 → 6H7/2 (magnetic dipole) and 6H5/2 → 4H9/2 (electric dipole) transition of the Sm3+ ion.32,33 The predominance of 600 nm i.e. magnetic dipole transition indicates that in our prepared samples, the Sm3+ ions occupy Sn4+ sites in the SnO2 lattice (having D2h or C2h symmetry) except magnetic dipole allowed transitions, all other transitions are forbidden.24
Fig. 5 shows the emission spectra of the 5 at% Sm3+ doped SnO2 nanoparticles at different annealing temperatures as-prepared, 500 and 900 °C (Fig. 5a) and at different concentrations of Sm3+ (2, 5, 7 and 10 at%) at 900 °C (Fig. 5b) excited at 300 nm. The intensity of the peaks increases with the increase of annealing temperature from as-prepared to 900 °C, which is related with the loss of −OH/dangling bonds present on the surface of the nanoparticles which acts as luminescence quencher through multiphonon relaxation.34–36 The emission peaks at 562, 600 and 644 nm, correspond to the 6G5/2 → 6H5/2, 6G5/2 → 6H7/2 and 6H5/2 → 4H9/2 transitions of Sm3+ ions. The magnetic dipole mechanism plays an important role in 6G5/2 → 6H7/2 transition. The transition 6G5/2 → 6H7/2 also contains some part of magnetic dipole component caused by the interaction between the Sm3+ ion and the magnetic field component. The 6G5/2 → 6H7/2 and 6H5/2 → 4H9/2 transitions are mainly induced by electric dipoles. The 6G5/2 → 6H7/2 electric-dipole transition is very sensitive to the local environment around Sm3+. The f–f electric dipole transitions are forbidden in nature and can be raised owing to the mixture of the 4fN transitional states with opposite-parity configurations.37 The intensity of the emissions peaks increases with the increase in the concentration of the Sm3+ ion from 2 to 10 at% (Fig. 5b) which is due to the increase in the number of active absorbing centers (Sm3+ ions). As the Sm3+ ion itself doesn't have an absorption peak around 300 nm, the observance of the emission spectra corresponding to the various transitions of Sm3+ ions when excited at 300 nm (which is the band edge absorption of SnO2) indicates that there is a good energy transfer from the host, SnO2 to the dopant, Sm3+ ion. Energy transfer occurs when the emission of the donor is equal to the absorption of the acceptor in resonance condition, as 420 nm emission of SnO2 is due to the defect levels and is almost overlapping the excitation peak of the Sm3+ ion at 406 nm, so energy transfer is possible from SnO2 to Sm3+ ions. The process of energy transfer is shown in Scheme S1 (see ESI†). The incoming radiation is absorbed by SnO2 nanoparticles and transferred to the Sm3+ ion and then the excited Sm3+ ion emits the characteristic emission peak of the Sm3+ ion at 652, 600 and 644 nm.
