Non-fullerene acceptors for organic photovoltaics: an emerging horizon
Yuze
Lin
ac and
Xiaowei
Zhan
*b
aBeijing National Laboratory for Molecular Sciences and Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
bDepartment of Materials Science and Engineering, College of Engineering, Peking University, Beijing 100871, China. E-mail: xwzhan@pku.edu.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 10th April 2014
Abstract
Although fullerenes and their derivatives, such as PCBM, have been the dominant electron-acceptor materials in organic photovoltaic cells (OPVs), they suffer from some disadvantages, such as weak absorption in the visible spectral region, limited spectral breadth and difficulty in variably tuning the band gap. It is necessary to explore non-fullerene electron acceptors that will not only retain the favorable electron-accepting and transporting properties of fullerenes but also overcome their insufficiencies. After a decade of mediocrity, non-fullerene acceptors are undergoing rapid development and are emerging as a hot area of focus in the field of organic semiconductors. Solution-processed bulk heterojunction (BHJ) OPVs based on non-fullerene acceptors have shown encouraging power conversion efficiencies of over 4%. This article reviews recent developments in several classes of solution-processable non-fullerene acceptors for BHJ OPVs. The remaining problems and challenges along with the key research directions in the near future are discussed.
 Yuze Lin | Yuze Lin received a BS degree in chemistry from Beijing Institute of Technology in 2009. Now he is a Ph.D. student at the Institute of Chemistry, Chinese Academy of Sciences. His research interests include the synthesis of conjugated small molecules and polymers and their application in solar cells. |
 Xiaowei Zhan | Xiaowei Zhan obtained a Ph.D. degree in chemistry from Zhejiang University in 1998. Dr. Zhan worked at the University of Arizona and Georgia Institute of Technology from 2002 to 2006 as a Research Associate and Research Scientist. He has been a full professor at the Institute of Chemistry, Chinese Academy of Sciences since 2006. In 2012, he moved to Peking University. His research interests are in the development of organic and polymeric materials for organic electronics and photonics. Prof. Zhan is an associate editor of Journal of Materials Chemistry C and a Fellow of the Royal Society of Chemistry. |
1. Introduction
Organic photovoltaic cells (OPVs) fabricated by simple solution-processing techniques are a promising cost-effective alternative for the utility of solar energy, and have attracted much attention due to their advantages of low-cost, light-weight, and the capability to fabricate flexible large-area devices.1–4 Nowadays, typical solution-processed OPVs are based on a bulk heterojunction (BHJ) active layer,5 which is a blend of bicontinuous and interpenetrating electron donor (D) and electron acceptor (A) components in a bulk volume. The absorption of solar photons creates excitons (hole–electron pairs), which diffuse to the D/A interface, where they dissociate into free holes and electrons, and opposite polarity carriers (holes and electrons) are transported through the donor and acceptor channels to the anode and cathode respectively, and subsequently charges are collected at the electrodes, resulting in the generation of electrical power. Over the last decade, the development of solution-processed BHJ OPVs has seen a dramatic rise in the power conversion efficiency (PCE) from less than 1% in the earliest reports to 10% in recent publications.6 This continuous and rapid improvement in BHJ OPV device performance has contributed to the enhanced understanding of materials and device fundamentals such as charge generation and transport, and better control of materials’ structure and blend morphology. Generally, the record efficiencies in OPVs directly result from the development of new electron donor materials that exhibit improved properties such as better spectral sensitivity, enhanced hole transport and favorably tuned HOMO/LUMO (highest occupied molecular orbital/lowest unoccupied molecular orbital) energy levels which match well with those of existing acceptors.1,3 In fact, the electron acceptors are of the same importance as the electron donors for high-performance OPVs. However, the development of electron acceptor materials has lagged far behind that of donor materials.Generally, electron acceptors should possess n-type semiconducting basic properties, including relatively low LUMO/HOMO energy levels and inherent electron transporting ability.7–9 Currently, a popular strategy for tailoring the properties of electron acceptors is through introducing electron-withdrawing building blocks. Typical electron-withdrawing groups are exemplified by fullerenes (C60 and C70), cyano, perfluoroalkyl, carbonyl, imide, amide groups and their analogues, etc. The introduction of the electron-withdrawing units into π-conjugated semiconductors can lower the LUMO energy levels, because the π* energy of the LUMO of the π-conjugated system is relatively close to that of the electron-withdrawing unit, and these orbitals can mix efficiently, resulting in the stabilization of the LUMO energy. The substitution of alkyl groups in π-systems with perfluoroalkyl units or their analogues directly affects the energy of the σ-orbitals and effectively reduces the electron density at the nuclei of the atoms in the π-systems, so that both the LUMO and HOMO levels tend to be lowered.7,10 Similarly, the replacement of one or more atoms in an extended π-system with more electronegative atoms, e.g. replacing one or two carbon atoms of thiophene with nitrogen atoms to yield thiazole or thiadiazole, will also tend to lower both the LUMO and HOMO levels.10
So far, fullerenes and their derivatives, particularly 6,6-phenyl C61 butyric acid methyl ester (PC61BM) and its C70-based homologue (PC71BM), are the most successful electron acceptor materials for solution-processed BHJ OPVs,11 owing to their (i) large electron affinity and strong tendency to accept electrons from donor semiconductors; (ii) high electron mobility (μe) even in composite form; (iii) ability to form favorable nanoscale morphological networks with donor materials; (iv) isotropy of charge transport; and (v) reversible electrochemical reduction. During the past decades, significant advances have been made on fullerene-based acceptors. Appropriate chemical modifications, such as bisadduct fullerenes and endohedral fullerenes, have up-shifted their LUMO energy levels, and the cost of fullerene production has also decreased along with an increase in their synthetic yields and material purity.
On the other hand, incentives remain to develop electron acceptors with other structures that will not only retain the favorable properties of fullerenes, but also overcome their insufficiencies, such as weak absorption in the visible spectral region, limited spectral breadth, and band gap variability, which are difficult to tune in fullerene systems by chemical modification. Recently, inspired by the rapid development of high-performance electron-transporting materials for organic field-effect transistors (OFETs), some research groups have begun to explore non-fullerene acceptors for solution-processed BHJ OPVs. BHJ OPV devices based on solution-processed non-fullerene acceptors have shown PCEs of over 3%, and even up to 4%,12,13 which are significantly improved relative to those (generally less than 1.5%) disclosed three years ago.14,15 More and more novel electron acceptor materials have been designed and synthesized. Some key physical issues on OPVs based on non-fullerene acceptors, such as exciton recombination and charge transport, D/A interface and morphology optimization,16 have also attracted attention. Non-fullerene acceptors are emerging as a hot area of focus in the field of organic semiconductors.
Non-fullerene acceptors include polymers and small molecules, most of which have been investigated by blending with polymer donors (Fig. 1). Widely used polymer donors include (a) typical linear polymers such as poly(3-hexylthiophene) (P3HT), poly(2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylene vinylene) (MEH-PPV) and poly(3-(4-n-octyl)-phenylthiophene) (POPT); (b) π-conjugated side chain polymers such as 2TV-PT, 3TV4-PT, 3TV6-PT and PT5TPA; and (c) high-performance narrow band gap polymers such as PBDTTT-C-T, PTB7, PTQ1, TTV7, PDSB and PSEHTT. Non-fullerene acceptors are also blended with small molecule donors, for example, the device based on a small molecule donor p-DTS(FBTTh2)2 and a perylene diimide (PDI) acceptor yielded a PCE of 3%.17
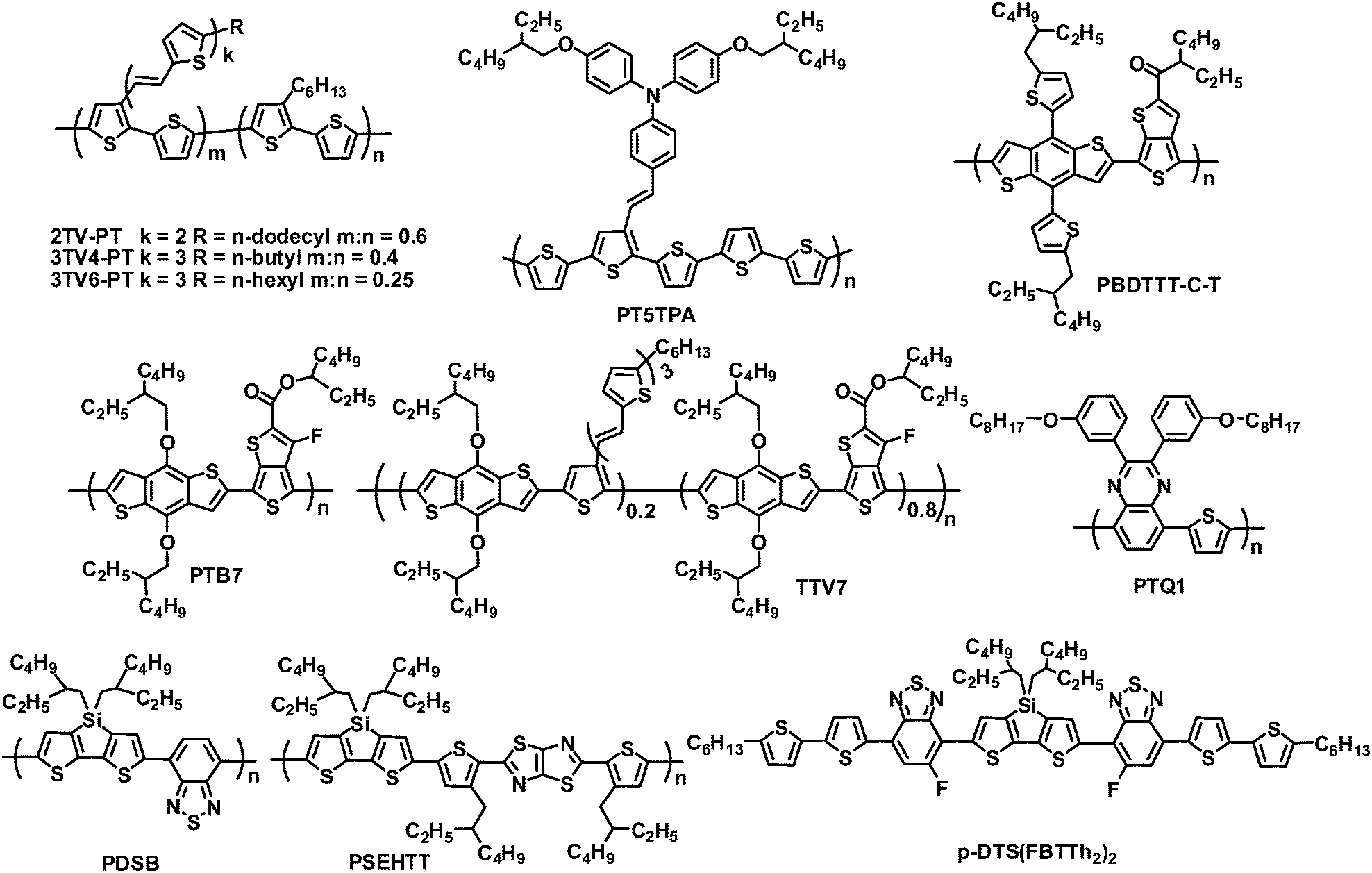 | ||
| Fig. 1 Chemical structures of several donor materials. | ||
There are several existing reviews on non-fullerene acceptor materials, which only covered small molecule materials14 or polymer materials,18 and/or did not update the latest progress and rapid development of materials and did not survey the most representative non-fullerene acceptors that have significant impacts in the field.14,15 In this review, we summarize the recent developments in several classes of solution-processable polymer and small molecule non-fullerene acceptors for solution-BHJ OPVs. Furthermore, the remaining problems and challenges and the key research directions in the near future are also discussed.
2. Polymer non-fullerene acceptors
Early in 1995, Friend,19 Heeger20 and coworkers investigated the application of n-type semiconducting polymers in OPVs by blending with polymer donors, and devices based on polymer donor/polymer acceptor blend systems are generally called all-polymer BHJ OPVs. Compared to fullerene systems, the energy levels of polymer acceptors can be tuned more efficiently by chemical tailoring. Furthermore, by combining matched electron-donating and electron-accepting building blocks, semiconducting polymers can exhibit broad absorption with high absorption coefficients in the visible spectral region due to intramolecular charge transfer (ICT), while it is quite hard to extend the absorption of fullerenes into the red and near-infrared regions, although C70 derivatives are better than their C60 counterparts. In addition, in all-polymer BHJ OPVs, polymer–polymer blends offer superior flexibility in controlling the solution viscosity, which is a critical factor for large scale OPV module production by solution processes. Some typical electron withdrawing building blocks such as PDI and benzothiadiazole (BT) were widely used in the main chain architecture of polymer acceptors. Meanwhile, cyano-containing π-conjugated polymers such as cyanated poly(phenylenevinylene)s (CN-PPVs) were also used as polymer acceptors.Polymers based on CN-PPV backbones were the first acceptors for all-polymer solar cells. Friend,19 Heeger20 and coworkers demonstrated that photogenerated excitons in the MEH-PPV and CN-PPV mixed layer can be efficiently dissociated into free carriers at the photoactive blend interface, and a PCE of 0.9% was achieved in these all-polymer BHJ systems. In contrast to laminated bilayer devices based on CN-PPV/POPT,21 Fréchet and coworkers reported BHJ OPV devices based on a CN-PPV:POPT blend, which afforded PCEs of 2.0% with a JSC of 5.5 mA cm−2 and a VOC of 1 V.22 However, in the last 5 years, there have been few reports on CN-PPV-type acceptors for OPVs. A few previous reviews have summarized CN-PPV acceptors,18 thus here we do not discuss CN-PPV acceptors, and instead focus on two other key polymeric acceptors: amide/imide-functionalized polymers and BT-based polymers.
2.1 Amide/imide-functionalized polymer acceptors
Rylene diimides have received considerable attention as alternative acceptor materials since they exhibit excellent photostability, easy alteration of their energy levels, strong optical absorption, high electron mobilities, and electron affinities similar to those of fullerenes, and each of these properties can be readily tailored through either variation of the substituents on the imide nitrogen atoms or on the rylene core.23–26 A number of polymer acceptors have been designed by incorporating rylene diimides such as PDI, naphthalene diimide (NDI) and their analogues. Rylene diimides can be polymerized via the imide or core positions. Although PDI-based polymer acceptors with connection at the imide position demonstrated promising performance in all-polymer OPVs,27,28 for example, all-polymer OPVs based on an alternating PDI–phenylenevinylene copolymer acceptor and poly(3-phenylhydrazone thiophene) donor exhibited PCEs of up to 2.32% under white light illumination calibrated to an AM 1.5 intensity of 30 mW cm−2,28 PDI polymer acceptors with connection at the bay position dominate (Fig. 2 and Table 1).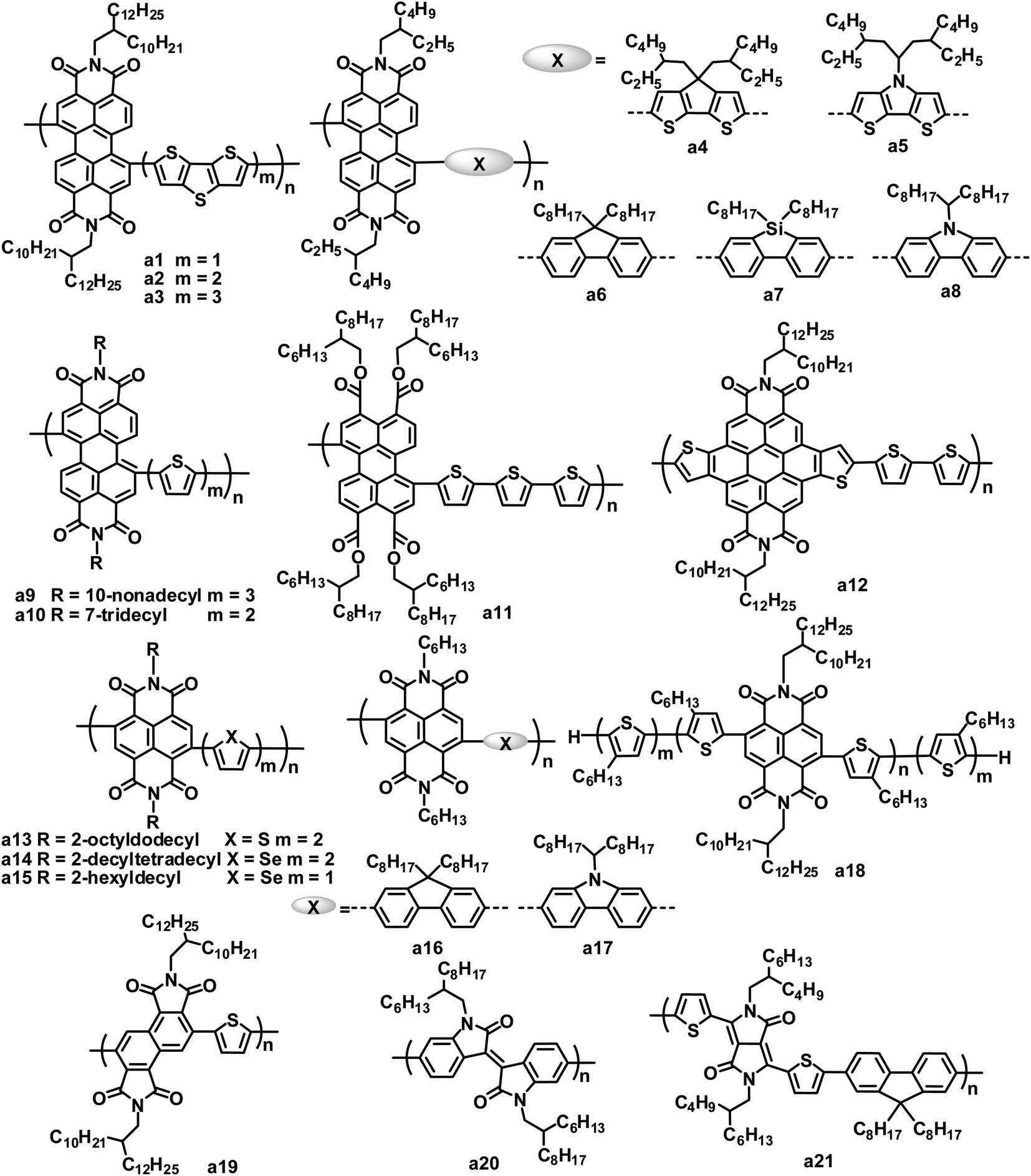 | ||
| Fig. 2 Chemical structures of amide/imide-functionalized polymers. | ||
| λ max /nm | E optg/eV | μ e /cm2 V−1 s−1 | HOMO/LUMO/eV | Donor | J SC/mA cm−2 | V OC/V | FF | PCEc (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| a In film. b O and S: measured by OFET or space charge limited current (SCLC) method; N and B: in neat or blend film. c AM 1.5G, 100 mW cm−2. | ||||||||||
| a1 | 630 | 1.7 | 0.06 (O,N) | −5.9/−3.9 | 2TV-PT | 4.2 | 0.63 | 0.39 | 1.03 | 29 |
| PBDTTT-C-T | 8.60 | 0.75 | 0.54 | 3.45 | 36 | |||||
| a2 | 647 | 1.5 | — | −5.7/−3.8 | 3TV4-PT | 5.02 | 0.69 | 0.43 | 1.48 | 34 |
| a3 | 678 | 1.5 | 7 × 10−4 (O,N) | −5.4/−4.0 | 3TV4-PT | 2.80 | 0.69 | 0.40 | 0.77 | 31 |
| a4 | 708 | 1.5 | — | −5.5/−4.0 | PDSB | 1.4 | 0.68 | 0.43 | 38 | |
| a5 | 715 | 1.32 | — | −5.5/−3.8 | 3TV6-PT | 3.05 | 0.66 | 0.46 | 0.93 | 39 |
| a6 | 560 | 1.9 | — | −5.93/−3.61 | 3TV6-PT | 1.77 | 0.76 | 0.43 | 0.58 | 40 |
| a7 | 550 | 1.9 | — | −6.01/−3.71 | 3TV6-PT | 2.80 | 0.72 | 0.40 | 0.81 | 40 |
| a8 | 550 | 1.9 | — | −5.83/−3.66 | 3TV6-PT | 6.35 | 0.70 | 0.50 | 2.23 | 40 |
| a9 | 620 | 1.6 | — | −5.94/−4.34 | P3HT | 2.81 | 0.56 | 0.51 | 0.8 | 43 |
| a10 | 594 | 1.68 | 5 × 10−4 (S,B) | −5.5/−3.8 | P3HT | 7.65 | 0.52 | 0.55 | 2.17 | 44 |
| a11 | 539 | 1.91 | 1.4 × 10−4 (S,N) | −5.60/−3.54 | P3HT | 2.74 | 0.83 | 0.34 | 0.76 | 45 |
| a12 | 642 | 1.78 | — | −5.70/−3.51 | PT5TPA | 2.14 | 0.92 | 0.426 | 0.84 | 49 |
| a13 | 701 | 1.45 | 0.85 (O,N) | −5.8/−4.0 | P3HT | 3.77 | 0.56 | 0.65 | 1.4 | 54 |
| PTB7 | 6.28 | 0.80 | 0.53 | 2.66 | 56 | |||||
| PTQ1 | 8.85 | 0.84 | 0.55 | 4.1 | 13 | |||||
| a14 | 700 | 1.4 | 0.07 (O,N) | −5.95/−3.94 | P3HT | 3.79 | 0.53 | 0.44 | 0.88 | 57 |
| a15 | 614 | 1.65 | 7 × 10−3 (O,N) | −5.65/−4.0 | PSEHTT | 7.78 | 0.76 | 0.55 | 3.26 | 58 |
| a16 | 520 | 2.1 | — | −5.93/−3.61 | 3TV6-PT | 3.63 | 0.68 | 0.66 | 1.63 | 59 |
| a17 | 530 | — | 1.7 × 10−4 (S,B) | −5.71/−3.68 | TTV7 | 7.71 | 0.88 | 0.54 | 3.68 | 61 |
| a18 | 546 | 1.46 | — | −5.60/−4.22 | P3HT | 4.57 | 0.56 | 0.50 | 1.28 | 62 |
| a19 | 513 | 1.99 | 2.15 × 10−6 (S,B) | −5.98/−3.77 | P3HT | 1.09 | 0.82 | 0.36 | 0.32 | 63 |
| a20 | 690 | 1.70 | 3.7 × 10−7 (S,N) | −5.44/−3.84 | P3HT | 1.91 | 0.62 | 0.41 | 0.47 | 64 |
| a21 | 650 | 1.72 | 5 × 10−6 (S,N) | −5.43/−3.71 | P3HT | 1.63 | 0.90 | 0.25 | 0.37 | 65 |
Studies on rylene diimide-based polymers began with the research of Zhan et al., using the PDI and dithienothiophene (DTT) alternating copolymer a1 as the electron acceptor.29 The LUMO/HOMO energy levels of a1 were estimated at −3.9/−5.9 eV by electrochemistry. The thin film of a1 showed significant absorption throughout the visible region and extending into the near infrared region, stemming from a strong ICT of the donor–acceptor arrangement of the main chain, along with a low band gap of 1.7 eV and a high electron mobility of up to 0.06 cm2 V−1 s−1.30 The blend film of the bis(thienylenevinylene)-substituted polythiophene (2TV-PT) donor and the a1 acceptor exhibited very broad absorption from 250 to 850 nm. An average PCE of 1.03% (JSC = 4.2 mA cm−2, VOC = 0.63 V and FF = 0.39) was obtained for the all-polymer BHJ OPVs based on 2TV-PT:a1.
Zhan and coworkers also explored a series of PDI and DTT copolymers with 1 to 3 DTT units, and investigated the effect of the DTT number on their electronic, optical and photovoltaic properties.31–33 With an increasing number of DTT units, the maximum absorption peak of the polymers red shifted from 630 to 678 nm and the band gap decreased from 1.7 to 1.5 eV; the HOMO level increased from −5.9 to −5.4 eV, while the LUMO levels were essentially insensitive. As the number of DTT units increased, the electron mobility of the polymers decreased by 2 orders of magnitude. All-polymer BHJ OPVs based on copolymer a2 with 2 DTT units gave a higher PCE than those based on a1 with 1 DTT unit and a3 with 3 DTT units. A blend of donor tris(thienylenevinylene)-substituted polythiophene (3TV4-PT) and a2 achieved a good device performance: a JSC of 5.02 mA cm−2, VOC of 0.69 V, FF of 0.43 and PCE of 1.48%.34
Zhan and coworkers further investigated the photovoltaic properties of the a1 acceptor blended with different polymer donors.35,36 The absorption, energy levels, hole mobility and crystalline properties of the polymer donors significantly affect the performance of all-polymer OPVs. An exciting PCE of 3.45% was obtained with a blend of PBDTTT-C-T and a1, which is the highest PCE value reported for PDI-based all-polymer OPVs.36 Since a dynamic Monte Carlo simulation of the all-polymer solar cells based on a1 indicates that a 5% PCE could be achieved with an optimum phase separation morphology,37 even higher PCE values could be expected for all-polymer OPVs based on PDI polymer acceptors. Very recently, Zhan and coworkers demonstrated the first example of large area roll-to-roll processed flexible all-polymer OPVs, prepared under ambient conditions without ITO, vacuum steps or the spin-coating method; blends of the a1 acceptor and a narrow band gap polymer donor (PDSB) yielded a PCE of 0.2%.35
Later, some other copolymers based on PDI and fused ring units, such as cyclopentadithiophene (CDT),38 dithienopyrrole (DTP), fluorene, dibenzosilole (DBS) and carbazole etc.,39–41 were synthesized and applied in BHJ OPVs as acceptor materials. PDI–CDT copolymer a4 and PDI–DTP copolymer a5 have similar structures and LUMO/HOMO energy levels to polymer a1; the devices based on blends of PDSB and a4 exhibited PCEs of 0.43%,38 and for a5, a PCE of 0.93% was achieved in a device blended with the 3TV6-PT donor.39 Hashimoto and coworkers replaced fused thiophene rings in polymers a1, a4 and a5 with fused benzene rings such as fluorene, DBS and carbazole to afford copolymers a6–a8.40 Relative to a1, a4 and a5, a6–a8 showed blue-shifted absorption maxima, larger band gaps (2.17 to 2.30 eV), higher LUMO levels (−3.61 to −3.71 eV) and lower HOMO levels (−5.83 to −6.01 eV), which are attributed to the relatively weak electron-donating ability of the fused benzene ring blocks. The VOC value of OPV devices mainly depends on the energy difference between the LUMO of the acceptor and the HOMO of the donor.42 In all-polymer OPVs using the same 3TV6-PT donor material, a6–a8 yielded VOC values of 0.70–0.76 V, higher than that (0.66 V) of a5. By careful BHJ morphology optimization, a PCE of up to 2.23% for 3TV6-PT:a8 was achieved using a mixed solvent of toluene and chloroform at a volume ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
Copolymers of PDI with thiophene,40 bithiophene or terthiophene43 were also used as acceptors in OPVs. On increasing the thiophene number in the polymer main chain, these polymers showed red-shifted absorption. All-polymer OPVs based on the blend of P3HT and a9 showed a PCE of 0.8%.43 In contrast, PDI and thiophene or bithiophene copolymers gave lower PCEs of 0.2 (ref. 40) to 0.4%,43 when blended with P3HT. Usually, the dibromination of PDI affords a mixture of 1,6- and 1,7-regioisomers of PDI dibromides. Since this mixture is difficult to purify, most PDI polymers were synthesized using the mixture monomer, which limited their photovoltaic properties to some extent. Zhao and coworkers isolated and purified the 1,7-dibromo PDI isomer via repeated column chromatography, and then synthesized a regio-regular 1,7-PDI–bithiophene copolymer a10.44 Compared to its irregular counterpart, polymer a10 exhibited nearly identical absorption, optical band gap (1.68 eV) and LUMO/HOMO energy levels (−3.8/−5.5 eV), but a higher electron mobility (5 × 10−4vs. 3 × 10−4 cm2 V−1 s−1). When blended with P3HT, the regio-regular polymer a10 yielded a higher PCE relative to its irregular counterpart (2.17% vs. 1.55%).
As stated above, PDI-based alternative copolymers generally have LUMOs of −3.7 to −4.0 eV, similar to that (−3.9 eV) of PC61BM. The relatively large LUMO energy gaps between P3HT (−2.7 to −3.0 eV) and PDI polymers leads to energy loss and low VOC values. To solve this problem, PDI analogues with raised LUMO levels have been developed. Replacing 2 imide groups with 4 carboxylic ester groups yields perylene tetracarboxylic tetraester (PTTE), leading to a negative 0.3 V shift in both the oxidative and reductive potentials of PDI. Thus, PTTE-based polymers have up-shifted LUMO levels relative to PDI-based polymers, which is beneficial to the improvement of the VOC of solar cells. PTTE–terthiophene copolymer a11 had a LUMO level (−3.54 eV) which matched with P3HT; all-polymer OPVs based on P3HT:a11 blends yielded a relatively high VOC of 0.83 V, resulting in a PCE of 0.76%.45
Annulation of PDI with a variety of cyclic units in the bay region is another way to up-shift the LUMO/HOMO energy levels of PDI. Recently, a product of thiophene annulation of PDI, dithienocoronene diimide (DTCDI), was used for the synthesis of organic semiconductors.46–49 Relative to PDI, DTCDI has a larger size core and weaker electron-deficient properties. There is a linear monomer linkage geometry with negligible dihedral angles in DTCDI-based polymer backbones, whereas PDI-based polymers have 1,6- and 1,7-regioisomers as well as two completely different backbone architectures: zigzag vs. linear geometry. DTCDI–oligothiophene copolymers showed high electron mobilities of up to 0.30 cm2 V−1 s−1,48 and LUMO energy levels of −3.5 to −3.7 eV. All-polymer OPVs based on a blend of the π-conjugated side chain polymer donor PT5TPA and the DTCDI–bithiophene copolymer acceptor a12 yielded a JSC of 2.14 mA cm−2, VOC of 0.92 V, FF of 0.426 and PCE of 0.84%.49
Compared to its large-sized analogue PDIs, NDIs are readily brominated to yield a pure 1,5-dibromoisomer, enabling the synthesis of a regioregular polymeric backbone. Due to the higher regioregularity, the resulting NDI copolymers possess enhanced π-delocalization and π-stacking, and consequently, better electron transport relative to their PDI counterparts. NDI and bithiophene copolymer a13 exhibited an exceptional electron mobility as high as 0.85 cm2 V−1 s−1,50 which can largely be attributed to the significant thin film crystallinity of a13.
The use of a13 as an acceptor in OPVs has attracted much attention due to its high electron mobility. Loi and coworkers firstly reported the photovoltaic properties of P3HT:a13 blend thin films.51 The FF values of the P3HT:a13 BHJ OPVs were very large (ca. 0.70), and benefited from the high charge separation efficiency and balanced carrier mobilities in the blend (hole mobility = 2 × 10−3 cm2 V−1 s−1 and electron mobility = 4 × 10−3 cm2 V−1 s−1).52 However, due to major charge carrier losses caused by geminate recombination, the devices exhibited very low JSC values of 0.34 to 0.49 mA cm−2, leading to very low PCEs (<0.16%).51 By improving the blend phase separation using xylene as the solvent, the PCEs increased to 0.62%, with a JSC of 2.39 mA cm−2 and FF of 0.54. Meanwhile, Sirringhaus and coworkers also investigated in detail the morphology, photophysics and device physics of the P3HT:a13 blends to further rationalize why, despite a high electron mobility and the near infrared absorption band of a13, the corresponding solar cells perform poorly.53 In the P3HT:a13 system, the rapid initial geminate recombination of the charge population and the morphology being far from ideal causes the low overall OPV efficiencies. Neher and coworkers clearly demonstrated that the NDI-based all-polymer OPV device performance was strongly enhanced when preventing the polymers from forming large and well-ordered crystallites in the BHJ layer. By adding cyanonaphthalene to the P3HT:a13 xylene solution to optimize the morphology, a marked performance improvement, with a best PCE of 1.4%, was achieved for a 1:1 xylene–cyanonaphthalene mixture.54
All-polymer OPVs based on blends of the low band gap polymer donor PTB7 and a13 yielded a PCE of 1.1%.55 Replacing P3HT with PTB7 as the donor resulted in a higher VOC along with a spectral response which was better matched with the solar spectrum. Recently, through careful selection of the solvent and film formation conditions, favorable PTB7:a13 blend morphologies were achieved, along with enlarged D/A interfacial areas and interpenetrating networks of both polymer domains. Such optimization strategies also afforded enhanced polymer/polymer π–π stacking as well as more efficient charge transport behavior both parallel and perpendicular to the substrates for the xylene-processed films. Finally, these PTB7:a13 blend films yielded PCEs of up to 2.7%.56 Meanwhile, polymer PTQ1 was also used in all-polymer OPVs by blending with acceptor a13, and the device showed a JSC of 8.85 mA cm−2, VOC of 0.84 V, FF of 0.55 and PCE of 4.1%, which is the highest value reported for all-polymer solar cells.13
Jenekhe and coworkers reported a new NDI-based crystalline polymer (a14) by replacing the bithiophene block in a13 with biselenophene.57 Polymer a14 has a high electron mobility of 0.07 cm2 V−1 s−1 and a broad visible and near infrared absorption band with a narrow optical band gap of 1.4 eV. All-polymer BHJ OPVs comprised of the P3HT donor and the a14 acceptor exhibited a PCE of 0.88%, along with a JSC of 3.79 mA cm−2, VOC of 0.53 V and FF of 0.44. They compared the NDI–thiophene copolymers with the NDI–selenophene copolymers as acceptors in OPVs by blending with PSEHTT, a narrow band gap polymer donor .58 The NDI–selenophene copolymers possessed smaller band gaps and higher electron mobilities than the NDI–thiophene copolymers. The devices based on PSEHTT and the NDI–selenophene copolymer a15 with 2-hexyldecyl side chains gave higher performance: a JSC of 7.78 mA cm−2, VOC of 0.76 V, FF of 0.55 and PCE of 3.26%. In comparison, the PSEHTT/NDI–thiophene copolymer gave a lower PCE of 1.30%.
Similar to PDI-based copolymers, fused ring electron donating units have also been used in the synthesis of NDI-based polymer acceptors.41,54,59–61 Hashimoto and coworkers synthesized an alternating copolymer (a16) of NDI and fluorene and investigated its photovoltaic properties as an acceptor in combination with a 3TV6-PT donor.59 By adding 1,8-diiodooctane (DIO) as a solvent additive to optimize the mixing morphology of the 3TV6-PT:a16 blend, the FF and PCE reached values of 0.66 and 1.63%, respectively. They also reported an NDI and carbazole alternating copolymer (a17), and investigated its photovoltaic properties by blending with a block polymer donor (TTV7) with broad absorption.61 The addition of DIO improved the PCE from 1.60% to 3.68%, which was mainly caused by the improvement in the crystallinity of a17 before significant phase separation occurred.
Nakabayashi and coworkers synthesized a block copolymer (a18) composed of P3HT and polyNDI segments. This polymer showed broad absorption in the range of 350 to 850 nm and exhibited a narrow optical band gap of 1.46 eV.62 All-polymer solar cells based on the P3HT:a18 blend film showed a PCE of 1.28%, with a JSC of 4.57 mA cm−2, VOC of 0.56 V and FF of 0.50.
Very recently, a series of n-type polymers were designed and synthesized by using angular-shaped NDI isomers as building blocks to tune the energy levels.63 The copolymers based on angular-shaped NDI and oligothiophene blocks have high-lying LUMO levels (ca. −3.7 eV) relative to those (ca. −4.0 eV) of their linear NDI counterparts. These polymers yielded relatively high VOC values up to 0.94 V when blended with the P3HT donor, and P3HT:a19 gave the highest PCE of 0.32%.
Parallel to the PDI and NDI-based polymeric acceptors, some other amide/imide functional blocks have been used to build polymer acceptors.64,65 The isoindigo homopolymer a20 showed a LUMO level of −3.84 eV and broad absorption throughout the visible light region.64 All-polymer OPVs based on the P3HT:a20 blend exhibited a PCE of 0.47%, with a JSC of 1.91 mA cm−2, VOC of 0.62 V and FF of 0.41. Janssen and coworkers developed diketopyrrolopyrrole (DPP)-based acceptor polymers for photovoltaic applications.65 Combining with P3HT, DPP and fluorene, copolymer a21 gave a PCE of 0.37%. Such a low efficiency resulted from the low electron mobility (5 × 10−6 cm2 V−1 s−1) of acceptor a21 combined with the inefficient generation of long-lived free charge carriers. DPP-based polymer acceptors have not shown good results in OPVs, whereas DPP-based small molecular acceptors have demonstrated promising PCEs (see Section 3.2).
2.2 Benzothiadiazole-based copolymers
The BT unit is a strong electron-withdrawing nitric heterocycle, used in electron acceptor polymers for OPVs (Fig. 3 and Table 2). Early in 2001, Friend and coworkers first investigated the photovoltaic properties of BT-based polymeric acceptor b1, which consists of BT and fluorene blocks and has a high electron affinity.66 Bradley and coworkers optimized OPVs based on P3HT:b1 blends by using LiF as an interlayer, and a PCE of 0.13% was achieved.67 This low efficiency was mainly attributed to poor charge generation and separation resulting from the failure of P3HT to reorganize.68 Kim and coworkers improved the all-polymer OPV performance by doping the acceptor b1 with an organosulfonic acid (4-ethylbezenesulfonic acid (EBSA)).69,70 The EBSA doping of b1 formed polymer b2 with various EBSA weight ratios, which exhibited an ionization potential shift towards lower energy, and a greatly enhanced electron mobility (b2vs.b1: 5 × 10−7 to 10−5vs. 10−8 cm2 V−1 s−1). Thus, the JSC of the solar cells was improved by over twofold (10 wt% EBSA doping), while the VOC increased by ca. 0.4 V. By using 1 wt% EBSA-doped P3HT as the donor and b2 (10 wt% EBSA doping) as the acceptor, the all-polymer devices showed a highest PCE of 0.4%, much higher than those (0.1%) of the control devices based on P3HT:b1 without doping.70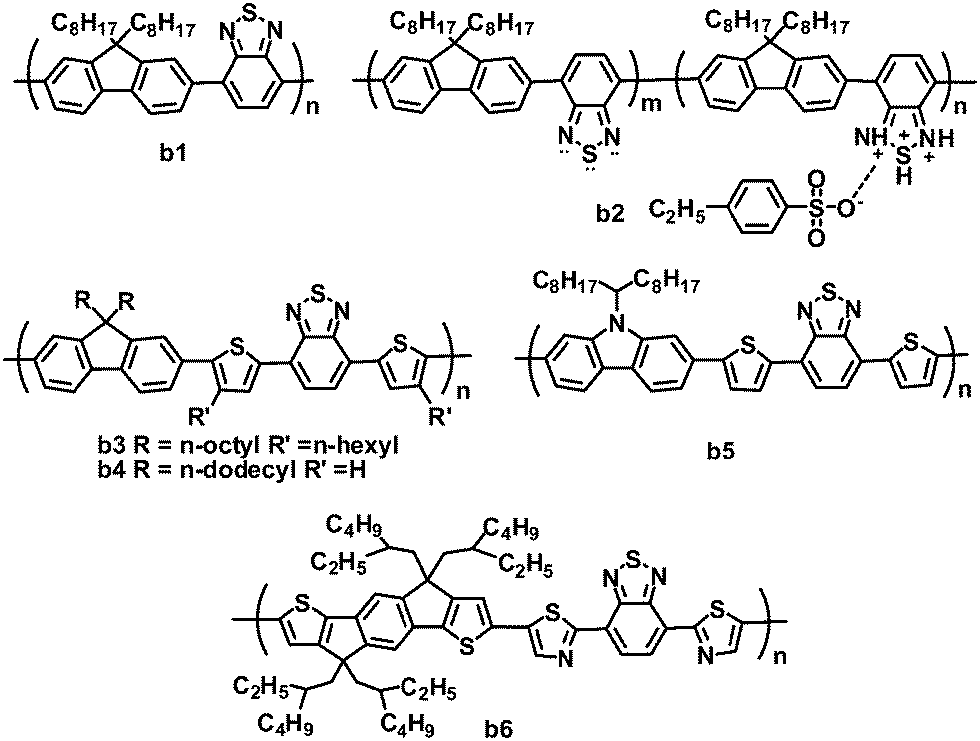 | ||
| Fig. 3 Chemical structures of BT-based polymers. | ||
| λ max /nm | E optg/eV | μ e /cm2 V−1 s−1 | HOMO/LUMO/eV | J SC/mA cm−2 | V OC/V | FF | PCEc (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a In film. b O and S: measured by OFET or SCLC method in neat film. c AM 1.5G, 100 mW cm−2. | |||||||||
| b1 | 480 | 2.3 | 10−8 (S) | −5.8/−3.7 | — | — | — | 0.13 | 67 |
| b2 | 480 | 2.3 | 5 × 10−7 (S) | −5.8/−3.5 | 1.07 | 1.26 | 0.297 | 0.40 | 70 |
| b3 | 570 | 2.0 | 8 × 10−5 (O) | −5.37/−3.15 | — | — | — | 1.8 | 71 |
| b4 | 580 | 2.0 | — | −5.5/−3.5 | 3.88 | 1.26 | 0.55 | 2.7 | 75 |
| b5 | 780 | — | — | — | 3.91 | 0.96 | 0.356 | 1.33 | 76 |
| b6 | 634 | 1.77 | 1.1 × 10−2 (O) | −5.43/−3.45 | 2.60 | 1.00 | 0.45 | 1.18 | 77 |
Inserting a thiophene bridge between BT and fluorene in polymer b1 formed polymer b3.71,72 Polymer b3 had a very high LUMO energy level of −3.15 eV and a field-effect electron mobility of 8 × 10−5 cm2 V−1 s−1, and OPV devices based on P3HT:b3 gave a highest PCE of 1.8%. Furthermore, the same blend system was investigated by Huck and coworkers. They used a novel approach to nanopattern the BHJ through a double nanoimprinting process that allows the formation of nanostructured polymer heterojunctions, the composition and morphology of which can be selected independently.73 The nanostructured devices showed a clear improvement in the PCE (up to 1.85%), 50% higher than the blend control devices. In this system, due to the large gap between the HOMO of P3HT and the LUMO of b3, the OPV devices gave a very high VOC of over 1.1 V.
Miyake and coworkers synthesized polymer acceptor b4 by modifying the molecular structure of polymer b3, removing the n-hexyl side chain on thiophene and replacing the n-octyl chain on the fluorene unit with a longer n-dodecyl chain.74 Compared to polymer b3, b4 possessed a smaller optical band gap and a larger absorption coefficient. The improved light absorption of b4 may be attributed to the absence of n-hexyl chains at the thiophene bridges, which reduced the steric hindrance and further increased the planarity of the polymer backbone. The morphologies of P3HT:b4 using different solvents were investigated in detail, and the blend film spin-coated from chloroform solution showed nanoscale phase-separated domains, suggesting well-mixed blend morphology. In contrast, blend films fabricated from o-dichlorobenzene (o-DCB) or chlorobenzene (CB) gave very clear phase-separated structures on the micro- or submicrometer scale. Thus, the devices fabricated from chloroform solution yielded a PCE of 2.0%. Furthermore, on increasing the molecular weight of b4 from 8500 to 78000 g mol−1, the device PCE increased from 1.9% to 2.7%, with a very high VOC of 1.26 V.75 The value of 2.7% is the highest PCE reported so far for all-polymer OPVs based on the P3HT donor.
Friend and coworkers fabricated all-polymer OPVs using the BT–carbazole copolymer b5 as the acceptor and P3HT as the donor, and optimized the morphology and crystallization of the polymer blends by solvent additive control.76 Due to its lower volatility than the principal solvent (chlorobenzene) and its selective solubility towards one blend component, 4-bromoanisole, as an effective and versatile solvent additive, promoted P3HT crystallization during the processing of P3HT:b5 blends, thereby forming pure, ordered and interpenetrating domains close to the ideal size. This improved morphology in turn led to enhanced OPV performance. The optimized devices showed PCEs of up to 1.33%, with a JSC of 3.91 cm−2, VOC of 0.96 and FF of 0.356.
Recently, the ladder-shaped indacenodithiophene (IDT) unit was incorporated into BT-based polymers,77 where IDT and BT blocks were linked by thiophene or thiazole bridges. These polymers showed relatively broad absorption with optical band gaps of ca. 1.77 eV. The polymer b6, containing electron-deficient thiazole units, had lower LUMO (−3.45 eV) and HOMO (−5.43 eV) levels than its thiophene-containing counterpart (LUMO/HOMO: −3.21/−5.28 eV), and b6 also exhibited a higher electron mobility (1.1 × 10−2vs. 2.9 × 10−4 cm2 V−1 s−1) and better miscibility with P3HT. As expected, polymer b6 is more suitable to blend with P3HT for all-polymer OPVs; P3HT:b6-based devices yielded a PCE of 1.18%, JSC of 2.6 mA cm−2, VOC of 1 V and FF of 0.44.
3. Small molecule non-fullerene acceptors
Small molecular non-fullerene acceptors for OPVs have attracted considerable attention due to their advantages over their polymer counterparts, which include a well-defined molecular structure, definite molecular weight, and high purity without batch to batch variations.1,78–80 Early in 1986, small molecular non-fullerene acceptors were used in bilayer heterojunction OPVs; Tang fabricated the first bilayer heterojunction solar cell by using a copper phthalocyanine as the donor and a PDI analogue as the acceptor, with the solar cell exhibiting an efficiency approaching 1%, which was a milestone in the development of OPVs.81 However, the performance of OPVs based on this bilayer structure is limited by the short exciton diffusion length in organic materials (typically 5–20 nm).82 Along with the rise of solution-processed BHJ OPVs, fullerene derivatives have gradually occupied a dominating position as electron acceptor materials. Nowadays, small molecule non-fullerene acceptors exhibit an uprising trend after a decade of mediocrity. In this section, we focus on recent promising examples of small molecule non-fullerene acceptors for solution-processed BHJ OPVs.3.1 Small molecular rylene diimides and their analogues
PDI small molecules are the earliest and most common non-fullerene acceptors investigated in OPVs. Since the first bilayer heterojunction OPVs in 1986,81 many early studies of OPVs incorporating PDIs consisted of layered structures fabricated by vacuum deposition. Since then, solution-processed BHJ OPVs based on blends of PDI acceptors with appropriate donor materials have attracted increasing attention (Fig. 4 and Table 3).23 PDI-based molecules can be solution-processed by introducing solubilizing groups such as alkyl groups on the imide nitrogen atoms. 3-Pentyl-substituted PDI (c1) was blended with typical donors such as P3HT and polycarbazole to fabricate OPVs by spin coating.83,84 By careful control of the solvent, annealing temperature and time, and the substrate temperature during deposition, the devices based on the P3HT:c1 (1:4) film showed a PCE of 0.25%, with a JSC of 1.65 mA cm−2, VOC of 0.45 V and FF of 0.34.84 Nonetheless, the strong tendency for c1 to form crystalline domains within polymer matrices limited the efficiencies of BHJ devices since the crystalline domains acted as electron traps which decreased the photocurrent.
 | ||
| Fig. 4 Chemical structures of PDI and NDI and related small molecules. | ||
| λ max /nm | E optg/eV | μ e /m2 V−1 s−1 | HOMO/LUMO/eV | Donor | J SC/mA cm−2 | V OC/V | FF | PCEc (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| a In film. b O and S: measured by OFET or SCLC method; N and B: in neat or blend film. c AM 1.5G, 100 mW cm−2. | ||||||||||
| c1 | 530 | — | — | −5.8/−3.8 | P3HT | 1.65 | 0.45 | 0.34 | 0.25 | 84 |
| c1 | — | — | 1.7 × 10−4 (S,B) | — | p-DTS(FBTTh2)2 | 7.4 | 0.78 | 0.52 | 3.0 | 17 |
| c2 | — | — | — | — | P3HT | 1.74 | 0.75 | 0.38 | 0.50 | 86 |
| c3 | 545 | — | 8 × 10−3 (O,N) | −5.9/−4.1 | PBDTTT-C-T | 9.0 | 0.77 | 0.46 | 3.20 | 88 |
| c4 | 522 | 1.88 | 1.2 × 10−3 (S,B) | −5.83/−3.95 | P3HT | 2.01 | 0.43 | 0.472 | 0.41 | 89 |
| c5 | 545 | 1.81 | 4.1 × 10−4 (S,B) | −5.65/−3.84 | P3HT | 2.89 | 0.59 | 0.448 | 0.76 | 89 |
| 1.0 × 10−3 (S,B) | — | PBDTTT-C-T | 8.86 | 0.85 | 0.541 | 4.03 | 12 | |||
| c6 | 574 | 1.75 | 7.1 × 10−4 (S,B) | −5.57/−3.84 | P3HT | 3.83 | 0.67 | 0.60 | 1.54 | 89 |
| c7 | 534 | — | 3.4 × 10−4 (S,B) | −5.48/−3.84 | P3HT | 5.83 | 0.68 | 0.49 | 1.95 | 90 |
| c8 | 550 | 2.0 | 7.1 × 10−5 (S,B) | −5.71/−3.71 | P3HT | 5.92 | 0.61 | 0.65 | 2.35 | 91 |
| c9 | 550 | 2.0 | 1.6 × 10−4 (S,B) | −5.71/−3.71 | P3HT | 6.27 | 0.61 | 0.60 | 2.28 | 91 |
| c10 | 533 | 2.1 | 5.11 × 10−4 (O,N) | −5.94/−3.84 | PBDTTT-C-T | 10.58 | 0.73 | 0.468 | 3.63 | 92 |
| c11 | 536 | 1.76 | 3.0 × 10−5 (S,N) | −5.40/−3.70 | PBDTTT-C-T | 11.27 | 0.87 | 0.328 | 3.22 | 93 |
| c12 | 400 | 1.6 | 0.12 (O,N) | −5.3/−3.7 | PSEHTT | 5.14 | 0.79 | 0.44 | 1.80 | 94 |
| c13 | 654 | 1.57 | 6.9 × 10−4 (O,N) | −5.5/−4.1 | P3HT | 3.51 | 0.82 | 0.52 | 1.50 | 97 |
| c14 | 433 | 2.20 | — | −5.81/−3.61 | P3HT | 4.87 | 0.58 | 0.57 | 1.6 | 98 |
The micrometer scale crystallization of PDIs in blends has an unfavorable impact on the performance of PDI-based OPVs. Therefore, disrupting the crystallinity without adversely affecting the charge transfer properties of PDIs is proposed as an important molecular design principle. Keeping this in mind, some studies have examined the impact of substituents on the imide nitrogen atoms or at the bay regions of the PDIs on device performance.85,86 Although PDIs have shown tunable LUMO energy levels over a range of ca. 0.7 eV, which affected the VOC of the devices, the introduction of longer alkyl substitutions on the imide nitrogen atoms and/or electron-rich or deficient substitutions at the bay region of PDIs has decreased device efficiencies to some extent when blended with P3HT, compared to the simple PDI c1.85 Laquai and coworkers demonstrated that the photovoltaic characteristics of blended films of P3HT and PDIs were improved upon using a non-bay-alkylated PDI derivative (c2) instead of the often-used N-alkylated PDIs.86 The alkyl substitution pattern affected the packing of the PDIs and improved the donor/acceptor mixing. The optimized OPV based on P3HT:c2 exhibited a PCE of 0.5%, with a JSC of 1.74 mA cm−2, VOC of 0.75 V and FF of 0.38. This moderate PCE of 0.5% is twice that of P3HT:c1.
Aside from modifying the PDI cores by alkyl side chain substitution to decrease the tendency of their crystallization, PDI dimers can also achieve similar effects. In molecule c3, in which two PDI units are brought together using hydrazine as a linker, the central N–N single bond is surrounded by four carbonyl groups, wherein the oxygen atoms carry a partial negative charge. To minimize the electronic repulsion between the oxygen atoms, the imide planes and hence the perylene units are oriented perpendicular to each other.87 The resulting loss of planarity results in lower phase segregation, reducing the formation of intermolecular states, expediting charge separation at the interface and finally improving the JSC and PCE. In combination with a low band gap polymer donor (PBDTTT-C-T), an efficiency of 3.2% has been achieved, along with a VOC of 0.77 V, JSC of 9.0 mA cm−2 and FF of 0.46.88 A ten-fold increase in JSC is observed in comparison with a control planar PDI monomer.87
Yao and coworkers designed and synthesized a series of PDI dimers (c4 to c6) with a thiophene bridge on their bay regions and investigated their photovoltaic properties by blending with P3HT.89 The planarity and aggregation ability of these PDI dimers are reduced by the rotatable thiophene bridge. Their compatibility with P3HT is tuned by changing the number of weakly solvophobic 2-methoxyethoxyl (EG) side chains. As increasing the number of EG side chains from 0 to 2, and then to 4, the device PCE was improved from 0.007% to 0.39%, and then to 0.88%. After using 1-chloronaphthalene as a solvent additive in o-DCB to further improve the compatibility and phase separation, the PCE was further increased up to 0.41% for PDI dimer c4 without EG, 0.76% for PDI dimer c5 with 2 EGs and 1.54% for PDI dimer c6 with 4 EGs. Replacing thiophene in c5 with 5-alkylthiophene-2-yl-substituted benzodithiophene (BDT) afforded PDI dimer c7.90 Molecule c7 has optimal conformations with a calculated dihedral angle of ca. 50° between the PDI–BDT–PDI planes; this highly twisted structure is expected to result in largely reduced aggregation. The best device based on a blend of P3HT:c7 exhibited a JSC of 5.83 mA cm−2, VOC of 0.68 V and FF of 0.49, leading to a PCE as high as 1.95%, which was higher than that (0.76%) of c5. The same group also blended c5 with the low band gap polymer donor PBDTTT-C-T. A solution-processed OPV based on PBDTTT-C-T:c5 showed a PCE of 0.77% without any post-treatment. After optimizing the blend morphology by adding 5% DIO solvent additive, the device exhibited a PCE as high as 4.03%, with a JSC of 8.86 mA cm−2, VOC of 0.85 V and FF of 0.541.12 This PCE of 4.03% is the highest reported for non-fullerene small molecule electron acceptors. In the control device, its monomeric counterpart PDI with 2 EGs yielded a poor PCE of 0.13%. The PBDTTT-C-T:c5 blend showed a significant reduction in the aggregation, leading to a JSC of up to 8.86 mA cm−2, 26 times higher than that (0.33 mA cm−2) of monomeric PDI.
In parallel, Zhao and coworkers also developed a series of PDI dimers featured with six different arylene bay linkers: para-phenylene, meta-phenylene, thiophene, bithiophene, spirobifluorene-2,7-diyl and spirobifluorene-2,2′-diyl, but no EG side chains.91 These PDI dimers showed strong absorption between 450 and 600 nm, with a perceptibly reduced optical band gap relative to that of monomer PDI. Thin films of these dimers had similar absorption spectra to those in solution, indicating minimal intermolecular aggregation in the thin films. Of these dimers, isomers c8 and c9 with non-coplanar spirobifluorene linkers gave almost identical absorption and energy levels, and moderate electron mobility (10−5 to 10−4 cm2 V−1 s−1) when blended with P3HT. Furthermore, the spirobifluorene groups facilitated the formation of a high-dimension electron transport network in the blend, by effectively discouraging one-dimensional stacking of the molecules. Finally, the inverted OPV devices based on P3HT:c8 or c9 yielded PCEs of 2.35% and 2.28%, respectively.
Wang and coworkers investigated the effect of different bay-linkages in well-defined singly-linked, chiral doubly-linked and graphene-like triply-linked PDI dimers.92 Compared to the locked twisted doubly-linked PDI dimers with nearly perpendicular PDI units and the rigid planar triply-linked PDI dimers, the singly-linked PDI dimers (c10) which have a flexibly twisted structure with an approximately 70° angle between the two PDI units, gave a much higher PCE (3.63% vs. 1.54% for doubly-linked PDIs and 1.36% for triply-linked PDIs) when blended with the polymer donor PBDTTT-C-T.
Very recently, Zhan and coworkers explored a novel, nonplanar, star-shaped PDI acceptor (c11) with a triphenylamine core.93 Compound c11 exhibited a quasi-3D structure, weak intermolecular interactions and molecular aggregation, and complementary absorption and an appropriate energy level match with PBDTTT-C-T. OPVs based on PBDTTTC-T:c11 blend films with 5% DIO solvent additive exhibited a PCE of 3.22%.
While PDIs gave some promising OPV results, several heterocyclic diimides, tetraazabenzodifluoranthene diimides (BFIs), were explored as novel n-type semiconductors for OFETs and OPVs.94 Inserting tunable tetraazaanthracenes into the PDI core yielded BFIs; these large rigid π-conjugation ladder-type structures possess high electron affinity by combining electron-deficient pyrazine with imide groups. All of the BFIs exhibited FET electron mobilities of 0.021 to 0.12 cm2 V−1 s−1. OPVs using a low band-gap polymer, PSEHTT, as the donor and thienyl-substituted BFI (c12) as the acceptor showed a PCE up to 1.8%, with a JSC of 5.14 mA cm−2, VOC of 0.79 V and FF of 0.44.
Compared to the PDI-based molecules, the smaller fused-ring unit naphthalene diimide (NDI)-based small molecules were less successful as electron acceptors in solution-processed OPVs, because they possess better planarity and stronger intermolecular interactions than their PDI analogues, along with a larger band gap and thus poorer absorption in the visible spectrum (generally the onset is less than 400 nm). Jenekhe and coworkers reported a series of molecules with NDI as the cores and oligothiophene as the arms for BHJ OPVs.95 These NDIs showed field-effect electron mobilities from 5.0 × 10−6 to 9.0 × 10−4 cm2 V−1 s−1 and LUMO levels from −3.97 to −4.14 eV. Compound c13 with terthiophene arms had a bicontinuous nanoscale morphology and a good photovoltaic response when blended with P3HT. In comparison, its homologues with bithiophene or tetrathiophene arms showed no photovoltaic response due to their microscale phase-separated morphologies. After annealing at 100 °C for 10 min and adding 0.2% DIO additive, the optimized P3HT:c13-based device showed a PCE of 1.5%, with a JSC of 3.51 mA cm−2, VOC of 0.82 V and FF of 0.52.96 P3HT:c13-based all-nanowire BHJ OPVs showed a PCE of 1.15%.97
The rylene diimides contain two electron-deficient imide groups that impart n-type character to an otherwise p-type polycyclic hydrocarbon. Wudl and coworkers focused their attention on a decacyclene structure that can support more than two imides. Multiple imides may increase the electron affinity of the bulk material, and thereby facilitate electron injection and charge transport.98 Decacyclene triimides (DTIs) with three electron-deficient imide groups, can be regarded as star-shaped trimers of naphthalene monoimides (NMIs). Some inherent communication between the three imide groups lowered the LUMO level of the DTIs compared to that (−2.69 eV) of the NMIs. According to valence bond theory, the π electrons of decacyclene cannot be delocalized over the entire core, implying that the three NMI units are partially independent moieties. Thus, the LUMO levels (−3.60 eV) of the DTIs were slightly higher than those (−3.62 to −3.70 eV) of the fully delocalized diimides PDI and NDI. BHJ OPVs fabricated using a P3HT:c14 blend had a JSC of 4.87 mA cm−2, VOC of 0.58 V and FF of 0.57, leading to a PCE of 1.60%, which greatly surpassed the efficiencies (generally less than 0.5%) of analogous P3HT:PDI monomer solar cells.
In addition to blending with polymer donors, rylene diimides have also been investigated in BHJ OPVs by blending with solution-processed small molecule donors.17,99–101 Bazan and coworkers used small molecule p-DTS(FBTTh2)2 as the donor and PDI c1 as the acceptor to fabricate BHJ solar cells, which gave a JSC of 7.4 mA cm−2, VOC of 0.78 V, FF of 0.52 and PCE of 3.0%.17 The PCE of 3.0% is promising, but still much lower than those (7–9%)102–104 of the previously reported p-DTS(FBTTh2)2:PC71BM. The reduced PCE of the p-DTS(FBTTh2)2:c1 system was primarily related to a significant reduction in the internal quantum efficiency (IQE). Bias-dependent IQE measurements showed a gradual increase in the IQE from ca. 50% at short-circuit conditions to ca. 60% at −10 V, and this 10% increase in the IQE could be explained by the detrapping of trapped charges, whereas the remaining 40% losses could be explained by some type of geminate recombination process and/or additional charge trapping. Furthermore, the measured electron mobility (1.7 × 10−4 cm2 V−1 s−1) of the p-DTS(FBTTh2)2:c1 system was one order of magnitude lower than that of the system utilizing fullerene acceptors, leading to the decreased FF value.
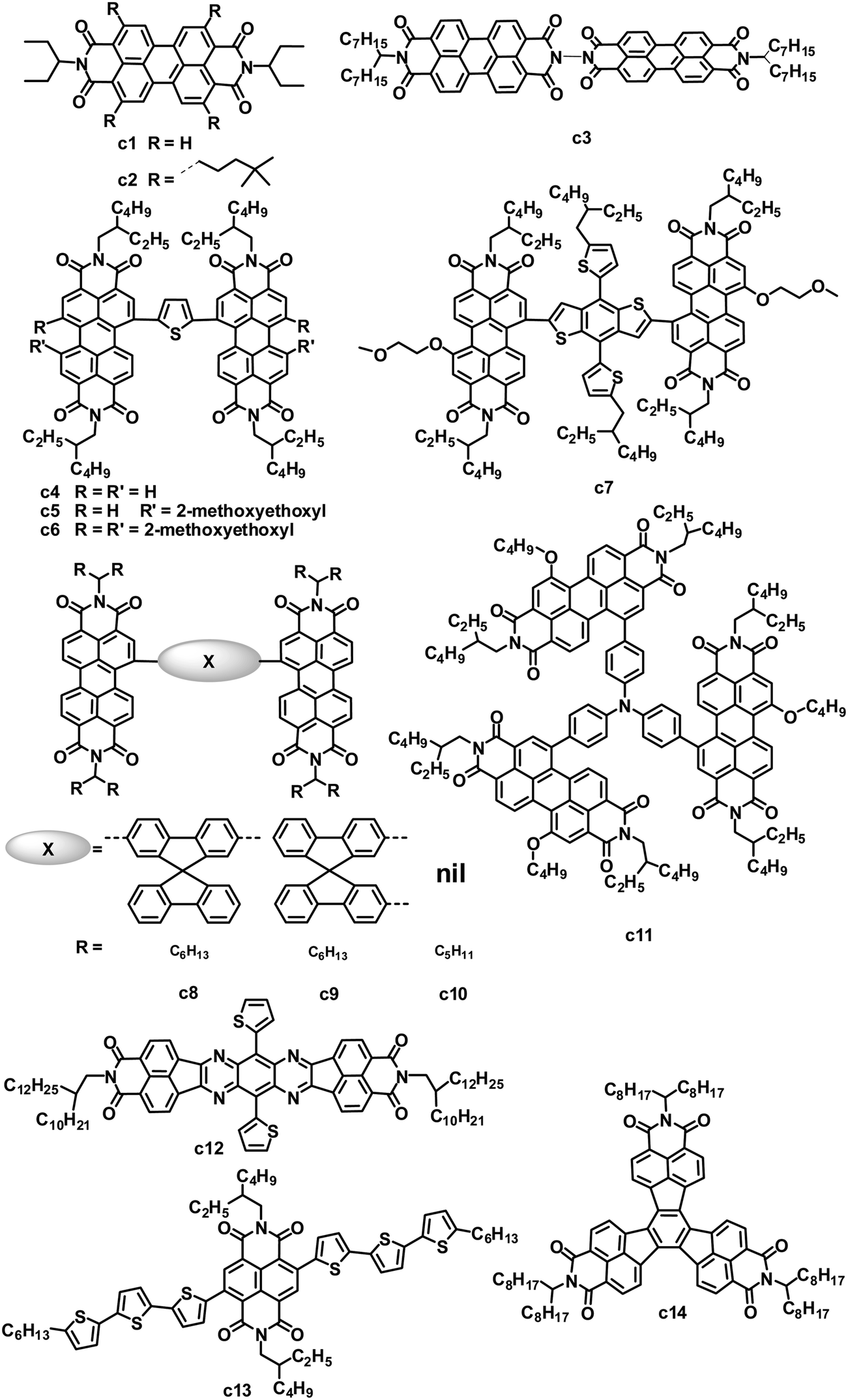
3.2 Other amide/imide-functionalized small molecular acceptors
Both amide and imide groups are commonly used in building n-type semiconductors for OPVs; among these units with amide or imide groups, rylene diimides and their analogues, as stated above, are the most successful so far. Some other amide/imide-functionalized small molecule electron acceptors have also exhibited promising results in solution-processed BHJ solar cells (Fig. 5 and Table 4).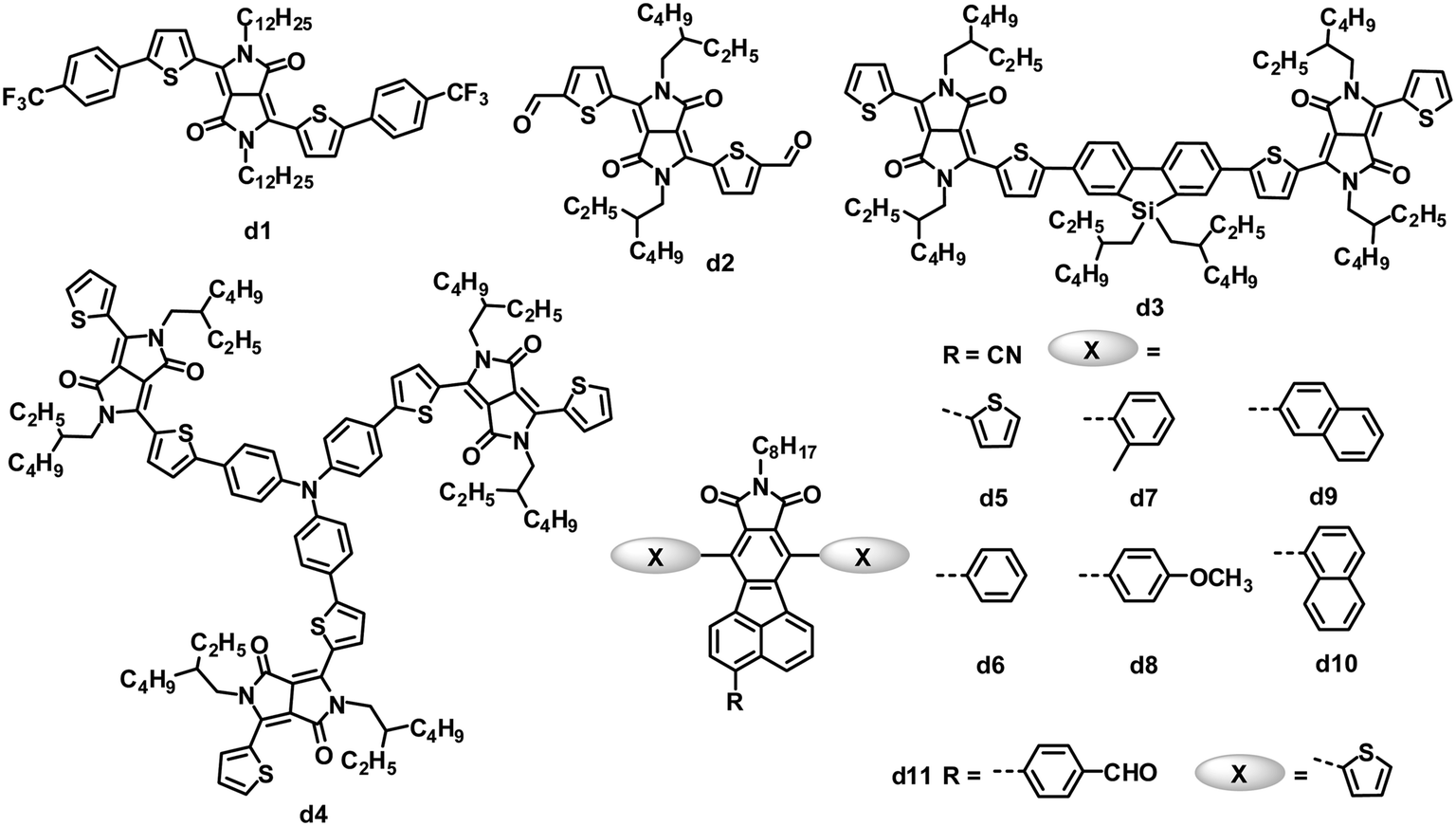 | ||
| Fig. 5 Chemical structures of DPP and FFI-based small molecules. | ||
| λ max /nm | E optg/eV | μ e /cm2 V−1 s−1 | HOMO/LUMO/eV | J SC/mA cm−2 | V OC/V | FF | PCEc (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a In film. b O and S: measured by OFET or SCLC method; N and B: in neat or blend film. c AM 1.5G, 100 mW cm−2. | |||||||||
| d1 | — | 1.81 | — | −5.26/−3.52 | 2.36 | 0.71 | 0.52 | 1.00 | 107 |
| d2 | 580 | 1.80 | 3 × 10−3 (O,N) | −5.9/−4.09 | 1.93 | 0.52 | 0.31 | 0.31 | 108 |
| d3 | 622 | 1.83 | 3.3 × 10−4 (S,N) | −5.30/−3.28 | 4.91 | 0.97 | 0.43 | 2.05 | 109 |
| d4 | 596 | 1.85 | 6.8 × 10−6 (S,B) | −5.26/−3.26 | 2.68 | 1.18 | 0.379 | 1.20 | 110 |
| d5 | — | 2.75 | 3 × 10−6 (S,B) | −6.23/−3.48 | 4.83 | 0.88 | 0.53 | 2.25 | 113 |
| d6 | — | 2.81 | 5 × 10−6 (S,B) | −6.24/−3.43 | 5.60 | 0.96 | 0.49 | 2.61 | 113 |
| d7 | — | 2.83 | 9 × 10−6 (S,B) | −6.27/−3.44 | 6.35 | 0.95 | 0.48 | 2.90 | 113 |
| d8 | — | 2.64 | 3 × 10−6 (S,B) | −6.04/−3.40 | 4.63 | 0.96 | 0.48 | 2.12 | 113 |
| d9 | — | 2.72 | 5 × 10−6 (S,B) | −6.15/−3.43 | 5.85 | 0.94 | 0.47 | 2.58 | 113 |
| d10 | — | 2.69 | 1 × 10−6 (S,B) | −6.12/−3.43 | 4.59 | 0.93 | 0.49 | 2.11 | 113 |
| d11 | 422 | — | 4.8 × 10−8 (S,B) | —/−3.30 | 5.14 | 1.03 | 0.45 | 2.40 | 114 |
Due to several attractive properties of DPP dyes containing amide groups, such as strong light absorption with a small band gap, easily tunable energy levels, good photochemical stability and facile synthetic modification,105,106 early in 2010, a variety of DPP derivatives with electron-withdrawing end capping groups (such as trifluoromethylphenyl, trifluorophenyl and aldehyde) were tested as acceptors in OPVs.107,108 The inherent electron affinity of DPP dyes was further improved by the introduction of electron-withdrawing groups. Among these acceptors, d1 provided the highest PCE of 1.0%, with a JSC of 2.36 mA cm−2, VOC of 0.71 V and FF of 0.52, when blended with the P3HT donor in a BHJ device.107 The aldehyde-substituted DPP d2 had a deeper LUMO level of −4.09 eV than that (−3.52 eV) of d1, and the P3HT:d2-based BHJ OPVs yielded a lower PCE of 0.31%, which was partly attributed to the lower VOC of 0.52 V.
Zhan and coworkers reported a DPP dimer (d3) with a weak electron-withdrawing dibenzosilole unit as a linker.109 Compound d3 was easily synthesized and purified, and exhibited significant absorption throughout the visible region (300–700 nm), appropriate energy level matching with P3HT (LUMO/HOMO: −3.28/−5.30 eV), and moderate electron mobility (3.3 × 10−4 cm2 V−1 s−1). Optimized solution-processed BHJ devices based on P3HT:d3 showed PCEs as high as 2.05%, which is the highest value reported for OPVs based on DPP acceptors so far. Meanwhile, a star-shaped DPP trimer d4, with triphenylamine as the core, was explored for BHJ OPVs as the first example of a 3D non-fullerene acceptor.110 Compared to 1D and 2D non-fullerene acceptor materials with high anisotropy in light absorption and charge transport, 3D materials are expected to have isotropic optical and charge-transporting properties. Solution-processed BHJ OPVs based on P3HT:d4 showed PCEs as high as 1.20%, with a very high VOC of up to 1.18 V.
Pei and coworkers have developed a novel series of fluoranthene-fused imide (FFI) derivatives and tuned their LUMO levels via introducing substituted groups on the FFI skeleton.111 Cyano-modified FFIs possess LUMO levels of −3.4 to −3.5 eV, which match with that of P3HT.112,113 OPVs based on a blend of P3HT and cyano-modified FFI (d5) with thiophene side chains showed an increased VOC compared to that (0.58 V) of the control device based on the P3HT:PC61BM blend,112 and a PCE of 2.14% was obtained.113 They further modified this acceptor structure by varying the side chains from thienyl to other aryl groups (d6–d10) perpendicular to the backbone of the acceptors. Acceptors d5–d10 had similar LUMO energy levels of −3.40 to −3.48 eV, and optical band gaps of 2.69 to 2.83 eV. The blends of these acceptors and P3HT also gave almost identical BHJ surface morphology and vertical phase separation. However, devices based on P3HT:d5–d10 yielded a maximum 37% difference in the PCEs (2.11 to 2.90%), similar to the trend in the JSC (4.59 to 6.35 mA cm−2). The JSC of these BHJ OPVs correlated well with the electron mobility (1 to 9 × 10−6 cm2 V−1 s−1) of the active films containing different acceptors.113 Pei and coworkers also modified FFIs by replacing the cyano group with other electron-withdrawing units; d11 with the p-formylphenyl group showed a higher LUMO level of −3.30 eV than those (−3.40 to −3.48 eV) of d5–d10, so when blended with P3HT, the OPV devices featured a higher VOC of 1.03 V and a PCE of 2.40%.114
3.3 Benzothiadiazole-based small molecules
As stated in Section 2.2, fluorene and BT-based copolymer acceptors have been systematically investigated in BHJ OPVs, and PCEs as high as 2.7% have been achieved. In 2011, Meredith and coworkers combined fluorene, BT and dicyanovinyl (DCV) groups together to synthesize a novel asymmetric electron acceptor (e1), which showed a LUMO level of −3.6 eV, lower than that of P3HT.115 When blended with P3HT, the photoinduced charge transfer in the BHJ film from P3HT to e1 was highly efficient. After annealing at 65 °C for 20 min, BHJ OPVs based on P3HT:e1 yielded a moderate PCE of 0.73%, with a JSC of 2.36 mA cm−2, VOC of 0.62 V and FF of 0.50 (Fig. 6 and Table 5).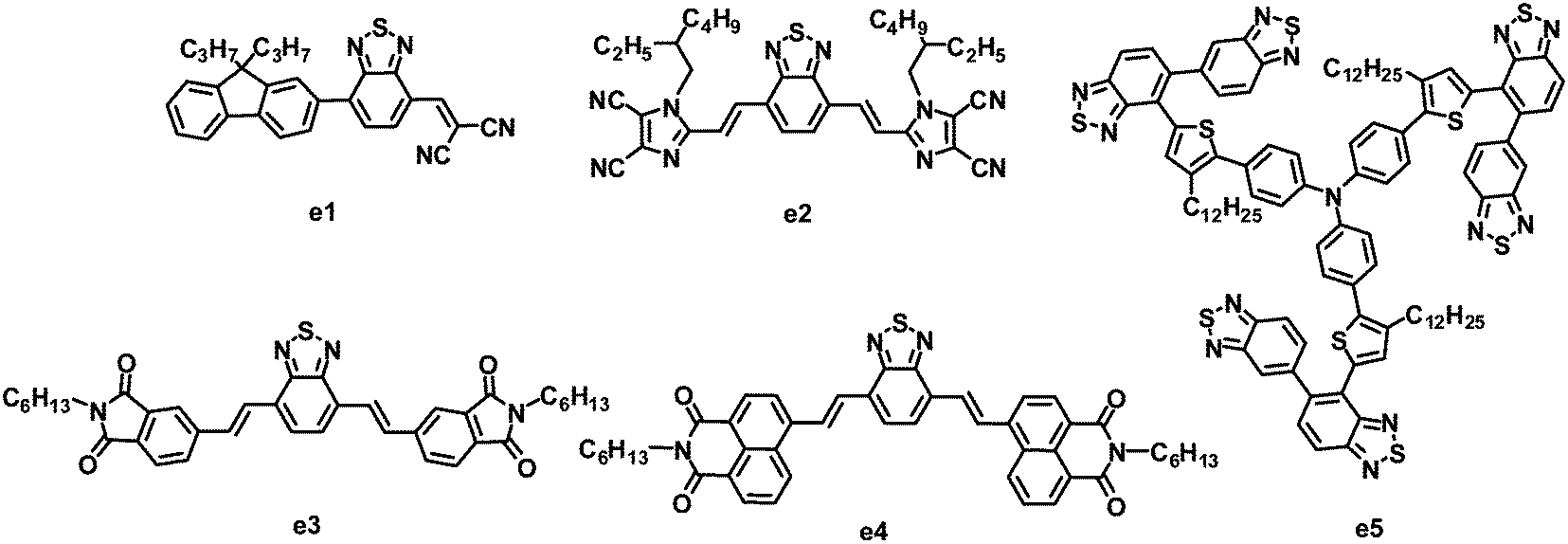 | ||
| Fig. 6 Chemical structures of BT-based small molecules. | ||
| λ max /nm | E optg/eV | μ e /cm2 V−1 s−1 | HOMO/LUMO/eV | Donor | J SC/mA cm−2 | V OC/V | FF | PCEc (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| a In film. b Measured by SCLC method in neat film. c AM 1.5G, 100 mW cm−2. | ||||||||||
| e1 | 475 | — | — | −6.2/−3.6 | P3HT | 2.36 | 0.62 | 0.50 | 0.73 | 115 |
| e2 | — | 2.43 | — | −6.0/−3.6 | P3HT | 3.00 | 0.76 | 0.48 | 1.10 | 121 |
| e2 | — | — | — | — | POPT | 5.50 | 0.62 | 0.40 | 1.40 | 121 |
| e3 | — | 2.34 | — | −5.77/−3.30 | P3HT | 4.7 | 0.96 | 0.56 | 2.54 | 123 |
| e4 | — | 2.16 | — | −5.66/−3.35 | P3HT | 0.5 | 0.65 | 0.40 | 0.12 | 123 |
| e5 | 334 | 2.18 | 2.6 × 10−6 | −5.48/−3.10 | P3HT | 1.81 | 0.99 | 0.452 | 0.81 | 128 |
Recently, another nitric heterocycle dicyanoimidazole known as vinazene has received attention as acceptors in solution-processed BHJ OPVs.116–122 This series of vinazene-based molecules displayed a wide range of electronic properties, with LUMO levels ranging from −2.76 to −3.60 eV.116,120 Among these acceptors, molecule e2 with BT as the core and Vinazene as the arms gave the most efficient photovoltaic performance when blended with P3HT. The P3HT:e2-based BHJ device showed a PCE of 1.1%, with a JSC of 3.0 mA cm−2, VOC of 0.76 V and FF of 0.48.121 An improved efficiency can be achieved by using POPT as a donor. The optimized device based on POPT:e2 yielded a JSC of 5.5 mA cm−2, VOC of 0.62 V, FF of 0.40 and PCE of 1.4%.121 The higher PCE from POPT may be explained by the ability of the conjugated phenyl substituent to twist out of planarity, resulting in a higher dissociation efficiency.
Sellinger and coworkers modified the molecular structure of e2 by replacing flanked vinazenes with phthalimides (e3) and naphthalimides (e4).123 These new acceptors showed strong absorption in the visible spectrum. Compared to e2, e3 had a higher LUMO energy level of −3.30 eV, hence a larger VOC (up to 0.96 V) was observed for the P3HT:e3-based device. This large VOC, along with a JSC of 4.7 mA cm−2 and FF of 0.56, resulted in a promising PCE as high as 2.54%. Relative to the highly planar e2 and e3, in the naphthalimide–BT analogue e4, steric interactions between neighboring hydrogen atoms in the naphthyl and vinyl moieties induce a 27.3° twist in the ground state of the isolated molecule; this twist can prohibit efficient π–π packing and crystal formation between neighboring molecules, and thus lower the electron mobility. Finally, the P3HT:e4-based devices afforded a PCE of only 0.1%.
5,5-Bibenzo[c][1,2,5]thiadiazole (BBT) is a 5,5-connected BT dimer. Due to several attractive properties of the BBT unit, such as easy molecular structure tailoring and facile electronic structure manipulation, Zhan and co-workers firstly synthesized BBT based organic semiconductors and used them in organic electronics.124–127 Relative to BT, BBT has a stronger electron-accepting ability due to the two BT units, therefore BBT could also be a promising building block for the synthesis of electron acceptors. Very recently, Zhan and co-workers designed and synthesized a star-shaped BBT trimer e5 similar to the DPP trimer d4.128 The BHJ OPVs based on the P3HT:e5 blend after annealing gave a PCE of 0.81%, with a JSC of 1.81 mA cm−2, VOC of 0.99 V and FF of 0.452.
3.4 Electron-deficient aromatic fused rings
Fused acenes, such as pentacene and tetracene, have been extensively studied as electron donor materials in OPVs, and especially, pentacene is well known to exhibit an extremely high hole mobility of over 1 cm2 V−1 s−1.8 Anthony and coworkers demonstrated that it is possible to switch fused acenes from electron donors to acceptors in OPVs by lowering their LUMO/HOMO levels through cyanation (Fig. 7 and Table 6).129 Cyanopentacenes with trialkylsilyl side chains have been developed; the energy levels can be tuned by changing the number of cyano groups, while the trialkylsilyl groups control the crystal packing and film morphology. The HOMO and LUMO levels can be down-shifted by ca. 0.14 eV for every cyano group introduced to the pentacene core. Relative to the 1D “slipped-stack” and 2D “brickwork” packing resulted from triisopropylsilylethynyl or triisobutylsilylethynyl side chains, a particularly strongly 1D “sandwich herringbone” crystal packing motif from the tricyclopentylsilyl chains yielded the highest OPV photocurrent. Tricyclopentylsilyl pentacene (f1) with one cyano group showed a LUMO energy level of −3.50 eV and a 1D “sandwich herringbone” crystal packing. OPVs based on P3HT:f1 exhibited the best performance: a JSC of 3.72 mA cm−2, VOC of 0.84 V, FF of 0.41 and PCE of up to 1.29%.130 Subsequently, several other monosubstituted pentacene acceptors were investigated; when blended with P3HT, chloropentacene f2 and trifluoromethylpentacene f3 showed PCEs of 1.00% and 1.26%, respectively, approaching that of P3HT:f1.130 Again, both acceptors f2 and f3 adopted a sandwich-type crystal packing arrangement, which yielded relatively high JSC and FF values.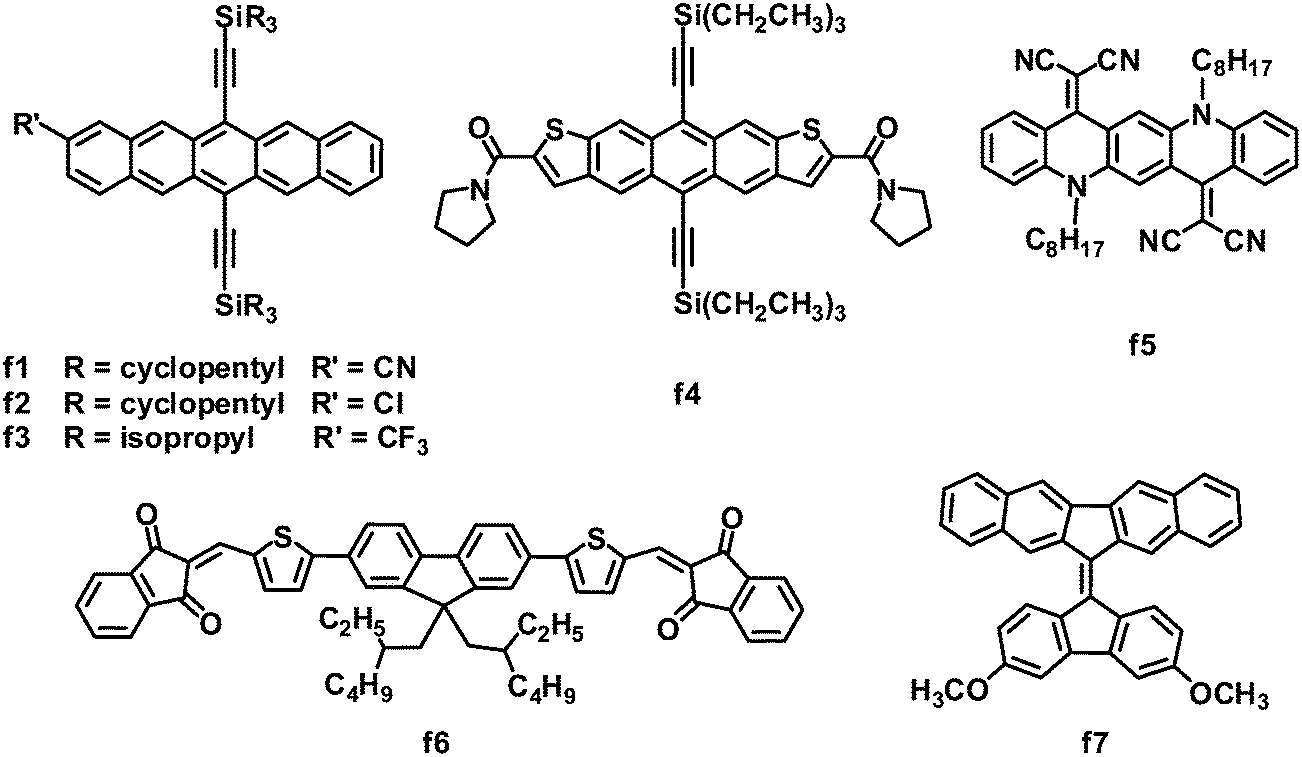 | ||
| Fig. 7 Chemical structures of fused-ring-based small molecules. | ||
| λ max /nm | E optg/eV | μ e /cm2 V−1 s−1 | HOMO/LUMO/eV | J SC/mA cm−2 | V OC/V | FF | PCEc (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a In film. b Measured by SCLC method in neat film. c AM 1.5G, 100 mW cm−2. | |||||||||
| f1 | — | 1.82 | — | −5.29/−3.50 | 3.72 | 0.84 | 0.41 | 1.29 | 130 |
| f2 | — | — | — | — | 2.44 | 0.95 | 0.43 | 1.00 | 130 |
| f3 | — | — | — | — | 3.17 | 0.80 | 0.50 | 1.26 | 130 |
| f4 | 620 | — | — | −5.34/−3.21 | 1.93 | 1.05 | 0.39 | 0.80 | 131 |
| f5 | 640 | 1.8 | 1.14 × 10−4 | −5.9/−4.1 | 5.72 | 0.48 | 0.57 | 1.57 | 132 |
| f6 | — | — | — | −5.95/−3.95 | 3.82 | 0.95 | 0.67 | 2.43 | 133 |
| f7 | 517 | 2.18 | — | −5.17/−3.24 | 3.9 | 1.1 | 0.4 | 1.7 | 134 |
Similar to pentacene, anthradithiophene was also converted into an electron acceptor for OPVs by flanking with electron-withdrawing groups. Because of the great challenge in the preparation of isomerically pure materials, anthradithiophene derivatives were usually studied as a mixture of syn- and anti-isomers. However, amide groups caused different self-assembly of the syn- and anti-isomers, allowing for the separation and property evaluation of isomerically pure anthradithiophenes. The photovoltaic properties of two pure anthradithiophene isomers were evaluated in BHJ OPVs with P3HT as the donor; the syn-isomer f4 yielded a PCE (0.8%) two orders of magnitude higher than that (0.008%) of the anti-isomer.131 This large difference in the PCE can be attributed to the difference in the BHJ film morphology; the P3HT:f4 blend showed a uniform coverage textured with small grains of the acceptor, whereas larger-scale aggregation was observed in the P3HT:anti-isomer blend film.
Quinacridone is a nitric pentacene analogue, which is widely used in organic pigments with high chemical and thermal stability, unique supramolecular assembly and optoelectronic properties. Quinacridone solid films usually absorb intensely in the visible light spectrum, due to the ICT feature and the strong intermolecular interactions in the aggregated state. By replacing the two carbonyl oxygens on quinacridone with malononitrile, Wang and coworkers developed DCV-substituted quinacridone derivatives as electron acceptors for BHJ OPVs.132 As expected, these acceptors exhibited remarkable absorption in the region from 400 to 700 nm, when blended with P3HT. In particular, molecule f5 exhibited a low band gap of 1.8 eV, a deep LUMO energy level of −4.1 eV and a moderate electron mobility of 1.14 × 10−4 cm2 V−1 s−1. A BHJ OPV device based on P3HT:f5 exhibited a JSC of 5.72 mA cm−2, VOC of 0.48 V and FF of 0.57, leading to a PCE of 1.57%. In this system, the relatively deep LUMO energy level of f5 led to a significant loss of the photovoltage, limiting further improvements in the device performance.
Compared to pentacene and quinacridone, fluorene is a smaller-size fused ring. Very recently, Winzenberg and coworkers introduced indan-1,3-dione end groups to lower the fluorene energy levels.133 Fluorene-based molecules end-capped with indan-1,3-diones exhibited LUMO energy levels of −3.75 to −3.95 eV and HOMO energy levels of −5.85 to −5.95 eV, which indicated that these molecules were suitable electron acceptors for BHJ solar cells in combination with the P3HT electron donor. Although the electron mobility of these acceptors has not been measured, these molecules showed good photovoltaic properties as acceptors, and the P3HT:f6 blend gave the best performance: a JSC of 3.82 mA cm−2, VOC of 0.95 V, FF of 0.67 and a PCE as high as 2.43%. The FF value of 0.67 is a scarce result in non-fullerene-based solution-processed BHJ OPVs.
Except for electron-deficient group-modified fused rings, the 9,9′-bifluorenylidene polycycle also possesses inherent potential as an electron acceptor in OPVs.134 In the ground state, the 9,9′-bifluorenylidene unit is forced to be coplanar due to the presence of the double bond bridge, but the interaction between protons twists its structure. The addition of one electron across the double bond is highly favorable because of steric strain relief and the gain in aromaticity to form a 14-π-electron system. Furthermore, the LUMO energy level of this system is easily tuned by substitution on the aromatic periphery. The asymmetric molecule f7 showed HOMO/LUMO levels of −5.17/−3.24 eV, a decent PCE of 1.7% and a high VOC of 1.10 V when blended with the P3HT donor.
4. Conclusions and outlook
In the past five years, significant progress has been made in the synthesis and application of polymer and small molecule non-fullerene acceptors for solution-processed BHJ OPVs, and the research on non-fullerene acceptors has become an emerging frontier in the field of OPVs. With the extensive research and accumulated practical experience that we have outlined in this progress report, the guidelines on pursuing and developing high-performance non-fullerene acceptors for solution-processed BHJ OPVs are becoming legible. The basic requirements of the specific intrinsic properties necessary for an ideal electron acceptor material for solution-processed BHJ OPVs include: (i) sufficient solubility and good film-forming properties; (ii) broad optical absorption range with a high extinction coefficient; for matching the solar spectrum and capturing sufficient solar energy, it would be best if the absorption ranges of the donor and acceptor materials are complementary; (iii) large electron affinity and a strong tendency to accept electrons from donor semiconductors and suitable LUMO/HOMO energy levels matched with existing donor materials to ensure a large VOC, a downhill energy offset for exciton dissociation and a lowest energy loss; (iv) high electron mobility balanced with the hole mobility of blending donors for efficient charge transport, which in turn allows a thick active layer required for efficient light harvesting and a roll-to-roll process, as well as reduces the charge recombination and series resistance; (v) good mixing with donor materials to form nanoscale phase separation and an ideal blend morphology. However, it is a big challenge to find a perfect combination of all of the above properties in one acceptor through molecular design. Thus, a far better understanding of the relationships between molecular design and the device performance of non-fullerene acceptors is necessary for their continuing development.Through molecular architecture tailoring, the tuning of the compounds’ solubility and optical absorption is straightforward. Generally, introducing alkyl side chains and varying their number and length can tune the compounds’ solubility. Strong absorption can also be achieved by incorporating organic dye blocks (e.g. PDI, DPP, etc.) with high extinction coefficients. ICT formed by push–pull structures (e.g. PDI, NDI or BT-based copolymers a1, a13 or b3, etc.) with electron-donating and electron-withdrawing units can extend the absorption to the long wavelength band, and optical band gaps can be adjusted by changing the electron-donating and/or withdrawing strength of the building units. For example, PDI-based copolymers (a6–a8) with benzo-fused rings have much larger optical band gaps and blue-shifted absorption relative to those with thiophene-fused rings (a1–a5).
Compared to the tuning of the solubility and optical absorption, it is a little more complex to obtain appropriate LUMO/HOMO energy levels of electron acceptors which are well matched with those of the blending donors. The above mentioned methods to tune optical band gaps can also roughly tune the molecular energy levels. Furthermore, in several systems, the careful molecular design and modification can accurately control the molecular energy levels; the energy levels can be tuned by changing the nature and number of the substituent groups. A typical example is cyano-modified pentacenes; the HOMO and LUMO levels can be down-shifted by 0.14 eV for every cyano group attached to the pentacene core.
Most existing research results confirm that the high electron mobility of the electron acceptors, balanced with the hole mobility of the blending donor, would help to achieve a high JSC and FF, and in turn a high PCE value. The π–π stacking in the film is a key factor determining the mobility of materials. The planarity of blocks affects the molecular π–π stacking. Compared to PDI-based polymers, NDI-based polymers generally showed higher electron mobilities, due to their better π–π stacking in thin films. In particular, polymer a13 gave a very high electron mobility of up to 0.85 cm2 V−1 s−1, and BHJ OPVs based on the PTQ1:a13 blend yielded a PCE of over 4%. Additionally, the regularity is also a key factor for molecular π–π stacking and in turn the charge mobility. In PDI-based polymers, 1,7-regio-isomer-pure a10 yielded a higher electron mobility and PCE than its mixed 1,6 and 1,7-regio-isomer counterpart.
The morphology and phase separation scale in the BHJ film have a significant impact on the device performance. It is critical for the BHJ active layer to form a bicontinuous interpenetrating network with the optimum morphology for building two distinct highways to transport electrons and holes. Moderate crystallinity of the acceptors can improve the electron mobility in the blend film, but high crystallinity will cause a large phase separation scale and a low JSC. Alkyl side chains affect not only the solubility and configuration, but also the intermolecular interactions and crystalline properties of the molecules, especially for small molecular acceptors. For example, in cyano-substituted pentacenes with trialkylsilyl side chains, the trialkylsilyl groups control the crystal packing and film morphology, and tricyclopentylsilyl side chains induced 1D “sandwich herringbone” crystal packing, leading to the highest device performance. Secondly, the molecular compatibility between the donors and acceptors, which can be optimized by changing the nature and number of solvophobic or solvophilic substituent groups (e.g. EG in c4–c6), can improve phase separation. Thirdly, twisted molecules caused by intramolecular steric hindrance tend to suppress molecular aggregation and facilitate mixing with donors, leading to enhanced device performance. For example, non-planar dimeric PDIs have shown promising PCEs of 2% to 4%. However, overly twisted structures are generally unfavorable for π–π stacking and charge transport. Finally, so far there are very few reports on high-performance OPV devices without any post-treatment; physical control during device fabrication is important for ideal BHJ morphology and nanoscale phase separation, and solvent additives, mixed solvents, thermal annealing and solvent annealing are effective methods.
The isotropy of charge transport in 3D fullerene derivatives renders them to dominate electron acceptors for OPVs. To a certain extent, twisted molecules and 3D molecules possess relatively isotropic charge transport. Several twisted acceptors have exhibited good OPV results, whereas rare 3D non-fullerene acceptors have been explored. In the view of success of isotropic fullerene acceptors, further attempts towards 3D non-fullerene acceptors are necessary.
So far the best PCE of solution-processed BHJ OPVs with non-fullerene acceptors is over 4% as reported in the literature, while a PCE as high as 6.4% was disclosed for all-PSCs by the Polyera company on the website http://www.polyera.com. The PCE of 6.4% is close to those (7–9%) of fullerene-based OPVs. We believe that non-fullerene electron acceptor-based OPVs will be very promising. An interdisciplinary approach, such as novel non-fullerene electron acceptors and new advanced device concepts, will probably bring high efficiencies even greater than their fullerene counterparts in the future.
Acknowledgements
This work was supported by the 973 Project (no. 2013CB834702 and 2011CB808401), the NSFC (no. 21025418, 51261130582), and the Chinese Academy of Sciences.References
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- R. Sondergaard, M. Hosel, D. Angmo, T. T. Larsen-Olsen and F. C. Krebs, Mater. Today, 2012, 15, 36 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- J. B. You, L. T. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C. C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728 RSC.
- Y. Z. Lin, H. J. Fan, Y. F. Li and X. W. Zhan, Adv. Mater., 2012, 24, 3087 CrossRef CAS PubMed.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970 RSC.
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791 CrossRef CAS PubMed.
- D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Adv. Energy Mater., 2014, 4, 1301006 Search PubMed.
- J. E. Anthony, Chem. Mater., 2011, 23, 583 CrossRef CAS.
- P. Sonar, J. P. Fong Lim and K. L. Chan, Energy Environ. Sci., 2011, 4, 1558 CAS.
- C. R. McNeill, Energy Environ. Sci., 2012, 5, 5653 CAS.
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2013, 16, 123 CrossRef CAS PubMed.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS.
- G. Yu and A. J. Heeger, J. Appl. Phys., 1995, 78, 4510 CrossRef CAS PubMed.
- M. Granstrom, K. Petritsch, A. C. Arias, A. Lux, M. R. Andersson and R. H. Friend, Nature, 1998, 395, 257 CrossRef CAS PubMed.
- T. W. Holcombe, C. H. Woo, D. F. J. Kavulak, B. C. Thompson and J. M. J. Fréchet, J. Am. Chem. Soc., 2009, 131, 14160 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613 CrossRef CAS PubMed.
- E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160 CrossRef CAS PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386 CrossRef CAS PubMed.
- M. R. Raj, S. Ramkumar and S. Anandan, RSC Adv., 2013, 3, 5108 RSC.
- J. A. Mikroyannidis, M. M. Stylianakis, G. D. Sharma, P. Balraju and M. S. Roy, J. Phys. Chem. C, 2009, 113, 7904 CAS.
- X. Zhan, Z. A. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246 CrossRef CAS PubMed.
- L. Zhang, C. A. Di, Y. Zhao, Y. L. Guo, X. N. Sun, Y. G. Wen, W. Y. Zhou, X. W. Zhan, G. Yu and Y. Q. Liu, Adv. Mater., 2010, 22, 3537 CrossRef CAS PubMed.
- X. Zhan, Z. A. Tan, E. Zhou, Y. Li, R. Misra, A. Grant, B. Domercq, X.-H. Zhang, Z. An, X. Zhang, S. Barlow, B. Kippelen and S. R. Marder, J. Mater. Chem., 2009, 19, 5794 RSC.
- S. M. Zhang, Y. G. Wen, W. Y. Zhou, Y. L. Guo, L. C. Ma, X. G. Zhao, Z. Zhao, S. Barlow, S. R. Marder, Y. Q. Liu and X. W. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1550 CrossRef CAS.
- J. Huang, Y. Wu, H. Fu, X. Zhan, J. Yao, S. Barlow and S. R. Marder, J. Phys. Chem. A, 2009, 113, 5039 CrossRef CAS PubMed.
- Z. A. Tan, E. J. Zhou, X. W. Zhan, X. Wang, Y. F. Li, S. Barlow and S. R. Marder, Appl. Phys. Lett., 2008, 93, 073309 CrossRef PubMed.
- Y. Liu, T. T. Larsen-Olsen, X. Zhao, B. Andreasen, R. R. Søndergaard, M. Helgesen, K. Norrman, M. Jørgensen, F. C. Krebs and X. Zhan, Sol. Energy Mater. Sol. Cells, 2013, 112, 157 CrossRef CAS PubMed.
- P. Cheng, L. Ye, X. Zhao, J. Hou, Y. Li and X. Zhan, Energy Environ. Sci., 2014, 7, 1351 CAS.
- L. Meng, Y. Shang, Q. Li, Y. Li, X. Zhan, Z. Shuai, R. G. E. Kimber and A. B. Walker, J. Phys. Chem. B, 2009, 114, 36 CrossRef PubMed.
- J. Hou, S. Zhang, T. L. Chen and Y. Yang, Chem. Commun., 2008, 6034 RSC.
- E. Zhou, K. Tajima, C. Yang and K. Hashimoto, J. Mater. Chem., 2010, 20, 2362 RSC.
- E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799 CrossRef CAS PubMed.
- X. Hu, L. Zuo, H. Pan, F. Hao, J. Pan, L. Fu, M. Shi and H. Chen, Sol. Energy Mater. Sol. Cells, 2012, 103, 157 CrossRef CAS PubMed.
- C. J. Brabec, A. Cravino, D. Meissner, N. S. Sariciftci, T. Fromherz, M. T. Rispens, L. Sanchez and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 374 CrossRef CAS.
- E. Kozma, D. Kotowski, F. Bertini, S. Luzzati and M. Catellani, Polymer, 2010, 51, 2264 CrossRef CAS PubMed.
- Y. Zhou, Q. Yan, Y.-Q. Zheng, J.-Y. Wang, D. Zhao and J. Pei, J. Mater. Chem. A, 2013, 1, 6609 CAS.
- Y. Jiang, L. Lu, M. Yang, C. Zhan, Z. Xie, F. Verpoort and S. Xiao, Polym. Chem., 2013, 4, 5612 RSC.
- H. Choi, S. Paek, J. Song, C. Kim, N. Cho and J. Ko, Chem. Commun., 2011, 47, 5509 RSC.
- W. Y. Zhou, F. Jin, X. B. Huang, X. M. Duan and X. W. Zhan, Macromolecules, 2012, 45, 7823 CrossRef CAS.
- H. Usta, C. Newman, Z. Chen and A. Facchetti, Adv. Mater., 2012, 24, 3678 CrossRef CAS PubMed.
- W. Zhou, Z.-G. Zhang, L. Ma, Y. Li and X. Zhan, Sol. Energy Mater. Sol. Cells, 2013, 112, 13 CrossRef CAS PubMed.
- H. Yan, Z. H. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679 CrossRef CAS PubMed.
- S. Fabiano, Z. Chen, S. Vahedi, A. Facchetti, B. Pignataro and M. A. Loi, J. Mater. Chem., 2011, 21, 5891 RSC.
- K. Szendrei, D. Jarzab, Z. H. Chen, A. Facchetti and M. A. Loi, J. Mater. Chem., 2010, 20, 1317 RSC.
- J. R. Moore, S. Albert-Seifried, A. Rao, S. Massip, B. Watts, D. J. Morgan, R. H. Friend, C. R. McNeill and H. Sirringhaus, Adv. Energy Mater., 2011, 1, 230 CrossRef CAS.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369 CrossRef CAS.
- Y. Tang and C. R. McNeill, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 403 CrossRef CAS.
- N. Zhou, H. Lin, S. J. Lou, X. Yu, P. Guo, E. F. Manley, S. Loser, P. Hartnett, H. Huang, M. R. Wasielewski, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Adv. Energy Mater., 2014, 4, 1300785 Search PubMed.
- Y.-J. Hwang, G. Ren, N. M. Murari and S. A. Jenekhe, Macromolecules, 2012, 45, 9056 CrossRef CAS.
- T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960 CrossRef CAS PubMed.
- E. Zhou, J. Cong, M. Zhao, L. Zhang, K. Hashimoto and K. Tajima, Chem. Commun., 2012, 48, 5283 RSC.
- M. Yuan, M. M. Durban, P. D. Kazarinoff, D. F. Zeigler, A. H. Rice, Y. Segawa and C. K. Luscombe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4061 CrossRef CAS.
- E. Zhou, J. Cong, K. Hashimoto and K. Tajima, Adv. Mater., 2013, 25, 6991 CrossRef CAS PubMed.
- K. Nakabayashi and H. Mori, Macromolecules, 2012, 45, 9618 CrossRef CAS.
- S.-C. Chen, Q. Zheng, Q. Zhang, D. Cai, J. Wang, Z. Yin and C. Tang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1999 CrossRef CAS.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303 CrossRef CAS.
- M.-F. Falzon, A. P. Zoombelt, M. M. Wienk and R. A. J. Janssen, Phys. Chem. Chem. Phys., 2011, 13, 8931 RSC.
- A. C. Arias, J. D. MacKenzie, R. Stevenson, J. J. M. Halls, M. Inbasekaran, E. P. Woo, D. Richards and R. H. Friend, Macromolecules, 2001, 34, 6005 CrossRef CAS.
- Y. Kim, S. Cook, S. A. Choulis, J. Nelson, J. R. Durrant and D. D. C. Bradley, Chem. Mater., 2004, 16, 4812 CrossRef CAS.
- C. R. McNeill, A. Abrusci, I. Hwang, M. A. Ruderer, P. Müller-Buschbaum and N. C. Greenham, Adv. Funct. Mater., 2009, 19, 3103 CrossRef CAS.
- S. Nam, M. Shin, H. Kim, C.-S. Ha, M. Ree and Y. Kim, Adv. Funct. Mater., 2011, 21, 4527 CrossRef CAS.
- S. Nam, M. Shin, S. Park, S. Lee, H. Kim and Y. Kim, Phys. Chem. Chem. Phys., 2012, 14, 15046 RSC.
- C. R. McNeill, A. Abrusci, J. Zaumseil, R. Wilson, M. J. McKiernan, J. H. Burroughes, J. J. M. Halls, N. C. Greenham and R. H. Friend, Appl. Phys. Lett., 2007, 90, 193506 CrossRef PubMed.
- C. R. McNeill, J. J. M. Halls, R. Wilson, G. L. Whiting, S. Berkebile, M. G. Ramsey, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2008, 18, 2309 CrossRef CAS.
- X. He, F. Gao, G. Tu, D. Hasko, S. Hüttner, U. Steiner, N. C. Greenham, R. H. Friend and W. T. S. Huck, Nano Lett., 2010, 10, 1302 CrossRef CAS PubMed.
- D. Mori, H. Benten, J. Kosaka, H. Ohkita, S. Ito and K. Miyake, ACS Appl. Mater. Interfaces, 2011, 3, 2924 CAS.
- D. Mori, H. Benten, H. Ohkita, S. Ito and K. Miyake, ACS Appl. Mater. Interfaces, 2012, 4, 3325 CAS.
- X. Liu, S. Huettner, Z. Rong, M. Sommer and R. H. Friend, Adv. Mater., 2012, 24, 669 CrossRef CAS PubMed.
- Y. Cao, T. Lei, J. Yuan, J.-Y. Wang and J. Pei, Polym. Chem., 2013, 4, 5228 RSC.
- J. Roncali, Acc. Chem. Res., 2009, 42, 1719 CrossRef CAS PubMed.
- B. Walker, C. Kim and T.-Q. Nguyen, Chem. Mater., 2011, 23, 470 CrossRef CAS.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020 CrossRef CAS PubMed.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183 CrossRef CAS PubMed.
- R. R. Lunt, N. C. Giebink, A. A. Belak, J. B. Benziger and S. R. Forrest, J. Appl. Phys., 2009, 105, 053711 CrossRef PubMed.
- J. L. Li, F. Dierschke, J. S. Wu, A. C. Grimsdale and K. Mullen, J. Mater. Chem., 2006, 16, 96 RSC.
- Z. Y. Xie, X. Y. Guo, L. J. Bu, Y. Zhao, Y. H. Geng and L. X. Wang, Thin Solid Films, 2009, 517, 4654 CrossRef PubMed.
- W. S. Shin, H.-H. Jeong, M.-K. Kim, S.-H. Jin, M.-R. Kim, J.-K. Lee, J. W. Lee and Y.-S. Gal, J. Mater. Chem., 2006, 16, 384 RSC.
- V. Kamm, G. Battagliarin, I. A. Howard, W. Pisula, A. Mavrinskiy, C. Li, K. Müllen and F. Laquai, Adv. Energy Mater., 2011, 1, 297 CrossRef CAS.
- S. Rajaram, R. Shivanna, S. K. Kandappa and K. S. Narayan, J. Phys. Chem. Lett., 2012, 3, 2405 CrossRef CAS.
- R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435 CAS.
- Z. Lu, X. Zhang, C. Zhan, B. Jiang, X. Zhang, L. Chen and J. Yao, Phys. Chem. Chem. Phys., 2013, 15, 11375 RSC.
- B. Jiang, X. Zhang, C. Zhan, Z. Lu, J. Huang, X.-L. Ding, S.-G. He and J. Yao, Polym. Chem., 2013, 4, 4631 RSC.
- Q. Yan, Y. Zhou, Y.-Q. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389 RSC.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024 RSC.
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014 DOI:10.1002/adma.201400525.
- H. Li, F. S. Kim, G. Ren, E. C. Hollenbeck, S. Subramaniyan and S. A. Jenekhe, Angew. Chem., Int. Ed., 2013, 52, 5513 CrossRef CAS PubMed.
- E. Ahmed, G. Ren, F. S. Kim, E. C. Hollenbeck and S. A. Jenekhe, Chem. Mater., 2011, 23, 4563 CrossRef CAS.
- G. Ren, E. Ahmed and S. A. Jenekhe, Adv. Energy Mater., 2011, 1, 946 CrossRef CAS.
- G. Ren, E. Ahmed and S. A. Jenekhe, J. Mater. Chem., 2012, 22, 24373 RSC.
- T. V. Pho, F. M. Toma, M. L. Chabinyc and F. Wudl, Angew. Chem., Int. Ed., 2013, 52, 1446 CrossRef CAS PubMed.
- G. D. Sharma, P. Balraju, J. A. Mikroyannidis and M. M. Stylianakis, Sol. Energy Mater. Sol. Cells, 2009, 93, 2025 CrossRef CAS PubMed.
- G. D. Sharma, P. Suresh, J. A. Mikroyannidis and M. M. Stylianakis, J. Mater. Chem., 2010, 20, 561 RSC.
- J. A. Mikroyannidis, P. Suresh and G. D. Sharma, Synth. Met., 2010, 160, 932 CrossRef CAS PubMed.
- V. Gupta, A. K. K. Kyaw, D. H. Wang, S. Chand, G. C. Bazan and A. J. Heeger, Sci. Rep., 2013, 3, 1965 Search PubMed.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, J. Zhang, S. Chand, G. C. Bazan and A. J. Heeger, Adv. Mater., 2013, 25, 2397 CrossRef CAS PubMed.
- T. S. van der Poll, J. A. Love, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646 CrossRef CAS PubMed.
- S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039 RSC.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684 CAS.
- P. Sonar, G.-M. Ng, T. T. Lin, A. Dodabalapur and Z.-K. Chen, J. Mater. Chem., 2010, 20, 3626 RSC.
- B. P. Karsten, J. C. Bijleveld and R. A. J. Janssen, Macromol. Rapid Commun., 2010, 31, 1554 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Adv. Energy Mater., 2013, 3, 724 CrossRef CAS.
- Y. Lin, P. Cheng, Y. Li and X. Zhan, Chem. Commun., 2012, 48, 4773 RSC.
- L. Ding, H.-Z. Ying, Y. Zhou, T. Lei and J. Pei, Org. Lett., 2010, 12, 5522 CrossRef CAS PubMed.
- Y. Zhou, L. Ding, K. Shi, Y.-Z. Dai, N. Ai, J. Wang and J. Pei, Adv. Mater., 2012, 24, 957 CrossRef CAS PubMed.
- Y. Zhou, Y.-Z. Dai, Y.-Q. Zheng, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Commun., 2013, 49, 5802 RSC.
- Y.-Q. Zheng, Y.-Z. Dai, Y. Zhou, J.-Y. Wang and J. Pei, Chem. Commun., 2014, 50, 1591 RSC.
- P. E. Schwenn, K. Gui, A. M. Nardes, K. B. Krueger, K. H. Lee, K. Mutkins, H. Rubinstein-Dunlop, P. E. Shaw, N. Kopidakis, P. L. Burn and P. Meredith, Adv. Energy Mater., 2011, 1, 73 CrossRef CAS.
- R. Y. C. Shin, T. Kietzke, S. Sudhakar, A. Dodabalapur, Z.-K. Chen and A. Sellinger, Chem. Mater., 2007, 19, 1892 CrossRef CAS.
- T. Kietzke, R. Y. C. Shin, D. A. M. Egbe, Z.-K. Chen and A. Sellinger, Macromolecules, 2007, 40, 4424 CrossRef CAS.
- Z. E. Ooi, T. L. Tam, R. Y. C. Shin, Z. K. Chen, T. Kietzke, A. Sellinger, M. Baumgarten, K. Mullen and J. C. deMello, J. Mater. Chem., 2008, 18, 4619 RSC.
- M. Schubert, C. H. Yin, M. Castellani, S. Bange, T. L. Tam, A. Sellinger, H. H. Horhold, T. Kietzke and D. Neher, J. Chem. Phys., 2009, 130, 094703 CrossRef PubMed.
- R. Y. C. Shin, P. Sonar, P. S. Siew, Z.-K. Chen and A. Sellinger, J. Org. Chem., 2009, 74, 3293 CrossRef CAS PubMed.
- C. H. Woo, T. W. Holcombe, D. A. Unruh, A. Sellinger and J. M. J. Fréchet, Chem. Mater., 2010, 22, 1673 CrossRef CAS.
- B. Walker, X. Han, C. Kim, A. Sellinger and T.-Q. Nguyen, ACS Appl. Mater. Interfaces, 2011, 4, 244 Search PubMed.
- J. T. Bloking, X. Han, A. T. Higgs, J. P. Kastrop, L. Pandey, J. E. Norton, C. Risko, C. E. Chen, J.-L. Brédas, M. D. McGehee and A. Sellinger, Chem. Mater., 2011, 23, 5484 CrossRef CAS.
- H. Wang, P. Cheng, Y. Liu, J. Chen, X. Zhan, W. Hu, Z. Shuai, Y. Li and D. Zhu, J. Mater. Chem., 2012, 22, 3432 RSC.
- H. Wang, T. Fukumatsu, Y. Liu, W. Hu, S. Seki and X. Zhan, J. Mater. Chem. C, 2013, 1, 414 RSC.
- Y. Liu, H. Wang, H. Dong, J. Tan, W. Hu and X. Zhan, Macromolecules, 2012, 45, 1296 CrossRef CAS.
- M. L. Jia, X. N. Ma, L. Y. Yan, H. F. Wang, Q. J. Guo, X. F. Wang, Y. Y. Wang, X. W. Zhan and A. D. Xia, J. Phys. Chem. A, 2010, 114, 7345 CrossRef CAS PubMed.
- Y. Lin, H. Wang, Y. Li, D. Zhu and X. Zhan, J. Mater. Chem. A, 2013, 1, 14627 CAS.
- Y.-F. Lim, Y. Shu, S. R. Parkin, J. E. Anthony and G. G. Malliaras, J. Mater. Chem., 2009, 19, 3049 RSC.
- Y. Shu, Y.-F. Lim, Z. Li, B. Purushothaman, R. Hallani, J. E. Kim, S. R. Parkin, G. G. Malliaras and J. E. Anthony, Chem. Sci., 2011, 2, 363 RSC.
- Z. Li, Y.-F. Lim, J. B. Kim, S. R. Parkin, Y.-L. Loo, G. G. Malliaras and J. E. Anthony, Chem. Commun., 2011, 47, 7617 RSC.
- T. Zhou, T. Jia, B. Kang, F. Li, M. Fahlman and Y. Wang, Adv. Energy Mater., 2011, 1, 431 CrossRef CAS.
- K. N. Winzenberg, P. Kemppinen, F. H. Scholes, G. E. Collis, Y. Shu, T. Birendra Singh, A. Bilic, C. M. Forsyth and S. E. Watkins, Chem. Commun., 2013, 49, 6307 RSC.
- F. G. Brunetti, X. Gong, M. Tong, A. J. Heeger and F. Wudl, Angew. Chem., Int. Ed., 2010, 49, 532 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |