
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity–activity relationships of oral anti-diabetic vanadium complexes in gastrointestinal media: an X-ray absorption spectroscopic study†
Aviva
Levina
a,
Andrew I.
McLeod
a,
Lauren E.
Kremer
a,
Jade B.
Aitken
ab,
Christopher J.
Glover
b,
Bernt
Johannessen
b and
Peter A.
Lay
*a
aSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: peter.lay@sydney.edu.au
bAustralian Synchrotron, 800 Blackburn Rd., Clayton, VIC 3168, Australia
First published on 28th July 2014
Abstract
The reactions of oral V(V/IV) anti-diabetic drugs within the gastrointestinal environment (particularly in the presence of food) are a crucial factor that affects their biological activities, but to date these have been poorly understood. In order to build up reactivity–activity relationships, the first detailed study of the reactivities of typical V-based anti-diabetics, Na3VVO4 (A), [VIVO(OH2)5](SO4) (B), [VIVO(ma)2] (C, ma = maltolato(−)) and (NH4)[VV(O)2(dipic)] (D, dipic = pyridine-2,5-dicarboxylato(2−)) with simulated gastrointestinal (GI) media in the presence or absence of food components has been performed by the use of XANES (X-ray absorption near edge structure) spectroscopy. Changes in speciation under conditions that simulate interactions in the GI tract have been discerned using correlations of XANES parameters that were based on a library of model V(V), V(IV), and V(III) complexes for preliminary assessment of the oxidation states and coordination numbers. More detailed speciation analyses were performed using multiple linear regression fits of XANES from the model complexes to XANES obtained from the reaction products from interactions with the GI media. Compounds B and D were relatively stable in the gastric environment (pH ∼ 2) in the absence of food, while C was mostly dissociated, and A was converted to [V10O28]6−. Sequential gastric and intestinal digestion in the absence of food converted A, B and D to poorly absorbed tetrahedral vanadates, while C formed five- or six-coordinate V(V) species where the maltolato ligands were likely to be partially retained. XANES obtained from gastric digestion of A–D in the presence of typical food components converged to that of a mixture of V(IV)–aqua, V(IV)–amino acid and V(III)–aqua complexes. Subsequent intestinal digestion led predominantly to V(IV) complexes that were assigned as citrato or complexes with 2-hydroxyacidato donor groups from other organic compounds, including certain carbohydrates. The absence of strong reductants (such as ascorbate) in the food increased the V(V) component in gastrointestinal digestion products. These results can be used to predict the oral bioavailability of various types of V(V/IV) anti-diabetics, and the effects of taking such drugs with food.
Introduction
The anti-diabetic activities of vanadium compounds have been known for over a century.1 In the early 1980s, a relationship was proposed1 between this activity and the role of vanadate ([VVO4]3−) as a powerful inhibitor of protein tyrosine phosphatases (PTPs),2 which act as negative regulators of the insulin signalling cascade.3 This finding spurred an interest in V-based oral drugs for the treatment of type 2 (non-insulin-dependent) diabetes, which is a growing health concern worldwide.1,4 Early studies on vanadium anti-diabetics concentrated on [VVO4]3− or [VIVO(OH2)5]2+ (A and B in Chart 1).5–7 However, more recent research has focussed on the design of neutral lipophilic V(IV) complexes of a general formula [VIVOL2] (where L is a monoanionic bidentate ligand), which have advantages of increased gastrointestinal absorption and reduced toxicity compared with inorganic V(V) or V(IV) salts.1,4,8,9 Archetypal examples of such ligands are maltol (see C in Chart 1) or its close analogue, ethyl maltol (both approved food additives);1,8,10 the V(IV) complex of the latter ligand had entered phase II clinical trials.10 Another widely studied class of potential anti-diabetics are anionic V(V) complexes of the dipicolinicato (pyridine-2,5-dicarboxylato(2−)) ligand (D in Chart 1) and its derivatives.9,11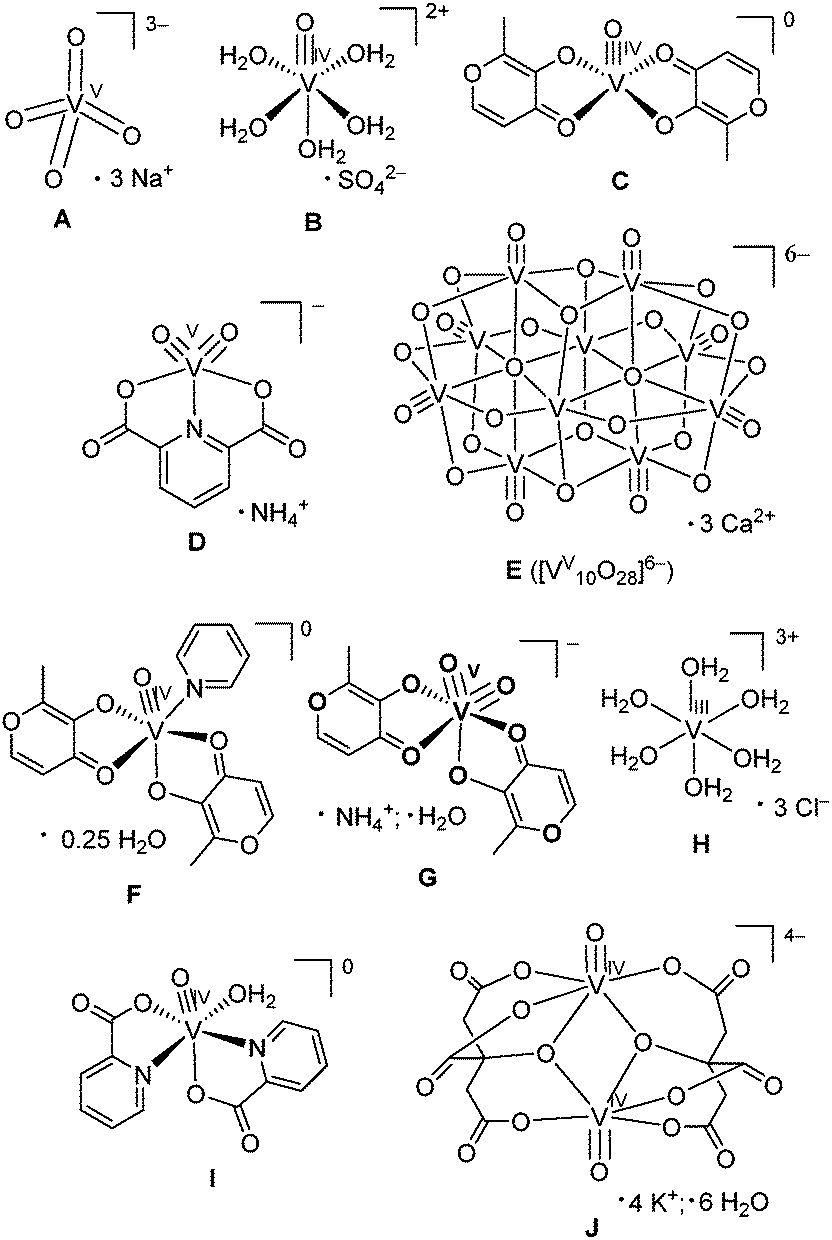 | ||
| Chart 1 Structures of anti-diabetic V(V) and V(IV) complexes used for the reactions with gastrointestinal media (A–D), and model complexes used for the data analysis (E–J). The V–oxido binding in V(IV) and V(V) complexes, except for A, is correctly represented by triple, rather than double, bonds (a combination of one σ and two π bonds).30,31 | ||
Significant issues were apparent when V(V) and V(IV) complexes were used as anti-diabetics in animal experiments and human phase I clinical trials including the following: (i) a distinct dichotomy of response (either achieved early in the treatment or not achieved at all);1 and (ii) a poor correlation between the glucose-lowering effect and V levels in the blood.12 In addition, administration together with food greatly reduced the oral bioavailability of such complexes, which suggested ligand substitution with food components.1,13 Animal studies using the 14C-labelled V(IV) ethylmaltolato complex showed that the compound dissociated within an hour after the ingestion (in fed animals), most likely in the stomach.1,12a These data indicated that the interactions with gastrointestinal media (including food components) were crucial for controlling the biological activities of V-based oral anti-diabetics.1,4,14,15 However, apart from some stability studies in acidic or neutral aqueous solutions (resembling gastric or intestinal environments, respectively),11,16 no reactivity studies of anti-diabetic V(V) or V(IV) complexes in gastrointestinal media have been performed, as yet.
Our group has previously applied XANES (X-ray absorption near-edge structure) spectroscopy in reactivity and speciation studies of toxic and medicinal metal ions in biological media,17–19 including the reactions of anti-diabetic Cr(III) and Mo(VI) complexes with artificial digestion systems.14,15 Such XANES analyses are particularly suitable for V complexes, due to the high sensitivity of their spectra to changes in oxidation states (V(V), V(IV), or V(III)) and coordination numbers (four, five or six) of their complexes that are typically encountered in biological systems.20–22 Recent analysis of XANES spectra of twenty-three biologically-relevant V(V), V(IV) and V(III) complexes showed that simple correlations, based on pre-edge and edge parameters, can be used for the reliable determination of oxidation states and coordination numbers of V species in complex biological and environmental samples.22 In the current research, this methodology has been applied to reactivity studies of V(V) and V(IV) complexes with known anti-diabetic properties (A–D in Chart 1)1,5–7,9 in simulated gastrointestinal media (in the presence or absence of food components).14,15
Experimental
Materials and sample preparation
The model V(V) and V(IV) complexes (Chart 1) were either purchased from Aldrich (A and B, purity >99%), or synthesized by modified literature procedures (C, D, F, G, I and J)23–29 and characterized by elemental analyses, infrared spectroscopy and electrospray mass spectrometry, as described previously.22 Note that V-oxido binding in V(IV) and V(V) complexes (except for A, Chart 1) is represented with triple, rather than double, bonds (contrary to the common convention). The triple bond arises from a combination of one σ and two π bonds.30,31 Published XANES spectra for E (the mineral pascoite, Ca3[VV10O28]·17H2O)32 and H (0.10 M solution of VIIICl3 in 1.0 M HCl)33 were also used in the fits. Other reagents of analytical or higher purity grade were purchased from Sigma-Aldrich or Merck, and used without further purification. Water was purified by the Milli-Q technique. The pH values of the reaction solutions were measured by a HI 9023 pH-meter (Hanna Instruments) equipped with a PHR-146 solid-state micro-pH electrode (Lazar Research Laboratories), and the instrument was calibrated daily with pH standard solutions (Sigma).A summary of the reaction conditions and designations of the samples are given in Table 1. Stock solutions of A, B and D (0.10 M in H2O) were prepared daily because these solutions are known to be stable for at least several days under ambient conditions.11,34 By contrast, C is sparingly water-soluble and undergoes significant oxidation to V(V) within hours in aerated aqueous solutions.23 Therefore, this compound was diluted with an inert solid14,15 by thorough mixing of C (6.30 mg, 0.200 mmol) with boron nitride (BN, 0.200 g). Aliquots of this mixture (10.0 mg) were used to prepare solutions of C in reaction media (1.0 mL of 1.0 mM V), the insoluble BN was then separated by gravity. Mixing of solid complexes with BN (∼10 mass parts BN per 1 part of V complex) was also used for the preparation of samples of model compounds for XAS analyses (A–D, F, G, I and G in Chart 1).22
| Samplea | Conditionsb | Modelc |
R
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
|---|---|---|---|
| a Designations of the initial compounds (A–D) correspond to those in Chart 1, and the numbers (1–6) designate the treatment conditions. The XANES spectra for A5 and C5 were collected at the ANBF, and all the other spectra were collected at the AS (see Experimental for details). b In all the experiments, the total V concentrations in the reaction mixtures were 1.0 mM. Details of the treatment conditions are described in the Experimental section. c A XANES spectrum or a combination of spectra of model V(V), V(IV) and V(III) complexes (Chart 1) that provide the best possible match for the XANES spectrum of the corresponding sample (see Fig. 3 and Fig. S1 and S2, ESI). Standard deviations were calculated using Origin software46 as a part of multiple linear regression procedure. d Regression coefficient for the superposition of the sample and model spectra (see Fig. S2, ESI for multiple linear regression results). e Since nearly identical XANES spectra were obtained from samples A3, B3, C3 and D3 (Fig. S1d and e, ESI), the averaged spectrum was used for the modelling. f Artificial meal 1 was a commercial semi-synthetic meal ("liquid breakfast");39 and artificial meal 2 was prepared from separate components;14,15 see ESI for the composition of the both meals. | |||
| A1 | Gastric digestion; no food | 100% E | 0.994 |
| B1 | 100% B | 0.999 | |
| C1 | 100% B | 0.990 | |
| D1 | 100% D | 0.993 | |
| A2 | Gastric and intestinal digestion; no food | 100% A | 0.994 |
| B2 | 100% A | 0.994 | |
| C2 | (30 ± 2)% D; (70 ± 2)% G | 0.998 | |
| D2 | 100% A | 0.996 | |
| (A3–D3)ave |
Gastric digestion; artificial meal 1f | (50 ± 4)% B; (22 ± 2)% H; (28 ± 2)% I | 0.999 |
| A4 | Gastric and intestinal digestion; artificial meal 1f | 100% J | 0.996 |
| B4 | 100% J | 0.999 | |
| C4 | 100% J | 0.999 | |
| D4 | (10 ± 1)% A; (90 ± 2)% J | 0.999 | |
| A5 | Gastric and intestinal digestion; artificial meal 2f | (66 ± 2)% A; (34 ± 2)% J | 0.998 |
| C5 | (48 ± 2)% A; (52 ± 2)% J | 0.997 | |
| C6 | pH = 7.4, no. O2 | 100% F | 0.997 |
Artificial gastrointestinal digestions of A–D in the absence of food components14,15 were performed by dissolving compounds A–D ([V]final = 1.0 mM) in artificial gastric juice (150 mM NaCl, acidified by HCl to pH = 1.8)35 and incubation of the resultant solutions for 1 h at 310 K in a rocking water bath with free access to air. In one series of experiments (A1–D1 in Table 1), the reaction solutions were then immediately frozen at ∼195 K and freeze-dried (217 K and 0.5 mbar for 16 h). Freeze-drying of the samples was used to stop the reactions, to increase the signal-to-noise ratio in XANES spectra, and to minimize the synchrotron radiation-induced chemical changes in the samples.36 In another series (A2–D2 in Table 1), simulated gastric digestion was followed by raising the pH value of the solutions to 7.535 by dropwise addition of aqueous NaHCO3 (1.0 M). The incubation was continued for further 2 h at 310 K (simulated intestinal digestion), after which the samples were freeze-dried, as described above. The chosen reaction times (1 h and 2 h) correspond to typical residence times (in humans) of ingested drugs in the stomach and small intestines, respectively.37,38
Modified literature procedures14,15,37,38 were used for artificial digestion of A–D in the presence of typical food components and digestive enzymes. For experiments A3–D3 and A4–D4 (Table 1), an aliquot (10 mL) of commercial liquid semi-synthetic meal ("liquid breakfast", Sanitarium, Australia; detailed composition is given in ESI†)39 was acidified to pH = 1.8 (from the initial value of 6.7) by dropwise addition of concentrated HCl (∼0.10 mL of 10 M solution), then pepsin solution (0.014 g porcine pepsin in 0.60 mL of 0.10 M HCl) was added. Aliquots (1.0 mL) of the resultant solution were immediately mixed with A–D to [V]final = 1.0 mM, and incubated for 1 h at 310 K in a rocking water bath with free access to air (simulated gastric digestion), followed either by freeze-drying of the samples (experiments A3–D3), or by simulated intestinal digestion (experiments A4–D4). For the latter experiments, NaHCO3 solution (1.0 M, ∼0.15 mL) and pancreatin-bile solution (0.010 g of porcine pancreatin and 0.050 g of porcine bile extract in 0.25 mL of 0.10 M NaHCO3) were added dropwise to the samples (to final pH = 7.5), followed by incubation for a further 2 h at 310 K, and freeze-drying of the resultant mixtures. Experiments A5 and C5 (Table 1) were performed in the same manner as A4–D4, except that the semi-synthetic meal was freshly prepared from separate components and did not contain vitamin additions (see ESI† for details). For experiment C6 (Table 1), compound C (in a mixture with BN, see above) was dissolved in HEPES-buffered saline (20 mM HEPES, 140 mM NaCl, pH = 7.40; where HEPES = 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)40 under a stream of Ar (to avoid V(IV) oxidation to V(V))23 to [V]final = 1.0 mM, BN was removed by centrifugation, and the resultant solution was immediately frozen at ∼195 K and freeze-dried.
XANES spectroscopy and data processing
Vanadium K-edge spectra of samples A–D (Chart 1), A1–D1, A2–D2, A3–D3, A4–D4 and C6 (Table 1) were recorded at the X-ray absorption spectroscopy beamline, Australian Synchrotron (AS, Melbourne, Australia). The beam energy was 3.0 GeV, and the maximal beam current was 200 mA. The beamline had a channel-cut Si[111] monochromator, an upstream collimating mirror, and a downstream sagittally focusing mirror; both mirrors were Rh-coated and also provided harmonic rejection. Mixtures of compounds A–D with BN or neat freeze-dried reaction products (see the Sample preparation section above) were pressed into 0.5 mm thick pellets that were supported within a polycarbonate spacer between two 63.5 μm Kapton tape windows (window size, 2 × 10 mm). The samples were placed in a He-filled box at ∼295 K, and the XANES data were collected in fluorescence detection mode, using a Ge planar detector (Eurisys; 100-element). Low-temperature XAS measurements were not used, since they led to a significant reduction in the signal-to-noise ratio, due to the strong absorption of photons at the 5–6 keV energy range by the cryostat windows and air between the sample compartment and the detector. For all the samples, only the XANES regions were recorded (5250–5700 eV range; step sizes, 10 eV below 5450 eV, 0.25 eV at 5450–5525 eV and 2 eV above 5525 eV). The energy scale was calibrated using a V foil as an internal standard (calibration energy, 5465.0 eV, corresponding to the first peak of the first derivative of V(0) edge).41 The XANES spectra of the model complexes, F, G, I and J (Chart 1), as well as those of samples A5 and C5 (Table 1), were recorded at ∼295 K using the fluorescence detection mode at the Australian National Beamline Facility (ANBF, beamline 20B, Photon Factory, KEK, Tsukuba, Japan), as described previously.22 No significant photodamage of V(V) samples occurred at ANBF (as shown by comparison of XANES data from sequential scans), while the relative V(IV) content increased by ∼10% in the second scans of the samples recorded at the AS.36 The extent of photodamage was higher at the AS compared with those at the ANBF because of the higher photon flux, but this also provided higher signal-to-noise ratios.36 Consistent results were obtained on both beamlines when only the first scans for each sample at the AS were used for the analyses.22,36Calibration, averaging and splining of XANES data were performed using the XFit software package.42 The spectra were normalized (using the Spline program within the XFit package) according to the method of Penner-Hahn and coworkers,43 to match the tabulated X-ray cross-section data44 for V (in a similar manner to the earlier work on Cr(III) XANES spectra).15 This normalization technique led to consistent XANES spectra for all of the samples, regardless of the beamline used and the signal-to-noise ratio. Published XANES spectra of E and H32,33 were digitized using WinDIG software,45 and the energy scales for these data were adjusted, as described previously.22 Multiple linear regression analyses of XANES data were performed using Origin software46 with previously described criteria for a successful fit.17
Results
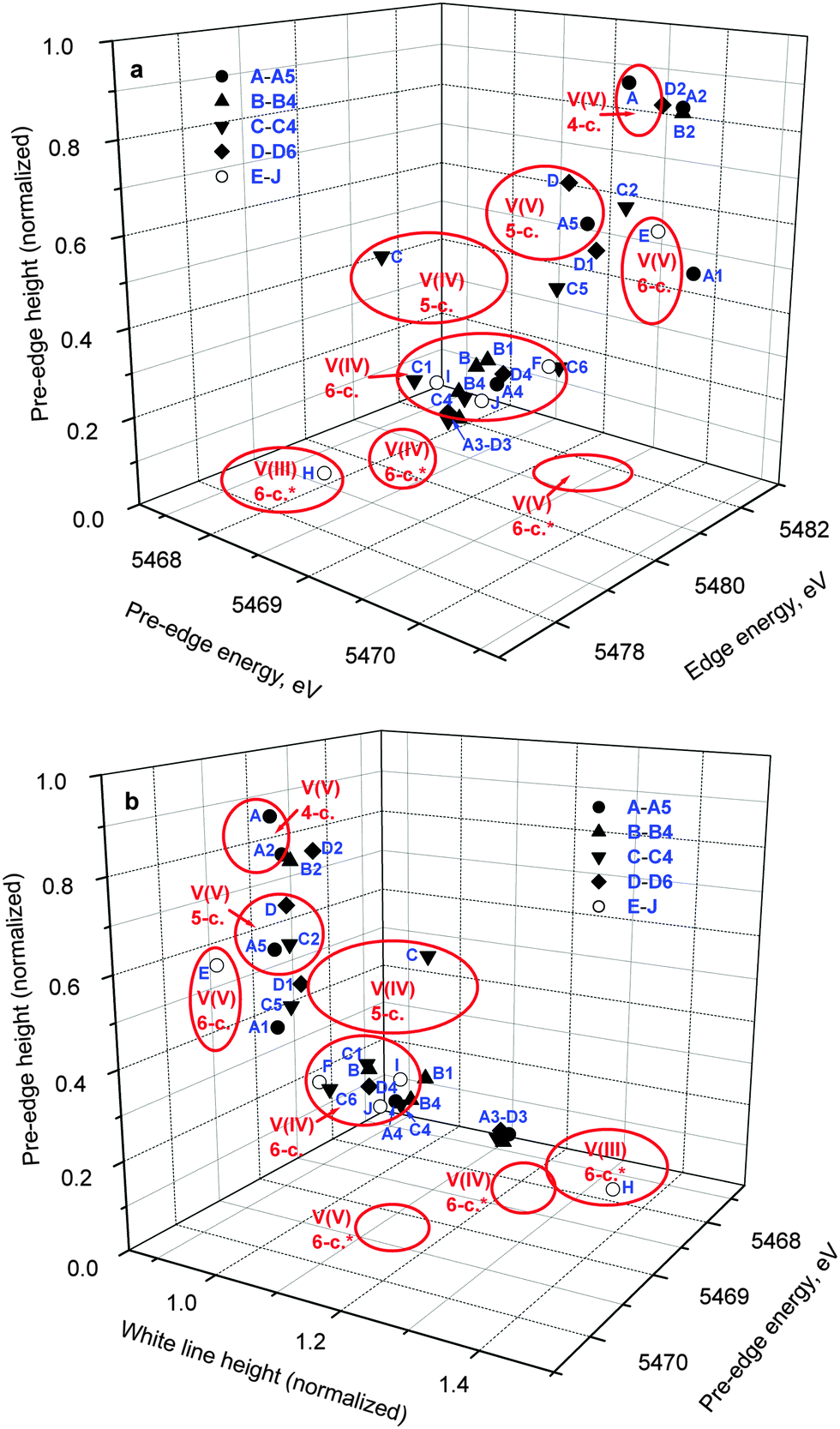
A comparison of XANES spectra (5460–5520 eV) of the parent complexes (A–D in Chart 1) with those of the reaction products in artificial gastrointestinal media (A1–D1, A2–D2, A3–D3 and A4–D4 in Table 1) is shown in Fig. 1. Key XANES parameters (position and intensity of the pre-edge absorbance, edge energy at the half-edge jump, and post-edge absorbance intensity) for these samples are plotted in Fig. 2, where they are compared with the corresponding parameters for model V(V/IV/III) complexes (designated with red ellipses in Fig. 2).22 Comparison of XANES of the reaction products (Table 1) with those of selected model complexes (Chart 1), or their linear combinations, are shown in Fig. 3 and Fig. S1 and S2 (ESI†), and the best fits to the XANES from each sample are summarized in Table 1.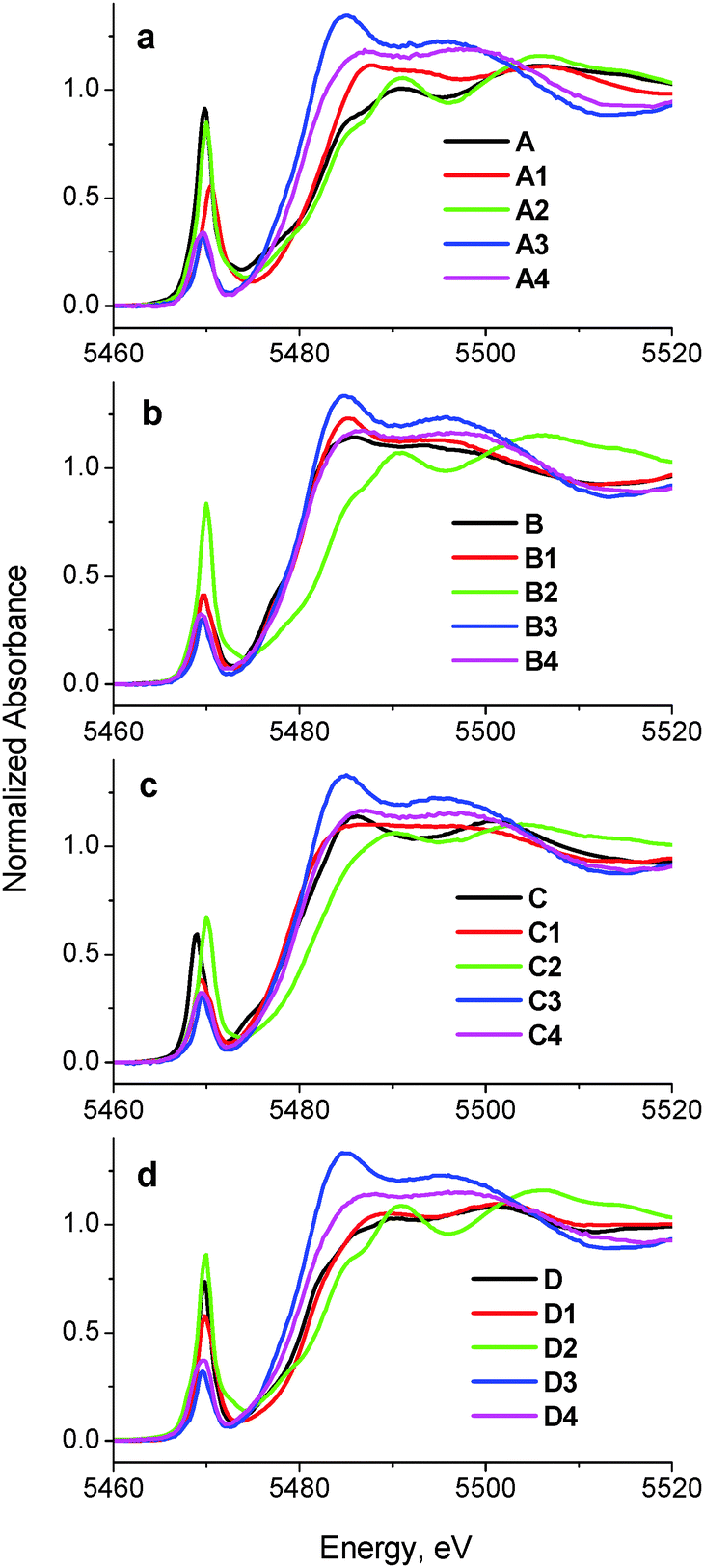 | ||
| Fig. 1 Comparison of XANES of initial complexes (A–D) with those of the reaction products with artificial digestion systems. Designations of the samples correspond to those in Chart 1 and Table 1. All the data were collected at ∼295 K in fluorescence detection mode for solid samples (mixtures of model complexes with BN or freeze-dried reaction mixtures; see Experimental for details). | ||
 | ||
| Fig. 2 Correlations of pre-edge and edge parameters in the XANES of the reaction mixtures (designations correspond to those in Table 1) and model complexes (Chart 1). The areas corresponding to various oxidation states and coordination numbers of V complexes (red ellipses) were drawn based on the XANES data for a library of twenty-three model V(V), V(IV) and V(III) complexes (asterisks designate non-oxido complexes).22 | ||
 | ||
| Fig. 3 Comparison of selected XANES spectra of the reaction products of A–D with those of model V(V), V(IV) and V(III) complexes (see also Fig. S1 and S2 in ESI;† designations correspond to those in Chart 1 and Table 1). All the data were collected at ∼295 K in fluorescence detection mode for solid samples (freeze-dried reaction mixtures or mixtures of model complexes with BN; see Experimental section for details). | ||
Treatment of A in artificial gastric juice in the absence of food components (pH ∼ 1.8; 1 h at 310 K; A1 in Table 1) led to a notable decrease in intensity of the pre-edge absorbance, but no significant changes in edge energy (Fig. 1a), which was consistent with the formation of six-coordinate V(V) species (Fig. 2).22,32,47 The XANES of A1 closely matched the literature data for [V10VO28]6− (E in Chart 1, the mineral pascoite),32 as shown in Fig. 3a. Decavanadate ([V10VO28]6−) is well-known to be the predominant form of V(V) in aqueous solutions at pH ∼ 2 and [V] ≥ 1 mM in the absence of strongly binding ligands.20,48 By contrast, treatment of B under the same conditions (B1 in Table 1) caused no significant changes in the pre-edge and edge parameters and a slight increase in post-edge absorbance (Fig. 1b and 2), which was likely to be caused by partial deprotonation of the aqua ligands in B.20,48 The reaction product of D under these conditions exhibited a similar XANES to that of the initial compound (D1 in Table 1 and Fig. 1d), apart from a slight decrease in pre-edge intensity, which pointed to the formation of a mixture of five- and six-coordinate V(V) species (Fig. 2). The relative stability of D in aqueous solutions at pH ∼ 2 compared to the other complexes has also been proposed previously from NMR spectroscopic studies.11
The XANES of C1 (Table 1) was distinguished from that of the initial compound, C, by a strong decrease in intensity and a shift in energy of the pre-edge absorbance (Fig. 1c), which was consistent with the formation of six-coordinate V(IV) species (Fig. 2). Of all the available model XANES spectra,22 that of B was the closest match for C1 (Table 1 and Fig. S1a, ESI†), which was in agreement with the literature data that the V(IV) maltolato complex was largely dissociated and converted to aquated V(IV) species at pH ≤ 2.16 By contrast, dissolution of C in neutral aqueous medium under an Ar atmosphere (to avoid oxidation to V(V))23 resulted in the formation of a different six-coordinate V(IV) species (C6 in Table 1 and Fig. 2 and Fig. S1b, ESI†). The XANES of C6 closely matched that of a model six-coordinate V(IV)-maltolato complex, F (Chart 1),23 as shown in Fig. S1b (ESI†) and in Table 1. This result was consistent with the addition of H2O as the sixth ligand to C in neutral aqueous solutions, similarly to the well-known addition of an aqua ligand to the V(IV) acetylacetonato complex (established by EPR spectroscopy).49
In summary, XANES data pointed to the relative stabilities of B and D in the acidic environment of the stomach (in the absence of food) compared with A and C, which converted predominantly to [V10VO28]6− and [VIVO(OH2)5]2+, respectively, under these conditions. These findings were consistent with the literature data on the reactivity of A–D in acidic aqueous solutions (obtained by NMR and EPR spectroscopies and by potentiometric titrations),11,16,20,48 which showed that the freeze-dried samples used in this work maintained the speciation of V(V/IV) reaction products that were formed in solutions.
Treatments of A, B, or D under the conditions that mimicked sequential gastric and intestinal digestions in the absence of food (pH ∼ 1.8 for 1 h and pH ∼ 7.5 for 2 h at 310 K; A2, B2 or D2 in Table 1) resulted in XANES that were close to each other and similar, but not equivalent, to that of A (Fig. 1a, b and d and Fig. S1c, ESI†). Data shown in Fig. 2 confirmed the formation of tetrahedral V(V) species under these conditions. Literature data on the pH- and concentration-dependent speciation of V(V) in aqueous solutions20,48 suggested that these products were likely to be the mixtures of [H2VVO4]− (predominant species) and oligomeric vanadates, such as [V3VO9]3−. The data for D2 (Fig. 1d and 2) point to complete dissociation of D under simulated intestinal conditions, which was consistent with the literature data on the low stability of D at pH > 7.11 Both V(IV) complexes studied, B and C, were completely oxidized to V(V) species under these conditions, but the differences in XANES of their reaction products (B2 and C2 in Table 1 and Fig. 1) indicated that the maltolato ligands were partially retained under conditions of C2. The XANES parameters of C2 were significantly different from those of A2, B2 and D2, and were consistent with the formation of a mixture of five- and six-coordinate V(V) species (Fig. 1c and 2). The XANES data for C2 were best matched by a combination of models D and G (V(V)-maltolato complex, Chart 1), as shown in Table 1 and in Fig. 3b and Fig. S2 (ESI†). The fit residues for C2 were small but significantly exceeded the experimental noise levels (Fig. S2, ESI†); however, the pre-edge and edge area (crucial determinants of V oxidation state and coordination number)22,50 were well reproduced. The differences in the post-edge area between the experimental and fitted spectra of C2 (Fig. S2, ESI†) were likely to be caused by the following factors: (i) the nature of V-bound ligands in the model compounds will not be an exact match to those in the reaction products and, as such, multiple-scattering contributions in this region of the XAS will be sensitive to non-coordinating atoms that are within ∼5 Å of the vanadium atom;51 and (ii) the XANES spectra of the reaction products and the model complexes were collected at different synchrotron sources, using different monochromator settings.22,50
Treatments of any of A–D under simulated gastric conditions in the presence of food components ("liquid breakfast", see ESI;† samples A3–D3 in Table 1) resulted in the formation of the same product, as shown in the XANES of Fig. 1, 2 and Fig. S1d (ESI†). Since the differences in XANES obtained amongst conditions A3–D3 were comparable to the experimental noise levels (Fig. S1e, ESI†), the average of these data was used for further processing (Fig. 3c). The edge and post-edge shapes for A3–D3 were close to those for [VIII(OH2)6]3+ (H in Chart 1),33 while the pre-edge intensity was close to that for [VIVO(OH2)5]2+(B in Chart 1), as shown in Fig. 3c. Clear separation of A3–D3 from all the other samples on the basis of XANES parameters is evident in Fig. 2b. The best match for XANES of A3–B3 was with a combination of the XANES from models B, H, and I (V(IV) picolinato complex, Chart 1; a model of V(IV)-amino acid or protein binding),26–28 as shown in Table 1 and in Fig. S2 (ESI†). These results show that V(III) species can be formed by reduction of either V(V) or V(IV) in acidic medium of the stomach in the presence of organic reductants (food components).
Treatments of A–D under the conditions of sequential gastric and intestinal digestion in the presence of food components ("liquid breakfast", A4–D4 in Table 1), resulted in the formation of similar products (Fig. 1 and Fig. S1f, ESI†), with XANES parameters that corresponded to those of six-coordinate V(IV) complexes (Fig. 2). The XANES of A4, B4 and C4 were closely matched by that of J (dimeric V(IV) citrate complex, Chart 1),29 while XANES of D4 was best fitted by a combination of the XANES of J (main component) and A (Table 1 and Fig. S2, ESI†). Similar treatments of A or C in the presence of a different semi-synthetic meal (not containing vitamin supplements, see ESI†) resulted in a higher proportion of V(V) vs. V(IV) species, which was best matched by combinations of XANES from models J and A (samples A5 and C5 in Table 1 and Fig. 3d and Fig. S2, ESI†). Close matches of XANES of the reaction products A4–C4 to that of the model J (Table 1 and Fig. 3d) suggest the binding of the resultant V(IV) species to hydroxido, alcoholato and/or carboxylato donor groups that most likely originate from citrate and/or other ligands, such as certain carbohydrate components of food, that contain 2-hydroxyacid moieties.4,52
Discussion
A summary of likely chemical transformations of anti-diabetic V(V/IV) complexes in gastrointestinal media, deduced from XANES spectra (Table 1), is shown in Scheme 1. The most striking result of these studies was the crucial role of the presence and composition of food components on V speciation. According to the results of phase 1 human clinical trials, V absorption from oral administration of 75 mg of V(IV)–ethylmaltolato complex (a close analogue of C) was ∼13-fold higher in the fasted state compared with the fed state.1 These data pointed to a low bioavailability of V(IV) complexes with combinations of hydroxido, alcoholato and/or carboxylato ligands that were likely to form in the intestines in the presence of food components (Scheme 1). These can form due to the high thermodynamic stabilities of V(V/IV) 2-hydrocarboxylato donor groups that can form monomeric and polynuclear complexes, while the formation of V(V/IV) 1,2-diolato complexes with carbohydrates is unlikely under biological conditions.52,53 Such 1,2-diolato complexes require strongly basic conditions to form in solution from reactions of V(V) or V(IV) with sugars.52 As is the case for Cr(IV), V(IV) requires 2-hydroxyacid groups rather than simple diols to stabilize this oxidation state under physiologically relevant conditions.53 Apart from small molecule ligands, sialoglycoproteins may also provide donor groups for strong V(V/IV) binding.54 The bioavailability of V-containing drugs is expected to decrease further if the consumed food is rich in strong reductants, such as ascorbate,55,56 as demonstrated by comparison of digestion products in the presence of two types of semi-synthetic meals (samples A4, C4, A5, C5 in Table 1; see ESI† for meal composition).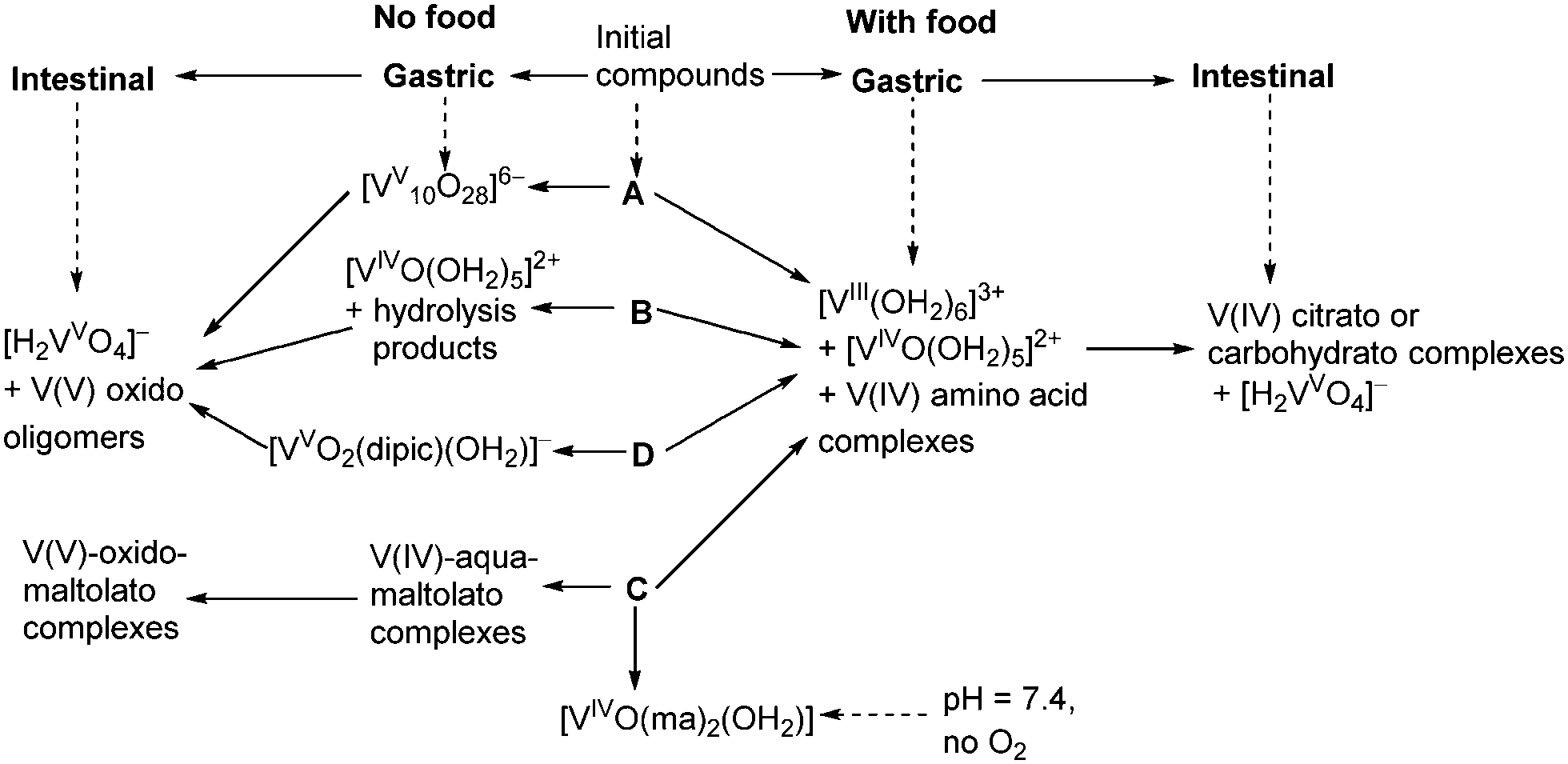 | ||
| Scheme 1 Proposed biotransformations of complexes A–D (Chart 1) in gastrointestinal environments based on the analyses of XANES data (Table 1). | ||
In the absence of food, V is likely to be absorbed from the intestines mainly in the form of V(V) oxido complexes, regardless of the oxidation state and chemical form of vanadium that was ingested (Scheme 1). However, maltolato and related ligands can remain partially bound to V(V) under these conditions (Scheme 1), as demonstrated by the difference in XANES of the sample C2 compared with A2, B2 and D2 (Table 1). This difference may explain the higher bioavailability of C and its analogues compared with that of B, which was demonstrated in both animal experiments and human clinical trials.1,57 Alternatively, V compounds can be partially absorbed from the oral cavity58 and the stomach, given the relatively rapid increases in V levels in the blood (which typically reached a maximum within 1–3 h after oral administration).1,12 Speciation of V in the gastric environment was more diverse compared with that in the simulated intestinal juices, particularly in the absence of food (Scheme 1). Some V anti-diabetic compounds, such as D (Scheme 1), are relatively stable in the gastric media11 and may be absorbed intact.9 Although the formation of [VV10O28]6− in simulated gastric media has been shown in the in vitro experiments ([V] = 1.0 mM; Table 1 and Scheme 1), it is unclear whether high enough V(V) concentrations can be reached for this process to take place in vivo. Absorbed [V10VO28]6− is expected to decompose slowly (within hours) in the blood and to show biological activities that are distinct from those of other V(V) or V(IV) species.59 Formation of V(III) in the stomach in the presence of reducing food components (Scheme 1) could facilitate V uptake through Fe(III) metabolic pathways.60 The close similarity between V(III) and Fe(III) in biochemical pathways has previously been demonstrated by high-affinity binding of V(III) to the main Fe(III) transport protein, transferrin.61 However, selective Fe(III) uptake by intestinal epithelial cells is preceded by its reduction to Fe(II).60 Although the possibility of formation of V(II) in biological systems has not yet been considered,20,62 stabilization of V(II) by biologically relevant imidazole ligands in vitro is known.63 However, given that V(III) only appears to be produced in the presence of food and there is decreased V absorption under these conditions, it is unlikely that these low-oxidation states are responsible for the main absorption pathways. Alternatively, these epithelial cells may take up V(IV) in the form of [VIVO]2+, as a chemical analogue of divalent metal ions.64 For instance, the recently discovered [VIVO]2+ transport system of ascidians (V-accumulating marine organisms) is also capable of transporting Fe(II).65
The usefulness of three-dimensional correlations of XANES parameters that were developed previously22 for the determination of oxidation states and coordination numbers of V in complex matrices has been confirmed in the current study (Fig. 2). However, caution is required when the samples are likely to contain V in various oxidation states, such as A3–D3, A5 and C5 (Table 1 and Fig. 2). Therefore, the initial assessment of the chemical state of V with the use of the diagrams in Fig. 2 has to be complemented by comparison of whole XANES spectra of the samples with those of model complexes (Fig. 3, Fig. S1 and S2, ESI†). On the whole, these data confirm the previous findings14,15 that metal-based anti-diabetics are likely to undergo complete chemical rearrangement in gastrointestinal media, particularly in the presence of food components. The current studies have enabled all oxidation states to be studied in the one experiment, which was not possible in previous studies and has provided many new insights in the speciation that is important in understanding efficacy and safety of V anti-diabetics. These changes determine the mode of gastrointestinal absorption and further metabolism of these drugs and have rationalized the differences in in vivo efficacies. Reactivity of oral V(V/IV) anti-diabetics in gastrointestinal media has to be taken into account when considering their further reactions in the blood,66 and the current work provides an entry point for such studies, which will be reported in the future.67 The current work illustrates how the XANES methodologies described herein can be used in studies of vanadium speciation in blood,18,67 and also in cells,18,68 and tissues.
Conclusions
While the importance of V speciation in controlling the activities of biological systems has been recognised for many years,69 the XANES results reported herein provide the most definitive studies to date on the speciation of all oxidation states vanadium under conditions that mimic oral administration. Typical anti-diabetic V(V) and V(IV) complexes undergo profound chemical changes in gastrointestinal media, including dissociation of the ligands, V(IV) oxidation to V(V) (in the absence of food), or V(V) reduction to V(IV) and even V(III) (in the presence of food). Formation of V(III) may be important for further metabolism via Fe(III) pathways, but the main absorption mechanisms appear to be associated with vanadate (poorly absorbed), VIVO2+ species via M2+ uptake mechanisms, and passive diffusion of neutral species. These data confirm the role of such complexes as pro-drugs that release the active components on the interactions with biological media. Three-dimensional diagrams of pre-edge and edge parameters in XANES spectra, developed on the basis of a library of model V(V/IV/III) complexes,22 have proven to be useful for the assessment of the chemical states of V in biological environments.Acknowledgements
The research was supported by Australian Research Council (ARC) Discovery Grants (DP0208409, DP0774173, DP0984722, DP1095310, and DP130103566), ARC Professorial Fellowships (DP0208409 and DP0984722) to P.A.L., and an ARC Linkage Infrastructure, Equipment and Facilities (LIEF) Program grant (LE0346515) for the 36-pixel Ge detector at ANBF. Initial X-ray absorption spectroscopy was performed partially at ANBF with support from the Australian Synchrotron Research Program (ASRP), which was funded by the Commonwealth of Australia under the Major National Research Facilities program (ANBF was operated by the Australian Synchrotron (AS) from 2009 until its closure in 2013). We acknowledge the LIEF program of the ARC for financial support (grant numbers LE0989759 and LE110100174) and the High Energy Accelerator Research Organisation (KEK) in Tsukuba, Japan, for operations support. We thank Drs Garry Foran, James Hester, Celesta Fong and Michael Cheah for the assistance with XAS experiments at ANBF, and Drs Glyn Devlin and Peter Kappen for those at the AS.Notes and references
- K. H. Thompson and C. Orvig, J. Inorg. Biochem., 2006, 100, 1925–1935 CrossRef CAS PubMed.
- M. Zhang, M. Zhou, R. L. Van Etten and C. V. Stauffacher, Biochemistry, 1997, 36, 15–23 CrossRef CAS PubMed.
- A. K. Srivastava and M. Z. Mehdi, Diabetic Med., 2005, 22, 2–13 CrossRef CAS PubMed.
- (a) A. Levina and P. A. Lay, Dalton Trans., 2011, 40, 11675–11686 RSC; (b) D. Rehder, Future Med. Chem., 2012, 4, 1823–1837 CrossRef CAS PubMed.
- C. E. Heyliger, A. G. Tahiliani and J. H. McNeill, Science, 1985, 227, 1474–1477 CAS.
- I. G. Fantus, G. Deragon, R. Lai and S. Tang, Mol. Cell. Biochem., 1995, 153, 103–112 CrossRef CAS.
- P. Poucheret, S. Verma, M. D. Grynpas and J. H. McNeill, Mol. Cell. Biochem., 1998, 188, 73–80 CrossRef CAS.
- K. H. Thompson and C. Orvig, Coord. Chem. Rev., 2001, 219–221, 1033–1053 CrossRef CAS.
- D. C. Crans, A. M. Trujillo, P. S. Pharazyn and M. D. Cohen, Coord. Chem. Rev., 2011, 255, 2178–2192 CrossRef CAS PubMed.
- K. H. Thompson, J. Lichter, C. LeBel, M. C. Scaife, J. H. McNeill and C. Orvig, J. Inorg. Biochem., 2009, 103, 554–558 CrossRef CAS PubMed.
- D. C. Crans, L. Yang, T. Jakusch and T. Kiss, Inorg. Chem., 2000, 39, 4409–4416 CrossRef CAS.
- (a) K. H. Thompson, B. D. Liboiron, Y. Sun, K. D. D. Bellman, I. A. Setyawati, B. O. Patrick, V. Karunaratne, G. Rawji, J. Wheeler, K. Sutton, S. Bhanot, C. Cassidy, J. H. McNeill, V. G. Yuen and C. Orvig, JBIC, J. Biol. Inorg. Chem., 2003, 8, 66–74 CrossRef CAS PubMed; (b) G. R. Willsky, K. Halvorsen, M. E. Godzala, L.-H. Chi, M. J. Most, P. Kaszynski, D. C. Crans, A. B. Goldfine and P. J. Kostyniak, Metallomics, 2013, 5, 1491–1502 RSC.
- J. B. Lichter, C. R. E. Orvig, M. C. Scaife and K. H. Thompson, WO Pat., 2009032591, 2009Chem. Abstr., 2009, 150, 322578 Search PubMed.
- A. Levina, A. I. McLeod, J. Seuring and P. A. Lay, J. Inorg. Biochem., 2007, 101, 1586–1593 CrossRef CAS PubMed.
- A. Nguyen, I. Mulyani, A. Levina and P. A. Lay, Inorg. Chem., 2008, 47, 4299–4309 CrossRef CAS PubMed.
- T. Kiss, T. Jakusch, D. Hollender, A. Doernyei, E. A. Enyedy, J. C. Pessoa, H. Sakurai and A. Sanz-Medel, Coord. Chem. Rev., 2008, 252, 1153–1162 CrossRef CAS PubMed.
- A. Levina, H. H. Harris and P. A. Lay, J. Am. Chem. Soc., 2007, 129, 1065–1075 CrossRef CAS PubMed.
- J. B. Aitken, A. Levina and P. A. Lay, Curr. Top. Med. Chem., 2011, 11, 553–571 CrossRef CAS.
- (a) A. Levina, J. B. Aitken, Y. Y. Gwee, Z. J. Lim, M. Liu, A. Mitra Singharay, P. F. Wong and P. A. Lay, Chem. – Eur. J., 2013, 19, 3609–3619 CrossRef CAS PubMed; (b) M. Liu, Z. J. Lim, Y. Y. Gwee, A. Levina and P. A. Lay, Angew. Chem., Int. Ed., 2010, 49, 1661–1664 CrossRef CAS PubMed.
- D. Rehder, Bioinorganic Vanadium Chemistry, John Wiley & Sons, Chichester, 2008 Search PubMed.
- P. Frank, E. J. Carlson, R. M. K. Carlson, B. Hedman and K. O. Hodgson, J. Inorg. Biochem., 2008, 102, 809–823 CrossRef CAS PubMed.
- A. Levina, A. I. McLeod and P. A. Lay, Chem. – Eur. J., 2014 DOI:10.1002/chem.201403993.
- P. Caravan, L. Gelmini, N. Glover, F. G. Herring, H. Li, J. H. McNeill, S. J. Rettig, I. A. Setyawati, E. Shuter, Y. Sun, A. S. Tracey, V. G. Yuen and C. Orvig, J. Am. Chem. Soc., 1995, 117, 12759–12770 CrossRef.
- B. Nuber, J. Weiss and K. Wieghardt, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1978, 33, 265–267 Search PubMed.
- K. Wieghardt, Inorg. Chem., 1978, 17, 57–64 CrossRef CAS.
- H. Sakurai, K. Fujii, H. Watanabe and H. Tamura, Biochem. Biophys. Res. Commun., 1995, 214, 1095–1101 CrossRef CAS PubMed.
- M. Melchior, K. H. Thompson, J. M. Jong, S. J. Rettig, E. Shuter, V. G. Yuen, Y. Zhou, J. H. McNeill and C. Orvig, Inorg. Chem., 1999, 38, 2288–2293 CrossRef CAS.
- E. Lodyga-Chruscinska, G. Micera and E. Garribba, Inorg. Chem., 2011, 50, 883–899 CrossRef CAS PubMed.
- M. Tsaramyrsi, M. Kaliva, A. Salifoglou, C. P. Raptopoulou, A. Terzis, V. Tangoulis and J. Giapinfzakis, Inorg. Chem., 2001, 40, 5772–5779 CrossRef CAS PubMed.
- C. J. Ballhausen and H. B. Gray, Inorg. Chem., 1962, 1, 111–122 CrossRef CAS.
- J. R. Winkler and H. B. Gray, Struct. Bonding, 2012, 142, 17–28 CrossRef CAS.
- P. Chaurand, J. Rose, V. Briois, M. Salome, O. Proux, V. Nassif, L. Olivi, J. Susini, J.-L. Hazemann and J.-Y. Bottero, J. Phys. Chem. B, 2007, 111, 5101–5110 CrossRef CAS PubMed.
- P. Frank and K. O. Hodgson, Inorg. Chem., 2000, 39, 6018–6027 CrossRef CAS.
- D. C. Crans and A. S. Tracey, ACS Symp. Ser., 1998, 711, 2–29 CrossRef CAS PubMed.
- B. Gammelgaard and O. Joens, in Handbook of Metal-Ligand Interactions in Biological Fluids: Bioinorganic Medicine, ed. G. Berthon, Marcel Dekker, New York, 1995, vol. 1, pp. 48–61 Search PubMed.
- G. N. George, I. J. Pickering, M. J. Pushie, K. Nienaber, M. J. Hackett, I. Ascone, B. Hedman, K. O. Hodgson, J. B. Aitken, A. Levina, C. Glover and P. A. Lay, J. Synchrotron Radiat., 2012, 19, 875–886 CrossRef CAS PubMed.
- R. P. Glahn, O. A. Lee, A. Yeung, M. I. Goldman and D. D. Miller, J. Nutr., 1998, 128, 1555–1561 CAS.
- S. Yun, J.-P. Habicht, D. D. Miller and R. P. Glahn, J. Nutr., 2004, 134, 2717–2721 CAS.
- Liquid breakfast, Sanitarium, Australia, http://www.sanitarium.com.au/products/breakfast/up-and-go/up-and-go-choc-ice, accessed 21.04.14.
- HEPES-buffered saline, Cold Spring Harbor Protocols., 2006, doi:10.1101/pdb.rec8786.
- A. C. Thompson, D. T. Attwood, E. M. Gullikson, M. R. Howells, J. B. Kortright, A. L. Robinson, J. H. Underwood, K.-J. Kim, J. Kirz, I. Lindau, P. Pianetta, H. Winick, G. P. Williams and J. H. Scofield, X-ray Data Booklet, University of California, Berkeley, CA, 2nd edn, 2001 Search PubMed.
- (a) P. J. Ellis and H. C. Freeman, J. Synchrotron Radiat., 1995, 2, 190–195 CrossRef CAS PubMed; (b) XFit for Windows, beta-version. Australian Synchrotron Research Program, Sydney, Australia, 2004.
- T.-C. Weng, G. S. Waldo and J. E. Penner-Hahn, J. Synchrotron Radiat., 2005, 12, 506–510 CrossRef CAS PubMed.
- W. H. McMaster, N. Kerr Del Grande, J. H. Mallett and J. H. Hubbell, Compilation of X-ray Cross Sections, Lawrence Livermore National Laboratory, Livermore, CA, 1969 Search PubMed.
- D. Lovy, WinDIG, University of Geneva, Geneva, Switzerland, 1996 Search PubMed.
- Microcal Origin Version 6.0, Microcal Software Inc., Northampton, MA, 1999.
- G. Giuli, E. Paris, J. Mungall, C. Romano and D. Dingwell, Am. Mineral., 2004, 89, 1640–1646 CAS.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849–902 CrossRef CAS PubMed.
- S. S. Amin, K. Cryer, B. Zhang, S. K. Dutta, S. S. Eaton, O. P. Anderson, S. M. Miller, B. A. Reul, S. M. Brichard and D. C. Crans, Inorg. Chem., 2000, 39, 406–416 CrossRef CAS.
- P. Chaurand, J. Rose, V. Briois, M. Salome, O. Proux, V. Nassif, L. Olivi, J. Susini, J.-L. Hazemann and J.-Y. Bottero, J. Phys. Chem. B, 2007, 111, 5101–5110 CrossRef CAS PubMed.
- A. Levina, R. S. Armstrong and P. A. Lay, Coord. Chem. Rev., 2005, 249, 141–160 CrossRef CAS PubMed.
- E. J. Baran, J. Inorg. Biochem., 2009, 103, 547–553 CrossRef CAS PubMed.
- (a) T. W. Hambley, R. J. Judd and P. A. Lay, Inorg. Chem., 1992, 31, 343–345 CrossRef CAS; (b) G. Barr-David, T. W. Hambley, J. A. Irwin, R. J. Judd, P. A. Lay, B. D. Martin, R. Bramley, N. E. Dixon, P. Hendry, J.-Y. Ji, R. S. U. Baker and A. M. Bonin, Inorg. Chem., 1992, 31, 4906–4908 CrossRef CAS; (c) R. Codd, T. W. Hambley and P. A. Lay, Inorg. Chem., 1995, 34, 877–882 CrossRef CAS; (d) R. Codd, P. A. Lay and A. Levina, Inorg. Chem., 1997, 36, 5440–5448 CrossRef CAS.
- (a) R. Codd, J. A. Irwin and P. A. Lay, Curr. Opin. Chem. Biol., 2003, 7, 213–219 CrossRef CAS; (b) C. Z. Soe, A. A. H. Pakchung and R. Codd, Inorg. Chem., 2014, 53, 5852–5861 CrossRef CAS PubMed.
- B. Song, N. Aebischer and C. Orvig, Inorg. Chem., 2002, 41, 1357–1364 CrossRef CAS PubMed.
- P. C. Wilkins, M. D. Johnson, A. A. Holder and D. C. Crans, Inorg. Chem., 2006, 45, 1471–1479 CrossRef CAS PubMed.
- I. A. Setyawati, K. H. Thompson, V. G. Yuen, Y. Sun, M. Battell, D. M. Lyster, C. Vo, T. J. Ruth, J. H. Zeisler, J. H. McNeill and C. Orvig, J. Appl. Physiol., 1998, 84, 569–575 CAS.
- K. Dralle Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef PubMed.
- (a) M. Aureliano and D. C. Crans, J. Inorg. Biochem., 2009, 103, 536–546 CrossRef CAS PubMed; (b) A. Chatkon, P. B. Chatterjee, M. A. Sedgwick, K. J. Haller and D. C. Crans, Eur. J. Inorg. Chem., 2013, 1859–1868 CrossRef CAS PubMed; (c) M. Aureliano, Inorg. Chim. Acta, 2014, 420, 4–7 CrossRef CAS PubMed.
- K. Pantopoulos, S. Kumar Porwal, A. Tartakoff and L. Devireddy, Biochemistry, 2012, 51, 5705–5724 CrossRef CAS PubMed.
- (a) I. Bertini, C. Luchinat and L. Messori, J. Inorg. Biochem., 1985, 25, 57–60 CrossRef CAS; (b) J. Costa Pessoa, G. Gonçalves, S. Roy, I. Correia, S. Mehtab, M. F. A. Santos and T. Santos-Silva, J. Inorg. Biochem., 2014, 420, 60–68 CAS.
- T. Ueki and H. Michibata, Coord. Chem. Rev., 2011, 255, 2249–2257 CrossRef CAS PubMed.
- M. Ciampolini and F. Mani, Inorg. Chim. Acta, 1977, 24, 91–95 CrossRef CAS.
- A. J. Ghio, C. A. Piantadosi, X. Wang, L. A. Dailey, J. D. Stonehuerner, M. C. Madden, F. Yang, K. G. Dolan, M. D. Garrick and L. M. Garrick, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2005, 289, L460–L467 CrossRef CAS PubMed.
- T. Ueki, N. Furuno and H. Michibata, Biochim. Biophys. Acta, 2011, 1810, 457–464 CrossRef CAS PubMed.
- (a) D. Sanna, M. Serra, G. Micera and E. Garriba, Inorg. Chem., 2014, 53, 1449–1464 CrossRef CAS PubMed; (b) D. Sanna, M. Serra, G. Micera and E. Garriba, Inorg. Chim. Acta, 2014, 420, 75–84 CrossRef CAS PubMed.
- A. Levina, A. I. McLeod, S. J. Gasparini, W. G. M. De Silva, J. B. Aitken, C. J. Glover, B. Johannessen and P. A. Lay, 2014, to be submitted.
- A. I. McLeod, A. Pulte, J. B. Aitken, A. Levina and P. A. Lay, 2014, to be submitted.
- (a) T. Kiss and A. Odani, Bull. Chem. Soc. Jpn., 2007, 80, 1691–1702 CrossRef CAS; (b) D. C. Crans, K. A. Woll, K. Prusinskas, M. D. Johnson and E. Norkus, Inorg. Chem., 2013, 52, 12262–12275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (i) Compositions of semi-synthetic meals; (ii) comparison of spectra of the reaction products in gastrointestinal media with those of the model complexes (Fig. S1, see also Fig. 1 and 2 in the main text); and (iii) overlays of experimental and fitted spectra for multiple linear regression analyses (Fig. S2, see also Table 1 in the main text). See DOI: 10.1039/c4mt00146j |
| This journal is © The Royal Society of Chemistry 2014 |