Organic semiconductor core–shell nanoparticles designed through successive solvent displacements†
Sylvain
Chambon
*ab,
Christophe
Schatz
cd,
Vivien
Sébire
ab,
Bertrand
Pavageau
efg,
Guillaume
Wantz
ab and
Lionel
Hirsch
ab
aUniv. Bordeaux, IMS, UMR 5218, F-33400 Talence, France. E-mail: sylvain.chambon@ims-bordeaux.fr
bCNRS, IMS, UMR 5218, F-33400 Talence, France
cUniv. Bordeaux, LCPO, UMR 5629, F-33600 Pessac, France
dCNRS, LCPO, UMR 5629, F-33600 Pessac, France
eUniv. Bordeaux, LOF, UMR 5258, F-33600 Pessac, France
fCNRS, LOF, UMR 5258, F-33600 Pessac, France
gSolvay-RHODIA, LOF, UMR 5258, F-33600 Pessac, France
First published on 24th February 2014
Abstract
This study reports for the first time the elaboration of core–shell organic nanoparticles through successive solvent displacements. The concept has been applied to generate donor–acceptor nanoparticles with the commonly used organic semiconductors P3HT and PCBM. The strategy is based on the sequential nanoprecipitation of P3HT and PCBM. Starting from a P3HT:PCBM solution in THF, the first solvent displacement with DMSO triggers the formation of the P3HT core while the second displacement with water generates the PCBM shell. The core–shell morphology is evidenced by both DLS and TEM. Efficient quenching of the P3HT photoluminescence is observed after the second solvent displacement procedure, confirming the formation of a well-defined PCBM shell around the P3HT core. Studying the emission of such core–shell nanoparticles leads to an estimation of the diffusion length of photogenerated excitons in P3HT of around ∼14 nm.
Nanoparticles based on organic semiconductors, conjugated polymers and small molecules, have been the subject of several studies in the past decade. Their use in organic electronics was motivated by the need to control the morphology of the active layer in bulk heterojunction organic solar cells and the desire to develop an environmentally friendly water-based process. Kietzke et al. developed the first such nanoparticles using the mini-emulsion technique1–3 and showed that they could be used to control the nano-phase morphology. Since then, the performances of photovoltaic devices with active layers based on nanoparticles have improved consistently4–7 to reach a record power conversion efficiency of 2.5% with poly(3-hexylthiophene) (P3HT):indene-C60-bisadduct (ICBA)8 nano-particle organic photovoltaic (NPOPV) devices. Charge transport in such assemblies of semi-conducting P3HT nanoparticles has also been deeply studied and mobilities comparable to drop-cast P3HT films have been demonstrated.9 These nano-objects have also been used as models to study the P3HT:[6,6]-phenyl-C61-butyric acid methyl ester (PCBM) morphology,10,11 the aggregation of P3HT,12,13 and photoluminescence dynamics.14 However, the ability to control the morphology of nano-particles (NPs) made from a binary blend has shown to be highly process dependent. For instance Kietzke et al. claim the formation of Janus like nanoobjects14 while Dastoor et al. showed evidence of a core–shell like morphology,4,5 both using the mini-emulsion approach. The fabrication of nanoparticles with well-defined morphologies is therefore not only of interest as a model for photo-physicists but also for device fabrication.
In this paper, we propose a novel methodology to fabricate core–shell nano-particles based on π-conjugated polymers and fullerene derivatives, with a P3HT core and a PCBM shell. An easy way to create nano-objects in solution is to use the solvent displacement method (also known as nanoprecipitation) which utilises the Ouzo effect.15 This technique has been widely applied to the manufacture of sub-micrometre particles of dyes, drugs and polymers. The process is simple: a hydrophobic material is first dissolved at a low concentration in a water-miscible solvent (the “good” solvent) and then a large amount of water (the “bad” solvent) is quickly added to the solution to induce the fast precipitation of the material at the nanometer scale.16 In the field of organic electronics, several groups have already worked with this technique, or equivalent processes. Yamamoto et al. did an extensive study on the π–π stacking in P3HT nano-colloids after solvent displacement from chloroform to methanol.17 Shimizu et al. and Setti et al. worked on P3HT-based NPs dispersed in aqueous media with and without surfactants.18,19 Gesquiere et al. have studied the formation of hybrid P3HT:PCBM colloids using tetrahydrofuran (THF) and water and have performed extensive spectroscopic and morphological analysis.10,11 Few electronic devices have been fabricated using surfactant-free colloidal solutions prepared via nanoprecipitation. Fréchet et al. synthesised and characterised P3HT-based nanoparticles using the solvent displacement method, displacing from chloroform to ethanol.20 In their study, Fréchet et al. tuned the sizes of the NPs in the range of 30 to 100 nm, simply by varying the initial polymer concentration. Organic field-effect transistors (OFETs) were fabricated with these P3HT-based NPs and the measured hole mobilities were similar to those obtained with thin P3HT films cast from chloroform. Using a similar method, Darwis et al. prepared surfactant-free P3HT:PCBM colloidal solutions which were used to fabricate organic solar cells. A power conversion efficiency of 1.09% was obtained with such a device.21
To the best of our knowledge, the fabrication of organic semiconductor-based donor–acceptor NPs featuring core–shell morphology via nanoprecipitation has not yet been reported. The present paper aims at describing the formation of such NPs using two successive solvent displacements. In the context of the bulk heterojunction,22 the sizes of the donor and acceptor domains have to match the exciton diffusion length in order to optimise the charge carrier generation. Several groups are working on the evaluation of these distances,23–25 knowledge of which is of utmost importance in the determination of the optimum domain size for efficient exciton dissociation and charge carrier generation. In this sense, the fabrication of donor–acceptor core–shell NPs may be of interest in furthering the understanding of the photo-physical processes that take place in an organic solar cell as they can be considered as building blocks of the active layer.
The process of core–shell NP fabrication is composed of two successive solvent displacements as can be seen in Scheme 1.
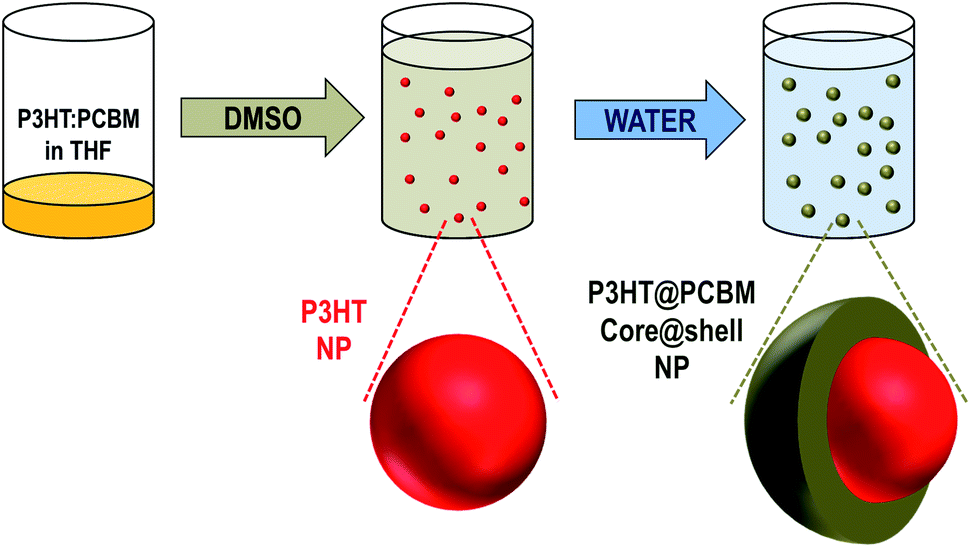 | ||
| Scheme 1 Fabrication of P3HT:PCBM core–shell NPs by successive solvent displacements. | ||
Starting from a solution, of P3HT and PCBM in THF, the first solvent displacement using dimethylsulfoxide (DMSO) as a non-solvent leads to the formation of P3HT NPs while PCBM remains dissolved in the medium. Then, water, which is a non-solvent of both materials (i.e. P3HT and PCBM), is used for the second solvent displacement. The strategy is to induce the formation of a PCBM-based shell around the P3HT-based NPs, the latter acting as nucleation centres for the PCBM. While this concept has already been used to wrap carbon-nanotubes (CNTs) with PMMA,26 the novelty here is that both components of the NPs, the core and the shell, are generated through a single process.
Generation of single component nanoparticles: THF was chosen as a solvent for both materials due to its miscibility with water and organic solvents. DMSO was used as a non-solvent for the first solvent displacement since P3HT is insoluble in DMSO while PCBM is slightly soluble in DMSO (Cmax = 0.2 mg mL−1).27 Even though this limit of solubility is low, it is sufficient for the purpose of our study. P3HT solutions in THF (CP3HT = 0.5 mg mL−1) have a single absorption band with a maximum located at 445 nm (Fig. 1A). This band is characteristic of the absorption of an isolated conjugated polymer chain in solution.17 The solvent displacement is applied using DMSO as a non-solvent via the procedure described in the Experimental section. New features appear in the UV-visible spectra of the dispersion, indicating the formation of P3HT aggregates in solution (Fig. 1A). The UV-visible absorption spectrum is no longer that of a single chain but corresponds to that of well organised P3HT, similar to that of thin films or nano-fibers with red-shifted vibronic peaks at 510, 550 and 610 nm assigned to A0–2, A0–1 and A0–0 peaks respectively. These bands are characteristic of well stacked P3HT chains13,17 and are often assigned to P3HT crystalline domains.28,29 It is interesting to note that the fast process of nanoprecipitation can lead to P3HT nanoparticles with organised domains. The hydrodynamic diameter of the colloidal particles was determined using dynamic light scattering (DLS) and was found to be 154 ± 57 nm (see Fig. S1 in ESI†).
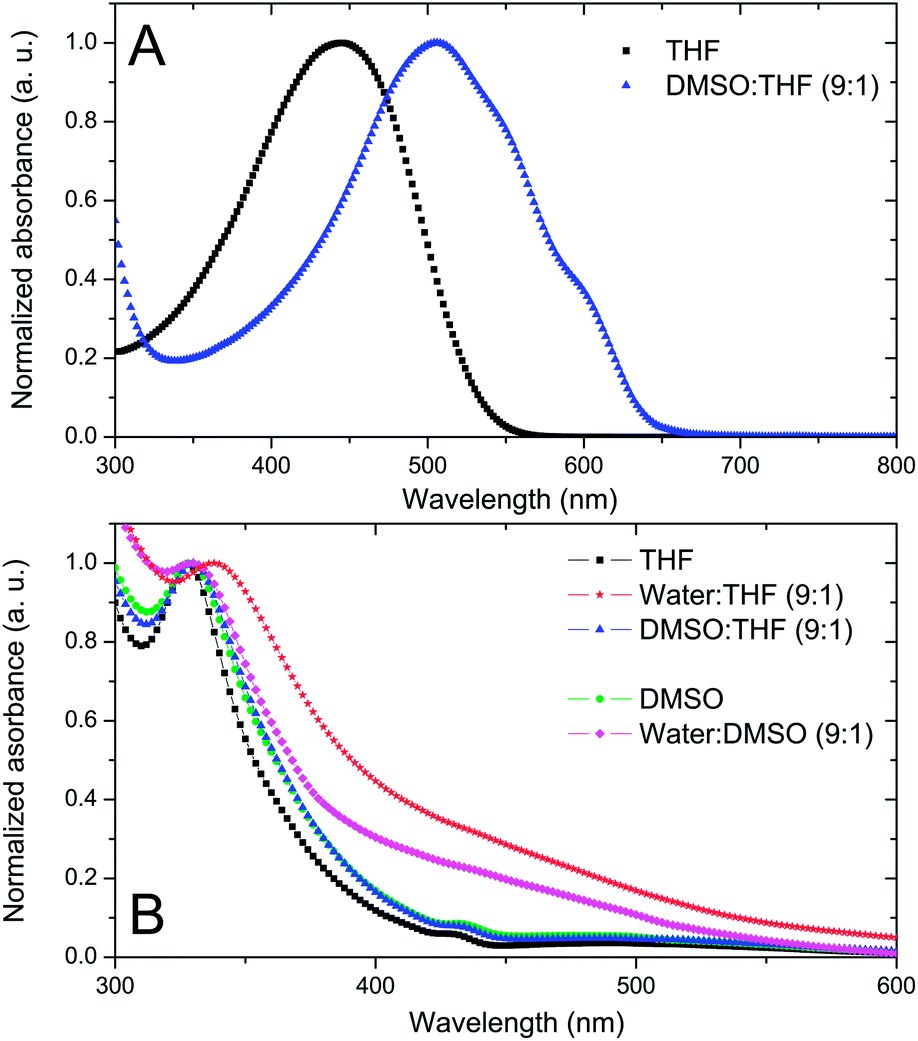 | ||
Fig. 1 (A) Normalised UV-visible spectra of P3HT dissolved in THF (C = 0.5 mg mL−1, black squares) and THF–DMSO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) dispersions of P3HT NPs (C = 0.05 mg mL−1, blue triangle). (B) Normalised UV-visible spectra of PCBM dissolved in THF (C = 0.05 mg mL−1; black squares), after solvent displacement with water (C = 0.05 mg mL−1; red stars) or DMSO (C = 0.05 mg mL−1; blue triangles) and PCBM dissolved in DMSO (C = 0.02 mg mL−1; green circles) and after solvent displacement with water (C = 0.02 mg mL−1; pink lozenge). :9) dispersions of P3HT NPs (C = 0.05 mg mL−1, blue triangle). (B) Normalised UV-visible spectra of PCBM dissolved in THF (C = 0.05 mg mL−1; black squares), after solvent displacement with water (C = 0.05 mg mL−1; red stars) or DMSO (C = 0.05 mg mL−1; blue triangles) and PCBM dissolved in DMSO (C = 0.02 mg mL−1; green circles) and after solvent displacement with water (C = 0.02 mg mL−1; pink lozenge). | ||
PCBM dissolved in THF has a UV band centred at 328 nm (Fig. 1B). When water is used for the solvent displacement, the PCBM UV band is red-shifted at 338 nm and a broad band appears between 400 and 500 nm. In C60, a similar band specific to C60 aggregates has been attributed to intermolecular charge transfer (CT) states.30,31 In accordance with the lack of solubility of PCBM in water, this kind of solvent displacement leads to the generation of nanoparticles detected by DLS with a size of 182 ± 35 nm (Fig. S2 in ESI†). On the other hand, when DMSO is used as a non-solvent, no clear difference is observed in the UV-visible spectrum of PCBM in THF before and after the addition of DMSO (Fig. 1B). Additionally, DLS measurements did not evidence the formation of any particles in THF–DMSO mixtures, indicating that at a low concentration (CPCBM = 0.05 mg mL−1) the PCBM is still soluble in the mixture. Fig. 1B shows also the absorption spectrum of PCBM in DMSO (CPCBM = 0.02 mg mL−1). At this concentration PCBM is still soluble in this solvent27 and, as expected, the general shape is very similar to that of PCBM dissolved in THF with a UV band centred at 328 nm and no spectral shoulder between 400 and 500 nm. When water is used for solvent displacement from the DMSO solution, new features appear in the UV-visible spectrum as shown in Fig. 1B. The PCBM UV band is slightly red-shifted and, more importantly, the large absorption between 400 and 500 nm indicates that aggregation of the PCBM has occurred. DLS data confirm the presence of PCBM-based NPs in the DMSO–water mixture (1:9 v/v) with a main population with hydrodynamic diameters of 49 ± 9 nm (Fig. S3 in ESI†).
Based on these results, it is expected that the successive solvent displacement, first with DMSO, and then with water, should account for the selective precipitation of P3HT and PCBM into core–shell NPs.
Generation of core–shell nanoparticles: in order to study the formation of core–shell NPs, three P3HT:PCBM solutions in THF varying in composition and concentrations were prepared (Blend 1, Blend 2 and Blend 3). A control experiment with only P3HT was also prepared (P3HT). For each condition (Blend 1, Blend 2, Blend 3 and P3HT), three kinds of dispersion were prepared following the solvent displacement procedure described in the Experimental section:
• xx-D. 1st solvent displacement with DMSO from THF solutions (9:1 v/v ratio).
• xx-D/W. 2nd solvent displacement with deionized water from xx-D dispersion (9:1 v/v ratio).
• xx-D/D. Addition of DMSO in the xx-D under the same conditions as water in order to conserve the same experimental protocol, i.e. heating, stirring and dilution (9:1 v/v ratio). Control experiment.
Where “xx” corresponds to the initial solution used to perform the solvent displacement procedure, i.e. “Blend 1”, “Blend 2”, “Blend 3” for P3HT:PCBM solutions and “P3HT” for P3HT solution. All these conditions are summarized in Table 1.
| Initial solution | P3HT:PCBM ratio | C P3HT (mg mL−1) | Solv. displ. procedure | Label | Hydro. diameter (nm) | sd (nm) |
|---|---|---|---|---|---|---|
| Blend 1 | 2:1 |
0.67 | DMSO–DMSO | Blend 1-D/D | 83 | 36 |
| DMSO–water | Blend 1-D/W | 82 | 14 | |||
| Blend 2 | 2:1 |
0.5 | DMSO–DMSO | Blend 2-D/D | 68 | 17 |
| DMSO–water | Blend 2-D/W | 77 | 17 | |||
| Blend 3 | 1:2 |
0.33 | DMSO–DMSO | Blend 3-D/D | 70 | 23 |
| DMSO–water | Blend 3-D/W | 85 | 21 | |||
| P3HT | 1:0 |
0.5 | DMSO–DMSO | P3HT-D/D | 132 | 47 |
| DMSO–water | P3HT-D/W | 96 | 16 | |||
The eight xx-D/W and xx-D/D dispersions were first analysed by UV-visible absorption spectroscopy (Fig. 2). After the first solvent displacement with DMSO, the shapes of the UV-visible spectra in the P3HT region appear to be very similar in all dispersions. P3HT vibronic bands at 510, 550 and 610 nm were detected in all the P3HT:PCBM dispersions (Blend 1-D/D, Blend 2-D/D, Blend 3-D/D) as well as in the P3HT dispersion (P3HT-D/D). This indicates that the nanoparticles generated during the first solvent displacement with DMSO contain well-organised P3HT domains whether they are generated from a P3HT:PCBM blend solution (Blend 1, Blend 2, Blend 3) or a pure P3HT solution (P3HT).
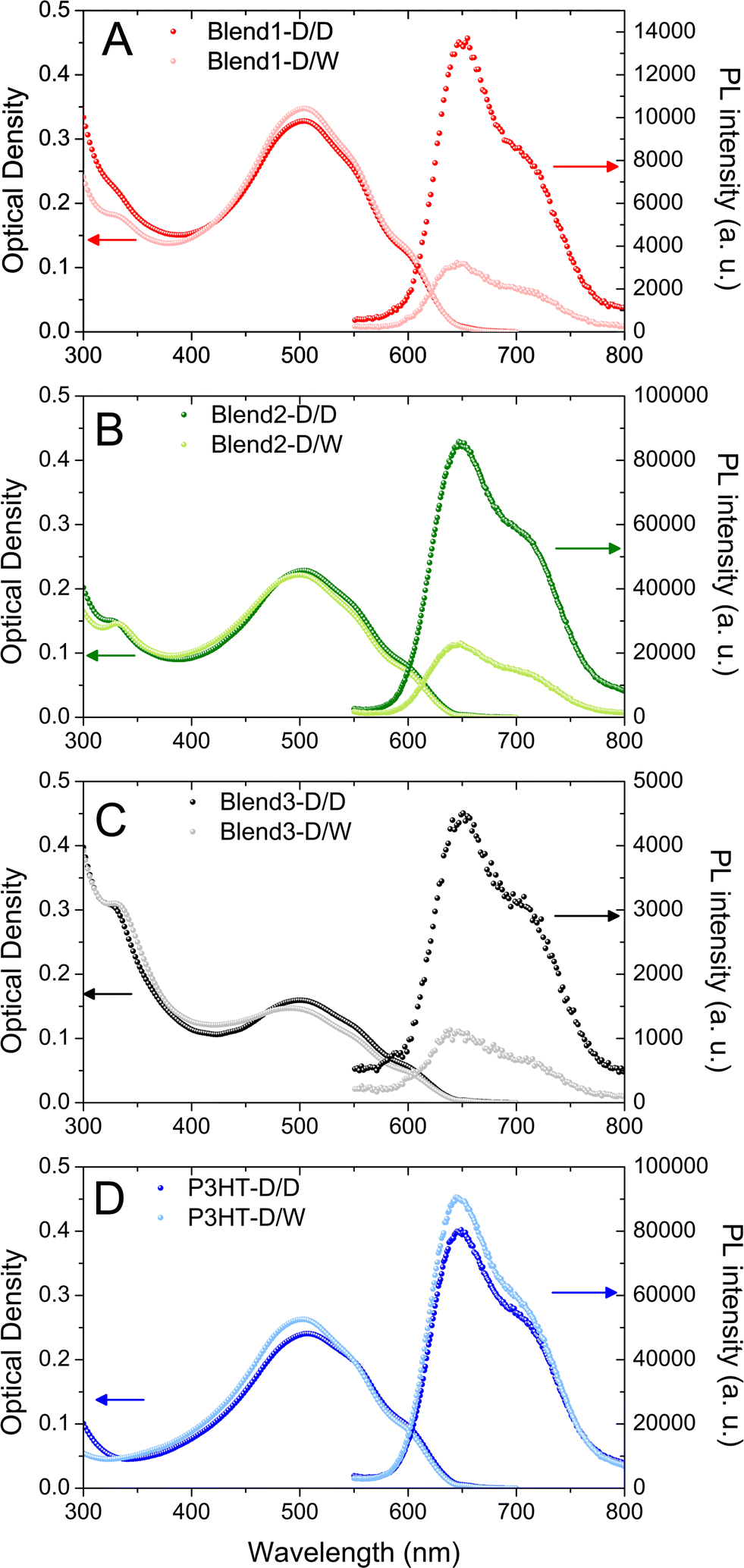 | ||
| Fig. 2 UV-visible absorbance and photoluminescence spectra of P3HT:PCBM (Blend 1 – red symbols, Blend 2 – green symbols and Blend 3 – black symbols) or P3HT (P3HT – blue symbols) dispersions after one solvent displacement with DMSO (dark colour) or two successive solvent displacements with DMSO–water (light colour). | ||
After the second solvent displacement with water, no significant change in the P3HT range is observed, indicating that the organisation of P3HT in the nanoparticles does not change upon addition of water. However, in the PCBM absorption region, the UV band centred at 328 nm is slightly red-shifted towards 338 nm for the Blend 1-D/W, Blend 2-D/W and Blend 3-D/W dispersions compared to their respective control experiments (Blend 1-D/D, Blend 2-D/D and Blend 3-D/D). As shown in Fig. 1, PCBM in its aggregated form presents slightly red-shifted UV-bands compared to the feature of dissolved PCBM. These data suggest that PCBM has aggregated after the second solvent displacement procedure with water and not with DMSO as expected from its respective solubility in these solvents. It is also an indication that, during the first solvent displacement with DMSO, no PCBM was trapped in the P3HT NPs and that it is still dissolved in the solvent.
The dispersions were also analysed by DLS which showed evidence of mono-modal size distributions of nanoparticles, no matter what solvent displacement procedure is used (Fig. 3). The P3HT-based NPs generated from the pure P3HT solution present the mean average hydrodynamic diameters of 132 and 96 nm for P3HT-D/D and P3HT-D/W respectively (Table 1 and Fig. 3D). The decrease of the mean diameter after the second solvent displacement in water (−36 nm) may be interpreted as the consequence of a better stabilization of the solid/liquid interface in a polar solvent like water.
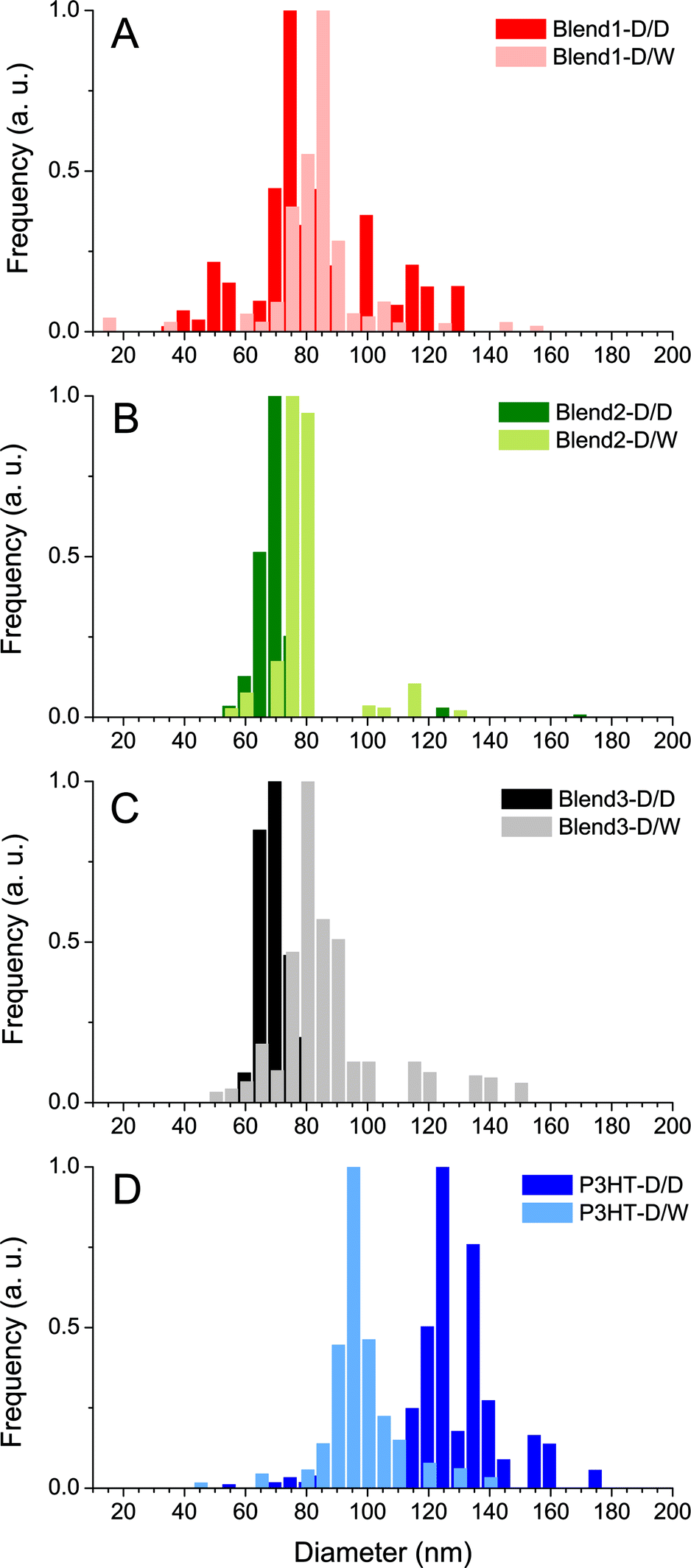 | ||
| Fig. 3 Intensity weighted size dispersion of the nanoparticles in Blend 1 (A), Blend 2 (B), Blend 3 (C) and P3HT (D) dispersions generated after one solvent displacement with DMSO (D/D) or two successive solvent displacements (D/W). | ||
For the P3HT:PCBM blends (Blend 1, Blend 2 and Blend 3), it was observed that the NPs generated after solvent displacement with DMSO are much smaller than those formed from the pure P3HT solution under same conditions, ∼75 nm and ∼130 nm respectively. The presence of diluted PCBM in the DMSO solution seems to change the nucleation/growth mechanisms involved in the nanoprecipitation phenomenon and may account for the difference in the diameters of P3HT NPs generated after solvent displacement with DMSO. After the second solvent displacement with water, the size distribution analysis evidenced the absence of new populations of NPs, which would have been the case if PCBM could nucleate separately. In contrast, the size distributions of the dispersions based on Blend 1-D/W, Blend 2-D/W and Blend 3-D/W shift to larger diameters compared to the control experiment with Blend 1-D/D, Blend 2-D/D and Blend 3-D/D (Fig. 3). The increase in the size in the 10–15 nm range is consistent with the formation of a PCBM shell around core NPs of P3HT rather than isolated PCBM nanoparticles. In contrast, PCBM forms small NPs of 49 nm in diameter under the same conditions of solvent displacement. Another indirect technique to observe the formation of the PCBM shell around P3HT nanoparticles is to perform photoluminescence (PL) experiments. Indeed, PCBM quenches the P3HT fluorescence efficiently due to exciton dissociation at the interface and the ultrafast electron transfer from the LUMO of the P3HT to that of the PCBM.32,33Fig. 2 shows the photoluminescence under excitation at 510 nm of the four dispersions based on only one solvent displacement with DMSO (Blend 1-D/D, Blend 2-D/D, Blend 3-D/D and P3HT-D/D) as well as that of the dispersions based on the successive solvent displacement (Blend 1-D/W, Blend 2-D/W, Blend 3-D/W and P3HT-D/W). The PL of P3HT-based nanoparticles generated from the pure P3HT solution (P3HT-D/D and P3HT-D/W) presents a similar PL intensity and shape. The solvent used (DMSO or water) does not seem to drastically change the PL response and the small difference in the PL intensity might come from the difference in the P3HT nanoparticle sizes, 132 and 96 nm for P3HT-D/D and P3HT-D/W respectively. Blend 1-D/W, Blend 2-D/W and Blend 3-D/W dispersions prepared after the addition of water present a weaker emission with respect to their respective control experiments, i.e. Blend 1-D/D, Blend 2-D/D and Blend 3-D/D. This difference cannot be attributed to some changes in the absorbance as for a given P3HT:PCBM ratio (2:1 or 1:2) and P3HT concentration (CP3HT = 3.3, 5 or 6.7 μg mL−1) the two resulting dispersions D/W and D/D present similar optical densities (see Fig. 2) and any possible solvent effect has already been ruled out. In the three cases, about 75% of the photoluminescence is quenched, indicating the proximity of PCBM and P3HT. The remaining fluorescence either comes from excitons which cannot reach a P3HT/PCBM interface to undergo the electronic transfer and which recombine radiatively, or from naked P3HT NPs.
Focusing on Blend 2-D/D, Blend 2-D/W, P3HT-D/D and P3HT-D/W gives some further insights into the NP morphology since all the dispersions were prepared from solutions with the same initial P3HT concentration (CP3HT = 0.5 mg mL−1). As a result, the optical densities of the four dispersions at 510 nm are almost identical (Fig. 2B and D) and, as the photoluminescence was performed under the same conditions, the PL intensities of such dispersions are directly comparable. The integrated PL intensity of the nanoparticles generated after the first solvent displacement procedure with DMSO is similar whether the initial solution is pure P3HT (P3HT-D/D) or a P3HT:PCBM mixture (Blend 2-D/D) of 9.1 × 106 and 9.7 × 106 arb. units respectively (see Fig. S4 in ESI† for direct comparison of the different PL spectra). Since no fluorescence quenching occurs in Blend 2-D/D, one can conclude that no PCBM was trapped in the P3HT NPs formed from the P3HT:PCBM solution (Blend 2) after the first solvent displacement with DMSO.
On the other hand, after the second solvent displacement, the integrated PL intensities of the dispersions prepared from the pure P3HT solution (P3HT-D/W) and from the P3HT:PCBM mixture (Blend 2-D/W) are very different, 10 × 106 and 2.5 × 106 arb. units respectively. Such a decrease in the PL intensity (−75%) unambiguously supports the formation of a PCBM coat layer around P3HT NPs generated in Blend 2-D/W. All these PL results indicate that PCBM has aggregated around P3HT-based nuclei after the second solvent displacement procedure. Furthermore, the fact that the decrease in the PL intensity is the same whatever the P3HT:PCBM ratio (1:2 and 2:1) shows that the PCBM concentration was high enough in the medium to fully coat P3HT-based NPs with a shell of PCBM.
TEM images of Blend 2-D/W dispersions after the 2nd solvent displacement procedure are shown in Fig. 4. At high magnification a layer surrounding the nano-object is clearly observed, confirming the core–shell morphology. According to the spectroscopic analysis and the respective solubility of PCBM in different solvents, the shell is composed of PCBM. The dimensions of the core and the shell of NPs are in accordance with the DLS results.
 | ||
| Fig. 4 High contrast (HC) TEM image of PCBM/P3HT NPs obtained from Blend 2-D/W with 200000× magnification. Inset: high resolution (HR) TEM images of PCBM/P3HT NPs obtained from Blend 2-D/W with 600000× magnification. | ||
Exciton diffusion length estimation: these nano-objects can be used as a model to estimate the exciton diffusion length in P3HT:PCBM blends. Since the photoluminescence is proportional to the volume of the emissive P3HT core, one can calculate the emissive P3HT volume after the PCBM shell formation, and then deduce the thickness of the quenched P3HT layer, which corresponds to the exciton diffusion length.
We will consider here the six dispersions prepared from P3HT:PCBM blends described in Table 1 (Blend 1, Blend 2, Blend 3). The three dispersions prepared with DMSO only led to the formation of pure P3HT NPs. P3HT nanoparticles in Blend 1-D/D, Blend 2-D/D and Blend 3-D/D have average radii of 42, 34 and 35 nm for integrated photoluminescence intensities of 1.38 × 106, 9.7 × 106 and 5.16 × 105 arb. units respectively. Their P3HT:PCBM core–shell counterparts, Blend 1-D/W, Blend 2-D/W and Blend 3-D/W, while presenting the same core size, show much lower integrated photoluminescence intensities, 3.4 × 105, 2.5 × 106 and 1.25 × 105 arb. units respectively, meaning that only the inner P3HT core, whose excitons cannot reach the P3HT:PCBM interface, emits light. The diameter of the inner fluorescent core is calculated assuming spherical particles and considering that the photoluminescence is proportional to the volume of emissive P3HT. For Blend 1-D/W, Blend 2-D/W and Blend 3-D/W, emissive radii of 26, 21 and 22 nm respectively were found for total core radii of 42, 34 and 35 nm. For the three varieties of P3HT:PCBM core–shell nanoparticles, the exciton diffusion lengths are then estimated at 16 ± 7, 13 ± 4 and 13 ± 5 nm for Blend 1-D/W, Blend 2-D/W and Blend 3-D/W respectively. Table 2 summarizes these results.
| Core radius (nm) | PL core (a.u.) | PL core–shell (a.u.) | Emissive radius (nm) | Exciton diffusion length (nm) | |
|---|---|---|---|---|---|
| Blend 1 | 42 ± 18 | 1.38 × 106 | 3.4 × 105 | 26 | 16 ± 7 |
| Blend 2 | 34 ± 9 | 9.7 × 106 | 2.5 × 106 | 21 | 13 ± 4 |
| Blend 3 | 35 ± 12 | 5.16 × 105 | 1.25 × 105 | 22 | 13 ± 5 |
Steady-state photoluminescence measurements enabled an estimation of the exciton diffusion length of around ∼14 nm. The discrepancy comes from the dispersion of particle sizes calculated from the statistical size distribution (Fig. 3). This value has to be compared with those determined with other techniques such as exciton–exciton annihilation (about 8 nm)25 or the exciton quenching layer (6.5–8.5 nm).34 Studies are on-going to (1) improve the process of core–shell NP fabrication in order to have a better control of the object size and (2) define an appropriate model for estimating the exciton diffusion length taking into account the side effects. Nevertheless, this study reports for the first time the elaboration of core–shell nanoparticles through successive solvent displacements. Such NPs are of interest as models for photophysical studies.
Conclusions
In this communication, we have presented a new method to generate P3HT:PCBM core–shell nano-particles based on the solvent displacement technique to induce the sequential nano-precipitation of P3HT and PCBM. By a careful choice of the appropriate non-solvent, it is possible to generate sequentially the P3HT core of the NP (first solvent displacement with DMSO) and then, the PCBM shell (second solvent displacement with water). The core–shell morphology was demonstrated and characterized both by DLS and TEM analysis. The presence of PCBM around the P3HT nanoparticles was further confirmed by photoluminescence experiments. Indeed, an important quenching of the P3HT photoluminescence was observed after the 2nd solvent displacement procedure, confirming the formation of a well-defined PCBM/P3HT interface. Steady-state photoluminescence measurements enabled an estimation of the exciton diffusion length around ∼14 nm. Studies to improve both the nanoparticle formation and the model for estimating the exciton diffusion length are in progress. Besides, we believe that the use of a successive solvent displacement procedure to generate core–shell NPs is a promising concept in polymer and colloid science. Especially, the ease and versatility of the process in addition to its potential applicability to a large range of polymers or organic compounds make it attractive for encapsulation purposes.Experimental section
Materials
Regio-regular poly(3-hexylthiophene) (P3HT) was provided by Solaris Chem Inc. Mw was determined at 40 kg mol−1 (GPC vs. polystyrene) with a PDI 1.7. Regio-regularity was determined by H-NMR at 95%. 1-(3-Methoxycarbonyl)propyl-1-phenyl[6,6]C61-(PCBM) was provided by Solaris Chem Inc. (purity: 99.5%).Solvent displacement procedure
P3HT or P3HT:PCBM was first dissolved in THF at concentrations ranging from 0.33 to 0.67 mg mL−1 and stirred for 24 h at 50 °C. 0.5 mL of the P3HT/P3HT:PCBM solution is transferred to a 6 mL vial and stirred at 50 °C at 250 rpm for 5 min and then 4.5 mL of a non-solvent (DMSO or water) are rapidly injected with a 5 mL micropipette. For the successive solvent displacement, 0.5 mL of the first dispersion generated with DMSO is transferred to a 6 mL vial, heated at 50 °C and stirred at 250 rpm for 5 min. Then 4.5 mL of deionized water (or DMSO for control experiments) are rapidly injected with a 5 mL micropipette.UV-visible spectroscopy
The dispersions were analysed with a UV-visible spectrophotometer (SAFAS UV mc2) in a double beam mode. Quartz cuvettes with 1 cm optical path were used. Spectra were recorded from 200 to 800 nm in steps of 2 nm and using an integration time of 1 s.Photoluminescence spectroscopy
Steady-state photoluminescence experiments were performed on a PTI (Photon Technology International) Quantmaster 40 setup with a Xenon arc lamp coupled with a monochromator for excitation and a photomultiplier detection system (model 810/814). The excitation wavelength was set at 510 nm and emission spectra were recorded at 90° from 525 nm to 800 nm by step of 1 nm and using an integration time of 1 s. The excitation and emission slits were all set at 2 mm. 4-sided quartz cuvettes with 1 cm optical path were used for these PL experiments.Size measurements
Hydrodynamic diameters of the nanoparticles were determined by dynamic light scattering using a Cordouan Particle Size Analyzer VASCO. The temperature of the dispersion was set at 25 °C. The refractive index and viscosity values for water–DMSO mixtures were taken from the literature.35 For THF–DMSO mixtures, the refractive indices were calculated using the following equation:in which n1, n2 and n12 are the refractive indices of solvent 1, solvent 2 and the mixture respectively and ϕ1 and ϕ2 are their volume fractions.36
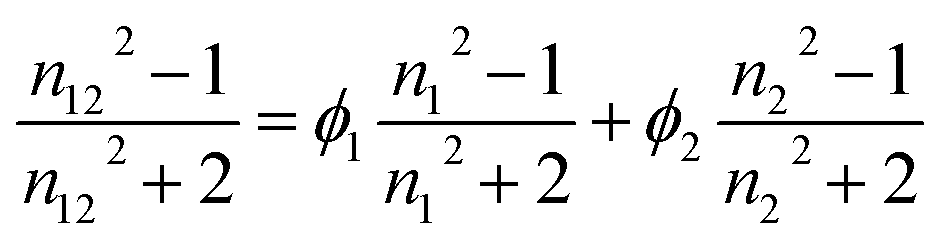
Viscosities of THF–DMSO mixtures were calculated according to the Arrhenius equation:
| logη12 = x1logη1 + x2logη2 |
Transmission electron microscopy
15 μl of the dispersions were deposited on carbon coated 200 mesh copper grids (positively charged, 20 mA, 2 min) and dried at room temperature. The grids were analyzed with a transmission electron microscope operated at 80 kV (H7650, HITACHI, Japan) in High Contrast (HC) and High Resolution (HR) mode.Acknowledgements
This work has been supported by Université Bordeaux 1, Idex and CNRS through PEPS “NPcoreshell” no. 130409.A80 project. The microscopy was done in the Bordeaux Imaging Center (UMS 3420 CNRS – Bordeaux University/US4 INSERM). The help from Sabrina Lacomme and Etienne Gontier is acknowledged. Finally, authors gratefully thank William Greenbank for his help in improving the language quality.References
- T. Kietzke, D. Neher, M. Kumke, R. Montenegro, K. Landfester and U. Scherf, Macromolecules, 2004, 37, 4882 CrossRef CAS
.
- T. Kietzke, D. Neher, K. Landfester, R. Montenegro, R. Guntner and U. Scherf, Nat. Mater., 2003, 2, 408 CrossRef CAS PubMed
- K. Landfester, R. Montenegro, U. Scherf, R. Güntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Adv. Mater., 2002, 14, 651 CrossRef CAS
- A. Stapleton, B. Vaughan, B. Xue, E. Sesa, K. Burke, X. Zhou, G. Bryant, O. Werzer, A. Nelson, A. L. David Kilcoyne, L. Thomsen, E. Wanless, W. Belcher and P. Dastoor, Sol. Energy Mater. Sol. Cells, 2012, 102, 114 CrossRef CAS PubMed
- S. Ulum, N. Holmes, D. Darwis, K. Burke, A. L. David Kilcoyne, X. Zhou, W. Belcher and P. Dastoor, Sol. Energy Mater. Sol. Cells, 2013, 110, 43 CrossRef CAS PubMed
- T. R. Andersen, T. T. Larsen-Olsen, B. Andreasen, A. P. Bottiger, J. E. Carle, M. Helgesen, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jorgensen and F. C. Krebs, ACS Nano, 2011, 5, 4188 CrossRef CAS PubMed
- T. T. Larsen-Olsen, B. Andreasen, T. R. Andersen, A. P. L. Bottiger, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jorgensen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2012, 97, 22 CrossRef CAS PubMed
- S. Ulum, N. Holmes, M. Barr, A. L. D. Kilcoyne, B. B. Gong, X. Zhou, W. Belcher and P. Dastoor, Nano Energy, 2013, 2, 897 CrossRef CAS PubMed
- M. Bag, T. S. Gehan, D. D. Algaier, F. Liu, G. Nagarjuna, P. Lahti, T. Russel and D. venkataraman, Adv. Mater., 2013, 25, 6411 CrossRef CAS PubMed
- Z. Hu and A. J. Gesquiere, Chem. Phys. Lett., 2009, 476, 51 CrossRef CAS PubMed
- Z. Hu, D. Tenery, M. S. Bonner and A. J. Gesquiere, J. Lumin., 2010, 130, 771 CrossRef CAS PubMed
- J. A. Labastide, M. Baghgar, I. Dujovne, B. H. Venkatraman, D. C. Ramsdell, D. Venkataraman and M. D. Barnes, J. Phys. Chem. Lett., 2011, 2, 2089 CrossRef CAS
- G. Nagarjuna, M. Baghgar, J. A. Labastide, D. D. Algaier, M. D. Barnes and D. Venkataraman, ACS Nano, 2012, 6, 10750 CAS
- T. Kietzke, D. Neher, M. Kumke, O. Ghazy, U. Ziener and K. Landfester, Small, 2007, 3, 1041 CrossRef CAS PubMed
- J. Aubry, F. Ganachaud, J.-P. Cohen Addad and B. Cabane, Langmuir, 2009, 25, 1970 CrossRef CAS PubMed
- B. K. Johnson and R. K. Prud'homme, Phys. Rev. Lett., 2003, 91, 1183021 Search PubMed
- T. Yamamoto, D. Komarudin, M. Arai, B.-L. Lee, H. Suganuma, N. Asakawa, Y. Inoue, K. Kubota, S. Sasaki, T. Fukuda and H. Matsuda, J. Am. Chem. Soc., 1998, 120, 2047 CrossRef CAS
- H. Shimizu, M. Yamada, R. Wada and M. Okabe, Polym. J., 2008, 40, 33 CrossRef CAS
- A. Fraleoni-Morgera, S. Marazzita, D. Frascaro and L. Setti, Synth. Met., 2004, 147, 149 CrossRef CAS PubMed
- J. E. Millstone, D. F. J. Kavulak, C. H. Woo, T. W. Holcombe, E. J. Westling, A. L. Briseno, M. F. Toney and J. M. J. Fréchet, Langmuir, 2010, 26, 13056 CrossRef CAS PubMed
- D. Darwis, N. Holmes, D. Elkington, A. L. D. Kilcoyne, G. Bryant, X. Zhou, P. Dastoor and W. Belcher, Sol. Energy Mater. Sol. Cells, 2014, 121, 99 CrossRef CAS PubMed
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS
- J. J. M. Halls, K. Pichler, R. H. Friend, S. C. Moratti and A. B. Holmes, Appl. Phys. Lett., 1996, 68, 3120 CrossRef CAS PubMed
- S. R. Scully and M. D. McGehee, J. Appl. Phys., 2006, 100, 034907 CrossRef PubMed
- P. E. Shaw, A. Ruseckas and I. D. W. Samuel, Adv. Mater., 2008, 20, 3516 CrossRef CAS PubMed
- P. Lucas, M. Vaysse, J. Aubry, D. Mariot, R. Sonnier and F. Ganachaud, Soft Matter, 2011, 7, 5528 RSC
- F. Machui, S. Langner, X. Zhu, S. Abbott and C. J. Brabec, Sol. Energy Mater. Sol. Cells, 2012, 100, 138 CrossRef CAS PubMed
- T. Erb, U. Zhokhavets, G. Gobsch, S. Raleva, B. Stühn, P. Schilinsky, C. Waldauf and C. J. Brabec, Adv. Funct. Mater., 2005, 15, 1193 CrossRef CAS PubMed
- C.-W. Chu, H. Yang, W.-J. Hou, J. Huang, G. Li and Y. Yang, Appl. Phys. Lett., 2008, 92, 103306 CrossRef PubMed
- S. Kazaoui, R. Ross and N. Minami, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 11665 CrossRef
- S. Sutty, G. Williams and H. Aziz, Org. Electron., 2013, 14, 2392 CrossRef CAS PubMed
- B. Kraabel, D. McBranch, N. S. Sariciftci, D. Moses and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 18543 CrossRef CAS
- N. S. Sariciftci and A. J. Heeger, Int. J. Mod. Phys. B, 1994, 8, 237 CrossRef CAS
- C. Goh, S. R. Scully and M. D. McGehee, J. Appl. Phys., 2007, 101, 114503 CrossRef PubMed
- R. G. LeBel and D. A. I. Goring, J. Chem. Eng. Data, 1962, 7, 100 CrossRef CAS
- R. Mehra, Proc. Indian Acad. Sci., 2003, 115, 147 CrossRef CAS
- S. A. Arrhenius, Z. Phys. Chem., 1887, 1, 631 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mh00021h |
| This journal is © The Royal Society of Chemistry 2014 |