Strategies for improving the efficiency of semiconductor metal oxide photocatalysis
Aleksandra B.
Djurišić
*a,
Yu Hang
Leung
a and
Alan Man
Ching Ng
ab
aDept. of Physics, the University of Hong Kong, Pokfulam Road, Hong Kong. E-mail: dalek@hku.hk; Fax: +852 2559 9152
bDept. of Physics, South University of Science and Technology of China, Shenzhen, China
First published on 17th April 2014
Abstract
Photocatalysis is of significant interest for a wide range of applications related to energy and environment, such as pollutant degradation and hydrogen production. We will provide a review of the relationship between photocatalyst properties and its photocatalytic performance, as well as the strategies for the enhancement of photocatalytic activity, in particular under solar/ambient/visible illumination. Common applications of photocatalysts will then be reviewed, and we will summarize existing problems and areas requiring further improvements.
 Alan Man Ching Ng, Aleksandra B. Djurišić, Yu Hang Leung | A. B. Djurišić obtained her PhD degree in electrical engineering from the School of Electrical Engineering at the University of Belgrade (now Serbia) in 1997. She joined the Department of Physics at the University of Hong Kong (HKU) in 2003. Her research interests include nanomaterials, wide-bandgap semiconductors, photocatalysts, and optoelectronic devices. Y. H. Leung obtained his PhD degree from the Department of Physics and Materials Science, City University of Hong Kong in 2011. He is currently a research assistant professor at Department of Physics, HKU. His current research interests include applications of nanostructured materials (solar cells, anti-bacterial applications, and photocatalysis). Alan Man-Ching Ng obtained his PhD degree in 2010 at HKU. He joined the South University of Science and Technology of China (SUSTC) in Aug 2011. His research interests include the fabrication and characterization of metal oxide nanostructures, and their applications in the field of energy and environment. |
1 Introduction
Photocatalysis has the potential to enable utilization of an abundant and clean energy source (natural sunlight) for the purpose of environmental remediation (pollutant degradation) and clean energy production (hydrogen production, CO2 reduction).1 Therefore, it is of significant practical interest, and there is intensive research effort to develop novel photocatalyst materials to improve the efficiency, in particular under visible/solar illumination. Semiconductor metal oxide photocatalysis is a process in which the absorption of light by a photocatalyst results in the creation of photogenerated electrons and holes which can then be transferred to other molecules at the photocatalyst surface. It is typically considered that an electron can be transferred to an acceptor molecule if its redox potential lies below the conduction band (CB) of the photocatalyst, while a hole can be transferred to a donor molecule if its redox potential lies above the valence band (VB) of the photocatalyst.2 An illustration of the mechanism of photocatalysis is shown in Fig. 1. However, it would be advisable to look into the energy levels of the adsorbed molecules instead of their redox potentials to establish whether electron or hole transfer is indeed possible in a given situation. For example, it has been shown that the energy levels of both water and adsorbed hydroxide groups on the stoichiometric TiO2 (101) surface have energy levels below the valence band edge.3 While the energy level of hydrated hydroxyl groups can be located above the valence band edge, this does not occur for molecular water.3 Thus, adsorbed molecular water on stoichiometric titania (101) surfaces cannot contribute to the generation of reactive oxygen species.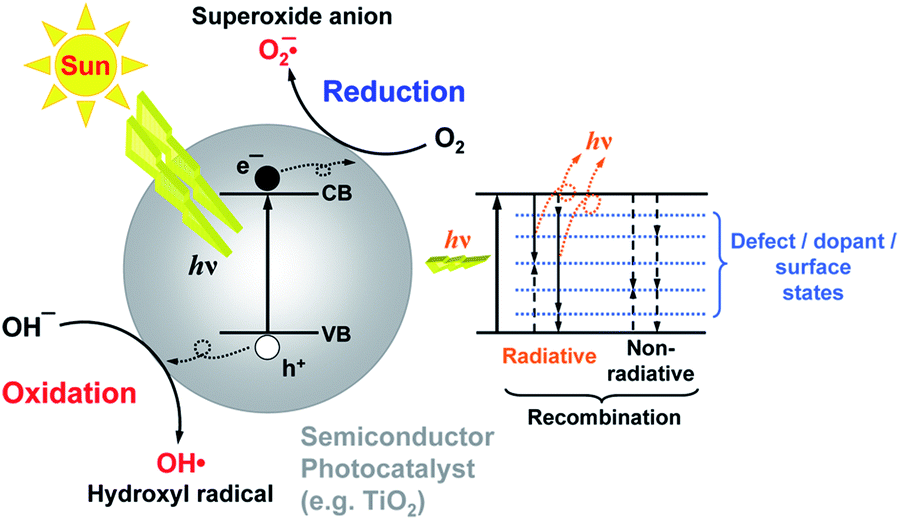 | ||
| Fig. 1 Schematic diagram of the principle of photocatalysis. | ||
Different reactive oxygen species (ROS) can be generated in the process of photocatalysis.2 Different studies in the literature have been proposing various ROS species as the dominant participant in the photocatalytic process, and the role of different reactive species generated by the photocatalyst upon illumination has also been studied.4 It was found that the photogenerated holes are the dominant species participating in the photocatalytic degradation since the degradation could occur in a vacuum environment, as illustrated in Fig. 2.4 The contribution of other reactive species, such as hydroxyl radicals, was dependent on the chemical structure of the molecule to be degraded.4 It has also been proposed that superoxide ions cannot destroy chemical bonds due to their negative charge, but they can be reactive to biological macromolecules.2 Nevertheless, attribution of photocatalytic reactions to different ROS in the literature is still common.
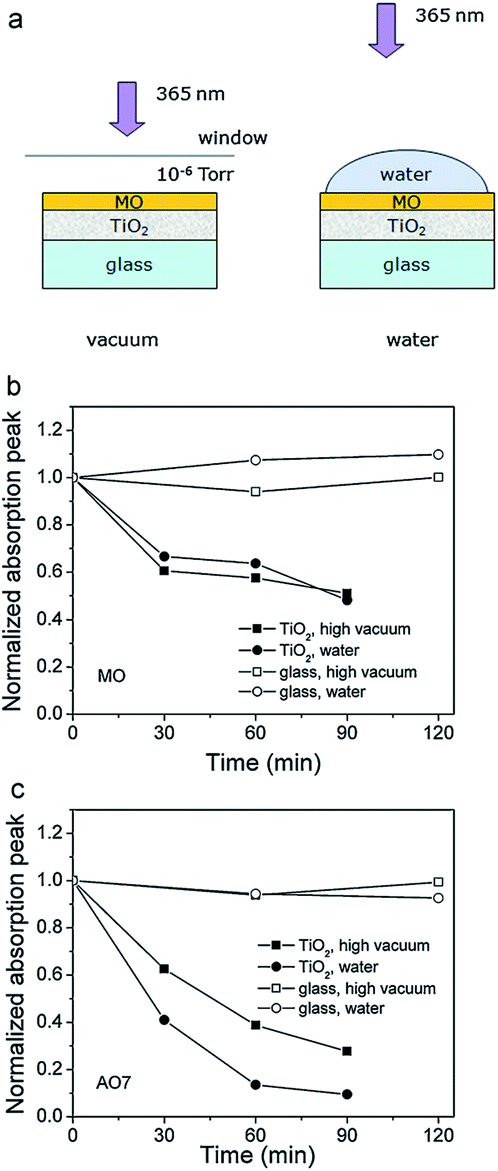 | ||
| Fig. 2 (a) Schematic diagram of the experiments studying the direct charge transfer between the metal oxide and the dye. Degradation curves of (b) methyl orange (MO) and (c) acid orange 7 (AO7) in high vacuum and water. Reprinted with permission from ref. 4. Copyright 2011 ©Elsevier. | ||
Various photocatalyst materials have been studied to date. Photocatalysts are generally nonselective, although some selectivity could be achieved by adjusting the properties of the photocatalyst or the reaction conditions and the use of composite materials.5 Among different photocatalysts, TiO2 has been the most extensively studied. There have been numerous reviews of its synthesis and photocatalytic applications.6–9 ZnO also has excellent photocatalytic activity under UV illumination, but it is less stable compared to titania.10 However, it can be suitable for photocatalytic degradation of pollutants in the gas phase rather than in liquid.11 It should also be noted that although titania is generally stable, dopants in doped titania may not necessarily be stable over a prolonged aging period.12 Both ZnO and TiO2 are wide bandgap semiconductors, although photocatalytic activity under visible illumination could be achieved by defect engineering or doping. Defect engineering implies controlled introduction of native defects, such as vacancies, interstitials, and antisites, while doping implies controlled introduction of impurities into the crystal lattice. Photocatalysis under illumination with light with energy lower than the bandgap of the semiconductor can also occur with the compound under degradation acting as a sensitizer and transferring electrons to the semiconductor.2 Other photocatalytic metal oxides include bismuth oxides (binary as well as ternary oxides with Fe, W, V, or Mo), copper oxides (cupric and cuprous), iron oxide, zinc germanium oxide,13 SrTiO3, and tungsten oxide WO3.1,14 Positions of the conduction band edge and valence band edge of some metal oxides are illustrated in Fig. 3. These materials can be used either alone, or in heterojunctions or composites with other materials. Some heterojunction structures, such as TiO2/Al2O3/WO3, were shown to be capable of storing photogenerated electrons for prolonged periods of time.15
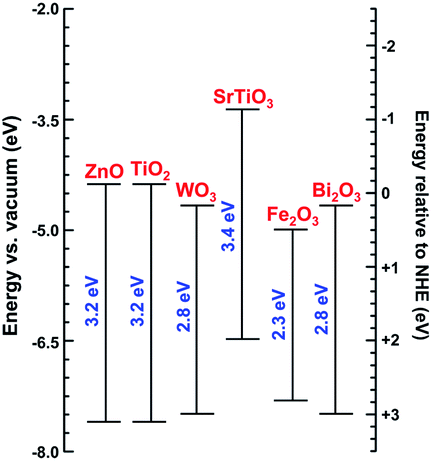 | ||
| Fig. 3 Band-edge energy positions of different semiconductors according to ref. 1, 16 and 17. | ||
In this review, we will discuss the material properties affecting the photocatalysis, strategies for enhancing the photocatalytic activity, and some specific photocatalytic applications, such as pollutant degradation, hydrogen generation, and antimicrobial activity. Our purpose is not to provide an exhaustive review of all the available photocatalyst materials, but rather to identify common research directions and point out unresolved issues, and for the sake of simplicity, convenience and brevity illustrate those on examples of commonly studied photocatalysts. We will mainly be discussing semiconductor metal oxide photocatalysts, with the majority of applications concerned with the photocatalyst use in aqueous solution. We will discuss in detail various controversies concerning the relationship between material properties and photocatalytic activity, and we will point out problems and unresolved issues concerning the enhancement of activity as well as specific applications.
2 Material properties affecting photocatalysis
Photocatalytic activity is affected by the properties of a photocatalyst, as well as the reaction conditions (pH, concentration, illumination power, etc.). Among the material properties affecting the photocatalytic activity, the most commonly considered are the surface area and the material quality (crystallinity and/or native defects). In Fig. 4, we can observe transmission electron microscopy (TEM) images of two common photocatalyst materials, anatase TiO2 and ZnO, from two different commercial suppliers.4 The samples with the same nominal particle size could have somewhat different aggregation properties,4 resulting in different effective surface areas. Surface area is considered important as a measure of available active surface sites for photocatalytic reactions. The rationale for this is the assumption that a greater surface area available for the adsorption of the compound to be degraded would result in faster degradation. Dye adsorption onto the photocatalyst surface is commonly considered essential for photocatalytic dye degradation.5,18 However, it has been shown that in ZnO there was no correlation between dye adsorption and dye degradation.4,19 The adsorption of anionic dyes onto ZnO was related to the presence of shallow donor centers exhibiting an EPR peak at g ≈ 1.957.19 The exact chemical nature of the defects was not known but the defects were likely not due to hydrogen impurities, and the dye adsorption was likely due to the electrostatic interaction between surface shallow donors and the sulfonate group in the dye molecule.19 Regardless of whether adsorption of pollutants is necessary for photocatalytic reaction or not, the surface area still provides some potentially useful information about the photocatalyst. However, an important question is how to determine the surface area relevant for photocatalytic experiments. | ||
| Fig. 4 Representative TEM images of (a) TiO2 nanoparticles from MKnano (division of M K Impex Corp.), the average particle size (APS) is 15 nm; (b) TiO2 nanoparticles from Nanostructured & Amorphous Materials Inc., APS is 15 nm; (c) ZnO nanoparticles from MKnano, APS is 20 nm; (d) ZnO nanoparticles from Nanostructured & Amorphous Materials Inc., APS is 20 nm. | ||
Brunauer–Emmett–Teller (BET) surface area analysis is a commonly used characterization technique to compare the available surface areas of different photocatalysts. However, BET measurements are normally conducted in vacuum, and involve subjecting the sample to baking and outgassing. An actual photocatalytic experiment on the other hand would typically involve the sample dispersed in a liquid, so that a more appropriate measure of the available surface area would be the aggregation size under the conditions under which the experiment is performed (the same solution composition if possible, concentration adjustment may be necessary in some cases to obtain an estimate of the aggregation size from dynamic light scattering techniques). Thus, the BET surface area obviously represents a maximum available surface area, while the surface area under realistic testing conditions can be much lower (and higher BET does not guarantee higher available surface area in solution). Similarly, the average particle size may not necessarily correlate well with the aggregation size in solution,20 due to the fact that smaller particles frequently have a large amount of native defects which may result in a large aggregation size in solution. There are numerous examples in the literature showing that a higher BET surface area does not necessarily imply higher photocatalytic activity.21–23 In fact, improved crystallinity can often result in higher photocatalytic activity despite the lower BET surface area,22,23 which is likely due to reduced recombination losses. Recombination is an opposite process from the generation of charge carriers under illumination, in which photogenerated electrons in the conduction band recombine with the photogenerated holes in the valence band. Thus, higher recombination means larger losses of photogenerated carriers and lower photocatalytic efficiency. Recombination can be radiative (involving the emission of a photon) or nonradiative (without light emission, typically involving phonons). These processes are illustrated in Fig. 1. The importance of minimizing recombination losses to improve photocatalytic efficiency is illustrated in Fig. 5, showing photoluminescence, ROS generation and degradation of two different dyes for ZnO nanostructures. It can be observed that the fastest dye degradation occurs for two samples exhibiting the best optical properties (the highest near band edge emission intensity, low defect emission), despite their lower BET surface area.
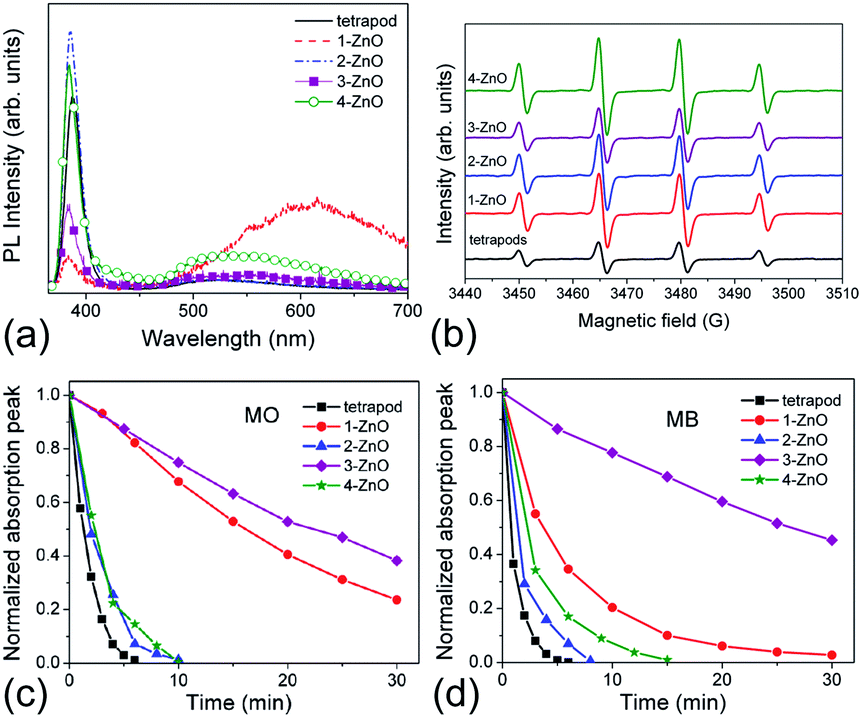 | ||
| Fig. 5 (a) Room-temperature PL spectra of different ZnO samples; (b) EPR spectra of ZnO samples with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) spin trap; (c) dye degradation curves for ZnO samples for methyl orange (MO), and (d) methylene blue (MB). Reprinted with permission from M. Y. Guo, A. M. C. Ng, F. Z. Liu, A. B. Djurišić, W. K. Chan, H. M. Su and K. S. Wong, J. Phys. Chem. C, 115, 11095 (2011).23 Copyright 2011 ©American Chemical Society. | ||
This illustrates the importance of the material quality (crystallinity and native defect types and concentrations). The important role of surface oxygen vacancies in photocatalysis was proposed for different materials. For example, In2O3 nanostructures with a higher concentration of oxygen vacancies were reported to have a higher photocatalytic activity.24 It is also well recognized that point defects affect the reactivity of the titania surface.25 However, studies on anatase surfaces, in particular the photocatalytically active (001) surface,25 have been scarce. The less active rutile crystal phase has been more commonly studied due to wide availability of rutile single crystal samples. It has been shown, however, that the reduction status of the anatase (001) surface affects its reactivity, which is similar to rutile surfaces.25 Surface oxygen vacancies were also proposed to play a significant role in the performance of ZnO photocatalysts.26 Improved photocatalytic activity of ZnO films prepared by dip-coating and cold plasma treatment was attributed to the presence of oxygen vacancies and interstitial zinc.27 Both metal and oxygen vacancies were reported to contribute towards high photocatalytic activity of ZnO/reduced graphene oxide (RGO) composites.28 However, the effect of oxygen vacancies on photocatalytic activity may also be dependent on the position of the vacancies. For example, surface oxygen vacancies can serve both as charge carrier traps and adsorption sites (so that charge transfer to adsorbed species can occur, resulting in a lower surface recombination), while oxygen vacancies in the bulk serve as charge carrier traps and sites for recombination of photogenerated carriers.29 It has been shown that samples having low defect concentrations and low specific surface areas can still exhibit excellent photocatalytic activity due to low losses of photogenerated charge carriers.23 The presence of nonradiative defects which could result in the loss of photogenerated carriers before they could be transferred to molecules on/near the surface of the photocatalyst would be detrimental to photocatalytic activity.20,23
Due to the important role and diverse possible effects of different defects in photocatalytic reactions, their comprehensive characterization is essential. However, frequently a conclusion on the oxygen vacancy concentration is made based on the measurement of optical properties, such as absorption spectra24 or photoluminescence.30 While these measurements can provide a general indication on the presence of defects (i.e. subbandgap absorption implies the existence of the defects in the bandgap, low intensity photoluminescence implies the existence of nonradiative defects, and visible photoluminescence in a wide bandgap materials implies the existence of radiative defects), unambiguous identification of defects solely based on optical measurements is unlikely. These measurements are not sufficient to make conclusive claims on the presence and concentration of a specific type of defect. Rarely, other measurements such as positron annihilation spectroscopy (PAS)11 or electron paramagnetic resonance spectroscopy (EPR)28,29 are used to confirm the presence of vacancies. XPS is also sometimes used to analyze the catalyst composition and determine the presence of vacancy defects.27 However, it should be noted that the oxygen signal detected in XPS may partly originate from any surface adsorbates, which would affect the obtained results. While surface adsorbates could be removed by sputtering, sputtering would result in the creation of defects and broadening of observed peaks, as well as in difficulties in calibrating the peak positions usually based on the position of the carbon peak, and the removal of surface adsorbates would remove carbon. While these techniques could be considered as more informative than optical spectroscopy alone, careful interpretation is still needed due to possible contribution of surface adsorbates in XPS (affecting the oxygen peak and its interpretation), as well as the lack of unambiguous assignment of EPR peaks to specific defects. Commercial ZnO samples can contain significant adsorbates, which can be determined from Fourier-transform infrared spectroscopy spectra.20
3 Strategies for enhancing photocatalytic activity
In principle, photocatalytic activity will be enhanced if the absorption of light is higher or if the recombination losses for photogenerated charge carriers are lower. Different strategies for enhancing the photocatalytic activity of TiO2 and ZnO under visible/solar illumination have been reviewed recently.7–9 Briefly, they include sensitization with different sensitizers (dyes, polymers, semiconductors, and surface-complex assisted), defect engineering (oxygen sub-stoichiometry), doping with different elements (metals and non-metals), spatial structuring,7–9 and morphology optimization for enhancing the proportion of active facets.8 In addition to these approaches, in recent studies the optical effects of the noble metal in photocatalyst composites have been recognised and there have been efforts to use plasmonic effects to enhance light absorption. The enhancement of photocatalytic activity using plasmonic effects and visible light active photocatalysts based on noble metal/metal oxide composites using surface plasmon resonance effects has been recently reviewed.16,31 It has been recognized that further work is needed in preparing photocatalysts with controlled spatial distribution of monodispersed metal nanoparticles in order to fully understand the plasmonic contributions.31 Nevertheless, there have been promising results on the use of plasmonic effects to enhance photocatalytic activity, for example SiO2–Ag–SiO2–TiO2 multishell structures.32 In such complex structures, multiple effects which affect photocatalytic activity can occur, with localised surface plasmon resonance, the scattering effect by silver nanoparticles, and the Schottky junction having a positive effect, while Förster resonant energy transfer and energetic electron transfer from Ag to N-doped titania have a negative effect on the photocatalytic activity.32 Au nanoparticles on anatase/rutile mixed titania nanotubes have also been reported, and the improved efficiency was attributed to the increased visible light absorption due to Au nanoparticles and improved charge separation at Au/rutile/anatase interfaces.33 There have also been other various strategies to enhance the absorption of visible light by tailoring the optical properties by changing the morphology of the photocatalyst. For example, photocatalytic dye degradation excited by monochromatic visible light was demonstrated for a 2D ZnO shell array.34 Enhanced photocatalytic activity under visible illumination due to enhanced dye sensitization was reported for 3D inverse opal ZnO photonic crystals.35 Improved photocatalytic properties under UV illumination have also been reported for hierarchical TiO2 photonic crystals, with the improvement attributed to both effects of photonic crystal structure as well as Mie scattering effects.36Among different comprehensively studied approaches to improve photocatalytic activity, a common approach is to modify the crystal structure and/or morphology of the photocatalyst. In some materials, such as perovskites with layered structure37,38 and nano-step-structured materials such as La:NaTaO3,39 oxidation and reduction sites are separated, which enhances the photocatalytic efficiency. Furthermore, it is well known that a combination of anatase and rutile crystal phases results in improved charge separation in P25 titania, resulting in excellent photocatalytic activity. In addition to improving the charge separation, manipulation of the crystal structure can also enhance photocatalytic activity under visible illumination. The surface phase with a reduced bandgap of ∼2.1 eV was demonstrated on (011) rutile titania surfaces.40 It was proposed that this surface phase was formed by oxidation of bulk titanium interstitial defects during annealing in a vacuum chamber at a low pressure of oxygen.40 However, rutile titania is typically less reactive than anatase, and any surface arrangements formed under UHV conditions may not necessarily be stable under ambient conditions. Annealing in general can be used as a method of improving crystallinity, enhancing the material properties, and improving photocatalytic activity. It should be noted, however, that the results of the same annealing conditions may be dependent on the starting properties of the material (ZnO nanoparticles).20 For titania nanostructures, crystal structures and native defects affected the performance of titania–carbon nanotube composites under different illumination conditions.41
While crystallinity and native defects can be affected by post-growth treatments such as annealing, the morphology of the photocatalyst is mainly determined by the growth/synthesis conditions (although in some cases it can be altered by annealing, in particular when the temperature is sufficiently high to result in a crystal phase transformation). It has been demonstrated that the photocatalytic activity is significantly affected by the photocatalyst morphology. For example, it has been reported that mesoporous hollow titania shells exhibit improved photocatalytic activity compared to P25 titania.42 Improvement in the activity was also observed for anatase titania nanowires43 and ultrafine titania nanofibers44 compared to P25. Morphology manipulations can affect the separation of photogenerated charge carriers,45 as well as the availability of active sites due to differences in terminating facets. In titania, (001) facets are often claimed to be more reactive compared to common (101) low energy facets.18,46,47 However, there have also been reports which find (001) less reactive than (101) facets.48,49 The facet reactivity may not necessarily be the same for different photocatalytic reactions, with low energy facets in some cases exhibiting higher activity for hydrogen evolution,48 decomposition of acetaldehyde,49 and noble metal deposition.50,51 Furthermore, different facets may have different roles in photocatalytic reactions, such as (101) participating in photocatalytic reduction while (001) participated in photocatalytic oxidation.51
Methods to synthesize titania nanostructures with a higher proportion of high energy facets have been recently reviewed.47 A common synthesis method involves the use of a capping agent to inhibit crystal growth in a certain direction and thus preserve more reactive facets.47 While some synthesis procedures to obtain a larger proportion of (001) facets use ammonium fluoride,18 in a number of reported synthesis procedures HF is used, which is highly toxic and corrosive (this applies in general to a number of fluorine- or amine-containing capping agents in anatase synthesis).46 Nevertheless, a high proportion of (001) facets in anatase titania has been recently obtained using H2SO4, which served both to control the crystal phase as well as the capping agent resulting in a large proportion of (001) facets.46 In addition to (001) facets, methods for synthesis of nanostructures with a higher proportion of (110) and (100) facets have also been reported.47 It has also been proposed that the correct ratio of high energy (001) facets and low energy (101) facets is needed for high photocatalytic activity to achieve reduced recombination of photogenerated charge carriers.52 Morphology manipulation can also lead to enhanced photocatalytic activity in other photocatalysts, such as ZnO.21 In the case of ZnO, different nanostructured morphologies could also display a significant difference in stability.21 It was proposed that the Zn-terminated polar (0001) surface is more reactive, and that surface defects which increase the adsorption of oxygen and OH− ions result in higher photocatalytic activity.53 However, another study attributed high photocatalytic activity of ZnO nanorods with a high aspect ratio due to better charge separation and the higher presence of unsaturated Zn2+ sites on (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) facets.54
0) facets.54
Another attractive strategy for enhancing the activity under visible/solar illumination is doping. Synthesis of mesoporous TiO2 doped with different elements has been recently reviewed.55 Hydrogen doping produces black titania with high absorption of solar radiation and high photocatalytic activity.56 This was attributed to the passivation of defect centres by hydrogen.56 The hydrogen doping was confined to an amorphous shell, and it was proposed that the localised surface plasmon resonance played a role in the obtained excellent properties.56 It should be noted, however, that the same dopant incorporated into the same metal oxide material by a different processing method can result in drastically different behavior. For example, in nitrogen doping of titania by wet chemical methods, different from heating in ammonia gas, β-substituted nitrogen is not responsible for visible light activity.57 In this case, visible light activity occurs due to crystal defects and distortion caused by incorporating NH3.57 It should also be noted that doping will inevitably result in the changes in the defect concentrations compared to undoped materials. Thus, if the doping results in the increase of defect induced trapping and recombination of photogenerated charge carriers, it may be difficult to achieve improved performance of the doped material.40
While the native defects complicate the analysis of the performance in the doped materials due to their influence on photocatalytic properties, they also enable manipulation of the photocatalyst properties without intentional introduction of dopants. Defect manipulation, such as the reduction of the catalyst material to tailor the native defect properties, is an alternative strategy to improve photocatalytic activity in general and to achieve photocatalytic activity under visible/solar illumination. For example, an improved carrier separation by defect engineering (the generation of surface oxygen vacancies) was previously reported.26,58 In addition, hydrogenated black titania with a disordered surface and efficient photocatalytic activity under solar illumination has been demonstrated.59 Black titania core/shell structures prepared by reduction of titania using Al were reported, and their absorption in the visible spectral range was attributed to Ti3+ 3d and O 2p (ref. 6) states within the gap in the band structure of an amorphous TiO2−x shell obtained by Al reduction.60 Blue titania consisting of a mixture of rutile and anatase phases with surface defects (VO–Ti3+ sites) has also been reported as an efficient photocatalyst.61 The surface defects contributed to higher activity under both UV and visible illumination.61 An extended response into the visible spectral region due to the presence of Ti3+ bulk defects was also reported.62 Narrowing of the bandgap with increased concentration of oxygen vacancies was also reported in ZnO.63,64
Thus, inducing oxygen deficiency is a common approach of defect engineering to alter the photocatalytic properties and extend the activity into the visible spectral range. Oxygen-deficient TiO2 for photocatalytic applications has been recently reviewed.65 An increase in the light absorption at longer wavelengths with increased concentration of oxygen vacancies has been observed.66 The methods to prepare titania with high oxygen vacancy content are hydrogen thermal treatment, thermal treatment under oxygen depleted conditions, high energy particle bombardment, doping with other elements, and tuning of the reaction conditions.65 Other less common approaches such as immersion into a solution of a reducing agent are also possible.29 However, it should be noted that many of these approaches would result in multiple types of defects. For example, hydrogen thermal treatment would result in an introduction of hydrogen impurities, high energy particle bombardment would result in the creation of multiple types of native defects, etc. In doped titania, care needs to be taken in attribution of observed behavior to the dopant or to the native defects, since the introduction of a dopant would change the types and concentrations of native defects present. Heating of ZnO in vacuum was also reported to result in the creation of surface oxygen vacancies.26 Also, in addition to native defect generation, lattice distortions can occur in titania samples subjected to heating and/or hydrogen treatment.67 Nevertheless, such treatments can result in enhanced photocatalytic activity.67 Defect manipulation can also be combined with other strategies, such as the modification of morphology, to enhance photocatalytic activity under white illumination.30
Other approaches include various heterostructures and composite materials, commonly involving carbon in different forms (carbon nanotubes, graphene, reduced graphene oxide).68–72 Heterojunctions enhance charge separation and thus improve photocatalytic efficiency, and have been demonstrated for a variety of materials, such as α − β phase Ga2O3,73 anatase/rutile TiO2,74 Ga2O3/ZnGa2O4,75 CaFe2O4/PbBi2Nb1.9W0.1O9,76etc. Improved charge carrier separation (and enhanced visible light absorption) was also achieved in SnO-nanocluster modified anatase titania.77 Visible light absorption and efficient photocatalysis were also achieved in titania with its surface modified by Co2O3 clusters,78 as well as iron oxide surface modification.79 Finally, another approach to improve photocatalytic activity is the use of up-conversion by a rare-earth doped luminescent material.80 Although improvements in photocatalytic efficiency using this approach have been reported,80 it is uncertain whether this strategy would give rise to large improvements in photocatalytic activity due to the fact that up-conversion is not a high yield process.
It should be noted that while the development of visible-light active photocatalysts is highly desirable from the point of view of practical applications, it is necessary to consider potential ecotoxicity of such a photocatalyst in the case of accidental release into the environment.81 Even the regular wide-bandgap photocatalysts could potentially exhibit detrimental environmental effects under ambient/solar illumination which contains small amounts of UV light. For example, phototoxic effects of titania nanoparticles under natural levels of UV light on marine phytoplankton have been recently demonstrated. On the other hand, in a natural environment the presence of natural organic matter may reduce the toxicity.82
4 Photocatalysis for pollutant degradation
Photocatalysis is of significant interest for the degradation of various pollutants. Photocatalytic degradation of different classes of pollutants, such as organochlorine compounds, polychlorinated bisphenols, organophosphorus compounds and phenolic compounds, including degradation pathways has been recently reviewed.83 Organic dyes are common probe molecules to study pollutant degradation.21,28,42,54,56,60,68–70,84–87 This is partly due to the fact that organic dyes represent significant pollutants due to their widespread use (for example in textile industry), large annual production and the fact that a significant portion (10–15%) is discharged in the waste water.22 Another reason for the common use of dyes as model pollutants is the ease of monitoring the degradation via simple UV-Vis absorption spectroscopy. Very commonly used dyes are anionic methyl orange (MO) and cationic methylene blue (MB). It should be noted, however, that the use of dyes as model pollutants creates a possible problem in the fundamental studies of photocatalytic mechanisms, since both the dye and photocatalyst can commonly be excited under illumination used.Generally, TiO2 and ZnO are the most commonly studied metal oxide materials for photocatalytic degradation of pollutants. However, there have been reports of successful photocatalytic decomposition of different compounds using other oxide materials. For example, In2O3 nanostructures resulted in significantly faster degradation of perfluorooctanic acid compared to P25 titania.24 Also, efficient degradation of nitrobenzene by NiTiO3 nanorods was reported.45
Many common approaches for the enhancement of photocatalytic activity have been implemented in pollutant degradation applications, such as doping, defect and morphology optimization, and the use of composites or heterostructures. The examples of efficient photocatalysts for dye degradation by optimization of native defects and/or morphology include ZnO nanoplate/nanowire architectures,88 hollow titania shells,89 and ZnO subjected to vacuum deoxidation.26 Efficient MO degradation and stable cycling properties were also reported for black titania under simulated solar illumination.56 Other examples of doped titania resulting in excellent photocatalytic activity for dye degradation include fluorine doped titania mesoporous spheres,90 iodine doped titania nanosheets,91 nitrogen doped titania nanoparticles,92 hollow rock-salt TiO1−xNx nanoparticles,93 as well as metal ion doped ordered mesoporous titania.94
Nanocomposites or core/shell structures or heterostructures of different materials for efficient dye degradation have also been reported, such as Au/TiO2,85 TiO2/graphene,68,69 halloysite–TiO2,84 TiO2/(ZnS)x(CuInS2)1−x nanocomposites,87 ZnO/CdS,95 hierarchical porous TiO2/C microspheres,70 TiO2/phenolic resol nanocomposites,96 WO3 nanoparticle/TiO2 nanotube/WO3 nanoparticle heterostructures,97 TiO2–SiO2,98 reduced graphene oxide (RGO)–TiO2 and RGO–SnO2 composites,71 TiO2/NiO,99 TiO2 nanoparticles/TiO2 nanobelts and Ag/TiO2 nanoparticles/TiO2 nanobelts,100 CdS quantum dots/porous TiO2 nanotubes,101 TiO2/carbon core/shell nanofibers with embedded Ag nanoparticles,72etc. Efficient photocatalysts consisting of heterojunctions of semiconductors exhibiting inferior photocatalytic activity when used alone have also been demonstrated, such as p-CuO/n-In2O3.86 In addition, successful combinations of different approaches have been achieved, such as doping and metal loading (N-doped Ag–TiO2),102 Cu-doped Zn/ZnO with carbon modification,103 Fe3O4/SiO2/Au/TiO2 with exposed (001) facets,18 and Ce3+ doped:TiO2(B)/anatase TiO2 core–shell structures.104
5 Photocatalysis for energy applications
Photocatalytic applications which are of interest for energy production are hydrogen evolution (by direct water splitting or by using sacrificial agents) and carbon dioxide reduction. Photocatalytic and photoelectrochemical water splitting for hydrogen production is of particular interest, and a number of different materials have been investigated.38,105 However, efficiency of photocatalytic water splitting (the splitting of water into H2 and O2 in a stoichiometric ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under illumination in the presence of a photocatalyst) is typically low due to the need to overcome a significant energy barrier.38 While the efficiency of hydrogen production can be improved by the addition of sacrificial agents, this approach is less interesting from the practical application point of view due to the cost of sacrificial agents.38 Thus, further work on the development of novel, more efficient photocatalyst materials is needed. To enhance the efficiency of photocatalytic hydrogen production, similar approaches to other photocatalysis applications, i.e. morphology and crystallinity optimization, defect engineering, doping, and heterostructures, have been employed. For this application, the achievement of high efficiency under solar/visible/ambient illumination is of particular interest. It has been shown that the changes in the morphology and/or reduction of contaminant incorporation can result in enhanced photocatalytic hydrogen production, as demonstrated on titania nanotubes prepared in ionic liquids compared to those prepared by conventional anodization in ethylene glycol.106 Defect engineering (Ti3+ defects) for improved hydrogen production under visible illumination was also reported.62 The use of reduced titania nanotube arrays was also demonstrated for photoelectrochemical water splitting under simulated solar illumination,29 while oxygen vacancy-rich titania demonstrated photocatalytic activity for photocatalytic hydrogen production under visible illumination.58 The narrowing of the bandgap was attributed to oxygen vacancies.29 Furthermore, it was demonstrated that black titania under simulated solar illumination produced 8.2 mmol h−1 g−1 of hydrogen gas, compared to 0.61 mmol h−1 g−1 for undoped titania.56 C:ZnO also exhibited significantly higher efficiency for photocatalytic hydrogen production compared to undoped ZnO.107 In addition to changing the efficiency, the dopants can also alter the selectivity of hydrogen or oxygen evolution during the photocatalytic hydrogen production. For example, the modulation of preference for hydrogen or oxygen evolution from photocatalytic hydrogen production due to band bending induced by interstitial boron has been demonstrated.108
:1 under illumination in the presence of a photocatalyst) is typically low due to the need to overcome a significant energy barrier.38 While the efficiency of hydrogen production can be improved by the addition of sacrificial agents, this approach is less interesting from the practical application point of view due to the cost of sacrificial agents.38 Thus, further work on the development of novel, more efficient photocatalyst materials is needed. To enhance the efficiency of photocatalytic hydrogen production, similar approaches to other photocatalysis applications, i.e. morphology and crystallinity optimization, defect engineering, doping, and heterostructures, have been employed. For this application, the achievement of high efficiency under solar/visible/ambient illumination is of particular interest. It has been shown that the changes in the morphology and/or reduction of contaminant incorporation can result in enhanced photocatalytic hydrogen production, as demonstrated on titania nanotubes prepared in ionic liquids compared to those prepared by conventional anodization in ethylene glycol.106 Defect engineering (Ti3+ defects) for improved hydrogen production under visible illumination was also reported.62 The use of reduced titania nanotube arrays was also demonstrated for photoelectrochemical water splitting under simulated solar illumination,29 while oxygen vacancy-rich titania demonstrated photocatalytic activity for photocatalytic hydrogen production under visible illumination.58 The narrowing of the bandgap was attributed to oxygen vacancies.29 Furthermore, it was demonstrated that black titania under simulated solar illumination produced 8.2 mmol h−1 g−1 of hydrogen gas, compared to 0.61 mmol h−1 g−1 for undoped titania.56 C:ZnO also exhibited significantly higher efficiency for photocatalytic hydrogen production compared to undoped ZnO.107 In addition to changing the efficiency, the dopants can also alter the selectivity of hydrogen or oxygen evolution during the photocatalytic hydrogen production. For example, the modulation of preference for hydrogen or oxygen evolution from photocatalytic hydrogen production due to band bending induced by interstitial boron has been demonstrated.108
In addition to defect engineering and doping, the successful use of metal oxide composites was also reported for hydrogen production under visible illumination, such as Fe2O3/TiO2 nanocomposites with Pt nanoparticles.109 In addition, Cu2O/CuO/TiO2 core shell nanowire arrays for water photoelectrolysis have been reported, where Cu2O functioned as a light absorber layer, CuO served to enhance activity and stability, and TiO2 served as a further protection from photocorrosion.110 Despite the progress, it is still necessary to substantially improve the quantum yield of photocatalytic hydrogen production using visible/solar illumination.1
Another energy related application of photocatalysis which is of significant interest is carbon dioxide reduction, or the conversion of CO2 and water into fuel (reaction products are methane and carbon monoxide).111 The rates of photocatalytic production of methane and CO for graphene:Ti0.91O2 were 8.91 μmol g−1 h−1 and 1.14 μmol g−1 h−1, respectively, which were significantly higher compared to P25 (0.69 μmol g−1 h−1 and 0.16 μmol g−1 h−1, respectively).111 Improved performance in carbon dioxide reduction has also been reported for a composite catalyst consisting of titania and copper indium sulfide.112 However, the efficiency of photocatalytic CO2 conversion still requires substantial improvements to become practical.1
6 Inactivation of microorganisms and other applications of photocatalysis
The inactivation of microorganisms is another application of photocatalysis that is of significant practical interest.113 Typically, photocatalysts for disinfection are based on either titania (with or without dopants and/or noble metal modification),114 or ZnO,115 with rutile titania commonly exhibiting lower toxicity compared to anatase.82 However, the same material can have different activities to different bacteria.116 Photocatalytic disinfection is frequently attributed to the oxidative stress and cell membrane damage due to generated reactive oxygen species, such as hydroxyl radicals.113,114 Other mechanisms can include the release of metal ions (if applicable, for titania the release of titanium is negligible, while for ZnO the release of zinc ions can be significant) and the interaction with the cell wall, as illustrated in Fig. 6. All of these mechanisms can be affected by illumination. While some metal oxides can produce ROS in the dark and upon sub-bandgap illumination, ROS production will be significantly higher upon illumination with light with energy higher than the bandgap. Metal ion release can also be affected if there is a photocorrosion effect, while the interaction between the cells and nanoparticles could potentially be affected by trapping of the photogenerated charges on the nanomaterial surface which could alter the electrostatic attraction to negatively charged bacteria cell walls. Antibacterial activity can be very sensitive to the medium in which the experiment is conducted.117 For example, the antibacterial activity in sodium phosphate medium was significantly lower compared to sodium chloride medium.117 This is in agreement with the proposed mechanism of antibacterial activity for lanthanide oxides, based on phosphate starvation.118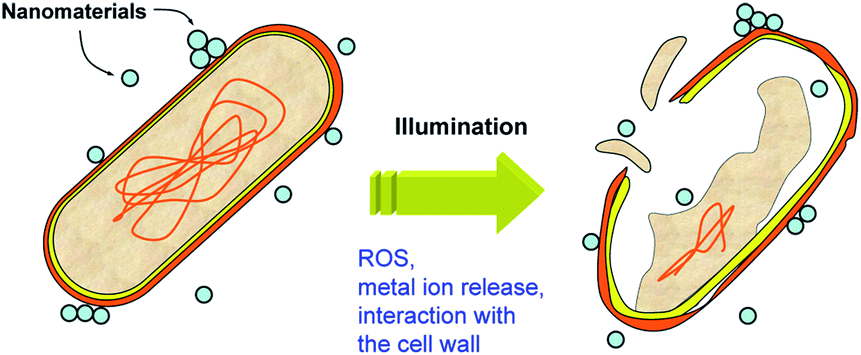 | ||
| Fig. 6 Schematic diagram of the possible mechanisms of antibacterial activity. | ||
Thus, the photocatalytic antibacterial activity of metal oxides is considerably more complex compared to pollutant degradation, due to multiple possible modes of action. In fact, there was no correlation between the photocatalytic antibacterial activity of ZnO and titania in two different suspension media and the photocatalytic dye degradation, indicating contribution of multiple mechanisms to the antibacterial effect of photocatalysts under illumination.117 It should be noted that although in some studies correlation between antibacterial activity and dye degradation is observed,116 the fact that this is not always the case117,119 clearly indicates differences in two processes. This is further illustrated by the fact that surface-modified ZnO retains antibacterial activity, while the activity for photocatalytic dye degradation is significantly reduced.120
The complexity of the bacterial inactivation process complicates the study of the mechanisms responsible for the observed cell membrane damage. Antibacterial activity of titania nanostructures was found to be morphology dependent, and it was attributed to the combined effects of photoactivity, aggregation of nano-titania, and the interactions between nano-titania and bacteria surfaces.119 The interaction between ZnO nanoparticles and bacteria cell walls was also found to play a significant role in the antibacterial effects of ZnO.120 These interactions can be electrostatic attraction, van der Waals forces, hydrophobic and receptor–ligand interactions121 and they can result in changes in cell wall permeability and cell wall damage.120 While many studies conclusively attribute the observed damage to one of the possible mechanisms of action (commonly ROS generation and lipid peroxidation), in other cases no clear correlation is observed. For example, the lack of correlation between ROS production and the level of antibacterial activity has been reported,116,120 although for some bacteria and some materials a correlation can be observed.116 It should also be noted that common methods of ROS detection, such as the use of luminescent probes, can result in experimental artefacts.120 This further complicates the study of the relationship between ROS generation and antibacterial activity.
In addition to the lack of correlation between ROS produced and antibacterial activity, no correlation was observed between Zn2+ ion release and antibacterial activity.120 Furthermore, in some metal oxides such as MgO, antibacterial activity was observed in the absence of ROS production, with the absence of lipid peroxidation and oxidative stress further confirmed by additional experiments as well as proteomics study.122 In addition, illumination affected the antibacterial activity of MgO even though it was in all cases sub-bandgap illumination demonstrating an important role of native defects in the process.122 All these indicate that the mechanisms of antibacterial activity are complex, and multiple phenomena likely contribute to the observed toxicity of metal oxide nanoparticles to bacteria under illumination.
Similar to other photocatalytic applications, it is of interest to achieve activity under not only UV illumination but also under visible and/or solar illumination. While ZnO can exhibit robust antibacterial activity under ambient illumination,120 titania typically requires UV illumination for efficient antibacterial activity. However, antibacterial activity under sunlight was reported for nitrogen doped titania.123 Methods for extending the photocatalytic antibacterial activity into the visible spectral range are similar to other photocatalytic applications, i.e. doping, composites, or defect engineering.115 There are also other less commonly studied applications of photocatalysis, such as wettability patterning and offset printing.124
7 Summary and outlook
Photocatalysis is of significant practical interest for a variety of applications, especially environmental remediation. Great progress has been made in the understanding of the process and improving the degradation efficiency for a number of different compounds, with studies of dye degradation being the most common, partly due to the convenience and ease of dye degradation experiments and partly due to environmental relevance of dyes as pollutants. Nevertheless, the role of native defects in the photocatalytic processes is still not fully clear. While recombination losses are obviously detrimental to photocatalytic efficiency, defects could also improve electron–hole separation and act as active centers on the photocatalyst surface. Thus, defects could in principle have both positive and negative effects on photocatalytic efficiency, depending on the type of defects as well as their location (surface vs. bulk). Further work is needed in conclusive identification of defects which have beneficial and defects which have detrimental effects on photocatalysis, especially considering the fact that in existing literature the identification of defects is commonly made based on optical measurements only which are insufficient for this purpose. Another area of significant interest is the enhancement of the photocatalytic activity, in particular under solar and/or visible light illumination. Although progress has been demonstrated using various approaches such as defect engineering, doping, and composite materials, further work in this area is needed. A related issue which to date has been receiving less attention is the separation of the photocatalyst from the treated wastewater and the studies of photocatalyst ecotoxicity. It has been shown that unpredictable effects of photocatalyst materials in the environment could be observed. For example, the degradation of surface coatings on pre-painted steel sheets was observed as a consequence of sunscreen residues (containing titania or ZnO nanoparticles).125 It is expected that materials with high activity under visible/solar illumination will have more significant detrimental environmental effects compared to conventional wide bandgap photocatalysts in the case of accidental release into the environment. While ecotoxicity under realistic environmental conditions may be mitigated by the presence of natural organic matter, this is still an important point to consider. In terms of practical applications of photocatalysts, pollutant degradation and antimicrobial action are the most relevant ones. Other applications such as photocatalytic hydrogen production, CO2 reduction, etc. require substantial improvement in efficiency before they can be practically implemented. For both pollutant degradation and antimicrobial action, important issues which need to be addressed are improving the activity under solar/visible/ambient illumination, as well as the study of the efficiency under realistic conditions (i.e. realistic wastewater composition for example).Acknowledgements
Financial support from the Strategic Research Theme, University Development Fund, and Seed Funding Grant (administrated by The University of Hong Kong) is acknowledged.References
- H. Tong, S. X. Ouyang, Y. P. Bi, N. Umezawa, M. Oshikiri and J. H. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- S. S. Shinde, C. H. Bhosale and K. Y. Rajpure, Catal. Rev.: Sci. Eng., 2013, 55, 79–133 CAS.
- H. Cheng and A. Selloni, Langmuir, 2010, 26, 11518–11525 CrossRef CAS PubMed.
- M. Y. Guo, A. M. C. Ng, F. Z. Liu, A. B. Djurišić and W. K. Chan, Appl. Catal., B, 2011, 107, 150–157 CrossRef CAS PubMed.
- M. A. Lazar and W. A. Daoud, RSC Adv., 2013, 3, 4130–4140 RSC.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS PubMed.
- S. Rehman, R. Ullah, A. M. Butt and N. D. Gohar, J. Hazard. Mater., 2009, 170, 560–569 CrossRef CAS PubMed.
- Y. Yang, H. Zhong and C. X. Tian, Res. Chem. Intermed., 2011, 37, 91–102 CrossRef CAS.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol., C, 2013, 15, 1–20 CrossRef CAS PubMed.
- M. Y. Guo, M. K. Fung, F. Fang, X. Y. Chen, A. M. C. Ng, A. B. Djurišić and W. K. Chan, J. Alloys Compd., 2011, 509, 1328–1332 CrossRef CAS PubMed.
- W. Xie, Y. Z. Li, W. Q. Shi, L. Zhao, X. J. Zhao, P. F. Fang, F. Zheng and S. J. Wang, Chem. Eng. J., 2012, 213, 218–224 CrossRef CAS PubMed.
- F. Spadavecchia, S. Ardizzone, G. Cappelletti, C. Oliva and S. Cappelli, J. Nanopart. Res., 2012, 14, 1301 CrossRef.
- L. K. Pan, X. J. Liu, Z. Sun and C. Q. Sun, J. Mater. Chem. A, 2013, 1, 8299–8326 CAS.
- H. D. Zheng, J. Z. Ou, M. S. Strano, R. B. Kaner, A. Mitchell and K. Kalantar-zadeh, Adv. Funct. Mater., 2011, 21, 2175–2196 CrossRef CAS.
- S. Kim and H. Park, RSC Adv., 2013, 3, 17551–17558 RSC.
- X. M. Zhang, Y. L. Chen, R.-S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- V. Stevanovic, S. Lany, D. S. Ginley, W. Tumas and A. Zunger, Phys. Chem. Chem. Phys., 2014, 16, 3706–3714 RSC.
- J. H. Shen, Y. H. Zhu, X. L. Yang and C. Z. Li, J. Mater. Chem., 2012, 22, 13341–13347 RSC.
- F. Z. Liu, Y. H. Leung, A. B. Djurišić, A. M. C. Ng and W. K. Chan, J. Phys. Chem. C, 2013, 117, 12218–12228 CAS.
- F. Z. Liu, M. Y. Guo, Y. H. Leung, A. B. Djurišić, A. M. C. Ng and W. K. Chan, Appl. Surf. Sci., 2013, 283, 914–923 CrossRef CAS PubMed.
- T.-J. Liu, Q. Wang and P. Jiang, RSC Adv., 2013, 3, 12662–12670 RSC.
- Y. Hong, C. G. Tian, B. J. Jiang, A. P. Wu, Q. Zhang, G. H. Tian and H. G. Fu, J. Mater. Chem. A, 2013, 1, 5700–5708 CAS.
- M. Y. Guo, A. M. C. Ng, F. Z. Liu, A. B. Djurišić, W. K. Chan, H. Su and K. S. Wong, J. Phys. Chem. C, 2011, 115, 11095–11101 CAS.
- Z. M. Li, P. Y. Zhang, T. Shao, J. L. Wang, L. Jin and X. Y. Li, J. Hazard. Mater., 2013, 260, 40–46 CrossRef CAS PubMed.
- Y. Wang, H. J. Sun, S. J. Tan, H. Feng, Z. W. Cheng, J. Zhao, A. D. Zhao, B. Wang, Y. Luo, J. L. Yang and J. G. Hou, Nat. Commun., 2013, 4, 2214 Search PubMed.
- Y. H. Lv, C. S. Pan, X. G. Ma, R. L. Zong, X. J. Bai and Y. F. Zhu, Appl. Catal., B, 2013, 138, 26–32 CrossRef PubMed.
- Z. X. Pei, L. Y. Ding, J. Hu, S. X. Weng, Z. Y. Zheng, M. L. Huang and P. Liu, Appl. Catal., B, 2013, 142, 736–743 CrossRef PubMed.
- T.-T. Chen, I.-C. Chang, M.-H. Yang, H.-T. Chiu and C.-Y. Lee, Appl. Catal., B, 2013, 142, 442–449 CrossRef PubMed.
- Q. Kang, J. Y. Cao, Y. J. Zhang, L. Q. Liu, H. Xu and J. H. Ye, J. Mater. Chem. A, 2013, 1, 5766–5774 CAS.
- Y. C. Liao, C. S. Xie, Y. Liu and Q. W. Huang, J. Alloys Compd., 2013, 550, 190–197 CrossRef CAS PubMed.
- X. M. Zhou, G. Liu, J. G. Yu and W. H. Fan, J. Mater. Chem., 2012, 22, 21337–21354 RSC.
- J. Zhou, F. Ren, S. F. Zhang, W. Wu, X. H. Xiao, Y. Liu and C. Z. Jiang, J. Mater. Chem. A, 2013, 1, 13128–13138 CAS.
- Y. Wen, B. T. Liu, W. Zeng and Y. H. Wang, Nanoscale, 2013, 5, 9739–9746 RSC.
- F. Wang, D. X. Zhao, Z. K. Xu, Z. K. Zheng, L. G. Zhang and D. Z. Shen, J. Mater. Chem. A, 2013, 1, 9132–9137 CAS.
- S. G. Meng, D. Z. Li, P. Wang, X. Z. Zheng, J. X. Wang, J. Chen, J. L. Fang and X. Z. Fu, RSC Adv., 2013, 3, 17021–17028 RSC.
- Q. Yang, M. Z. Li, J. Liu, W. Z. Shen, C. Q. Ye, X. D. Shi, L. Jiang and Y. L. Song, J. Mater. Chem. A, 2013, 1, 541–547 CAS.
- P. Liu, J. Nisar, B. Sa, B. Pathak and R. Ahuja, J. Phys. Chem. C, 2013, 117, 13845–13852 CAS.
- J. Xing, W. Q. Fang, H. J. Zhao and H. G. Yang, Chem.–Asian J., 2012, 7, 642–657 CrossRef CAS PubMed.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- J. G. Tao, T. Luttrell and M. Batzill, Nat. Chem., 2011, 3, 296–300 CrossRef CAS PubMed.
- M. Y. Guo, F. Z. Liu, Y. H. Leung, A. M. C. Ng, A. B. Djurišić and W. K. Chan, Curr. Appl. Phys., 2013, 13, 1280–1287 CrossRef PubMed.
- J. B. Joo, I. Lee, M. Dahl, G. D. Moon, F. Zaera and Y. D. Yin, Adv. Funct. Mater., 2013, 23, 4246–4254 CrossRef CAS.
- H. B. Wu, H. H. Hng and X. W. D. Lou, Adv. Mater., 2012, 24, 2567–2571 CrossRef CAS PubMed.
- D. K. Chacko, A. A. Madhavan, T. A. Arun, S. Thomas, G. S. Anjusree, T. G. Deepak, A. Balakrishnan, K. R. V. Subramanian, N. Sivakumar, S. V. Nair and A. S. Nair, RSC Adv., 2013, 3, 24858–24862 RSC.
- Y. Qu, W. Zhou, Z. Y. Ren, S. C. Du, X. Y. Meng, G. H. Tian, K. Pan, G. F. Wang and H. G. Fu, J. Mater. Chem., 2012, 22, 16471–16476 RSC.
- Z. Zhao, Z. C. Sun, H. F. Zhao, M. Zheng, P. Du, J. L. Zhao and H. Y. Fan, J. Mater. Chem., 2012, 22, 21965–21971 RSC.
- C. Z. Wen, H. B. Jiang, S. Z. Qiao, H. G. Yang and G. Q. M. Lu, J. Mater. Chem., 2011, 21, 7052–7061 RSC.
- J. Pan, G. Liu, G. Q. M. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- N. Murakami, Y. Kurihara, T. Tsubota and T. Ohno, J. Phys. Chem. C, 2009, 113, 3062–3069 CAS.
- W. Farneth, R. McLean, J. Bolt, E. Dokou and M. Barteau, Langmuir, 1999, 15, 8569–8573 CrossRef CAS.
- W. Xiang, L. Ren-Gui, X. Qian, H. Hong-Xian and L. Can, Acta Phys.-Chim. Sin., 2013, 29, 1566–1571 Search PubMed.
- N. Roy, Y. Sohn and D. Pradhan, ACS Nano, 2013, 7, 2532–2540 CrossRef CAS PubMed.
- J. H. Yang, J. Wang, X. Y. Li, J. H. Lang, F. Z. Liu, L. L. Yang, H. J. Zhai, M. Gao and X. T. Zhao, J. Alloys Compd., 2012, 528, 28–33 CrossRef CAS PubMed.
- A. Leelavathi, G. Madras and N. Ravishankar, Phys. Chem. Chem. Phys., 2013, 15, 10795–10802 RSC.
- A. A. Ismail and D. W. Bahnemann, J. Mater. Chem., 2011, 21, 11686–11707 RSC.
- Z. Wang, C. Y. Yang, T. Q. Lin, H. Yin, P. Chen, D. Y. Wan, F. F. Xu, F. Q. Huang, J. H. Lin, X. M. Xie and M. H. Jiang, Adv. Funct. Mater., 2013, 23, 5444–5450 CrossRef CAS.
- T. Sano, N. Mera, Y. Kanai, C. Nishimoto, S. Tsutsui, T. Hirakawa and N. Negishi, Appl. Catal., B, 2012, 128, 77–83 CrossRef CAS PubMed.
- X. X. Zou, J. K. Liu, J. Su, F. Zuo, J. S. Chen and P. Y. Feng, Chem.–Eur. J., 2013, 19, 2866–2873 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- Z. Wang, C. Y. Yang, T. Q. Lin, H. Yin, P. Chen, D. Y. Wan, F. F. Xu, F. Q. Huang, J. H. Lin, X. M. Xie and M. H. Jiang, Energy Environ. Sci., 2013, 6, 3007–3014 CAS.
- M. S. Hamdy, R. Amrollahi and G. Mul, ACS Catal., 2012, 2, 2641–2647 CrossRef CAS.
- F. Zuo, L. Wang, T. Wu, Z. Y. Zhang, D. Borchardt and P. Y. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- J. P. Wang, Z. Y. Wang, B. B. Huang, Y. D. Ma, Y. Y. Liu, X. Y. Qin, X. Y. Zhang and Y. Dai, ACS Appl. Mater. Interfaces, 2012, 4, 4024–4030 CAS.
- S. A. Ansari, M. M. Khan, S. Kalathil, A. Nisar, J. Lee and M. H. Cho, Nanoscale, 2013, 5, 9238–9246 RSC.
- X. Y. Pan, M.-Q. Yang, X. Z. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- C. H. Chen, J. Shieh, S. M. Hsieh, C. L. Kuo and H. Y. Liao, Acta Mater., 2012, 60, 6429–6439 CrossRef CAS PubMed.
- S. Horikoshi, Y. Minatodani, H. Tsutsumi, H. Uchida, M. Abe and N. Serpone, J. Photochem. Photobiol., A, 2013, 265, 20–28 CrossRef CAS PubMed.
- G. S. Anjusree, A. S. Nair, S. V. Nair and S. Vadukumpully, RSC Adv., 2013, 3, 12933–12938 RSC.
- J. S. Lee, K. H. You and C. B. Park, Adv. Mater., 2012, 24, 1084–1088 CrossRef CAS PubMed.
- J. D. Zhuang, Q. F. Tian, H. Zhou, Q. Liu, P. Liu and H. M. Zhong, J. Mater. Chem., 2012, 22, 7036–7042 RSC.
- J. T. Zhang, Z. G. Xiong and X. S. Zhao, J. Mater. Chem., 2011, 21, 3634–3640 RSC.
- P. Zhang, C. L. Shao, Z. Y. Zhang, M. Y. Zhang, J. B. Mu, Z. C. Guo, Y. Y. Sun and Y. C. Liu, J. Mater. Chem., 2011, 21, 17746–17753 RSC.
- X. Wang, Q. Xu, M. Li, S. Shen, X. Wang, Y. Wang, Z. Feng, J. Shi, H. Han and C. Li, Angew. Chem., Int. Ed., 2012, 51, 13089–13092 CrossRef CAS PubMed.
- T. Kawahara, Y. Konishi, H. Tada, N. Tohge, J. Nishii and S. Ito, Angew. Chem., Int. Ed., 2002, 41, 2811 CrossRef CAS.
- X. Wang, S. Shen, S. Jin, J. Yang, M. Li, X. Wang, H. Han and C. Li, Phys. Chem. Chem. Phys., 2013, 15, 19380–19386 RSC.
- H. Kim, P. Borse, W. Choi and J. Lee, Angew. Chem., Int. Ed., 2005, 44, 4585–4589 CrossRef CAS PubMed.
- A. Iwaszuk and M. Nolan, J. Mater. Chem. A, 2013, 1, 6670–6677 CAS.
- Q. L. Jin, H. Yamamoto, K. Yamamoto, M. Fujishima and H. Tada, Phys. Chem. Chem. Phys., 2013, 15, 20313–20319 RSC.
- Q. L. Jin, M. Fujishima and H. Tada, J. Phys. Chem. C, 2011, 115, 6478–6483 CAS.
- J. Mendez-Ramos, P. Acosta-Mora, J. C. Ruiz-Morales, T. Hernandez, M. E. Borges and P. Esparza, RSC Adv., 2013, 3, 23028–23034 RSC.
- A. B. Djurišić, X. Chen, Y. H. Leung and A. M. C. Ng, J. Mater. Chem., 2012, 22, 6526–6535 RSC.
- T. Z. Tong, C. T. T. Binh, J. J. Kelly, J.-F. Gaillard and K. A. Gray, Water Res., 2013, 47, 2352–2362 CrossRef CAS PubMed.
- G. Rammohan and M. N. Nadagouda, Curr. Org. Chem., 2013, 17, 2338–2348 CrossRef CAS.
- C. P. Li, J. Q. Wang, S. Q. Feng, Z. L. Yang and S. J. Ding, J. Mater. Chem. A, 2013, 1, 8045–8054 CAS.
- X. D. Wang, T. Dornom, M. Blackford and R. A. Caruso, J. Mater. Chem., 2012, 22, 11701–11710 RSC.
- L. H. Yu, Y. Huang, G. C. Xiao and D. Z. Li, J. Mater. Chem. A, 2013, 1, 9637–9640 CAS.
- Y. H. Lin, F. Zhang, D. C. Pan, H. X. Li and Y. F. Lu, J. Mater. Chem., 2012, 22, 8759–8763 RSC.
- F. Xu, Y. T. Shen, L. T. Sun, H. B. Zeng and Y. N. Lu, Nanoscale, 2011, 3, 5020–5025 RSC.
- J. Ming, Y. Q. Wu, S. Nagarajan, D.-J. Lee, Y.-K. Sun and F. Y. Zhao, J. Mater. Chem., 2012, 22, 22135–22141 RSC.
- J. H. Pan, Z. Y. Cai, Y. Yu and X. S. Zhao, J. Mater. Chem., 2011, 21, 11430–11438 RSC.
- G. Liu, C. H. Sun, L. Z. Wang, S. C. Smith, G. Q. M. Lu and H.-M. Cheng, J. Mater. Chem., 2011, 21, 14672–14679 RSC.
- M. Gohin, I. Maurin, T. Gacoin and J.-P. Boilot, J. Mater. Chem., 2010, 20, 8070–8077 RSC.
- S. G. Seo, C.-H. Park, H.-Y. Kim, W. H. Nam, M. Jeong, Y. n.-N. Choi, Y. S. Lim, W.-S. Seo, S.-J. Kim, J. Y. Lee and Y. S. Cho, J. Mater. Chem. A, 2013, 1, 3639–3644 CAS.
- S. Liu, E. Y. Guo and L. W. Yin, J. Mater. Chem., 2012, 22, 5031–5041 RSC.
- M. Villani, D. Calestani, L. Lazzarini, L. Zanotti, R. Mosca and A. Zappettini, J. Mater. Chem., 2012, 22, 5694–5699 RSC.
- Y. J. Jiang, L. Q. Meng, X. D. Mu, X. T. Li, H. S. Wang, X. F. Chen, X. C. Wang, W. Wang, F. Wu and X. Y. Wang, J. Mater. Chem., 2012, 22, 23642–23649 RSC.
- B. A. Lu, X. D. Li, T. H. Wang, E. Q. Xie and Z. Xu, J. Mater. Chem. A, 2013, 1, 3900–3906 CAS.
- M. Fujishima, H. Takatori and H. Tada, J. Colloid Interface Sci., 2011, 361, 628–631 CrossRef CAS PubMed.
- J. J. Lin, J. X. Shen, R. J. Wang, J. J. Cui, W. J. Zhou, P. G. Hu, D. O. Liu, H. Liu, J. Y. Wang, R. I. Boughton and Y. Z. Yue, J. Mater. Chem., 2011, 21, 5106–5113 RSC.
- W. J. Zhou, G. J. Du, P. G. Hu, G. H. Li, D. Z. Wang, H. Liu, J. Y. Wang, R. I. Boughton, D. Liu and H. D. Jiang, J. Mater. Chem., 2011, 21, 7937–7945 RSC.
- F.-X. Xiao, J. W. Miao, H.-Y. Wang and B. Liu, J. Mater. Chem. A, 2013, 1, 12229–12238 CAS.
- J. W. Lu, F. L. Su, Z. Q. Huang, C. X. Zhang, Y. Liu, X. B. Ma and J. L. Gong, RSC Adv., 2013, 3, 720–724 RSC.
- H. C. Ma, L. X. Yue, C. L. Yu, X. L. Dong, X. X. Zhang, M. Xue, X. F. Zhang and Y. H. Fu, J. Mater. Chem., 2012, 22, 23780–23788 RSC.
- D. J. Yang, J. Zhao, H. W. Liu, Z. F. Zheng, M. O. Adebajo, H. X. Wang, X. T. Liu, H. J. Zhang, J.-C. Zhao, J. Bell and H. Y. Zhu, Chem.–Eur. J., 2013, 19, 5113–5119 CrossRef CAS PubMed.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS PubMed.
- H. Q. Li, J. Qu, Q. Z. Cui, H. B. Xu, H. M. Luo, M. F. Chi, R. A. Meisner, W. Wang and S. Dai, J. Mater. Chem., 2011, 21, 9487–9490 RSC.
- S. T. Kochuveedu, Y. H. Jang, Y. J. Jang and D. H. Kim, J. Mater. Chem. A, 2013, 1, 898–905 CAS.
- G. Liu, J. Pan, L. C. Yin, J. T. S. Irvine, F. Li, J. Tan, P. Wormald and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 3233–3238 CrossRef CAS.
- K. E. deKrafft, C. Wang and W. Lin, Adv. Mater., 2012, 24, 2014–2018 CrossRef CAS PubMed.
- Q. Huang, F. Kang, H. Liu, Q. Li and X. D. Xiao, J. Mater. Chem. A, 2013, 1, 2418–2425 CAS.
- W. G. Tu, Y. Zhou, Q. Liu, Z. P. Tian, J. Gao, X. Y. Chen, H. T. Zhang, J. G. Liu and Z. G. Zou, Adv. Funct. Mater., 2012, 22, 1215–1221 CrossRef CAS.
- V. Singh, I. J. Castellanos Beltran, J. C. Ribot and P. Nagpal, Nano Lett., 2014, 14, 597–603 CrossRef CAS PubMed.
- S. Noimark, C. W. Dunnill and I. P. Parkin, Adv. Drug Delivery Rev., 2013, 65, 570–580 CrossRef CAS PubMed.
- P. Wu, J. A. Imlay and J. K. Shang, Biomaterials, 2010, 31, 7526–7533 CrossRef CAS PubMed.
- A. Lipovsky, A. Gedanken and R. Lubart, Photomed. Laser Surg., 2013, 31, 526–530 CrossRef CAS PubMed.
- R. J. Barnes, R. Molina, J. Xu, P. J. Dobson and I. P. Thompson, J. Nanopart. Res., 2013, 15, 1432 CrossRef.
- A. M. C. Ng, C. M. N. Chan, M. Y. Guo, Y. H. Leung, A. B. Djurišić, X. Hu, W. K. Chan, F. C. C. Leung and S. Y. Tong, Appl. Microbiol. Biotechnol., 2013, 97, 5565–5573 CrossRef CAS PubMed.
- L. C. Gerber, N. Moser, N. A. Luechinger, W. J. Stark and R. N. Grass, Chem. Commun., 2012, 48, 3869–3871 RSC.
- T. Z. Tong, A. Shereef, J. S. Wu, C. T. T. Binh, J. J. Kelly, J.-F. Gaillard and K. A. Gray, Environ. Sci. Technol., 2013, 47, 12486–12495 CrossRef CAS PubMed.
- Y. H. Leung, C. M. N. Chan, A. M. C. Ng, H. T. Chan, M. W. L. Chiang, A. B. Djurišić, Y. H. Ng, W. Y. Jim, M. Y. Guo, F. C. C. Leung, W. K. Chan and D. T. W. Au, Nanotechnology, 2012, 23, 475703 CrossRef CAS PubMed.
- W. Jiang, H. Mashayekhi and B. S. Xing, Environ. Pollut., 2009, 157, 1619–1625 CrossRef CAS PubMed.
- Y. H. Leung, A. M. C. Ng, X. Xu, Z. Shen, L. A. Gethings, M. T. Wong, C. M. N. Chan, M. Y. Guo, Y. H. Ng, A. B. Djurišić, P. K. H. Lee, W. K. Chan, L. H. Yu, D. L. Phillips, A. P. Y. Ma and F. C. C. Leung, Small, 2014, 10, 1171–1183 CrossRef CAS PubMed.
- N. C. Raut, T. Mathews, P. K. Ajikumar, R. P. George, S. Dash and A. K. Tyagi, RSC Adv., 2012, 2, 10639–10647 RSC.
- K. Nakata, T. Ochiai, T. Murakami and A. Fujishima, Electrochim. Acta, 2012, 84, 103–111 CrossRef CAS PubMed.
- P. J. Barker and A. Branch, Prog. Org. Coat., 2008, 62, 313–320 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |