 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress on graphene-based hybrid electrocatalysts
BaoYu
Xia
,
Ya
Yan
,
Xin
Wang
* and
Xiong Wen (David)
Lou
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459. E-mail: wangxin@ntu.edu.sg; xwlou@ntu.edu.sg; davidlou88@gmail.com
First published on 31st March 2014
Abstract
Graphene-based hybrid nanostructures have attracted increasing interest worldwide. Benefiting from their remarkable electrochemical catalytic properties derived from chemical compositions and synergetic effects of multi-functionalities, these graphene-based hybrid nanomaterials will play a significant role in cutting-edge innovation for novel electrocatalysts. In this review, we summarize recent progress in the design and synthesis of graphene-based hybrid nanomaterials with controlled shape, size, composition and structure, and their application as efficient electrocatalysts for energy related systems. We conclude this article with some future trends and prospects which are highlighted for further investigations on graphene-based nanomaterials as advanced electrocatalysts.
 Bao Yu Xia | Bao Yu Xia received his Ph.D. degree in Materials Science and Engineering from Shanghai JiaoTong University (China) in 2010. He then moved to Nanyang Technological Univerisity, where he is a research fellow under the supervision of Prof. X. Wang and Prof. X. W. Lou. His research interests are in the areas of nanostructured electrocatalysts and their application in energy conversion. |
 Ya Yan | Ya Yan earned her bachelor's degree in Chemical Engineering from Northwest University (China) in 2010. She is currently pursuing a doctoral degree under the supervision of Prof. Xin Wang in the School of Chemical and Biomedical Engineering, Nanyang Technological University. Her research interests focus on the development of non-noble metal based nanocatalysts for electrochemical hydrogen evolution. |
 Xin Wang | Xin Wang received his Bachelor (1994) and Master (1997) degrees in Chemical Engineering from Zhejiang University, and Ph. D. degree (2002) in Chemical Engineering from Hong Kong University of Science and Technology. He joined Nanyang Technological University as an assistant professor in 2005 and was promoted to associate professor in 2010. He has been working on electrocatalysis and electrochemical technologies for energy harvesting. He has authored more than 120 papers in refereed journals. He also holds six patents on novel nanomaterials for energy applications. |
 Xiong Wen (David) Lou | Xiong Wen (David) Lou received his B.Eng. (1st class honours) (2002) and M.Eng. (2004) degrees from the National University of Singapore. He obtained his Ph.D. degree in chemical engineering from Cornell University in 2008. He is currently an Associate Professor at Nanyang Technological University. As the lead author, he has published about 160 papers with total citation of ∼10 |
1. Introduction
Stimulated by the increasing demand for energy and pressing need for fossil fuel alternatives, researchers worldwide have been actively exploring new sustainable energy sources and technologies.1 Amongst the various possibilities, electrochemical energy conversion technology is considered one of the most viable solutions, in view of the fact that it is environmentally friendly, affords high power density and high stability. Furthermore, some electrochemical energy conversion technologies such as fuel cells can also provide high energy densities, making them promising technologies for the generation of sustainable and green energy.2Various electrochemical reactions are involved in these electrochemical technologies. In fuel cells, electrochemical fuel (hydrogen and alcohols) oxidation reactions (HOR and AOR) and oxygen reduction reaction (ORR) take place at the anode and the cathode, respectively.3 ORR and oxygen evolution reaction (OER) are also the two half-reactions in the discharging–charging process of rechargeable metal–air batteries.2 Besides OER, hydrogen evolution reaction (HER) is the other half-reaction involved in water splitting.4 All these electrochemical reactions require the presence of efficient electrocatalysts, which play an important role in determining the conversion efficiency of electrochemical systems.5 Pt-based electrocatalysts are most widely used in these electrochemical devices. However, high cost and low stability of noble metals limit their practical application in a large scale. Therefore, design and preparation of advanced alternative electrocatalysts with excellent performance and low cost are of great importance for the development of these electrochemical systems.6
In view of their abundance and stability, different carbon materials have been investigated for their potential application in electrochemical devices.7 Of particular interest is graphene, a new carbon material, which consists of a single atomic layer of carbon atoms.8 Its planar two-dimensional (2D) structure offers high surface area for the attachment of nanocrystals with good distribution, therefore preventing aggregation. Several highly desirable properties of graphene further favor their potential application. First, good electrical and optical properties enable potentially high performance as electron carriers in electrochemical applications.9 Second, good mechanical and thermal properties endow graphene-based composites with excellent thermal and mechanical stability which may be important in electronic devices or electrocatalytic systems involving heat release, such as fuel cells, metal–air batteries and the water splitting process.10,11 Lastly, development of low cost and simple preparation methods also makes the processing of graphene-based nanocomposites simple and hence favors their practical applications. These nanocomposites have the potential to manifest not only unique properties of their individual components but also novel physical and chemical properties arisen from the synergetic effect between graphene and coupled nanostructures, eventually resulting in further improvement in performance.15 These promising properties together with the ease of processibility and functionalization make graphene-based functional materials ideal candidates for a variety of energy applications.12–14
Some recent reviews have summarized the progress of graphene-based nanocomposites in energy storage and conversion fields such as solar cells, lithium-ion batteries, and supercapacitors.16–22 Graphene-based nanocomposites are highly promising as advanced catalysts with excellent performance.23–25 However, only a handful of reviews have focused on graphene-based hybrid nanomaterials for electrocatalysts.26,27 In this review, we focus on the synthesis of graphene-based hybrid nanomaterials and their electrocatalytic applications in fuel cells, Li–O2 batteries and water splitting. To facilitate our discussion, a wide variety of graphene-based hybrid nanocomposites is grouped into three main categories: doped/modified graphene, noble metal/graphene hybrids, and graphene/non-metal composites. For each category, a few important aspects will be discussed including advantages of structural characteristics, interaction between attached components and graphene, and their electrochemical performance as electrocatalysts. Further understanding and development of graphene-based hybrid electrocatalysts could therefore help to address the demand for new electrochemical energy devices and challenges for the widespread use of graphene-based electrocatalysts.
2. Graphene as electrocatalysts
2.1. Graphene doped with non-metals as electrocatalysts
Graphene is intrinsically inert, but starts possessing electrocatalytic activity after chemical modification or doping.28,29 Chemical doping involves replacement of carbon atoms in the hexagonal lattice by heteroatoms. Thus, the composition and the lattice structure of graphene are changed during the doping process.30–32Doping graphene with nitrogen (N) atoms has made exceptional progress over the past decade and many promising material concepts have been demonstrated.33 Three types of N species including graphitic N, pyridinic and pyrrolic N can be introduced into graphene, each of which affects its atomic charge distribution differently. Theoretical results on the effect of N doping into graphene have revealed that both atomic charge and spin density determine the material's catalytic properties.35 Consequently, the mechanisms through which N-doped graphene achieves activity enhancement also differ depending on the nature of N species present.34 It is thus important to determine the correlation between nitrogen bonding configuration in graphene and electrocatalytic activity.
Pyridinic and quaternary nitrogen species are generally considered as the active components of N-doped graphene, but some doubts still exist.36,37 Selective doping of graphene has hence been undertaken to elucidate the ORR activity of different nitrogen species. To this end, Ruoff's group concluded that the electrocatalytic activity of N-doped graphene is highly dependent on the graphitic-N content while pyridinic-N species improve the onset potential of ORR.34 On the other hand, Yasuda's work has recently shed some light on the origin of ORR activity in N-doped graphene. More importantly, the authors also presented a method for synthesizing highly efficient N-doped graphene electrocatalysts with tunable nitrogen active sites.38 Knights further determined that a low nitrogen doping level of 2.8% is sufficient to obtain high ORR activity via a 4e process (Fig. 1a–c).39 This implies that the synergistic effect among N-containing species present contributes significantly to the enhanced ORR activity even at low doping levels. Two different ORR pathways (associative and dissociative) have been proposed for N-doped graphene electrocatalysts in an alkaline medium.28 However, the detailed mechanism should be further explored.
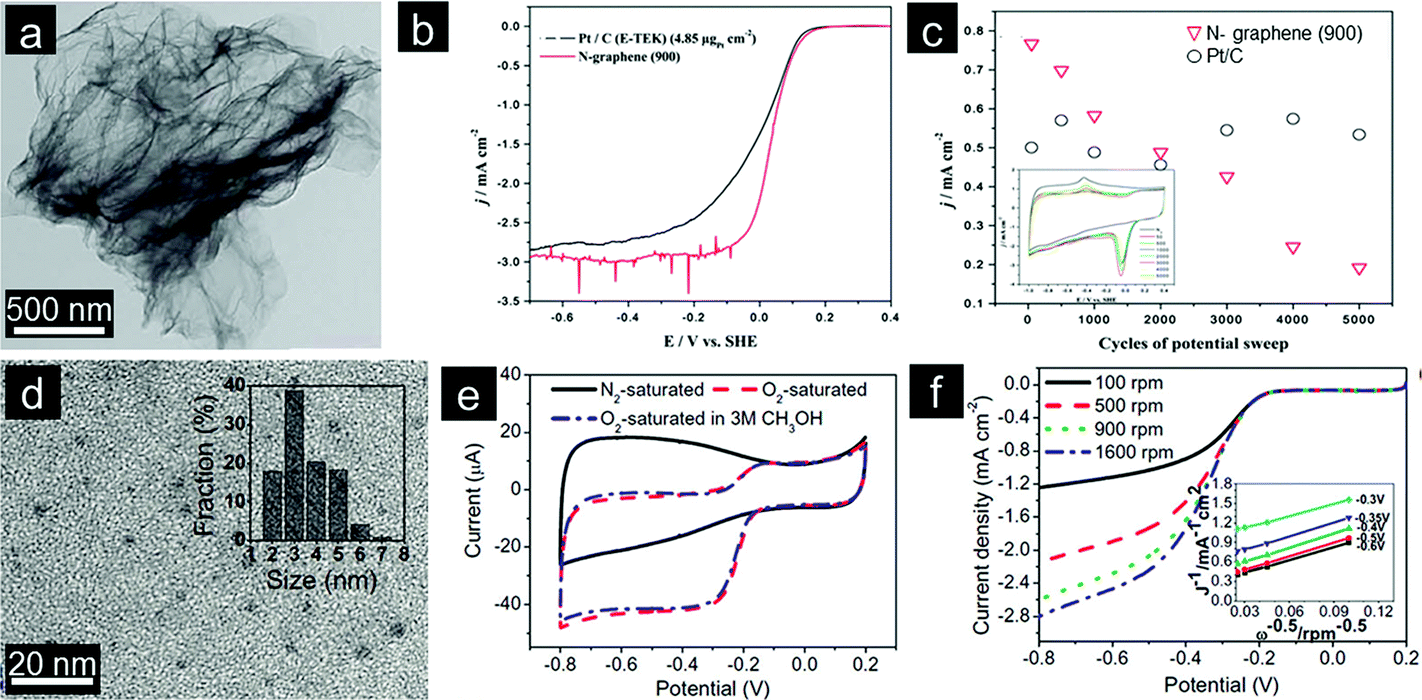 | ||
| Fig. 1 (a) Typical TEM image and (b and c) electrochemical properties of N-doped graphene. (Reprinted with permission from ref. 39. Copyright© 2011, Royal Society of Chemistry.) (d) TEM image with an inset showing the size distribution, (e) CVs and (f) rotating disk electrode (RDE) curves of GQDs in 0.1 M KOH. (Reprinted with permission from ref. 49. Copyright© 2012, American Chemical Society.) | ||
In addition to the doped N content, the morphology of final graphene structures also plays a significant role in the electrochemical performance. For instance, Dai's recent work clearly indicates that the conjugation size of graphene is an important parameter.40,41 On the other hand, the presence of abundant edges and functional groups is necessary for N-doped colloidal graphene quantum dots (GQDs) to exhibit electrocatalytic activity. GQDs also provide unique opportunities for systematic investigation of mechanisms of ORR (Fig. 1d–f).42 Porous structures are also beneficial, given that such structures can possibly tune N-doped graphene's band gap energy.43
The exfoliated graphene nanosheets produced by chemical methods have many edge and defect sites.44 Such edge and defect sites are considered to serve as active sites for catalyzing chemical reactions. Furthermore, the planar structure of graphene nanosheets allows reactant access from both sides of the sheet.45 With these characteristics, graphene nanosheets are expected to be efficient cathode electrocatalysts for Li–O2 batteries.46–49 N doping of graphene can modify the electronic property and provide more active sites for enhanced interaction between the carbon structure and other molecules, thus helping to improve the electrochemical performance in the Li–O2 battery.50 Sun and coworkers first applied N-doped graphene as the cathode electrocatalyst for Li–O2 cells. N-doped graphene has been shown to exhibit higher electrochemical activity compared to graphene in a non-aqueous electrolyte due to the defects and functional groups introduced by nitrogen doping.51
Besides the highly electronegative N atom, graphene has also been doped with other heteroatoms and subsequently investigated as metal-free electrocatalysts for ORR.52 Interestingly, graphene also exhibits significantly improved ORR catalytic activity when doped with heteroatoms including S, Se and I with similar electronegativity as C.53,54 Huang and coworkers have prepared S-doped graphene by annealing graphene oxide (GO) and benzyl disulfide in argon, and determined that under specific annealing conditions, S-doped graphene exhibits similar ORR onset potential and higher current density compared to commercial Pt/C electrocatalysts.55 S has electronegativity close to that of C, thus the enhanced ORR activity of S-doped graphene is attributed to the modifications in spin density instead of changes in atomic charge density as in B and N doping.56 Furthermore, the authors also speculated that the presence of C–S bonds, formed mostly at the edges or defects of the graphene sheet, plays a crucial role as catalytic active sites for promotion of ORR. Explanations outlined above for S-doped graphene can also be extended to Se-doped graphene supported on carbon nanotubes (CNTs).57 The latter's improved ORR activity is also contributed by its evidently larger atomic size of Se compared to C. This larger atomic size may cause higher strain at the graphene edges where heteroatoms are predominantly located; this may in turn facilitate charge localization and associated O2 chemisorption. Furthermore, Se has high polarizability and its electron lone pairs can easily interact with molecules in the surrounding electrolyte. These factors thus enable Se-doped graphene supported on CNTs (1 wt% Se) to possess comparable ORR activity to N-doped graphene at higher doping levels (4–8 wt% N). For I-doped graphene prepared by annealing GO and I2, the content and more importantly the nature of I species present are crucial in determining electrochemical performance. Structures with a greater percentage of more negative I3− species result in higher ORR activity compared to those containing I5− species, owing to a greater degree of positive charge induced by the former.58
Graphene has also been doped by heteroatoms which are more electropositive (or more electron deficient) compared to carbon. In these cases, the positive charge is localized on the dopant itself which serves as the catalytic site for O2 chemisorption. P-doped (0.26 at%) graphene has been prepared via pyrolysis of toluene and triphenylphosphine. Such P-doped graphene shows high electrocatalytic ORR activity, long-term stability, and excellent tolerance to cross-over effects of methanol in an alkaline medium.59 Additionally, a higher P doping level (1.16 at%) improves ORR electrocatalytic activity and the ORR undergoes a 4e process.60 Boron is another prime candidate for doping of graphene, owing to its strong electron withdrawing capability.61 The resultant B-doped graphene exhibits excellent electrocatalytic activity towards ORR in alkaline electrolytes due to its unique structure and electronic properties.62
Considering the different electronegativities of various doping elements, a dual doping approach will be able to induce a unique electronic structure in graphene and achieve synergistic coupling between heteroatoms.29,63 Dual-doped graphene catalysts are thus expected to be catalytically more active than their singly doped counterparts.64,65 In this regard, dual doping of graphene with two heteroatoms is currently one important strategy for tailoring chemical and physical properties of graphene for specific electrocatalytic applications.66,67 Dai and coworkers synthesized B, N co-doped graphene with tunable composition by thermal annealing GO in the presence of boric acid and ammonia.68 The resultant B, N co-doped graphene demonstrates superior electrocatalytic activity over commercial Pt/C electrocatalysts for ORR in alkaline medium.69 Qiao and coworkers prepared B, N co-doped graphene with high purity and well-defined doping sites by a two-step doping method. Qiao's dual-doped-B, N graphene electrocatalysts exhibit better ORR electrocatalytic efficiency and greater stability compared to singly doped graphene. The improved performance is attributed to an enhanced synergistic coupling between B and N which facilitates the electrocatalytic ORR (Fig. 2a–c).70 Feng and Mullen prepared a three-dimensional (3D) B, N co-doped graphene aerogel in order to increase possible interactions between electrode materials and the electrolyte. This 3D B, N dual-doped graphene exhibits excellent electrochemical performance as a result of its high surface area, 3D macroporosity and high electrical conductivity.69,70 The synergistic effect between heteroatoms was also investigated for N and S dual-doped graphene with the porous structure. Again, the competitive ORR catalytic activity displayed by the dual-doped catalyst with pyrrolic/graphitic N-dominant structures was attributed to the synergistic effect of N and S co-doping.71,72 Qiao and coworkers also reported the synthesis of S and N dual-doped graphene with a mesoporous structure which shows excellent catalytic activity, comparable to that of the commercial Pt/C catalyst, including a highly positive onset potential and very high kinetic limiting current.73 Outstanding performance of dual-doped graphene is explained by its dual activation of C atoms, as evidenced by both experimental measurements and quantum chemistry calculations (Fig. 2d–f).73
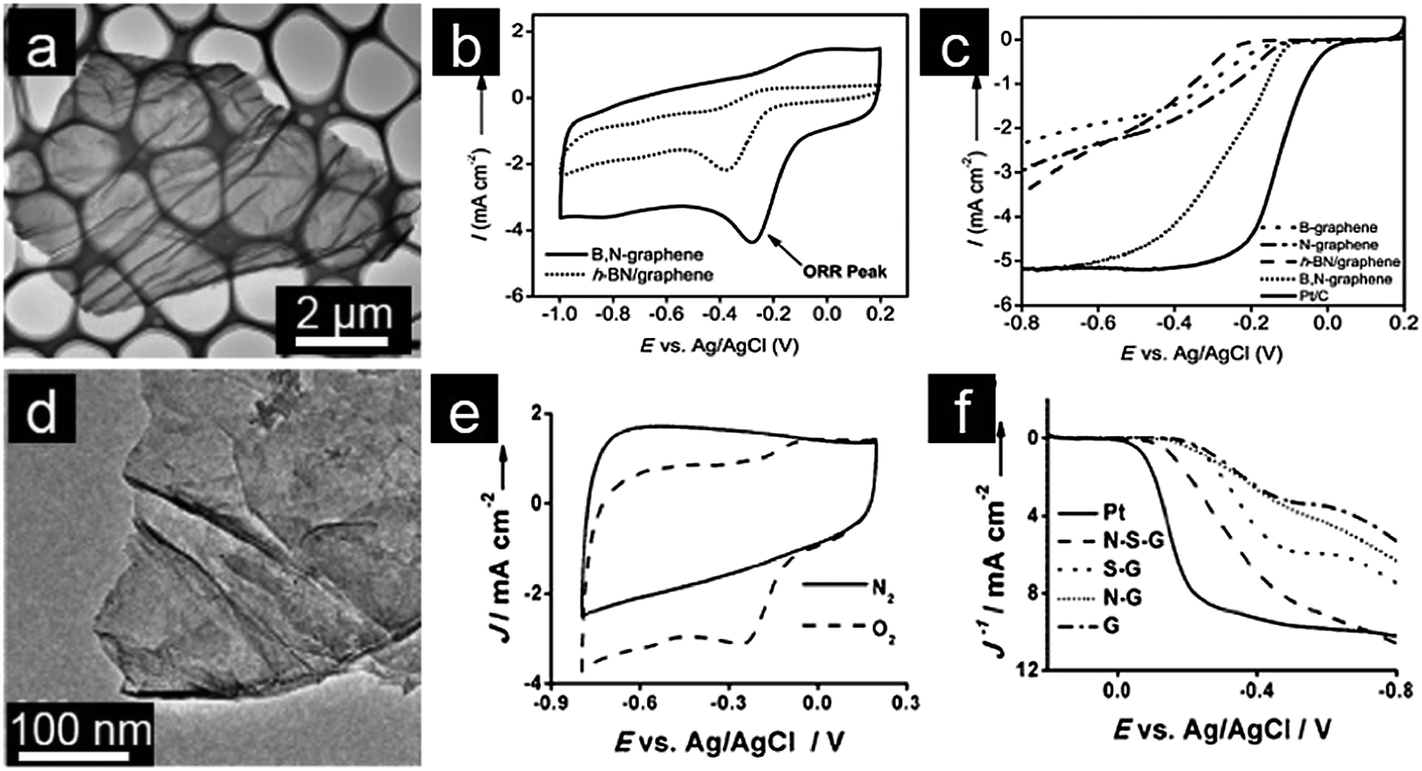 | ||
| Fig. 2 (a) TEM image, (b) CVs and (c) RDE curves of B, N co-doped graphene. (Reprinted with permission from ref. 70. Copyright© 2013, WILEY-VCH.) (d) TEM image, (e) CVs and (f) RDE curves of S, N co-doped graphene. (Reprinted with permission from ref. 73. Copyright© 2012, WILEY-VCH.) | ||
2.2. Graphene doped with transition metals as electrocatalysts
Despite the superior ORR activity in alkaline media, electrocatalytic performance of graphene doped with non-metals is generally poor in acidic media, perhaps because of the presence of inactive surface sites. Some previous studies show that non-graphene carbon materials are able to exhibit high ORR activity in acidic media via the addition of metal species and formation of transition metal–N coordination structures.74–77 This idea was then extended to graphene with active sites for coordination with transition metals such as Fe and Co. It was expected that transition metal–graphene hybrid materials would lead to enhanced electrocatalytic performance.78 For example, Feng and Mullen synthesized Fe incorporated N-doped graphene which displays significantly enhanced electrocatalytic performance of high current density and stability. This Fe–N-graphene catalyst features a 4e transfer process and outperforms the commercial Pt/C electrocatalyst toward ORR in both acidic and alkaline media.79 Similarly, Fe-doped graphene structures were also reported to be efficient electrocatalysts for ORR in acid media.80,81 Chen and coworkers further designed 3D Fe–N coordination graphene/CNT structures to facilitate mass exchange and diffusion during the electrochemical ORR. Optimized doping level and synergistic coupling arising from the coordination between N and Fe favor ORR and boost electrochemical performance. Dong and coworkers reported the coordination of Co with N-doped graphene as ORR electrocatalysts in alkaline media, showing high selectivity for the reaction.82 Co, Fe or even bimetallic (Co–Fe) embedded graphene hybrids can be prepared via thermal treatment of these salts with graphene. The transition metal–graphene hybrids prepared by this method also show promising ORR activity in acidic media.83–852.3. Functionalized graphene as electrocatalysts
Although N-containing graphene is considered to be a promising candidate for ORR, significant challenges still impede its commercialization for fuel cell technology. A deeper understanding of the roles played by various nitrogen states during electrocatalysis and improvement on chemical doping procedures are some of the key issues which need to be solved before commercialization is feasible.86 In the latter case, most chemical doping approaches reported so far are complex and performed under harsh conditions, thus severely limiting their large scale utilization.29,87As aforementioned, the enhanced ORR catalytic activity of N-doped graphene is attributed to the electron-accepting ability of N atoms present. Given this understanding, researchers thus extended this concept to the functionalization of graphene with polyelectrolytes as metal-free electrocatalysts.88–90 For example, Wang and coworkers reported that physical adsorption of poly-diallyldimethylammonium chloride (PDDA), a positively charged polyelectrolyte with electron-withdrawing ability, onto N-free graphene creates positive charges on the graphene surface via intermolecular charge transfer.91 The resultant PDDA-functionalized/adsorbed graphene manifests dramatically enhanced electrocatalytic activity toward ORR. This nanocomposite also exhibits better fuel selectivity and improved stability compared to the commercial Pt/C catalyst. A similar observation was made for tridodecylmethylammonium chloride (TDMAC), another quaternary ammonium salt.93On the other hand, Hou and coworkers designed amino-functionalized graphene from graphene oxide in the presence of ammonia solution via a solvothermal approach.92 Their results show that the graphitic- and amino-type of nitrogen components determine the onset potential and the electron transfer number respectively, while the total content of graphitic and pyridinic N atoms is the key factor in enhancing current density for ORR. Dong and coworkers prepared neutral red covalently-functionalized graphene to improve the solubility and stability of graphene.94 Electrochemical impedance results further confirmed that neutral red functionalization can effectively accelerate electron transfer. The nanocomposites also exhibit excellent electrocatalytic activity toward uric acid oxidation due to the synergistic effect between neutral red and graphene. Transition metal phthalocyanine is also a promising candidate often used for functionalization of graphene as efficient ORR electrocatalysts.95–97 These nanocomposites are constructed by the strong π–π interaction and exhibit high activity for ORR with excellent stability and selectivity.98,99
3. Graphene nanostructures as supports for noble metals
Although graphene-based electrocatalysts have the advantage of significantly reducing the cost, their activity remains relatively low compared to noble metal based electrocatalysts.100 Noble metals and their alloys are still the best performing electrocatalysts for electrochemical reactions. Thus developing a new generation of low cost and highly active electrocatalysts is considered urgent for future electrochemical energy technologies. Moreover, majority of previous reports for metal-free electrocatalysts are related to ORR reaction at the cathode in basic media, probably due to faster reaction kinetics in alkaline media. But many electrochemical reactions are also performed in acidic media, such as the anode reactions in HOR and AOR fuel cells, and the research of novel electrocatalysts in acidic media is equally important.101Despite the great advances in unsupported nanostructured noble metals for electrocatalysts,102 the relatively low efficiency and high usage of noble metals in such unsupported noble metal electrocatalysts still limit their practical applications. This has triggered much research into improving Pt based electrocatalysts. Two important strategies are commonly employed, sometimes combined, to improve the electrochemical performance, stability and durability of these catalysts. The first is to develop specific morphologies. These shaped noble metal nanoparticles can lead to improved performance and also, in some cases, eliminate the need for supports thus avoiding the carbon corrosion problem. Another strategy is to design supports for better anchorage and enhanced interaction with metals. In this respect, graphene has emerged as a particular favorite as it possesses high surface area, low cost and enhanced conductivity compared to other traditional carbon supports for electrochemical applications.103,104 Furthermore, when alloy nanostructures are supported on graphene, it not only results in enhanced electrocatalytic activity and stability, but also allows for synergetic effects between metals and graphene, thereby reducing the noble metal content.105–107
3.1. Graphene supported Pt-based electrocatalysts
Pt-based electrocatalysts are the most popular choice for fuel oxidation at the anode and oxygen reduction at the cathode.5 Since around 2004, many efforts have been made towards obtaining Pt/graphene electrocatalysts with high electrochemical performance. Pt/graphene composites are generally prepared by the direct reduction of Pt precursors in the presence of reducing agents, such as H2, ethylene glycol, and NaBH4. Nature of Pt nanoparticles and the π–d hybridization interaction between Pt nanoparticles and graphene together determine the final catalytic performance.108 Yoo and coworkers reported that strong interaction between Pt nanoparticles and graphene nanosheets (GNS) enables formation of subnanometer sized (below 0.5 nm) Pt clusters (Fig. 3a). This allows the Pt nanoparticles to acquire specific electronic structures which in turn modifies the catalytic activity and gives unusually high methanol electro-oxidation activity.110 Therefore, controllable deposition of Pt nanocrystals on graphene is highly desirable for enhanced performance.109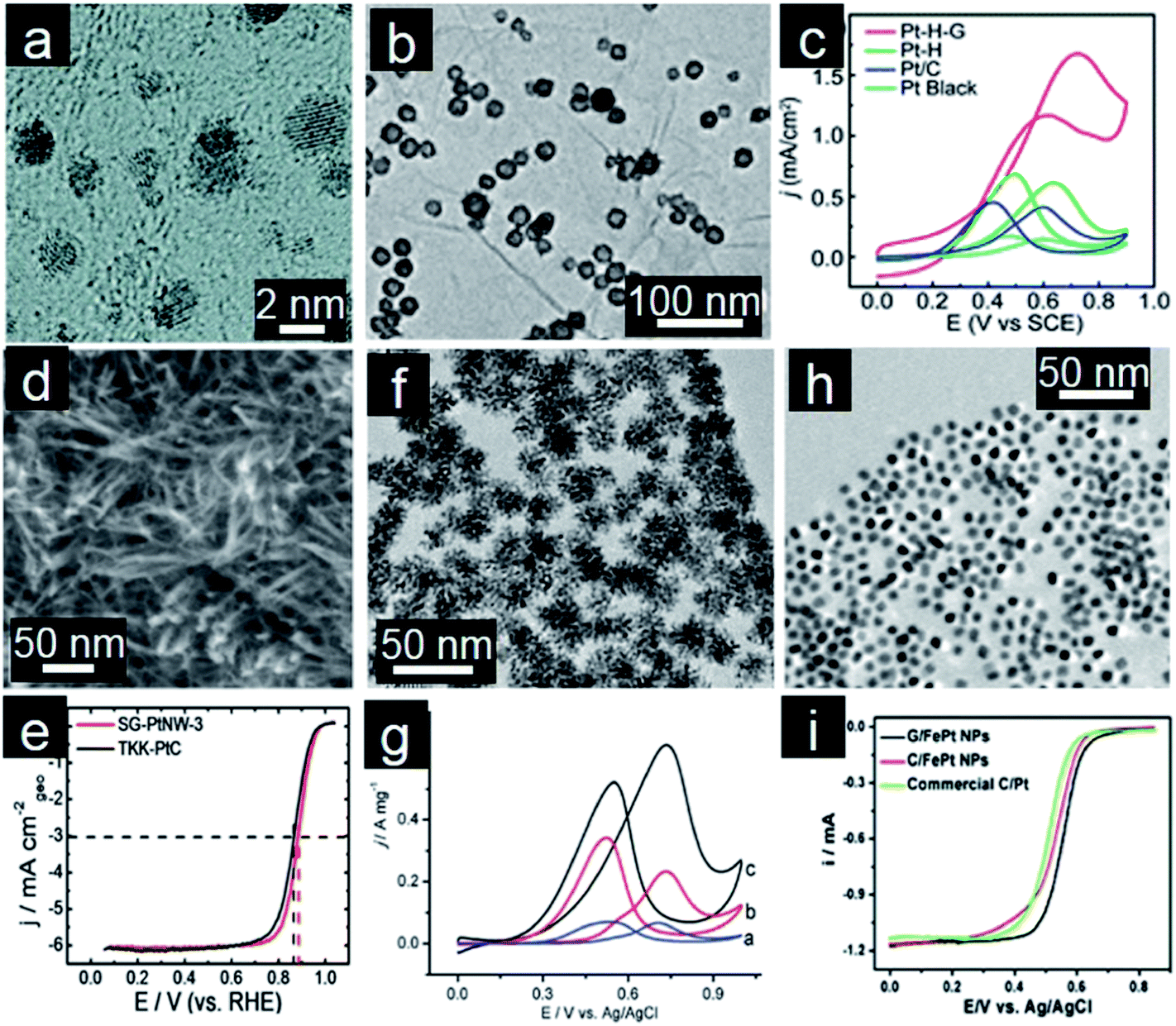 | ||
| Fig. 3 (a) TEM image of Pt subnanoclusters/graphene. (Reprinted with permission from ref. 110. Copyright© 2009, American Chemical Society.) (b) TEM image and (c) CVs for methanol oxidation of hollow Pt/graphene. (Reprinted with permission from ref. 112. Copyright© 2012, Royal Society of Chemistry.) (d) TEM image, (e) ORR polarization curves in O2-saturated 0.1 M HClO4 solution (10 mV s−1; 1600 rpm) of Pt nanowire arrays/S-doped graphene. (Reprinted with permission from ref. 114. Copyright© 2013, Nature Publishing Group.) (f) TEM image, (g) CVs for methanol oxidation of 3D PtPd dendrites/graphene. (Reprinted with permission from ref. 127. Copyright© 2010, American Chemical Society.) (h) TEM image and (i) RDE curves of FePt nanocubes assembled on graphene. (Reprinted with permission from ref. 137. Copyright© 2012, American Chemical Society.) | ||
The morphology of Pt nanocrystals also affects the performance of these Pt/graphene catalysts.111 Wan and coworkers developed in situ preparation of hollow Pt nanocrystals decorated on graphene (Fig. 3b). Both the merits of hollow nanoparticles and the synergetic effect between Pt nanoparticles and graphene supports contribute to the enhanced electrocatalytic activity for methanol oxidation reaction (MOR, Fig. 3b and c).112 Furthermore, one-dimensional (1D) Pt nanowires have also drawn much attention owing to their unique anisotropic structures, surface properties and excellent electrocatalytic activities.113 Ultrathin Pt nanowire arrays prepared on S-doped graphene (Fig. 3d), lead to 2–3 times higher catalytic activities toward the oxygen reduction and methanol oxidation reaction compared with the commercial Pt/C catalyst (Fig. 3e).114 A branched Pt nanowire/graphene hybrid was also prepared by a HCOOH reduction method.115 However, monometallic Pt is susceptible to deactivation or poisoning by some reaction intermediates especially CO molecules. Therefore, Pt-based alloys have been investigated as alternatives for improved catalyst stability. Alloying also results in the concomitant reduction of cost due to lowered noble metal content. Because of that, much effort has been directed towards the development of bimetallic (PtPd,116,117 PtAu,118,119 PtRu,120 PtFe,121 PtCo,122 PtNi,123 PtCu,124 PtSn,125etc.) and even ternary (PtPdAu126) nanocatalysts. Typically, bimetallic nanodendrites with controllable size and numbers of Pt branches exhibit larger electrochemical active surface areas (Fig. 3f), thus leading to higher electrocatalytic activity compared to Pt/C electrocatalysts for MOR (Fig. 3g).127
As discussed above, electrochemical performance of catalysts is strongly related to not only the size and composition but also to the shape and morphology of the nanoparticles.128 Considering the strong interaction between graphene supports and the formed noble metal nanoparticles, the supports will have a significant impact on the resultant nanoparticles with specific morphology due to different surface chemistries present.129,130 To exert greater control over the particle formation process and thus reduce the influence of support surface chemistry, self-assembly techniques are employed to attach nanoparticles onto graphene.131–133 This involves assembly of pre-prepared nanocrystals and graphene supports, thus the morphology and shape of nanocrystals can be efficiently controlled and improvement of electrochemical performance can be realized.134,135 Wang and coworkers reported self-assembly of mixed Pt and Au nanoparticles with controlled molar ratios and loadings onto PDDA-functionalized graphene and these hybrid materials were used as highly effective electrocatalysts for formic acid oxidation.136 Sun's group developed a facile solution phase self-assembly of FePt nanocubes on the graphene surface (Fig. 3h).137 The desired size, composition and shape of FePt nanocrystals could be controlled during the preparation process. These graphene supported FePt nanocubes are more active and durable catalysts for ORR in 0.1 M HClO4 than unsupported nanoparticles or commercial Pt particles deposited on conventional carbon supports (Fig. 3i).
3.2. Graphene supported other noble metals as electrocatalysts
Pt and Pt-based alloys usually suffer from several disadvantages such as poisoning effects, corrosion by some electrolytes, and high costs.138,139 These problems might be in part mitigated in other noble metals and alloys. Further exploration of other noble metals and their alloys on graphene is thus of importance for both fundamental research and practical applications.140Pd nanocatalysts are more favorable for methanol and formic acid oxidation than Pt due to greater abundance and higher catalytic activity.141 Xie and coworkers prepared Pd/graphene by the redox reaction between K2PdCl4 and GO. Oxygenated functional groups present on the GO surface may take part in the formation of Pd/graphene. The clean surface of Pd/graphene, as a result of a surfactant-free synthesis procedure, alongside good dispersion on graphene, allows this nanocatalyst to show high electrocatalytic ability toward formic acid and ethanol oxidation (Fig. 4a).142 Using Ag/graphene as the support and the template, Chen and coworkers designed hollow PdAg nanorings decorated graphene nanosheets by a facile galvanic displacement reaction (Fig. 4b). More importantly, this PdAg/graphene nanohybrid exhibits not only high catalytic activity for the ORR but also remarkably high tolerance towards methanol crossover.143 Kim and coworkers designed a layer-by-layer (LBL) assembly to create 3D electrocatalytic thin films for methanol oxidation.144 Graphene supported ultrafine Au nanoclusters without protecting agents exhibit surprisingly excellent electrocatalytic activity toward ORR and superior methanol tolerance (Fig. 4c and d).145 The authors concluded that this observation could be ascribed to small size of Au nanoparticles with a clean surface and the synergetic coupling effect between Au nanoclusters and graphene. Ag nanowires have been proven to be a good cathode catalyst in alkaline media.146 Thus, the combination of Ag nanowires and graphene also results in a better electrocatalyst than graphene or Ag nanowires because of the synergistic effect and creation of porous nanochannels.147
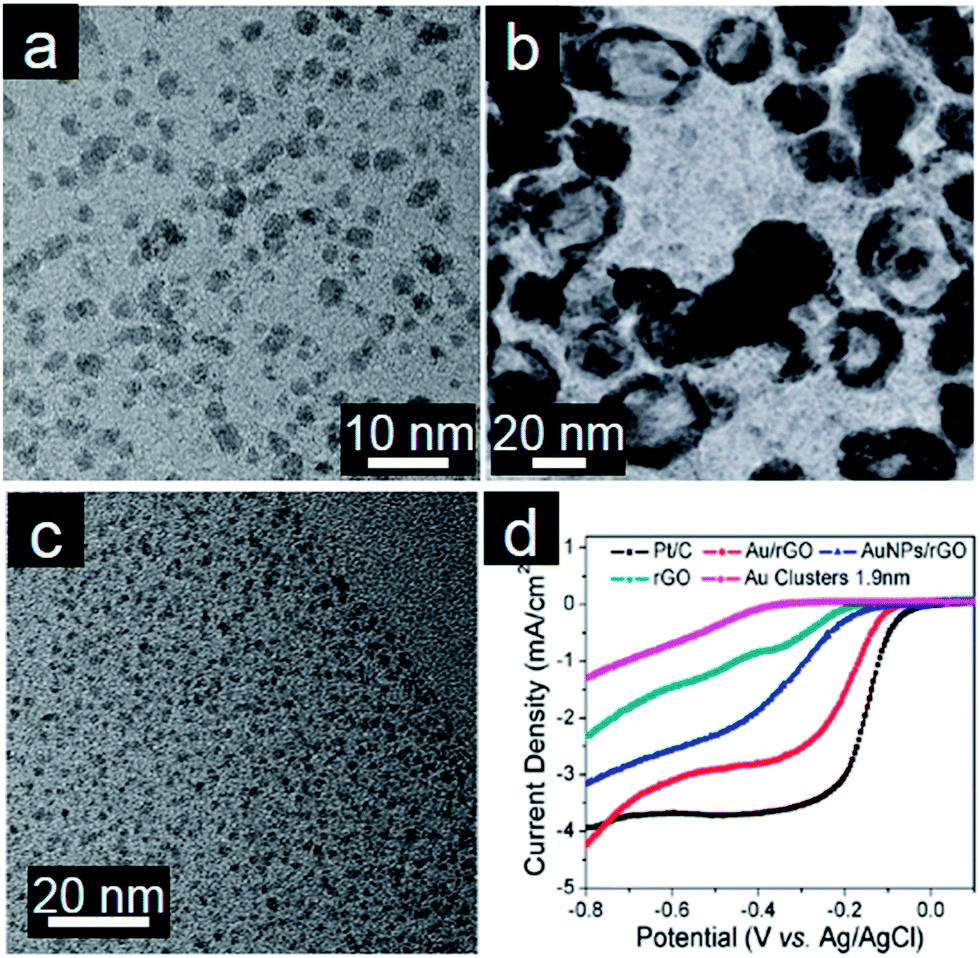 | ||
| Fig. 4 (a) TEM image of Pd/graphene. (Reprinted with permission from ref. 142. Copyright© 2011, American Chemical Society.) (b) TEM image of PdAg nanorings/graphene. (Reprinted with permission from ref. 143. Copyright© 2013, WILEY-VCH.) (c) TEM image of assembled Au/graphene and (d) RDE curves of different electrocatalysts in 0.1 M KOH (50 mV s−1; 1600 rpm). (Reprinted with permission from ref. 145. Copyright© 2011, American Chemical Society.) | ||
3.3. Noble metals attached on functionalized graphene
As mentioned above, noble metal nanocrystals are deposited on graphene oxides with abundant functional groups.148 These functional groups and defects present on the graphene surface act as active sites and nucleation sites for the formation of noble metals. They also strengthen the interaction between noble metal nanocrystals and graphene, thus enhancing the electrocatalytic activity and durability of the hybrid nanocatalysts.149 Moreover, these oxygen-containing functional groups allow for good dispersion of graphene oxide in water and other organic solvents, which is a key factor for electrocatalytic application to prevent aggregation during catalyst preparation.150 However, reduced graphene oxides obtained from chemical and thermal reduction of graphene oxides inevitably possess defects and small defective graphene fragments. These defects include structural imperfections and chemical impurities and serve to disrupt the desirable electronic properties of reduced graphene oxide.151 Reduced graphene oxides are also easily oxidized and become unstable under harsh electrochemical conditions due to the corrosion of graphene.152 Thus graphene with few defects is often required in the preparation of nanocatalysts. Graphene with high quality is obtained mostly by chemical vapor deposition (CVD) and other methods.153 However, graphene prepared by these methods possesses hydrophobic surfaces, which make it difficult to anchor metal nanoparticles directly. Therefore, surface modification is an efficient way to tune the surface chemistry of graphene, which in turn affects its dispersion ability and affinity for metal nanoparticles attachment.88Various methods have been developed to prepare functionalized graphene.154,155 The resultant Pt/functionalized graphene catalysts typically exhibit improved catalytic performance and properties with respect to their specific applications compared to Pt/graphene. For instance, Wang and coworkers prepared Pt/N-doped graphene via a mild, plasma assisted approach which involves reduction of GO in ammonia plasma with subsequent simultaneous reduction of these N-doped graphene sheets and H2PtCl6 in hydrogen plasma.156 This Pt/N-doped graphene hybrid exhibits high catalytic activity as a result of homogenous dispersion of 2–4 nm Pt nanoparticles onto N-doped graphene. The presence of N- and O-containing functional groups is credited for enabling this strong interaction. Zheng and coworkers prepared Pt/graphene composites in an ionic liquid environment which is more oxytropic and less methanol-philic than the exterior aqueous solution. The resultant Pt/graphene hybrid exhibits both enhanced electrocatalytic activity and excellent methanol tolerance for ORR.157 Graphene can also be functionalized by genomic double stranded DNA (gDNA) which serves two critical roles. First, gDNA bridges adjacent GO sheets via strong non-covalent interactions, thereby preventing aggregation. Second, purine bases in gDNA also serve as nucleation sites for Pt2+ owing to strong cation–π interaction between Pt2+ and the purine ring. Pt/gDNA–GO composites exhibit significantly improved ORR performance compared to Pt/GO and Pt/C electrocatalysts. Again, the improved performance is likely due to the benefits of graphene functionalization with gDNA which helps in the efficient positioning of Pt nanoclusters onto GO.158
Graphene can also be functionalized with molecules or charged polyelectrolytes via π–π stacking or hydrophobic interactions. Formation of these non-covalent bonds enables the intrinsic electronic and structural properties of graphene to be preserved. Non-covalent functionalization of graphene by PDDA, a linear cationic polyelectrolyte, is commonly reported to stabilize supports and metal nanoparticles.159,160 Other non-covalent bonding agents such as cetyltrimethylammonium bromide (CTAB) are also used to functionalize graphene for electrocatalytic application.161 Additionally, introduction of conductive polymers also provides a way to improve the conductivity of graphene supports and reduce interfacial resistance of hybrid catalysts.162 Li and coworkers reported the preparation of the Pt/polyaniline (PANI) coated graphene composite which exhibits significantly enhanced electrocatalytic activity for methanol oxidation owing to the strong interaction between Pt and PANI–graphene.163 As the conducting bridge, polypyrrole (PPy) coating can also increase the hydrophilicity of pristine graphene nanosheets which are hydrophobic. These Pt/graphene–polymer nanocomposites have a higher surface area with more active sites leading to better electrocatalytic performance when compared to Pt/graphene.164,165
3.4. Noble metals supported on graphene nanostructures
The prominent effects of graphene supports on the activity and stability of electrocatalysts have been demonstrated.166,167 Aggregation of graphene sheets can also be resolved by using GQDs, a newly developed nanosized graphene.168,169 Abundant structural defects of GQDs can manipulate the dissociative adsorption of oxygen and binding of reaction intermediates O* and HO* on Pt surfaces.170 Recently, the covalent interaction between Pd particles and GQDs was proven by infrared spectroscopy.171 Generally, an electrode with a proper and well-designed nanoarchitecture can achieve excellent performance.172,173 On the other hand, the planar stacking of 2D graphene results in the formation of irreversible agglomerates which in turn leads to a drastic loss of electroactivity during the electrocatalyst assembly.174 3D porous graphene structures have also been designed and could lead to significantly enhanced electrochemical performance.175 As a consequence, 3D graphene frameworks are desirable for high catalyst loading to facilitate the mass transfer and maximize accessibility of the catalyst surface.176–180 Dong and coworkers reported the assembly of Pt nanoparticles on graphene to form 3D nanocomposites as ORR electrocatalysts. The electrocatalytic activity of resultant Pt/graphene nanocomposites can be tailored by simply changing the layering processes.181 Dai and coworkers prepared shape-defined ternary Pt/PdCu nanoboxes anchored onto a 3D graphene framework by a solvothermal process (Fig. 5a). As a new catalyst system for ethanol electrooxidation, the ternary Pt/PdCu nanoboxes/graphene electrocatalyst demonstrates significantly higher electrochemical performance when normalized to the total mass loading of hollow structures of active particles. Again it is attributed to the synergetic effect among components as well as the 3D interconnected graphene framework (Fig. 5b).182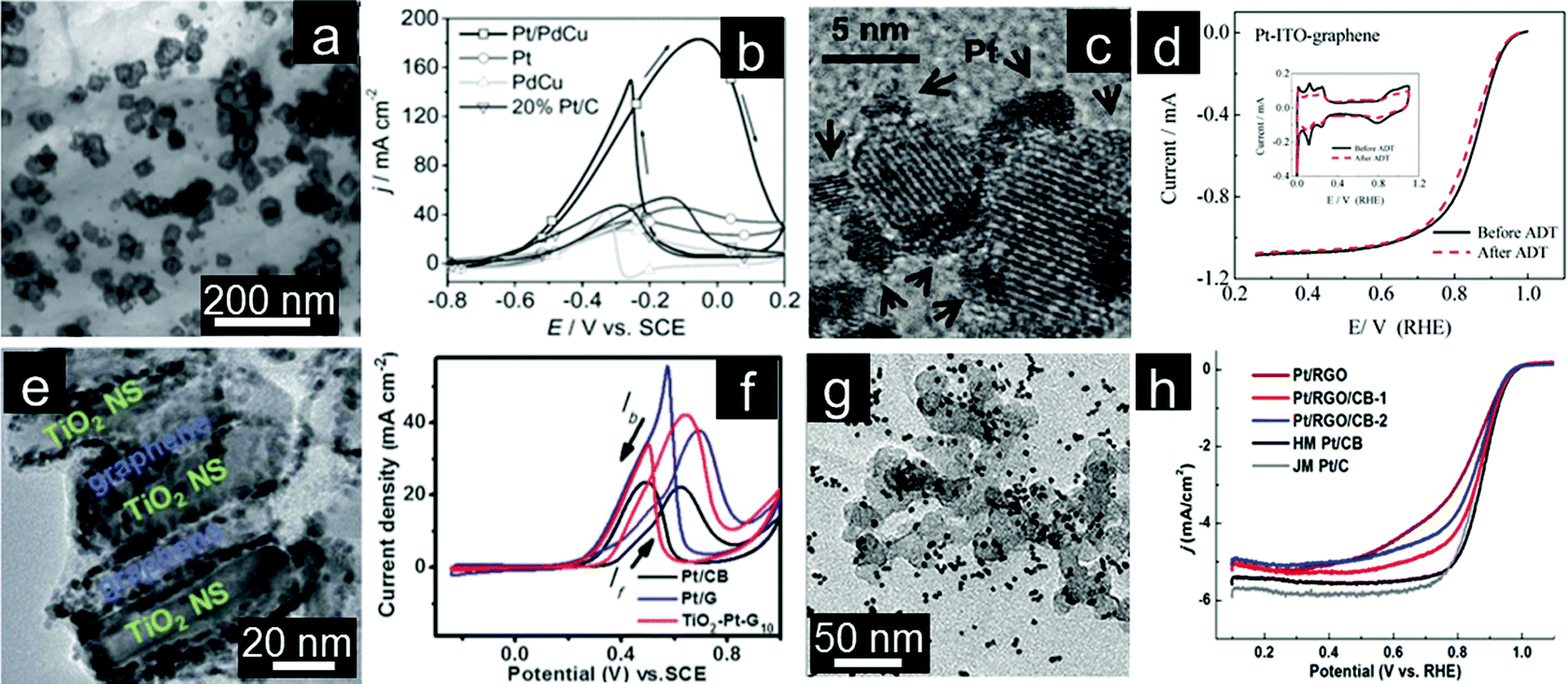 | ||
| Fig. 5 (a) TEM image and (b) CVs for ethanol oxidation of Pt/PdCu nanocubes assembled on the 3D graphene framework. (Reprinted with permission from ref. 182. Copyright© 2012, WILEY-VCH.) (c) TEM image and (d) RDE curves of the ITO–Pt–graphene electrocatalyst. Inset of (d) shows stability tests. (Reprinted with permission from ref. 191. Copyright© 2011, American Chemical Society.) (e) TEM image and (f) CVs for methanol oxidation of sandwich structured TiO2–Pt–graphene. (Reprinted with permission from ref. 192. Copyright© 2012, Royal Society of Chemistry.) (g) TEM image and (h) RDE curves of different Pt/graphene–carbon black composites for electrocatalysts. (Reprinted with permission from ref. 242. Copyright© 2012, American Chemical Society.) | ||
Re-stacking of 2D individual graphene sheets can be partially overcome by intercalating nanoparticles between the graphene layers.183 Moreover, migration of noble metal nanoparticles and CO poisoning also limit the efficiency of electrocatalysts.184 To enhance the electrochemical performance in terms of activity and durability, graphene coupled with metal oxides have proved to be effective supports.185–190 For instance, Liu and coworkers reported the in situ formation of ITO–Pt–graphene triple junctions that enhances the interaction and contact among the components for the improved electrochemical activity and stability (Fig. 5c and d).191 TiO2 nanosheets are introduced into Pt–graphene catalysts to form sandwich structures which enable maximum interaction among TiO2, Pt and graphene (Fig. 5e), hence improving the electrocatalytic performance (Fig. 5f).192 Incorporation of SnO2, CeO2 and MnO2 into graphene-based hybrid electrocatalysts has also been investigated.187,193,194 It is important to note that simultaneous incorporation of different components onto graphene with homogenous distribution will play a critical role on the eventual electrochemical performance of the catalyst. Ideally, components should be incorporated with maximum interaction with each other to generate synergetic effects. Density functional theory (DFT) study has proved that the enhanced interaction of three components present is responsible for the enhanced electrochemical performance.191
Re-stacking of individual graphene sheets can be effectively inhibited by introducing carbon materials to form 3D hierarchical structures.195–199 For example, Huang and coworkers reported an approach to design a highly active and durable ORR catalyst by loading Pt nanoparticles on graphene nanosheets that are supported on high surface area carbon black (CB) (Fig. 5g). Placing CB nanoparticles between the Pt/graphene sheets not only enhances the catalytic activity but also dramatically improves the durability of the catalyst (Fig. 5h).200 The authors concluded that the unique 2D structure of graphene functions as a barrier to prevent leaching of Pt into the electrolyte and CB nanoparticles within the vicinity act as active sites to recapture/renucleate the dissolved Pt species.
4. Non-metal/graphene hybrid nanostructures as electrocatalysts
Driven by the economical consideration, electrocatalysts based on graphene and non-metals have attracted great interest in the past few years.201,202 Incorporation of inorganic nanocrystals onto graphene would reduce dissolution, sintering and aggregation of nanoparticles during the electrochemical operation. At the same time, the low intrinsic electric conductivity of inorganic composites is also significantly improved.203 Tremendous progress has been made in exploring these non-metal/graphene electrocatalysts.204,205 These novel nanostructures exhibit superior electrocatalytic activity as well as selectivity and durability, which can act as promising electrodes for electrochemical reactions.2064.1. Metal oxides/graphene hybrids as electrocatalysts
Transition metal (Co, Mn, Fe, Cu, etc.) oxides with mixed valences are reportedly able to serve as donor–acceptor chemisorption sites for the reversible adsorption of oxygen and hence have the potential to replace noble metal based electrocatalysts.207,208 However, metal oxides, which are poorly conductive, frequently suffer from dissolution and aggregation during the electrochemical reaction.208–210 A suitable strategy for overcoming the abovementioned problems is to anchor these transition metal oxides onto graphene. Graphene-based transition metal oxide electrocatalysts thus show remarkable improvements in electrochemical activity, durability and dispersion.4,15 Dai and coworkers deposited Co3O4 nanoparticles on a graphene support and investigated their electrocatalytic activity.211 Using N-doped graphene leads to more uniform distribution of Co3O4 nanoparticles with smaller sizes (Fig. 6a). The resultant Co3O4/graphene hybrid exhibits higher ORR activity than either Co3O4 or graphene alone and favors a 4e process (Fig. 6b). Moreover, the ORR activity decreases monotonically when the Co loading on graphene decreases from 20% to 3%. Importantly, the hybrid is also a highly active OER catalyst for water oxidation (Fig. 6c). The less oxidized CoO nanoparticles possibly make the electrocatalyst more electron rich, which might be beneficial for enhanced ORR activity.212,213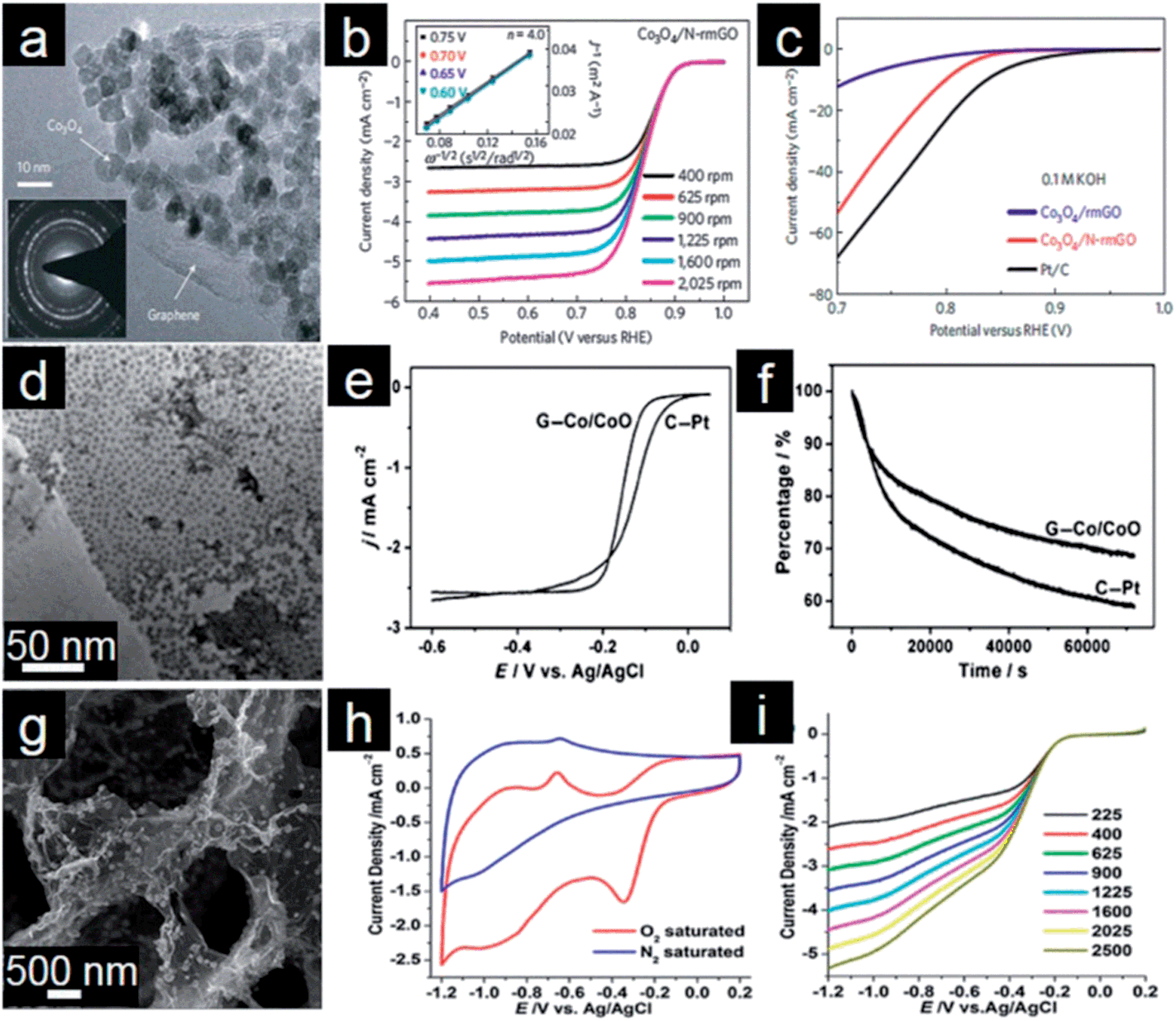 | ||
| Fig. 6 (a) TEM image with an inset showing a selected-area electron diffraction (SAED) pattern of Co3O4/graphene. (b) RDE curves in 0.1 M KOH with different rotation rates; inset of (b) shows corresponding Koutecky–Levich (K–L) plots. (c) RDE curves of different electrocatalysts in 0.1 M KOH. (Reprinted with permission from ref. 211. Copyright© 2011, Nature Publishing Group.) (d) TEM of Co/CoO/graphene composites, (e) RDE curves of Pt/C and Co/CoO/graphene (10 mV s−1; 400 rpm) and (f) chronoamperometric responses at −0.3 V in O2 saturated 0.1 M KOH solution. (Reprinted with permission from ref. 215. Copyright© 2012, WILEY-VCH.) (g) SEM image, (h) CVs and (i) RDE curves of Fe3O4/3D N-doped graphene aerogel composites. (Reprinted with permission from ref. 230. Copyright© 2012, American Chemical Society.) | ||
Metal oxide nanoparticles are typically loaded on graphene with little control in size and morphology.128 This makes tuning the interaction between nanoparticles and graphene difficult.214 To address this issue, Sun and coworkers assembled pre-synthesized monodisperse Co/CoO core/shell nanoparticles on the graphene surface (Fig. 6d).215 The Co core size and thickness of the CoO shell in Co/CoO core/shell nanoparticles can be tuned by controlling the oxidation conditions. They also demonstrated the importance of Co/CoO dimension and graphene as a support in tuning electrocatalysts for efficient ORR with a selective 4e process (Fig. 6e). The optimized Co/CoO–graphene electrocatalyst shows comparative activity and better stability than the commercial Pt/C catalyst and thus might serve as a promising alternative to Pt/C catalysts for the ORR in alkaline solutions (Fig. 6f). It was further shown that the ORR activity of cobalt oxide–graphene hybrid electrocatalysts is dramatically improved by increasing the Co content and coupling with N-doped graphene.216 Cobalt oxides/graphene hybrids were also used as the ORR catalysts in Li–O2 batteries. A bi-functional catalyst composed of Co3O4 nanofibers immobilized on both sides of graphene sheets was proposed for the oxygen electrode.217 The excellent electrochemical performance could mainly be attributed to the structure and interaction between Co3O4 nanofibers and graphene for the facile electron transport and fast O2 diffusion through the ultrathin graphene layer and porous Co3O4 nanofiber networks.
Manganese oxides supported on graphene have also been employed as active, stable and low-cost cathode electrocatalysts for fuel cells and Li–air batteries.218–220 The results show that the catalytic performance of metal oxides is associated with morphology, size and oxide–graphene coupling.221,222 For example, Qiao and coworkers reported a mesoporous architecture of Mn3O4/graphene hybrids which demonstrates a competitive ORR activity and excellent durability with high selectivity.223 Kim and coworkers also found that the oxygen reduction pathway of these composites is tunable with the relative amount of Mn3O4 supported on graphene sheets. For example, the ORR mechanism of the system with a lower content (19.2%) of Mn3O4 nanoparticle is similar to that of the Pt/C electrode with a 4e transfer, whereas the hybrid with a higher Mn3O4 content (52.5%) undergoes a classical 2e process.224 Other graphene supported metal oxides such as Fe3O4,225 Fe2O3,226 Cu2O,227,228 and Ru2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 229 were also investigated as electrocatalysts for ORR in fuel cells and Li–air batteries. Feng and Mullen demonstrated the controllable assembly of Fe3O4 nanoparticles on 3D N-doped graphene aerogels.230 As shown in Fig. 6g, the graphene hybrid shows an interconnected macroporous framework with uniform deposition of Fe3O4 nanoparticles. Due to unique structural features, the resulting Fe3O4/N–graphene aerogels show excellent electrocatalytic activity for the ORR in alkaline electrolytes, including a higher current density, lower ring current, lower H2O2 yield, higher electron transfer number, and better durability (Fig. 6h and i).
229 were also investigated as electrocatalysts for ORR in fuel cells and Li–air batteries. Feng and Mullen demonstrated the controllable assembly of Fe3O4 nanoparticles on 3D N-doped graphene aerogels.230 As shown in Fig. 6g, the graphene hybrid shows an interconnected macroporous framework with uniform deposition of Fe3O4 nanoparticles. Due to unique structural features, the resulting Fe3O4/N–graphene aerogels show excellent electrocatalytic activity for the ORR in alkaline electrolytes, including a higher current density, lower ring current, lower H2O2 yield, higher electron transfer number, and better durability (Fig. 6h and i).
Substitution of Co by Mn and Ni in the spinel lattice can further improve electrocatalytic performance of the resulting MnCo2O4–graphene and NiCo2O4–graphene hybrid catalysts.231 Dai and coworkers reported MnCo2O4/N-doped graphene hybrids as highly efficient ORR electrocatalysts in alkaline conditions (Fig. 7a). Mn substitution increases activity of catalytic sites of the hybrid materials, further boosting the ORR activity compared with the pure Co3O4/graphene hybrid (Fig. 7b).232 The MnCo2O4–graphene hybrid material was also employed as the cathode catalyst for Li–O2 battery in a non-aqueous electrolyte. It is suggested that the excellent catalytic activity originates mainly from the strong coupling effect between MnCo2O4 nanoparticles and N-doped graphene. The high catalytic activity of the MnCo2O4–graphene hybrid for the ORR could facilitate the discharging reaction in Li–O2 cells.233 Moreover, incorporation of Ni atoms into the octahedral sites of the spinel lattice not only creates new active sites but also significantly improve the electrical conductivity.234 Therefore, the NiCo2O4 nanoplates–graphene hybrid can be used as a highly active bi-functional catalyst for ORR and OER.235 There are also some reports on preparation of graphene–Co(OH)2 composites as cathode electrocatalysts for ORR.236,237 These nanocomposites show good catalytic activity, in which graphene decreases the reaction overpotential and Co(OH)2 catalyzes the disproportionation of intermediates.
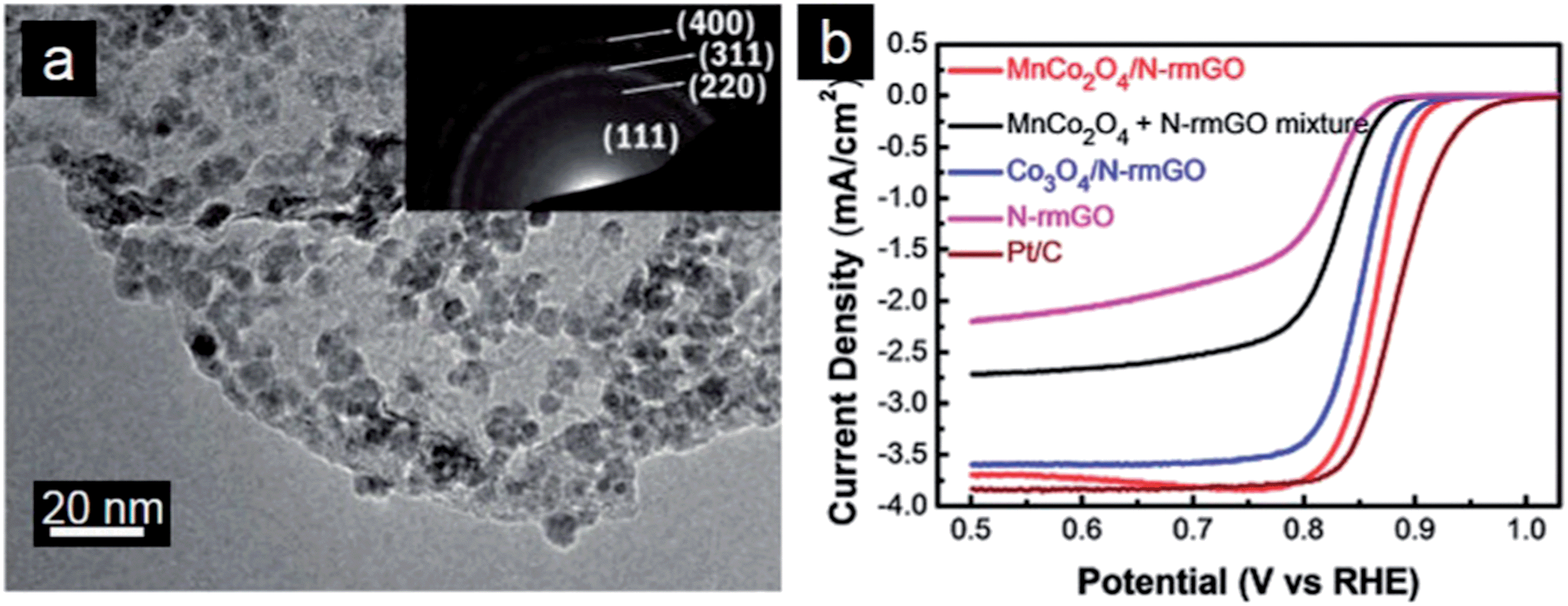 | ||
| Fig. 7 (a) TEM image of MnCo2O4/N-doped graphene hybrids with the inset showing the SAED pattern and (b) RDE curves of different electrocatalysts in O2-saturated 1 M KOH (5 mV s−1; 1600 rpm). (Reprinted with permission from ref. 232. Copyright© 2012, American Chemical Society.) | ||
4.2. Metal chalcogenides/graphene hybrids as electrocatalysts
The metal oxide/graphene electrocatalysts discussed above are mainly active in a basic environment. However, many electrochemical systems, such as proton exchange membrane fuel cells, also involve an acidic environment; thus it is necessary to explore other non-metal electrocatalysts which exhibit high activity and stability under acidic conditions.238,239 Sulfides of non-precious transition metals have attracted intensive interest owing to their noble metal-like catalytic properties.240 Cobalt sulfides have shown great potential as electrocatalysts because they are thermally and chemically more stable than metal oxides, especially in acidic media. Therefore, cobalt sulfides are regarded as one of the most active ORR electrocatalysts in both acidic and basic electrolytes.241–243 However, low conductivity of these sulfides and the dominance of the 2e pathway limit their electrochemical performance for ORR.4 To overcome these limitations, Dai and coworkers employed graphene as a conducting support for controlling the size of catalytically active Co1−xS particles (Fig. 8a).244 The designed Co1−xS/graphene hybrid shows improved electrocatalytic activity and stability compared to Co1−xS particles, and it undergoes the 4e ORR process in both acidic and alkaline media (Fig. 8b and c). Therefore, incorporation of Co1−xS particles onto graphene affords an efficient way to render electrochemical active sites for unprecedented ORR catalytic performance. Subsequently, Hou and coworkers investigated the multi-functionality of Co3S4/graphene composites to enhance the thermal stability and conductivity.245 The resultant cobalt sulfide/graphene composites similarly exhibit high durability in acidic and basic media and also follow the 4e process. Yu and coworkers reported that the electrochemical ORR activity of ZnSe could be enhanced by coupling with N-doped graphene.246 With the further Ni doping into cobalt sulfides, the designed NiCo2S4/graphene composites manifest not only higher ORR activity but also much better durability toward OER than the CoS/graphene system (Fig. 8d–f).247 Different from its widely accepted role as electron reservoir, a novel role of graphene for ORR as a peroxide cleaner has been proposed in these sulfides/graphene composites.248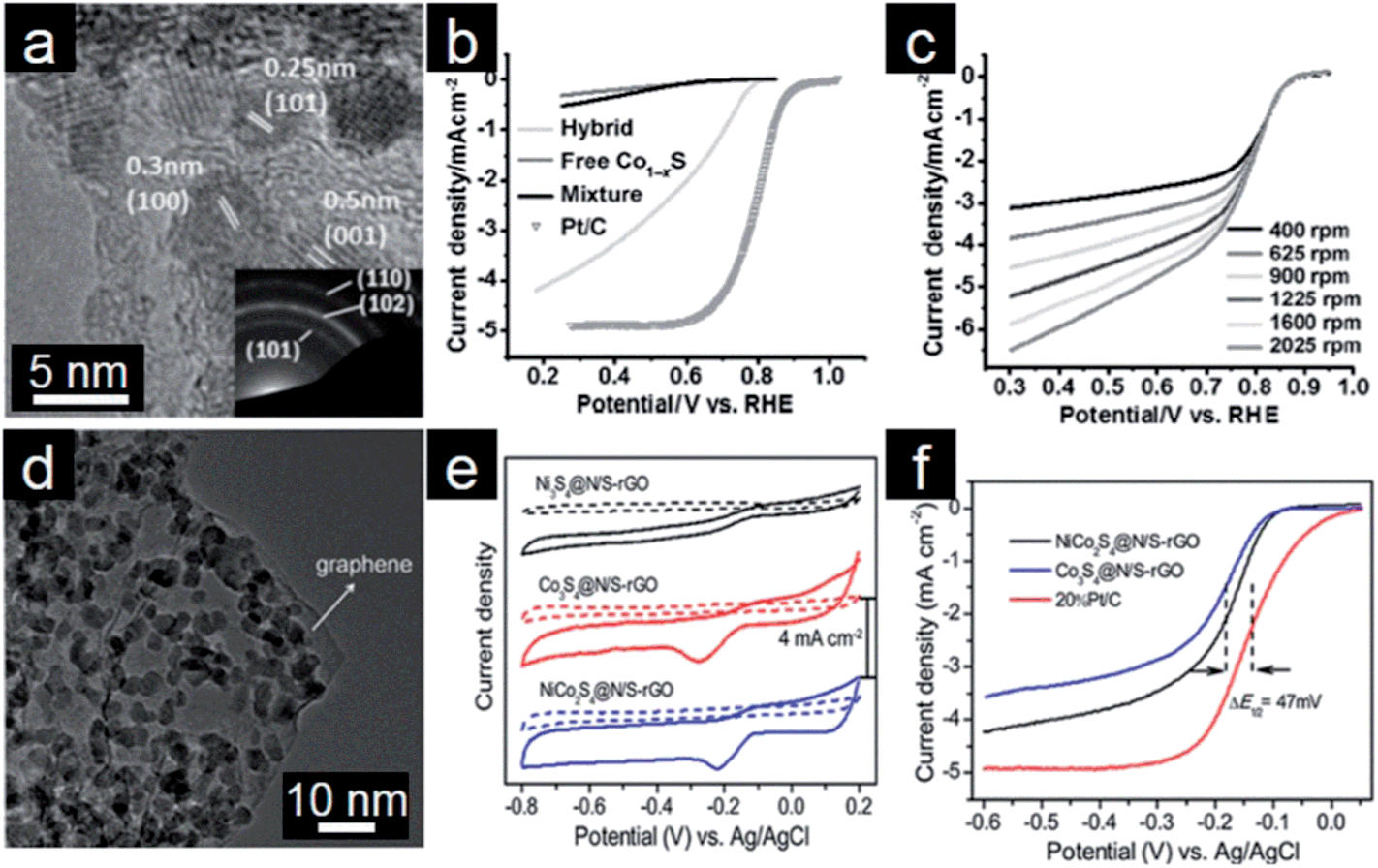 | ||
| Fig. 8 (a) TEM image of the Co1−xS/rGO hybrid with the inset showing the SAED pattern. (b) RDE curves of different catalysts in O2-saturated 0.5 M H2SO4 (5 mV s−1; 1600 rpm). (c) RDE curves of the Co1−xS/rGO hybrid in O2 saturated 0.1 M KOH solution with a scanning rate of 5 mV s−1. (Reprinted with permission from ref. 244. Copyright© 2011, WILEY-VCH.) (d) TEM image of NiCo2S4/graphene, (e) CVs and (d) RDE curves of different cobalt sulfides/graphene hybrids in O2-saturated 0.1 M KOH solution (5 mV s−1; 1600 rpm). (Reprinted with permission from ref. 247. Copyright© 2013, American Chemical Society.) | ||
The hydrogen evolution reaction (HER, 2H+ + 2e → H2), which involves proton reduction and concomitant hydrogen evolution, is an important reaction involved in water splitting. Efficient electrocatalysts for HER are required to reduce overpotential and enable HER to be carried out with high energy efficiency.249 Under these considerations, Pt-based noble metals are considered as the most effective HER electrocatalysts.250 However, the search for new HER electrocatalysts with low cost and high activity has attracted a great deal of interest due to their important role in clean energy generation.240 MoS2 nanoparticles demonstrate good potential as an inexpensive alternative to Pt for HER.251,252 The catalytic activity of MoS2 is mainly localized to some small fraction of surface sites, whereas the bulk part is relatively inert.253 Therefore, increasing the amount of active sites for MoS2-based electrocatalysts is necessary to enable greater activity for HER. Inspired by the great enhancement observed by coupling inorganic nanocrystals with graphene, Dai and coworkers introduced MoS2 nanoparticles on graphene (Fig. 9a).254 Strong chemical and electronic coupling between MoS2 and graphene allows for good dispersion of MoS2 nanoparticles and rapid electron transport from the less conductive MoS2 nanoparticles to the electrode. The MoS2/graphene hybrid catalyst exhibits smaller overpotential of ∼0.1 V and much improved HER current density (Fig. 9b). The subsequent Tafel analysis showed a Tafel slope of ∼41 mV per decade (Fig. 9c). The large amount of exposed edges (presumably active sites) from these small MoS2 nanoparticles uniformly dispersed on graphene leads to significantly enhanced activity.255 Furthermore, the interconnected conductive network with porous nanostructure (from 3D graphene framework) was anticipated to enhance the activity of embedded MoS2 nanocatalysts (Fig. 9d).256 Thanks to its 3D architectural characteristics, this MoS2/graphene composite shows high activity for HER with a Tafel slope of ∼42 mV per decade and a small overpotential of ∼0.1 V (Fig. 9e and f). 3D graphene based electrocatalysts are further developed by growing MoSx onto graphene-protected 3D Ni foam to provide robust protection and efficiently increase their stability in acidic media. The 3D porous structure also effectively increases the MoSx catalyst loading, leading to enhanced electrocatalytic HER efficiency. The hydrogen evolution rate is as high as 302 mL g−1 cm−2 h−1 at an overpotential of 0.2 V.257
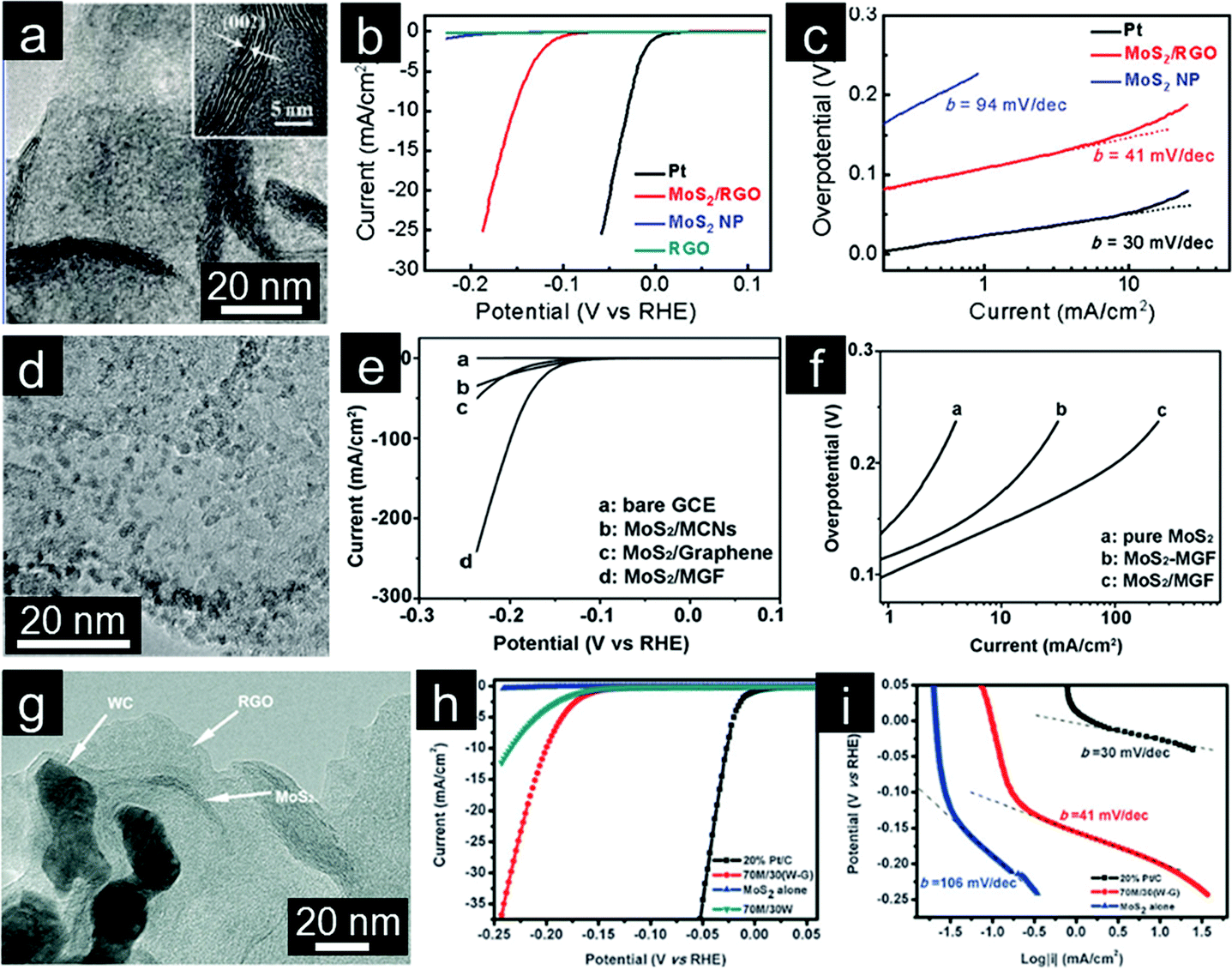 | ||
| Fig. 9 (a) TEM image with the inset showing the folded edge of the MoS2/rGO hybrid, (b) polarization curves, and (c) corresponding Tafel plots of different catalysts for HER. (Reprinted with permission from ref. 254. Copyright© 2011, American Chemical Society.) (d) TEM image of the MoS2/mesoporous graphene hybrid, (e) polarization curves, and (f) corresponding Tafel plots of different catalysts. (Reprinted with permission from ref. 256. Copyright© 2013, WILEY-VCH.) (g) TEM image of the MoS2/WC/graphene composite, (h) polarization curves, and (i) corresponding Tafel plots of different catalysts. (Reprinted with permission from ref. 263. Copyright© 2013, Royal Society of Chemistry.) | ||
2D MoS2 nanosheets with rich exposed edges demonstrate improved activity for HER.258 However, curling of sheets and aggregation to form tubes or fullerene-like structures will lower the activity of MoS2 nanosheets.259 As such, GO has been employed as a highly selective substrate for the synthesis of layered MoS2 hybrid electrocatalysts.260,261 However, both GO and MoS2 tend to re-stack and aggregate. Thus, the introduction of other conductive and active nanoparticles has been used as a method to improve the H2 production performance.262 Wang and coworkers introduced WC nanoparticles as the spacers to create void for the growth of MoS2 nanosheets (Fig. 9g). The as-prepared MoS2/WC/graphene hybrid material displays high HER activity with a Tafel slope of ∼41 mV per decade and a small overpotential of ∼0.15 V (Fig. 9h and i). Synergistic effects among the components and enhanced electron transfer are suggested responsible for the enhanced electrochemical performance.263
4.3. Nitrides/graphene hybrids as electrocatalysts
As previously mentioned, pyridinic and graphitic N atoms are generally accepted as the active sites in graphene-based electrocatalysts. However, the limited N doping content (2–5%) and leaching of N active sites on graphene composites result in low and unstable electrocatalytic activity. Therefore, carbon or transition metal nitrides with stable structures and high N content such as those containing pyridinic and graphitic N moieties may serve as potential non-noble metal electrocatalysts.264–266 However, the low conductivity of pure nitrides will strongly restrict electron transfer during electrochemical reactions and also reduce the electrocatalytic activity.267 Therefore, researchers focused on incorporation of conductive components such as graphene nanosheets into the nitride matrix to improve the electron transfer efficiency in the resulting hybrids.268,269 Considering the excellent conductivity of graphene and layered structures of both graphene and carbon nitride nanosheets, Feng and coworkers developed a nanocasting technique to fabricate graphene-based carbon nitride nanosheets (Fig. 10a).265 The resulting composites possess beneficial features such as high N content, small thickness, high surface area, large aspect ratio as well as enhanced electrical conductivity. Such features are favorable for ease of oxygen access to the catalyst surface and facilitate rapid transfer of electrons in the electrode during the oxygen–reduction process. Consequently, the composites exhibit excellent electrocatalytic performance for ORR (Fig. 10b), including high electrocatalytic activity, long-term durability, and high selectivity, all of which are superior to those observed in nitride sheets without graphene and commercial Pt/C catalysts.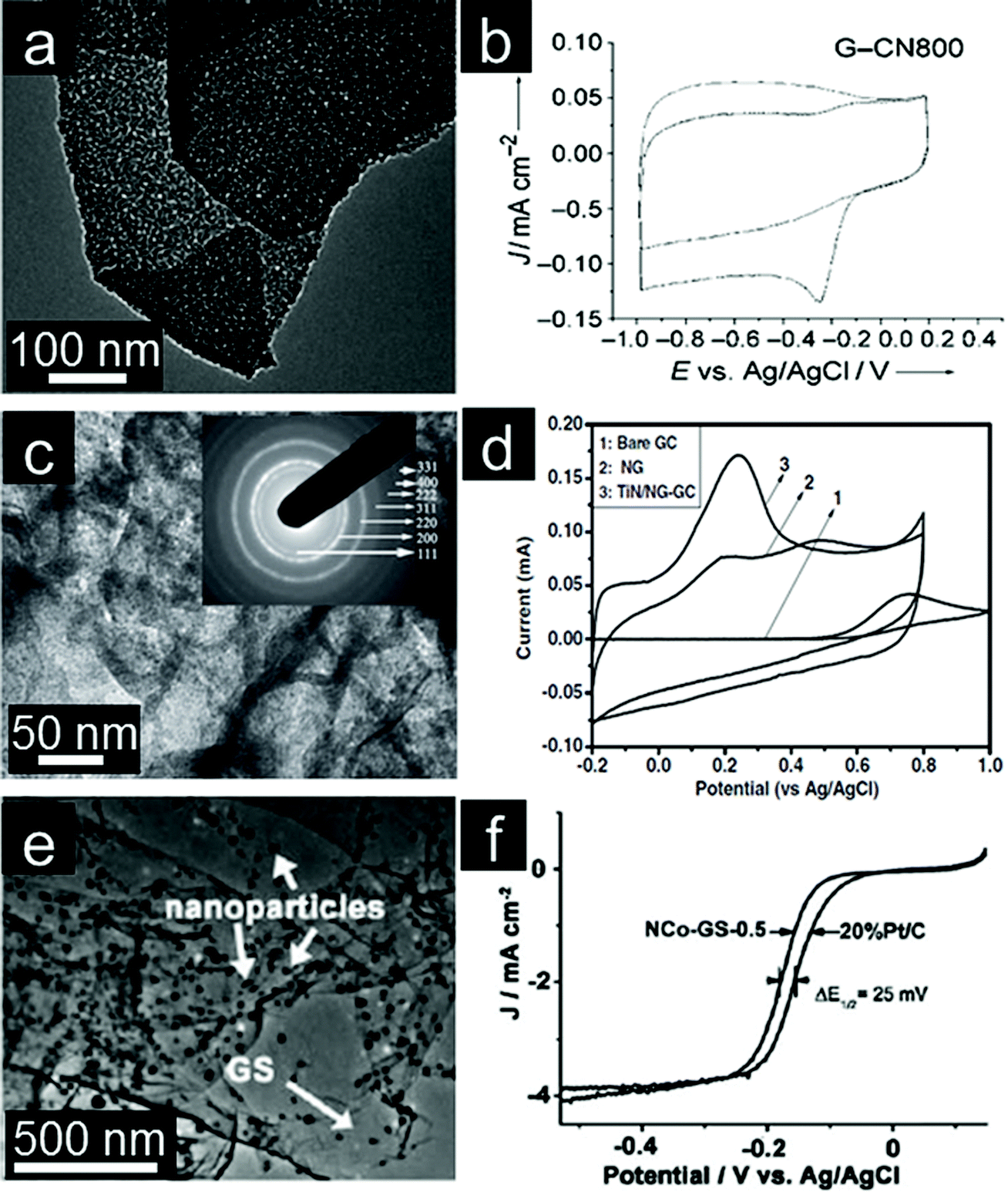 | ||
| Fig. 10 (a) TEM image of graphene/carbon nitride composites and (b) corresponding CVs in O2- and Ar-saturated 0.1 M KOH solution. (Reprinted with permission from ref. 265. Copyright© 2011, WILEY-VCH.) (c) TEM image of TiN/graphene hybrids with the inset showing the SAED pattern and (d) corresponding CVs. (Reprinted with permission from ref. 278. Copyright© 2011, WILEY-VCH.) (e) TEM image of Co–N–graphene and (f) corresponding RDE curves in 0.1 M KOH solution. (Reprinted with permission from ref. 279. Copyright© 2013, Royal Society of Chemistry.) | ||
Graphitic carbon nitride (g-C3N4) based materials are also becoming significant due to their unusual properties and promising applications as electrocatalysts.270 The combination of graphene and g–C3N4 integrates their respective merits together, and the resulting composites show enhanced or unique properties.269,271 Furthermore, the large amount of pyridine-like nitrogen in g–C3N4 can trap transition metals to form potential active sites.272 Hence, assemblies consisting of transition metals (Ti,273 Co,274 Fe,275,276etc.) and graphene/g-C3N4 manifest improved electrocatalytic activity.277 Feng and coworkers demonstrated this concept by preparing titanium nitride/graphene nanocomposites, in which C3N4 acts as both the nitrogen source and the template (Fig. 10c and d).278 With the aid of templates, uniform TiN nanoparticles can be attached with good dispersion on the graphene/CNT hybrid (Fig. 10e). The resultant hybrid exhibits better activity for ORR than bare TiN nanoparticles and comparable performance as the Pt/C catalyst (Fig. 10f). In addition, good tolerance to methanol was also demonstrated. This dramatic enhancement is ascribed to a synergistic effect between TiN and the CNT/graphene hybrid, whose formal roles are to provide active sites for ORR and electron pathway respectively.279 On the other hand, there is evidence that metal (Fe or Co) coordination in N-doped graphene will lead to far superior electrocatalytic activity.280
4.4. Other carbon materials/graphene hybrids as electrocatalysts
While metal/metal oxides–graphene hybrids electrocatalysts exhibit significantly higher electrocatalytic activity, they are often costly and more severely suffer from poor stability under strong acidic conditions.281 As a result, numerous research efforts have been made to develop efficient and durable electrocatalysts in acidic media.282 N-doped carbon materials possess good electrocatalytic activity for ORR because of their increased active sites, good conductivity, and excellent stability in acidic media.267,283 To further improve the performance, CNTs have been investigated for their important role as the promoter in graphene-based electrocatalysts.284,285 In one embodiment, graphene, produced by partial unzipping of the outer walls of CNTs, is attached to the mostly intact inner walls (Fig. 11a).286 Upon the subsequent annealing process in NH3, the abundant edges and defect sites on graphene give rise to high ORR catalytic activity. The resultant N-doped graphene/CNTs complex exhibits good activity and durability for ORR in both acidic and alkaline media (Fig. 11b). It is proposed that both iron impurities and the N doping effect contribute to the high electrocatalytic activity for ORR, similar to the above discussed heteroatoms or transition metal doping into graphene. In order to avoid the use of harsh conditions and sophisticated instruments, Yu and coworkers reported a hydrothermal method to prepare N-doped graphene/CNTs nanocomposites (Fig. 11c).287 The electrochemical results show that the nanocomposites undergo a 4e pathway for ORR with high positive onset potential, large peak current and high durability (Fig. 11d). The presence of graphene and CNTs synergistically enhances electrochemical activity for ORR. Furthermore, graphene/CNTs self-assemblies are also synthesized through electrostatic interaction between graphene and CNTs. These materials are applied as electrocatalysts for ORR in acidic media after N doping.285 In the assembled structure, CNTs act as circuits for facile electron transfer and spacers for preventing re-stacking of graphene layers. At the same time, the inter-graphene space introduced by CNTs facilitates the effective transport of reactants.288 Interestingly, ORR electrocatalysts made from N-doped graphene/CNTs frameworks retain their electrocatalytic activity even in the non-aqueous environment when employed as cathode electrocatalysts for Li–O2 batteries.289 High surface area, porous structure, and good conductivity are considered as important factors for high electrocatalytic activity. As such, Qiao and coworkers recently reported the in situ growth of N-doped graphene on hierarchical ordered porous carbon (Fig. 11e). As expected, synergistically enhanced ORR performance was observed (Fig. 11f).290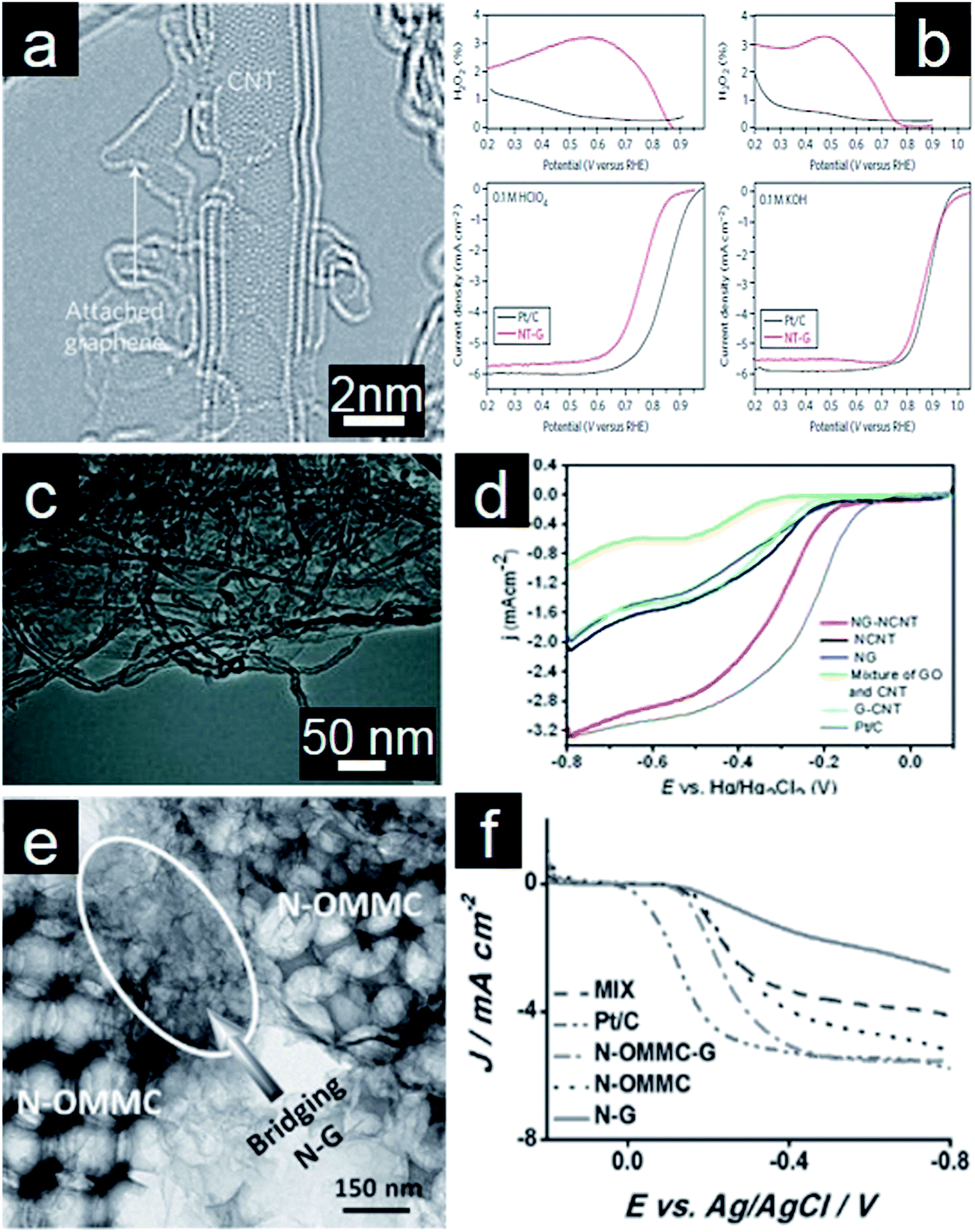 | ||
| Fig. 11 (a) TEM image of N-doped graphene/CNT composites and (b) their corresponding rotating ring-disk electrode (RRDE) polarization curves in O2 saturated 0.1 M HClO4 and 0.1 M KOH solution. (Reprinted with permission from ref. 286. Copyright© 2012, Nature Publishing Group.) (c) TEM of N-doped graphene/CNT nanocomposites and (d) RDE curves of different catalysts for ORR in O2-saturated 0.1 M KOH solution (20 mV s−1; 1600 rpm). (Reprinted with permission from ref. 287. Copyright© 2013, WILEY-VCH.) (e) TEM image of N-doped graphene on ordered porous carbon and (f) RDE curves of different catalysts for ORR in O2-saturated 0.1 M KOH (1600 rpm). (Reprinted with permission from ref. 290. Copyright© 2013, WILEY-VCH.) | ||
5. Other novel graphene-based electrocatalysts
Metal–organic frameworks (MOFs), a new class of porous crystalline materials, may serve as an outstanding candidate in this field based on their high surface areas, controllable structures and excellent electrochemical properties.291 For example, Loh and coworkers used pyridine-functionalized graphene as a support for MOF particles. The linkage between graphene and pyridine ligands in MOF indeed increases the electrocatalytic activity of MOF/graphene hybrids and facilitates ORR via 4e reaction. In addition, methanol crossover is also minimized in this MOF/graphene hybrid because of its inactivity to methanol oxidation.292 The integration of GO and a Cu-centered MOF shows good performance as a tri-functional electrocatalyst for HER, OER and ORR. In polymer electrolyte membrane fuel cell testing, the GO incorporated Cu–MOF is able to deliver 76% of power density of the commercial Pt catalyst. The enhanced electrocatalytic properties and stability under acidic conditions of the MOF/graphene composite are attributed to the unique porous scaffold structure, improved charge transport and synergistic interactions between graphene and MOF.293,2946. Conclusions and perspectives
The search for highly efficient electrocatalysts with excellent stability is of great importance for sustainable energy technologies including fuel cells, Li–air batteries and water splitting. Graphene has emerged as a unique material because of its 2D structure, good electrical conductivity and high surface area. Graphene is therefore expected to be an important component for developing alternative electrocatalysts. In this review, we have systematically summarized the recent progress in preparation of graphene-based hybrids with controlled size, composition, shape/morphology and structure, and highlighted their superior electrochemical activity, excellent stability and selectivity. This review is intended to provide valuable insights for researchers to speed up innovation in this area.Although significant advance has been achieved in the area of graphene-based hybrid electrocatalysts, this field is still in its early stage from the practical perspective, especially in the direction of non-metal/graphene nanocomposites. Further development of graphene-based hybrid electrocatalysts is being impeded by the challenges listed below. Doping of heteroatoms is an efficient way to tune properties of graphene and increase the active sites on the graphene surface. However, the electrocatalytic performance of these doped catalysts is still far from satisfactory, mainly due to issues such as low doping content and the lack of control over the doping process. Selective and tunable coordination of heteroatoms into graphene may help to increase the number of exposed active sites and improve their activity, thus enhancing the electrochemical performance. Combined experimental and theoretical studies are thus required to develop a method for controlled doping of heteroatoms into designated sites, and to understand the proposed mechanism on the catalytic activity of modified graphene.
Graphene is a unique support for noble metal nanocatalysts. Graphene supported noble metal nanoparticles possessing controlled size, shape, morphology and composition can enhance electrochemical performance for fuel oxidation, oxygen reduction and water splitting reactions. However, the migration, aggregation and dissolution in harsh electrochemical environments as well as the weak CO tolerance would greatly affect their practical exploitation. Hybrid structures constructed from modified graphene and alloyed metal nanoparticles could be alternative efficient electrocatalysts with designed stability and selectivity. Coupling graphene with non-metals leads to superior activity and lowers the cost. But electrochemical performance of such hybrid materials is strongly dependent on individual components and their interactions with each other. Also, mechanisms of these graphene based nanohybrids working in energy related systems remain largely unclear. Thus, further studies are required to understand the underlying complex interactions. Finally, exploration of new graphene-based hybrids with novel structures and appropriate electronic properties such as MOF/graphene can further expand the materials database for efficient electrocatalysts.
Undoubtedly, graphene-based nanohybrids are likely to find real significance for sustainable electrochemical energy conversion and storage. Research efforts devoted to these directions will surely pay off in the near future.
References
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 1388 CAS
.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736 CAS
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013 CrossRef CAS PubMed
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780 CrossRef CAS PubMed
- P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 3404 CrossRef CAS
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782 CrossRef CAS PubMed
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed
- X. Wan, Y. Huang and Y. Chen, Acc. Chem. Res., 2012, 45, 598 CrossRef CAS PubMed
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC
- M. Pumera, Chem. Soc. Rev., 2010, 39, 4146 RSC
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876 CrossRef CAS PubMed
- N. G. Sahoo, Y. Pan, L. Li and S. H. Chan, Adv. Mater., 2012, 24, 4203 CrossRef CAS PubMed
- S. Guo and S. Dong, Chem. Soc. Rev., 2011, 40, 2644 RSC
- H. Wang and H. Dai, Chem. Soc. Rev., 2013, 42, 3088 RSC
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983 RSC
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873 CrossRef CAS
- M. Pumera, Energy Environ. Sci., 2011, 4, 668 CAS
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113 CAS
- H.-J. Choi, S.-M. Jung, J.-M. Seo, D. W. Chang, L. Dai and J.-B. Baek, Nano Energy, 2012, 1, 534 CrossRef CAS
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805 CrossRef CAS PubMed
- T. Kuila, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Nanoscale, 2013, 5, 52 RSC
- B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54 CAS
- C. C. Huang, C. Li and G. Q. Shi, Energy Environ. Sci., 2012, 5, 8848 CAS
- S. H. Hur and J.-N. Park, Asia-Pac. J. Chem. Eng., 2013, 8, 218 CrossRef CAS
- J. Hou, Y. Shao, M. W. Ellis, R. B. Moore and B. Yi, Phys. Chem. Chem. Phys., 2011, 13, 15384 RSC
- C. Zhu and S. Dong, Nanoscale, 2013, 5, 1753 RSC
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS
- L. S. Panchakarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726 CAS
- Y. Y. Shao, S. Zhang, M. H. Engelhard, G. S. Li, G. C. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. H. Lin, J. Mater. Chem., 2010, 20, 7491 RSC
- D. Deng, X. Pan, L. Yu, Y. Cui, Y. Jiang, J. Qi, W.-X. Li, Q. Fu, X. Ma, Q. Xue, G. Sun and X. Bao, Chem. Mater., 2011, 23, 1188 CrossRef CAS
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421 CrossRef CAS
- Z. Yang, H. G. Nie, X. Chen, X. H. Chen and S. M. Huang, J. Power Sources, 2013, 236, 238 CrossRef CAS
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936 CAS
- C. H. Choi, M. W. Chung, S. H. Park and S. I. Woo, RSC Adv., 2013, 3, 4246 RSC
- K. A. Kurak and A. B. Anderson, J. Phys. Chem. C, 2009, 113, 6730 CAS
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038 RSC
- S. Yasuda, L. Yu, J. Kim and K. Murakoshi, Chem. Commun., 2013, 49, 9627 RSC
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760 CAS
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2011, 134, 15 CrossRef PubMed
- Q. Li, S. Zhang, L. Dai and L.-s. Li, J. Am. Chem. Soc., 2012, 134, 18932 CrossRef CAS PubMed
- Y. Liu and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 3362 CAS
- T. Palaniselvam, H. B. Aiyappa and S. Kurungot, J. Mater. Chem., 2012, 22, 23799 RSC
- X. Jia, J. Campos-Delgado, M. Terrones, V. Meunier and M. S. Dresselhaus, Nanoscale, 2011, 3, 86 RSC
- S. Xin, Y.-G. Guo and L.-J. Wan, Acc. Chem. Res., 2012, 45, 1759 CrossRef CAS PubMed
- Y. Li, J. Wang, X. Li, D. Geng, R. Li and X. Sun, Chem. Commun., 2011, 47, 9438 RSC
- E. Yoo and H. Zhou, ACS Nano, 2011, 5, 3020 CrossRef CAS PubMed
- J. Xiao, D. Mei, X. Li, W. Xu, D. Wang, G. L. Graff, W. D. Bennett, Z. Nie, L. V. Saraf, I. A. Aksay, J. Liu and J.-G. Zhang, Nano Lett., 2011, 11, 5071 CrossRef CAS PubMed
- Z.-L. Wang, D. Xu, J.-J. Xu, L.-L. Zhang and X.-B. Zhang, Adv. Funct. Mater., 2012, 22, 3699 CrossRef CAS
- E. Yoo, J. Nakamura and H. Zhou, Energy Environ. Sci., 2012, 5, 6928 CAS
- Y. L. Li, J. J. Wang, X. F. Li, D. S. Geng, M. N. Banis, R. Y. Li and X. L. Sun, Electrochem. Commun., 2012, 18, 12 CrossRef CAS
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 CAS
- M. Seredych, J.-C. Idrobo and T. J. Bandosz, J. Mater. Chem. A, 2013, 1, 7059 CAS
- I.-Y. Jeon, S. Zhang, L. Zhang, H.-J. Choi, J.-M. Seo, Z. Xia, L. Dai and J.-B. Baek, Adv. Mater., 2013, 25, 6138 CrossRef CAS PubMed
- Z. Yang, Z. Yao, G. F. Li, G. Y. Fang, H. G. Nie, Z. Liu, X. M. Zhou, X. Chen and S. M. Huang, ACS Nano, 2012, 6, 205 CrossRef CAS PubMed
- Y. L. Li, J. J. Wang, X. F. Li, D. S. Geng, M. N. Banis, Y. J. Tang, D. N. Wang, R. Y. Li, T. K. Sham and X. L. Sun, J. Mater. Chem., 2012, 22, 20170 RSC
- Z. P. Jin, H. G. Nie, Z. Yang, J. Zhang, Z. Liu, X. J. Xu and S. M. Huang, Nanoscale, 2012, 4, 6455 RSC
- Z. Yao, H. Nie, Z. Yang, X. Zhou, Z. Liu and S. Huang, Chem. Commun., 2012, 48, 1027 RSC
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. A. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257 CrossRef PubMed
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932 CrossRef CAS PubMed
- H. Wang, Y. Zhou, D. Wu, L. Liao, S. Zhao, H. Peng and Z. Liu, Small, 2013, 9, 1316 CrossRef CAS PubMed
- Z.-H. Sheng, H.-L. Gao, W.-J. Bao, F.-B. Wang and X.-H. Xia, J. Mater. Chem., 2012, 22, 390 RSC
- S.-A. Wohlgemuth, R. J. White, M.-G. Willinger, M.-M. Titirici and M. Antonietti, Green Chem., 2012, 14, 1515 RSC
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135, 1201 CrossRef CAS PubMed
- C. H. Choi, M. W. Chung, H. C. Kwon, S. H. Park and S. I. Woo, J. Mater. Chem. A, 2013, 1, 3694 CAS
- S. Wang, E. Iyyamperumal, A. Roy, Y. Xue, D. Yu and L. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756 CrossRef PubMed
- C. H. Choi, M. W. Chung, S. H. Park and S. I. Woo, Phys. Chem. Chem. Phys., 2013, 15, 1802 RSC
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J.-B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209 CrossRef CAS PubMed
- Z.-S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130 CrossRef CAS PubMed
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110 CrossRef CAS PubMed
- J. X. Xu, G. F. Dong, C. H. Jin, M. H. Huang and L. H. Guan, ChemSusChem, 2013, 6, 493 CrossRef CAS PubMed
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS PubMed
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63 CrossRef CAS PubMed
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71 CrossRef PubMed
- X. Yuan, X. Zeng, H.-J. Zhang, Z.-F. Ma and C.-Y. Wang, J. Am. Chem. Soc., 2010, 132, 1754 CrossRef CAS PubMed
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed
- R. J. Toh, H. L. Poh, Z. Sofer and M. Pumera, Chem.–Asian J., 2013, 8, 1295 CrossRef CAS PubMed
- K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng and K. Müllen, ACS Nano, 2012, 6, 9541 CrossRef CAS PubMed
- K. Kamiya, K. Hashimoto and S. Nakanishi, Chem. Commun., 2012, 48, 10213 RSC
- H. Peng, Z. Mo, S. Liao, H. Liang, L. Yang, F. Luo, H. Song, Y. Zhong and B. Zhang, Sci. Rep., 2013, 3, 1765 Search PubMed
- S. Jiang, C. Zhu and S. Dong, J. Mater. Chem. A, 2013, 1, 3593 CAS
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Mater. Chem. A, 2013, 1, 1450 CAS
- X. Fu, Y. Liu, X. Cao, J. Jin, Q. Liu and J. Zhang, Appl. Catal., B, 2013, 130–131, 143 CrossRef CAS
- K. Niu, B. Yang, J. Cui, J. Jin, X. Fu, Q. Zhao and J. Zhang, J. Power Sources, 2013, 243, 65 CrossRef CAS
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335 RSC
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752 CrossRef CAS PubMed
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed
- L. Dai, Acc. Chem. Res., 2012, 46, 31 CrossRef PubMed
- Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541 RSC
- S. Wang, D. Yu, L. Dai, D. W. Chang and J.-B. Baek, ACS Nano, 2011, 5, 6202 CrossRef CAS PubMed
- C. Z. Zhang, R. Hao, H. B. Liao and Y. L. Hou, Nano Energy, 2013, 2, 88 CrossRef CAS
- M. S. Ahmed and S. Jeon, J. Power Sources, 2012, 218, 168 CrossRef CAS
- J. P. Song, J. Qiao, S. M. Shuang, Y. J. Guo and C. Dong, J. Mater. Chem. A, 2012, 22, 602 RSC
- Y. Zhang, G. Mo, X. Li and J. Ye, J. Power Sources, 2012, 197, 93 CrossRef CAS
- G. Lv, L. Cui, Y. Wu, Y. Liu, T. Pu and X. He, Phys. Chem. Chem. Phys., 2013, 15, 13093 RSC
- H. Tang, H. Yin, J. Wang, N. Yang, D. Wang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 5585 CrossRef CAS PubMed
- C. Zhang, R. Hao, H. Yin, F. Liu and Y. Hou, Nanoscale, 2012, 4, 7326 RSC
- Y. Jiang, Y. Lu, X. Lv, D. Han, Q. Zhang, L. Niu and W. Chen, ACS Catal., 2013, 3, 1263 CrossRef CAS
- M. Giovanni, H. L. Poh, A. Ambrosi, G. J. Zhao, Z. Sofer, F. Sanek, B. Khezri, R. D. Webster and M. Pumera, Nanoscale, 2012, 4, 5002 RSC
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18 CrossRef CAS PubMed
- B. Y. Xia, W. T. Ng, H. B. Wu, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 7213 CrossRef CAS PubMed
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990 CAS
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157 RSC
- W. Qian, R. Hao, J. Zhou, M. Eastman, B. A. Manhat, Q. Sun, A. M. Goforth and J. Jiao, Carbon, 2013, 52, 595 CrossRef CAS
- L. N. Gao, W. B. Yue, S. S. Tao and L. Z. Fan, Langmuir, 2013, 29, 957 CrossRef CAS PubMed
- E. Antolini, Appl. Catal., B, 2012, 123–124, 52 CrossRef CAS
- R. Siburian, T. Kondo and J. Nakamura, J. Phys. Chem. C, 2013, 117, 3635 CAS
- J. D. Qiu, G. C. Wang, R. P. Liang, X. H. Xia and H. W. Yu, J. Phys. Chem. C, 2011, 115, 15639 CAS
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255 CrossRef CAS PubMed
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 12337 CrossRef CAS PubMed
- Y. P. Xiao, S. Wan, X. Zhang, J. S. Hu, Z. D. Wei and L. J. Wan, Chem. Commun., 2012, 48, 10331 RSC
- B. Y. Xia, H. Bin Wu, Y. Yan, X. W. Lou and X. Wang, J. Am. Chem. Soc., 2013, 135, 9480 CrossRef CAS PubMed
- R. Wang, D. C. Higgins, M. A. Hoque, D. Lee, F. Hassan and Z. Chen, Sci. Rep., 2013, 3, 2431 Search PubMed
- Z. M. Luo, L. H. Yuwen, B. Q. Bao, J. Tian, X. R. Zhu, L. X. Weng and L. H. Wang, J. Mater. Chem., 2012, 22, 7791 RSC
- X. Yang, Q. Yang, J. Xu and C.-S. Lee, J. Mater. Chem. A, 2012, 22, 8057 RSC
- W. He, H. J. Jiang, Y. Zhou, S. D. Yang, X. Z. Xue, Z. Q. Zou, X. G. Zhang, D. L. Akins and H. Yang, Carbon, 2012, 50, 265 CrossRef CAS
- S. Zhang, Y. Y. Shao, H. G. Liao, J. Liu, I. A. Aksay, G. P. Yin and Y. H. Lin, Chem. Mater., 2011, 23, 1079 CrossRef CAS
- Z. Y. Huang, H. H. Zhou, C. H. Li, F. Y. Zeng, C. P. Fu and Y. F. Kuang, J. Mater. Chem., 2012, 22, 1781 RSC
- H. J. Kim, S. M. Choi, M. H. Seo, S. Green, G. W. Huber and W. B. Kim, Electrochem. Commun., 2011, 13, 890 CrossRef CAS
- G. Wei, Y. Zhang, S. Steckbeck, Z. Su and Z. Li, J. Mater. Chem., 2012, 22, 17190 RSC
- Q. Yue, K. Zhang, X. Chen, L. Wang, J. Zhao, J. Liu and J. Jia, Chem. Commun., 2010, 46, 3369 RSC
- B. P. Vinayan and S. Ramaprabhu, Nanoscale, 2013, 5, 5109 RSC
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 13934 CrossRef CAS PubMed
- F. Han, X. M. Wang, J. Lian and Y. Z. Wang, Carbon, 2012, 50, 5498 CrossRef CAS
- Y. Z. Zhang, Y. E. Gu, S. X. Lin, J. P. Wei, Z. H. Wang, C. M. Wang, Y. L. Du and W. C. Ye, Electrochim. Acta, 2011, 56, 8746 CrossRef CAS
- S. J. Guo, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 547 CrossRef CAS PubMed
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526 CrossRef CAS PubMed
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602 CrossRef CAS PubMed
- S. Guo and S. Dong, J. Mater. Chem., 2011, 21, 16704 RSC
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 242 CrossRef CAS
- S. Guo, D. Li, H. Zhu, S. Zhang, N. M. Markovic, V. R. Stamenkovic and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3465 CrossRef CAS PubMed
- M. T. M. Koper, Nanoscale, 2011, 3, 2054 RSC
- T. Jin, S. Guo, J.-l. Zuo and S. Sun, Nanoscale, 2013, 5, 160 RSC
- S. Y. Wang, X. Wang and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 7187 Search PubMed
- S. Guo and S. Sun, J. Am. Chem. Soc., 2012, 134, 2492 CrossRef CAS PubMed
- B. P. Vinayan, R. Nagar and S. Ramaprabhu, J. Mater. Chem., 2012, 22, 25325 RSC
- D. P. He, K. Cheng, T. Peng, X. L. Sun, M. Pan and S. C. Mu, J. Mater. Chem., 2012, 22, 21298 RSC
- J. H. Shim, J. E. Kim, Y.-B. Cho, C. Lee and Y. Lee, Phys. Chem. Chem. Phys., 2013, 15, 15365 RSC
- H. Huang and X. Wang, J. Mater. Chem., 2012, 22, 22533 RSC
- X. Chen, G. Wu, J. Chen, X. Chen, Z. Xie and X. Wang, J. Am. Chem. Soc., 2011, 133, 3693 CrossRef CAS PubMed
- M. M. Liu, Y. Z. Lu and W. Chen, Adv. Funct. Mater., 2013, 23, 1289 CrossRef CAS
- Y. Choi, M. Gu, J. Park, H. K. Song and B. S. Kim, Adv. Energy Mater., 2012, 2, 1510 CrossRef CAS
- H. Yin, H. Tang, D. Wang, Y. Gao and Z. Tang, ACS Nano, 2012, 6, 8288 CrossRef CAS PubMed
- S. M. Alia, K. Duong, T. Liu, K. Jensen and Y. Yan, ChemSusChem, 2012, 5, 1619 CrossRef CAS PubMed
- D. Yu, J. Yao, L. Qiu, Y. Wu, L. Li, Y. Feng, Q. Liu, D. Li and H. Wang, RSC Adv., 2013, 3, 11552 RSC
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed
- I. Fampiou and A. Ramasubramaniam, J. Phys. Chem. C, 2012, 116, 6543 CAS
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711 CrossRef CAS PubMed
- L. Yan, Y. B. Zheng, F. Zhao, S. Li, X. Gao, B. Xu, P. S. Weiss and Y. Zhao, Chem. Soc. Rev., 2012, 41, 97 RSC
- B. Xiong, Y. K. Zhou, Y. Y. Zhao, J. Wang, X. Chen, R. O'Hayre and Z. P. Shao, Carbon, 2013, 52, 181 CrossRef CAS
- D. Wei and Y. Liu, Adv. Mater., 2010, 22, 3225 CrossRef CAS PubMed
- R. Kou, Y. Y. Shao, D. H. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. M. Wang, Y. H. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954 CrossRef CAS
- A. Ghosh, S. Basu and A. Verma, Fuel Cells, 2013, 13, 355 CrossRef CAS
- Q. Wang, M. M. Song, C. L. Chen, W. P. Hu and X. K. Wang, ChemPlusChem, 2012, 77, 432 CrossRef CAS
- Y. M. Tan, C. F. Xu, G. X. Chen, N. F. Zheng and Q. J. Xie, Energy Environ. Sci., 2012, 5, 6923 CAS
- J. N. Tiwari, K. Nath, S. Kumar, R. N. Tiwari, K. C. Kemp, N. H. Le, D. H. Youn, J. S. Lee and K. S. Kim, Nat. Commun., 2013, 4, 2221 Search PubMed
- J. J. Shi, G. H. Yang and J. J. Zhu, J. Mater. Chem. A, 2011, 21, 7343 RSC
- Z. H. Wang, J. F. Xia, X. M. Guo, Y. Z. Xia, S. Y. Yao, F. F. Zhang, Y. H. Li and L. H. Xia, Adv. Mater., 2013, 5, 483 CAS
- K. W. Nam, J. Song, K. H. Oh, M. J. Choo, H. Park, J. K. Park and J. W. Choi, Carbon, 2012, 50, 3739 CrossRef CAS
- A. Y. W. Sham and S. M. Notley, Soft Matter, 2013, 9, 6645 RSC
- Z. Cui, C. X. Guo and C. M. Li, J. Mater. Chem. A, 2013, 1, 6687 CAS
- S. Yang, C. Shen, Y. Liang, H. Tong, W. He, X. Shi, X. Zhang and H.-j. Gao, Nanoscale, 2011, 3, 3277 RSC
- Y. C. Zhao, L. Zhan, J. N. Tian, S. L. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967 CrossRef CAS
- D. H. Deng, L. Yu, X. L. Pan, S. Wang, X. Q. Chen, P. Hu, L. X. Sun and X. H. Bao, Chem. Commun., 2011, 47, 10016 RSC
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027 CrossRef CAS PubMed
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J.-J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844 CrossRef CAS PubMed
- J. Shen, Y. Zhu, X. Yang and C. Li, Chem. Commun., 2012, 48, 3686 RSC
- G. He, Y. Song, K. Liu, A. Walter, S. Chen and S. Chen, ACS Catal., 2013, 3, 831 CrossRef CAS
- X. Yan, Q. Li and L.-s. Li, J. Am. Chem. Soc., 2012, 134, 16095 CrossRef CAS PubMed
- C. Wu, X. Huang, G. Wang, L. Lv, G. Chen, G. Li and P. Jiang, Adv. Funct. Mater., 2013, 23, 506 CrossRef CAS
- J. Zhang, F. Zhao, Z. Zhang, N. Chen and L. Qu, Nanoscale, 2013, 5, 3112 RSC
- P. V. Kamat, J. Phys. Chem. Lett., 2009, 1, 520 CrossRef
- D. Wu, F. Zhang, H. Liang and X. Feng, Chem. Soc. Rev., 2012, 41, 6160 RSC
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020 CrossRef CAS PubMed
- K. Chen, L. Chen, Y. Chen, H. Bai and L. Li, J. Mater. Chem., 2012, 22, 20968 RSC
- D. A. C. Brownson, L. C. S. Figueiredo-Filho, X. Ji, M. Gomez-Mingot, J. Iniesta, O. Fatibello-Filho, D. K. Kampouris and C. E. Banks, J. Mater. Chem. A, 2013, 1, 5962 CAS
- S. Nardecchia, D. Carriazo, M. L. Ferrer, M. C. Gutierrez and F. del Monte, Chem. Soc. Rev., 2013, 42, 794 RSC
- S. Sattayasamitsathit, Y. Gu, K. Kaufmann, W. Jia, X. Xiao, M. Rodriguez, S. Minteer, J. Cha, D. B. Burckel, C. Wang, R. Polsky and J. Wang, J. Mater. Chem. A, 2013, 1, 1639 CAS
- S. Guo, D. Wen, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 3959 CrossRef CAS PubMed
- C. G. Hu, H. H. Cheng, Y. Zhao, Y. Hu, Y. Liu, L. M. Dai and L. T. Qu, Adv. Mater., 2012, 24, 5493 CrossRef CAS PubMed
- Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792 CrossRef CAS
- Y. Tang, Z. Yang and X. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566 RSC
- C. S. Chen and F. M. Pan, Appl. Catal., B, 2009, 91, 663 CrossRef CAS
- B. Y. Xia, H. B. Wu, J. S. Chen, Z. Wang, X. Wang and X. W. Lou, Phys. Chem. Chem. Phys., 2012, 14, 473 RSC
- Z. L. Wen, S. D. Yang, Y. Y. Liang, W. He, H. Tong, L. A. Hao, X. G. Zhang and Q. J. Song, Electrochim. Acta, 2010, 56, 139 CrossRef CAS
- H. Huang, Q. Chen, M. He, X. Sun and X. Wang, J. Power Sources, 2013, 239, 189 CrossRef CAS
- X. Li, W. Qi, D. Mei, M. L. Sushko, I. Aksay and J. Liu, Adv. Mater., 2012, 24, 5136 CrossRef CAS PubMed
- J. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J. M. Gibson and J. Huang, ACS Nano, 2011, 5, 8943 CrossRef CAS PubMed
- R. Kou, Y. Shao, D. Mei, Z. Nie, D. Wang, C. Wang, V. V. Viswanathan, S. Park, I. A. Aksay, Y. Lin, Y. Wang and J. Liu, J. Am. Chem. Soc., 2011, 133, 2541 CrossRef CAS PubMed
- B. Y. Xia, B. Wang, H. B. Wu, Z. Liu, X. Wang and X. W. Lou, J. Mater. Chem., 2012, 22, 16499 RSC
- X. Wang, X. Y. Li, D. P. Liu, S. Y. Song and H. J. Zhang, Chem. Commun., 2012, 48, 2885 RSC
- R. Liu, H. H. Zhou, J. Liu, Y. Yao, Z. Y. Huang, C. P. Fu and Y. F. Kuang, Electrochem. Commun., 2013, 26, 63 CrossRef CAS
- D. Yu and L. Dai, J. Phys. Chem. Lett., 2009, 1, 467 CrossRef
- A. Marinkas, F. Arena, J. Mitzel, G. M. Prinz, A. Heinzel, V. Peinecke and H. Natter, Carbon, 2013, 58, 139 CrossRef CAS
- V. C. Tung, L.-M. Chen, M. J. Allen, J. K. Wassei, K. Nelson, R. B. Kaner and Y. Yang, Nano Lett., 2009, 9, 1949 CrossRef CAS PubMed
- Y. Z. Chang, G. Y. Han, M. Y. Li and F. Gao, Carbon, 2011, 49, 5158 CrossRef CAS
- S. Y. Yang, K. H. Chang, Y. F. Lee, C. C. M. Ma and C. C. Hu, Electrochem. Commun., 2010, 12, 1206 CrossRef CAS
- Y. J. Li, Y. J. Li, E. B. Zhu, T. McLouth, C. Y. Chiu, X. Q. Huang and Y. Huang, J. Am. Chem. Soc., 2012, 134, 12326 CrossRef CAS PubMed
- L. De Rogatis, M. Cargnello, V. Gombac, B. Lorenzut, T. Montini and P. Fornasiero, ChemSusChem, 2010, 3, 24 CrossRef CAS PubMed
- W. I. Park, C.-H. Lee, J. M. Lee, N.-J. Kim and G.-C. Yi, Nanoscale, 2011, 3, 3522 RSC
- N. Li, M. Cao and C. Hu, Nanoscale, 2012, 4, 6205 RSC
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550 CrossRef CAS PubMed
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878 CrossRef CAS PubMed
- S. Cui, S. Mao, G. Lu and J. Chen, J. Phys. Chem. Lett., 2013, 4, 2441 CrossRef CAS
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed
- M. Hamdani, R. N. Singh and P. Chartier, Int. J. Electrochem. Sci., 2010, 5, 556 CAS
- Y.-g. Wang, L. Cheng, F. Li, H.-m. Xiong and Y.-y. Xia, Chem. Mater., 2007, 19, 2095 CrossRef CAS
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2009, 22, 898 CrossRef
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed
- J. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333 CAS
- Y. Liang, H. Wang, P. Diao, W. Chang, G. Hong, Y. Li, M. Gong, L. Xie, J. Zhou, J. Wang, T. Z. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2012, 134, 15849 CrossRef CAS PubMed
- J. Xiao, Q. Kuang, S. Yang, F. Xiao, S. Wang and L. Guo, Sci. Rep., 2013, 3, 2300 Search PubMed
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770 CrossRef CAS PubMed
- Q. He, Q. Li, S. Khene, X. Ren, F. E. López-Suárez, D. Lozano-Castelló, A. Bueno-López and G. Wu, J. Phys. Chem. C, 2013, 117, 8697 CAS
- W.-H. Ryu, T.-H. Yoon, S. H. Song, S. Jeon, Y.-J. Park and I.-D. Kim, Nano Lett., 2013, 13, 4190 CrossRef CAS PubMed
- J. Feng, Y. Liang, H. Wang, Y. Li, B. Zhang, J. Zhou, J. Wang, T. Regier and H. Dai, Nano Res., 2012, 5, 718 CrossRef CAS
- T. N. Lambert, D. J. Davis, W. Lu, S. J. Limmer, P. G. Kotula, A. Thuli, M. Hungate, G. Ruan, Z. Jin and J. M. Tour, Chem. Commun., 2012, 48, 7931 RSC
- Y. Cao, Z. K. Wei, J. He, J. Zang, Q. Zhang, M. S. Zheng and Q. F. Dong, Energy Environ. Sci., 2012, 5, 9765 CAS
- Y. Cao, Z. Wei, J. He, J. Zang, Q. Zhang, M. Zheng and Q. Dong, Energy Environ. Sci., 2012, 5, 9765 CAS
- J. Zhang, C. Guo, L. Zhang and C. M. Li, Chem. Commun., 2013, 49, 6334 RSC
- J. Duan, Y. Zheng, S. Chen, Y. Tang, M. Jaroniec and S. Qiao, Chem. Commun., 2013, 49, 7705 RSC
- J.-S. Lee, T. Lee, H.-K. Song, J. Cho and B.-S. Kim, Energy Environ. Sci., 2011, 4, 4148 CAS
- J. Xiao, G. Xu, S.-G. Sun and S. Yang, Part. Part. Syst. Charact., 2013, 30, 893 CrossRef CAS
- W. Zhang, Y. Zeng, C. Xu, H. Tan, W. Liu, J. Zhu, N. Xiao, H. H. Hng, J. Ma, H. E. Hoster, R. Yazami and Q. Yan, RSC Adv., 2012, 2, 8508 RSC
- X.-Y. Yan, X.-L. Tong, Y.-F. Zhang, X.-D. Han, Y.-Y. Wang, G.-Q. Jin, Y. Qin and X.-Y. Guo, Chem. Commun., 2012, 48, 1892 RSC
- S. Qiao, R. Zhou, Y. Zheng and D. Jurcakova, J. Mater. Chem. A, 2013, 1, 13179 Search PubMed
- H.-G. Jung, Y. S. Jeong, J.-B. Park, Y.-K. Sun, B. Scrosati and Y. J. Lee, ACS Nano, 2013, 7, 3532 CrossRef CAS PubMed
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082 CrossRef CAS PubMed
- V. Neburchilov, H. Wang, J. J. Martin and W. Qu, J. Power Sources, 2010, 195, 1271 CrossRef CAS
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517 CrossRef CAS PubMed
- H. L. Wang, Y. Yang, Y. Y. Liang, G. Y. Zheng, Y. G. Li, Y. Cui and H. J. Dai, Energy Environ. Sci., 2012, 5, 7931 CAS
- G. Zhang, B. Y. Xia, C. Xiao, L. Yu, X. Wang, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 8643 CrossRef CAS PubMed
- D. U. Lee, B. J. Kim and Z. Chen, J. Mater. Chem. A, 2013, 1, 4754 CAS
- Y. Wang, D. Zhang and H. Liu, J. Power Sources, 2010, 195, 3135 CrossRef CAS
- J. Wu, D. Zhang, Y. Wang, Y. Wan and B. Hou, J. Power Sources, 2012, 198, 122 CrossRef CAS
- L. Zhang, J. Zhang, D. P. Wilkinson and H. Wang, J. Power Sources, 2006, 156, 171 CrossRef CAS
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421 CrossRef CAS
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986 RSC
- Y.-X. Zhou, H.-B. Yao, Y. Wang, H.-L. Liu, M.-R. Gao, P.-K. Shen and S.-H. Yu, Chem. –Eur. J., 2010, 16, 12000 CrossRef CAS PubMed
- J. S. Jirkovský, A. Björling and E. Ahlberg, J. Phys. Chem. C, 2012, 116, 24436 Search PubMed
- C. Zhao, D. Li and Y. Feng, J. Mater. Chem. A, 2013, 1, 5741 CAS
- H. Wang, Y. Liang, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2011, 50, 10969 CrossRef CAS PubMed
- N. Mahmood, C. Z. Zhang, J. Jiang, F. Liu and Y. L. Hou, Chem. –Eur. J., 2013, 19, 5183 CrossRef CAS PubMed
- P. Chen, T.-Y. Xiao, H.-H. Li, J.-J. Yang, Z. Wang, H.-B. Yao and S.-H. Yu, ACS Nano, 2011, 6, 712 CrossRef PubMed
- Q. Liu, J. T. Jin and J. Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 5002 CAS
- Z.-G. Zhao, J. Zhang, Y. Yuan, H. Lv, Y. Tian, D. Wu and Q.-W. Li, Sci. Rep., 2013, 3, 2263 Search PubMed
- W.-F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896 RSC
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed
- A. B. Laursen, S. Kegnaes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577 CAS
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308 CrossRef CAS PubMed
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed
- P. Ge, M. D. Scanlon, P. Peljo, X. Bian, H. Vubrel, A. O'Neill, J. N. Coleman, M. Cantoni, X. Hu, K. Kontturi, B. Liu and H. H. Girault, Chem. Commun., 2012, 48, 6484 RSC
- L. Liao, J. Zhu, X. Bian, L. Zhu, M. D. Scanlon, H. H. Girault and B. Liu, Adv. Funct. Mater., 2013, 23, 5326 CrossRef CAS
- Y.-H. Chang, C.-T. Lin, T.-Y. Chen, C.-L. Hsu, Y.-H. Lee, W. Zhang, K.-H. Wei and L.-J. Li, Adv. Mater., 2013, 25, 756 CrossRef CAS PubMed
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698 CrossRef CAS PubMed
- H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059 CrossRef PubMed
- Y. Shi, W. Zhou, A.-Y. Lu, W. Fang, Y.-H. Lee, A. L. Hsu, S. M. Kim, K. K. Kim, H. Y. Yang, L.-J. Li, J.-C. Idrobo and J. Kong, Nano Lett., 2012, 12, 2784 CrossRef CAS PubMed
- E. G. S. Firmiano, M. A. L. Cordeiro, A. C. Rabelo, C. J. Dalmaschio, A. N. Pinheiro, E. C. Pereira and E. R. Leite, Chem. Commun., 2012, 48, 7687 RSC
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed
- Y. Yan, B. Xia, X. Qi, H. Wang, R. Xu, J.-Y. Wang, H. Zhang and X. Wang, Chem. Commun., 2013, 49, 4884 RSC
- A.-M. Alexander and J. S. J. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388 RSC
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339 CrossRef CAS PubMed
- J. Tian, Q. Liu, C. Ge, Z. Xing, A. M. Asiri, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 8921 RSC
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith, M. Jaroniec, G. Q. Lu and S. Z. Qiao, J. Am. Chem. Soc., 2011, 133, 20116 CrossRef CAS PubMed
- X.-H. Li, J.-S. Chen, X. Wang, J. Sun and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074 CrossRef CAS PubMed
- Y. Zhang, T. Mori, L. Niu and J. Ye, Energy Environ. Sci., 2011, 4, 4517 CAS
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 CAS
- A. Du, S. Sanvito, Z. Li, D. Wang, Y. Jiao, T. Liao, Q. Sun, Y. H. Ng, Z. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 134, 4393 CrossRef CAS PubMed
- Y. Di, X. Wang, A. Thomas and M. Antonietti, ChemCatChem, 2010, 2, 834 CrossRef CAS
- D. H. Youn, G. Bae, S. Han, J. Y. Kim, J.-W. Jang, H. Park, S. H. Choi and J. S. Lee, J. Mater. Chem. A, 2013, 1, 8007 CAS
- Q. Liu and J. Zhang, Langmuir, 2013, 29, 3821 CrossRef CAS PubMed
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609 CrossRef CAS
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658 CrossRef CAS PubMed
- M. Chisaka and H. Muramoto, ChemElectroChem, 2013, 1, 544 CrossRef
- Z. Wen, S. Cui, H. Pu, S. Mao, K. Yu, X. Feng and J. Chen, Adv. Mater., 2011, 23, 5445 CrossRef CAS PubMed
- J. Jin, X. Fu, Q. Liu and J. Zhang, J. Mater. Chem. A, 2013, 1, 10538 CAS
- C.-W. Tsai, M.-H. Tu, C.-J. Chen, T.-F. Hung, R.-S. Liu, W.-R. Liu, M.-Y. Lo, Y.-M. Peng, L. Zhang, J. Zhang, D.-S. Shy and X.-K. Xing, RSC Adv., 2011, 1, 1349 RSC
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 CAS
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593 CrossRef CAS PubMed
- Q. Liu, H. Zhang, H. Zhong, S. Zhang and S. Chen, Electrochim. Acta, 2012, 81, 313 CrossRef CAS
- Y. J. Tong, Chem. Soc. Rev., 2012, 41, 8195 RSC
- C. H. Choi, M. W. Chung, H. C. Kwon, J. H. Chung and S. I. Woo, Appl. Catal., B, 2014, 144, 760 CrossRef CAS
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394 CrossRef CAS PubMed
- P. Chen, T.-Y. Xiao, Y.-H. Qian, S.-S. Li and S.-H. Yu, Adv. Mater., 2013, 25, 3192 CrossRef CAS PubMed
- J.-S. Lee, K. Jo, T. Lee, T. Yun, J. Cho and B.-S. Kim, J. Mater. Chem. A, 2013, 1, 9603 CAS
- G. Wu, N. H. Mack, W. Gao, S. G. Ma, R. Q. Zhong, J. T. Han, J. K. Baldwin and P. Zelenay, ACS Nano, 2012, 6, 9764 CrossRef CAS PubMed
- J. Liang, X. Du, C. Gibson, X. W. Du and S. Z. Qiao, Adv. Mater., 2013, 25, 6226 CrossRef CAS PubMed
- S.-L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656 CAS
- M. Jahan, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707 CrossRef CAS PubMed
- M. Jahan, Z. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363 CrossRef CAS
- T. K. Kim, J. Y. Cheon, K. Yoo, J. W. Kim, S.-m. Hyun, H. S. Shin, S. H. Joo and H. R. Moon, J. Mater. Chem. A, 2013, 1, 8432 CAS
| This journal is © The Royal Society of Chemistry 2014 |