 | ||
| Fig. 5 Emission spectra of SnO2:Sm3+ (5 at%) nanoparticles at (a) different annealing temperatures and (b) at different doping concentrations (2, 5, 7, 10 at%). | ||
Fig. 6 shows the photocatalytic behavior of as-prepared pure (Fig. 6a and b) and Sm3+ (5 at%) doped (Fig. 6c and d) SnO2 nanoparticles on the degradation reaction of the selected dyes. The maximum absorption (λmax) of the ethyl violet dye is found to be 593 nm and that of BG is 625 nm. The intensity of the dyes decreases with the addition of SnO2 nanoparticles when irradiated indicating the decolourisation of dyes in the presence of the pure and doped SnO2 (catalyst), the decolourisation of dyes increase with the increase of concentration of catalyst. The phenomenon of photocatalysis can be explained as follows. By applying UV-visible light irradiation with a photon of energy equal to or higher than the band gap energy (3.6 eV), the SnO2 catalyst generates electron–hole pairs with free electrons in the empty conduction band. The electron–hole pairs thus produced, can go across the band gap to the conduction band, and holes (h+) stay in the valence band as formulated in eqn (i). In the presence of aqueous solution, the h+ is scavenged by surface hydroxyl groups or H2O molecules to produce highly reactive and non hydroxyl radicals (OH˙), as shown in eqn (ii) and (iii). Then, the photo-generated electrons in the conduction band could react with O2 acceptors to produce a superoxide radical anion (O2−˙) of oxygen in eqn (iv). The OH˙ are considered to be the dominant oxidizing agent contributing to the destruction of organic dyes.38 Degradation mechanisms of the dye molecules under the contact with light irradiation and photocatalyst are summarized in eqn (v)–(vii).18
| SnO2 + hν → SnO2 (e−CB + h+VB) | (i) |
| SnO2 (h+VB) + H2O → SnO2 + H+ + OH˙ | (ii) |
| SnO2 (h+VB) + OH− → SnO2 + OH˙ | (iii) |
| SnO2 (e−CB) + O2 → SnO2 + O2−˙ | (iv) |
| Dye + OH˙ → degradation products | (v) |
| Dye + h+VB → oxidation products | (vi) |
| Dye + e−CB → reduction products | (vii) |
 | ||
| Fig. 6 Photocatalysis of dyes (EV and BG) in presence of (a, b) pure SnO2 and (c, d) SnO2:Sm3+ (5 at%) nanoparticles. | ||
From the above discussion we observed that e−CB and h+VB are responsible for photocatalysis of dyes. The e−CB and h+VB have a chance for recombination which reduces the efficiency of the photocatalyst or undergo redox reaction (eqn (iii) and (iv)) which increase the efficiency of photocatalyst by reactions with any organics dyes which adsorbed on the surface of SnO2 through weak van der Waals forces in dye contaminated waters,39 and consequently decomposed to harmless products. The introduction of lanthanide/metal ions reduced the width of the band gap energy levels between the conduction band and valence band allows the catalyst to be active under visible light.40 When lanthanide is introduced as a doping agent for SnO2, these energy levels can be incorporated into the band gap of SnO2. These new energy levels can either accept electrons from the valence band or donate electrons to the conduction band. Owing to the lower energy separation between the new energy levels and the valence band or conduction band and thereby suppressing the e−CB and h+VB, recombination and efficiency of photocatalysis increase. Moreover oxygen vacancy found in photoluminescence studies also increases the degradation of dyes which acted as an electron trap and delay the recombination of electron–hole pairs.41 Therefore, the degradation efficiency of dyes increase in the case of Sm3+ ion doped than undoped SnO2 nanoparticles (Fig. 7a and b).
 | ||
| Fig. 7 Absorbance spectra of dyes (a) EV and (b) BG in presence of Sm3+ doped and undoped SnO2 nanomaterials. | ||
BET surface area measurements were performed in order to get a better understanding of the surface active site of the catalyst for the photodegradation of the dyes under study. The specific surface areas are found to be 137.41 and 167.56 m2 g−1 for pure SnO2 and Sm3+ doped SnO2 nanomaterials respectively. The increase in the surface area of the Sm3+ doped materials further confirms the enhanced photocatalytic behavior over the undoped samples. Considering the recent SnO2 nanomaterials in the literature42–45 the observed surface area of undoped and doped SnO2 nanomaterials in this work is extraordinarily high. For example, porous SnO2 nanowire,43 mesoporous SnO2 hollows45 with surface areas up to 101 m2 g–1 were reported and applied as photocatalysts. It is believed that the high specific surface areas and reduction of the width of the band gap energy levels between the conduction band and valence band of the Sm3+ doped SnO2 nanomaterials are beneficial for the diffusion of reactant molecules to the active sites, resulting in an enhanced loading of dye molecules leading to the photodegradation of the dye.
Conclusions
A spherical shape is exhibited for pure and Sm3+ ion doped SnO2 nanoparticles, prepared using ethylene glycol as a capping agent and its photoluminescence properties have been studied at different annealing temperatures (as-prepared, 500 and 900 °C). The crystallinity of the samples was found to be increasing with the increase in annealing temperature, which is due to the evaporation of water and organic moiety from the surface of the nanoparticles. There are efficient energy transfers from host SnO2 to the Sm3+ ion. The photocatalytic behavior of the prepared samples was investigated on selected triphenylmethane dyes, viz. Ethyl Violet and Brilliant Green. The degradation of dyes was found to be enhanced in the presence of Sm3+ ions.References
- M. Law, H. Kind, B. Messer, F. Kim and P. Yang, Angew. Chem., Int. Ed., 2002, 41, 2405 CrossRef CAS.
- S. C. Lee, J. H. Lee, T. S. Oh and Y. H. Kim, Sol. Energy Mater. Sol. Cells, 2003, 75, 481 CrossRef CAS.
- I. T. Weber, A. Valentini, L. F. D. Probst, E. Longo and E. R. Leite, Mater. Lett., 2008, 62, 1677 CrossRef CAS.
- B. Wang, D. Su, J. Park, H. Ahn and G. Wang, Nanoscale Res. Lett., 2012, 7, 215 CrossRef PubMed.
- J. S. Lee, S. K. Sim, B. Min, K. Cho, S. W. Kim and S. Kim, J. Cryst. Growth, 2004, 267, 145 CrossRef CAS.
- D. B. Pablo, M. R. S. Luisa, W. L. A. Horacio, F. S. J. Eronides and V. C. A. Lucy, Nanoscale Res. Lett., 2012, 7, 540 CrossRef PubMed.
- Y. Chen, H. K. Y. Jong, W. Chung, B. K. Moon, H. Choi and J. H. Jeong, J. Korean Phys. Soc., 2010, 6, 1760 Search PubMed.
- Y. Yu, D. Chen, P. Huang, H. Lin, A. Yang and Y. Wang, J. Solid State Chem., 2011, 184, 236 CrossRef CAS.
- J. P. Arago, B. J. Pez, E. Cordoncillo, P. Escribano, F. Pelle, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 5193 RSC.
- V. Venkatramu, P. Babu, C. K. Jayasankar, T. T. Ster, W. Sievers and G. Wortmann, Opt. Mater., 2007, 29, 1429 CrossRef CAS.
- J. P. Wu, D. M. Newman and I. Viney, J. Lumin., 2002, 99, 237 CrossRef CAS.
- M. Wang, Y. Gao, L. Dai, C. Cao and X. Guo, J. Solid State Chem., 2012, 189, 49 CrossRef CAS.
- F. Gu, S. F. Wang, M. K. Liu, Y. X. Qi, G. J. Zhou, D. Xu and D. R. Yuan, Opt. Mater., 2004, 25, 59 CrossRef CAS.
- M. Nogami, T. Enomoto and T. Hayakawa, J. Lumin., 2002, 97, 147 CrossRef CAS.
- J. Wu and J. L. Coffer, J. Phys. Chem. C, 2009, 113, 12 CAS.
- A. Kar, S. Kundu and A. Patra, J. Phys. Chem. C, 2011, 115, 118 CAS.
- M. N. Luwang, R. S. Ningthoujam, N. S. Singh, R. Tewari, S. K. Srivastava and R. K. Vatsa, J. Colloid Interface Sci., 2010, 349, 27 CrossRef CAS PubMed.
- J. Chanathaworn, C. Bunyakan, W. Wiyaratn and J. Chungsiriporn, J. Sci. Technol., 2012, 34, 203 CAS.
- Y. Zhu, X. Su, C. Yang, X. Gao, F. Xiao and J. Wang, J. Mater. Chem., 2012, 22, 13914 RSC.
- J. Santhanalakshmi and R. Komalavalli, Res. J. Chem. Sci., 2012, 2, 64 CAS.
- S. Bingham and W. A. Daoud, J. Mater. Chem., 2011, 21, 2041 RSC.
- F. Meng, J. Li, S. K. Cushing, M. Zhi and N. Wu, J. Am. Chem. Soc., 2013, 135, 10286 CrossRef CAS PubMed.
- F. Meng, J. Li, S. K. Cushing, J. Bright, M. Zhi, J. D. Rowley, Z. Hong, A. Manivannan, A. D. Bristow and N. Wu, ACS Catal., 2013, 3, 746 CrossRef CAS.
- R. S. Ningthoujam, V. Sudarsan and S. K. Kulshreshtha, J. Lumin., 2007, 127, 747 CrossRef CAS.
- Z. Hou, P. Yang, C. Li, L. Wang, H. Lian, Z. Quan and J. Lin, Chem. Mater., 2008, 20, 6686 CrossRef CAS.
- J. D. CaStillo, V. D. Rodriguez, A. C. Yanes, J. Mendez-Ramos and M. E. Torres, Nanotechnology, 2005, 16, S300 CrossRef.
- A. C. Yanes, J. D. CaStillo, M. Torres, J. Peraza, V. D. Rodriguez and J. Mendez-Ramos, Appl. Phys. Lett., 2004, 85, 2343 CrossRef CAS.
- L. Abello, B. Bochu, A. Gaskov, S. Koudryavtseva, G. Lucazeau and M. Roumyantseva, J. Solid State Chem., 1998, 135, 78 CrossRef CAS.
- W. Kemp, Organic Spectroscopy, Macmillan Educational Ltd, Houldmills, Hampshire, 1987 Search PubMed.
- F. Gu, S. F. Wang, M. K. Liu, Y. X. Qi, G. J. Zhou, D. Xu and D. R. Yuan, J. Cryst. Growth, 2003, 255, 357 CrossRef CAS.
- F. Gu, S. F. Wang, M. K. Liu, Y. X. Qi, G. J. Zhou, D. Xu and D. R. Yuan, J. Phys. Chem. B, 2004, 108, 8119 CrossRef CAS.
- H. Wang, J. Yang, C. M. Zhang and J. Lin, J. Alloys Compd., 2009, 182, 2716 CAS.
- M. Sobczyk, P. Starynowicz, R. Lisiecki and W. R. Romanowski, Opt. Mater. (Amsterdam), 2008, 30, 1571 CrossRef CAS.
- M. N. Luwang, R. S. Ningthoujam, S. Jagannath, S. K. Srivastava and R. K. Vatsa, J. Am. Chem. Soc., 2010, 132, 2759 CrossRef CAS PubMed.
- M. N. Luwang, R. S. Ningthoujam, S. K. Srivastava and R. K. Vatsa, J. Am. Chem. Soc., 2011, 133, 2998 CrossRef CAS PubMed.
- M. N. Luwang, R. S. Ningthoujam, S. K. Srivastava and R. K. Vatsa, J. Mater. Chem., 2011, 21, 5326 RSC.
- M. B. Reddy, S. Sailaja, P. Giridhar, C. N. Raju and B. S. Reddy, Ferroelectr., Lett. Sect., 2011, 38, 40 CrossRef CAS.
- M. Bekbolet, A. S. Suphandag and C. S. Uyguner, J. Photochem. Photobiol., A, 2001, 148, 121 CrossRef.
- C. Bauer, P. Jacques and A. Kalt, J. Photochem. Photobiol., A, 2001, 140, 87 CrossRef CAS.
- W. C. Hung, S. H. Fu, J. J. Tseng, H. Chu and T. H. Ko, Chemosphere, 2007, 66, 2142 CrossRef CAS PubMed.
- A. W. Xu, Y. Gao and H. Q. Liu, J. Catal., 2002, 207, 15 Search PubMed.
- Y. Zhao, J. Li, N. Wang, C. Wu, G. Dong and L. Guan, J. Phys. Chem. C, 2012, 116, 18612 CAS.
- Y. Han, X. Wu, Y. Ma, L. Gong, F. Qu and H. Fan, CrystEngComm, 2011, 13, 3506 RSC.
- S. Ding and X. W. Lou, Nanoscale, 2011, 3, 3586 RSC.
- J. Choi, S. Y. Han, J. Jin, J. Kim, J. H. Park, S. M. Lee, H. J. Kim and S. U. Son, J. Mater. Chem. A, 2013, 1, 8609 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nj00759f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |