Nucleobase peptide amphiphiles†
Christopher J.
Serpell
a,
Maciej
Barłóg
b,
Kaustuv
Basu
c,
Johans F.
Fakhoury
a,
Hassan S.
Bazzi
b and
Hanadi F.
Sleiman
*a
aDepartment of Chemistry and Center for Self-Assembled Chemical Structures (CSACS), McGill University, 801 Sherbrooke Street West, Montreal, QC H3A-2K6, Canada. E-mail: hanadi.sleiman@mcgill.ca
bDepartment of Chemistry, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar
cFacility for Electron Microscopy Research, McGill University, Strathcona Anatomy & Dentistry Building, 3640 University Street, Montreal, QC, Canada H3A 2B2
First published on 17th January 2014
Abstract
A new class of peptide materials is introduced, integrating orthogonal aspects of peptide, nucleoside, and amphiphile chemistry. In solution, species such as rod-like or helical micelles are formed, which can lead to nanoribbons under lateral or longitudinal hierarchical growth regimes. Gelation of a wide range of solvents can be induced, including water and aqueous buffer, providing new avenues for nucleobase-specific electrophoresis, oligonucleotide delivery and bioactive cell growth media.
Peptide self-assembly is a rapidly developing field in which the structures found in native proteins are taken, adapted, expanded upon, and integrated into synthetic systems. While total de novo design of discrete proteins has recently reached important milestones,1–4 the exploitation of smaller peptide self-assembly motifs to construct larger-scale materials is of particular importance. The similarity of the products to certain misfolded protein systems responsible for diseases such as Parkinson's and Alzheimer's5 aids the understanding and treatment of such conditions, while the ability to generate order from small building blocks over a large range of length scales had led to interesting new materials with applications such as nanoelectronics,6 water purification,7,8 and therapeutic drug delivery.9 Even systems as small as dipeptides are capable of forming a wide diversity of structures,10 due to the combination of peptide secondary structure motifs with orthogonal assembly features such as π–π stacking,11 and amphiphile-based phase separation.12 These results have led to two broadly defined sub-fields: dipeptides as low molecular weight gelators (LMWGs);13 and peptide-amphiphile (PA) self-assembly of micellar structures12,14,15 (although these topics may interweave16,17). LMWGs are small molecules which aggregate through specific supramolecular interactions to create nanoscale fibres, the intertwining of which creates a network which entraps solvent, forming a gel. The high levels of interest in LMWGs arise because the supramolecular nature of the linkages endows the materials with dynamic molecule-responsive character,16,18–20 in contrast to polymer-based gels. Moreover, since the tuneable rheological properties of gels make them outstanding for 3D cell culture purposes, biomolecule-based gels are especially topical for their potential to influence cell growth.21 Conversely, PA systems are often proposed as drug delivery vehicles, since they permit the construction of micelles with tailored geometries capable of encapsulating drug molecules and incorporating bioactive targeting moieties.
We here report a new class of dipeptide-based materials (‘nucleobase peptide amphiphiles’, NPAs), which uses simple modifications to assemble into a range of novel and distinct hierarchical structures: from rod-like micelles, to nano- and microribbons, to bulk gel phases. These building blocks are based on a bis-tyrosine dipeptide (Fig. 1), where one tyrosine is substituted with the nucleobase adenine, while the other carries either a hexyl, or an oligoethylene glycol chain. As such, they are capable of three degrees of orthogonal association: (i) their aromatic peptide units can assemble via hydrogen bonding and π–π stacking into higher-order structures, such as β-sheets; (ii) their adenine units are self-complementary via hydrogen-bonding, and capable of interacting with other DNA strands; (iii) their hexyl or oligoethylene glycol substituents and benzoyl protection status of the adenine can modulate amphiphilic character and hydrophobic properties. Each of the four molecules discussed herein exhibits its own characteristic self-assembly properties, and each of the three potential levels of self-assembly are observed.
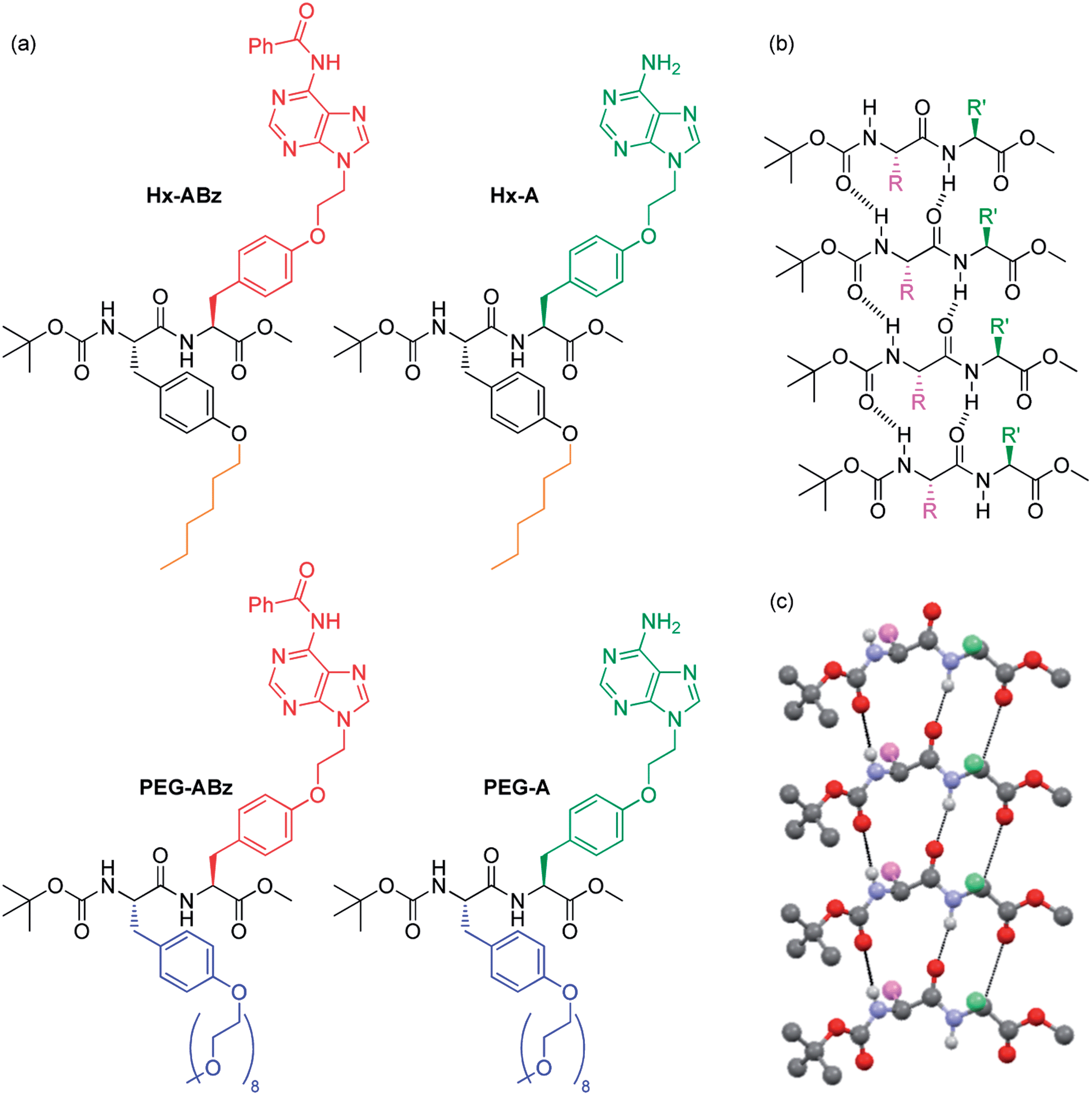 | ||
| Fig. 1 NPAs discussed in this manuscript (a), and self-assembly of Boc-methoxy dipeptides into parallel β-sheets: chemical representation (b) and crystallographically determined 3D structure of peptide backbone of Boc/methyl ester protected diphenylalanine taken from the literature.22 | ||
On the level of molecular structure, NPAs are substantially distinct from established dipeptide LMWGs. Classic peptide LMWGs rely on aromatic N-terminal protection and C-terminal deprotection to drive aggregation by π–π stacking and hydrophobic collapse, often in specific pH ranges: our NPAs, being protected at both termini using non-aromatic groups, demonstrate that this aromatic N-protection/C-terminal deprotection is not necessary for dipeptide assembly. To our knowledge only one example of a dipeptide gelator with this protection scheme has been reported previously.23 Side-chain based hydrophilicity modulation also distinguishes our NPAs from existing examples of PAs in which a linear topology is employed.14 Xu and coworkers have previously replaced the classic N-terminal aromatic group of dipeptide gelators with a nucleobase.24–26 Our NPAs are capable of gelation of a greater range of solvents at concentrations an order of magnitude lower. They form a library of large-scale hierarchical nanostructures in the solution phase not seen with nucleopeptides, and undergo structure modification by interaction with DNA. Finally, the combination of nucleobases and amphiphilic units on the side chains with a selective terminal protection scheme gives them the potential as building blocks for sequence-defined DNA-mimetic peptides via Merrifield synthesis. In the modular incorporation of three different self-assembly regimes – nucleobase pairing, peptide folding, and amphiphile aggregation – dityrosine NPAs constitute a distinct new class of self-assembling molecule.
Results and discussion
Tyrosine was selected as the basis for the NPAs because of the ease of selective alkylation at the phenolic hydroxyl group, and its enantiopurity and chemical robustness. Facile functionalization of protected tyrosine monomers by alkylation of the phenyl hydroxyl group was employed to create amino acids with protected adenine groups (Scheme S1, ESI†): firstly an ethylene spacer was inserted by reaction of adenine with ethylene carbonate, then Boc-methoxy-tyrosine was attached under Mitsunobu conditions, followed by benzoyl protection of the adenine exocyclic amine with benzoyl chloride. Finally the tyrosine amine was deprotected using trifluoroacetic acid, giving the amino acid (labelled ABz) ready for coupling with a second amino acid in 29% overall yield. The amino acids for amphilicity modulation consisted of an n-hexyl chain (Hx, hydrophobic) or an oligoethylene glycol chain (PEGMw = 350, n ≈ 8, hydrophilic) attached to the phenolic oxygen, and were synthesised by simple alkylation or Mitsunobu reaction respectively followed by carboxyl deprotection (85% and 64% overall yields for Hx and PEG respectively). After coupling of ABz with Hx or PEG tyrosines, the dipeptides Hx-ABz and PEG-ABz were obtained and could be deprotected on the adenine unit by stirring with methanolic potassium carbonate to give NPAs Hx-A and PEG-A (Fig. 1) respectively (89% and 82% yields). The N- and C-termini were left protected in this instance to minimise acid/base related charge effects upon self-assembly.The self-assembly of both free adenine and benzoyl protected analogues was studied in order to make comparisons regarding the effect of nucleobase interactions. Solution-phase self-assembly of each NPA was studied by circular dichroism (CD) spectroscopy and light scattering measurements, and morphological analysis was performed by atomic force microscopy (AFM) and transmission electron microscopy (TEM) imaging.
In methanol, Hx-ABz (2 mM) was found to display an unusual CD spectrum with two positive peaks at 205 and 229 nm (Fig. 2a). While the positions correspond to those of a β-sheet, the higher wavelength peak, arising from the n–π* amide transition,27 has been inverted, suggesting a deformation of the structure. The overall intensity was modest, and no CD signal was seen at longer wavelengths, indicating that the level of assembly is low, and that the chiral environment does not extend to the nucleobase region. The limited extent of assembly in methanol was confirmed by measuring the light scattering intensity, which was low, at 8200 counts per second (8.2 kcps; on our instrument < 4 kcps corresponds to molecular solutions). Tenfold dilution with water or pH 8 TAEMg buffer (a system commonly used for DNA hybridization) was used to encourage aggregation of the hydrophobic residues while maintaining peptide-like secondary structure, however in this case a thick precipitate was observed, which hindered examination of the system by CD or light scattering. Analysis of particle morphology by TEM revealed a system of rods extending from nodules in both cases, although in water the rods were more prominent (Fig. 2b), whereas in buffer the nodules dominated (Fig. 2c). Although this NPA demonstrated incompatibility with aqueous media (unsurprising given the weak hydrophilicity of the polar components), it was capable of gelling organic solvents. Benzene can be gelled by heating powdered Hx-ABz followed by slow cooling (Fig. 2d). Gels of other organic solvents could be obtained using Hx-ABz by slow diffusion of a second solvent into the mixture, resulting in chloroform–hexane (Fig. 2e) and methanol–diisopropyl ether gels (Fig. 2f). TEM analysis of the gels displayed their constituent fibres; in the case of the methanolic gel, these were found to exhibit helicity, demonstrating the levels of hierarchical assembly of which the single molecule is capable.
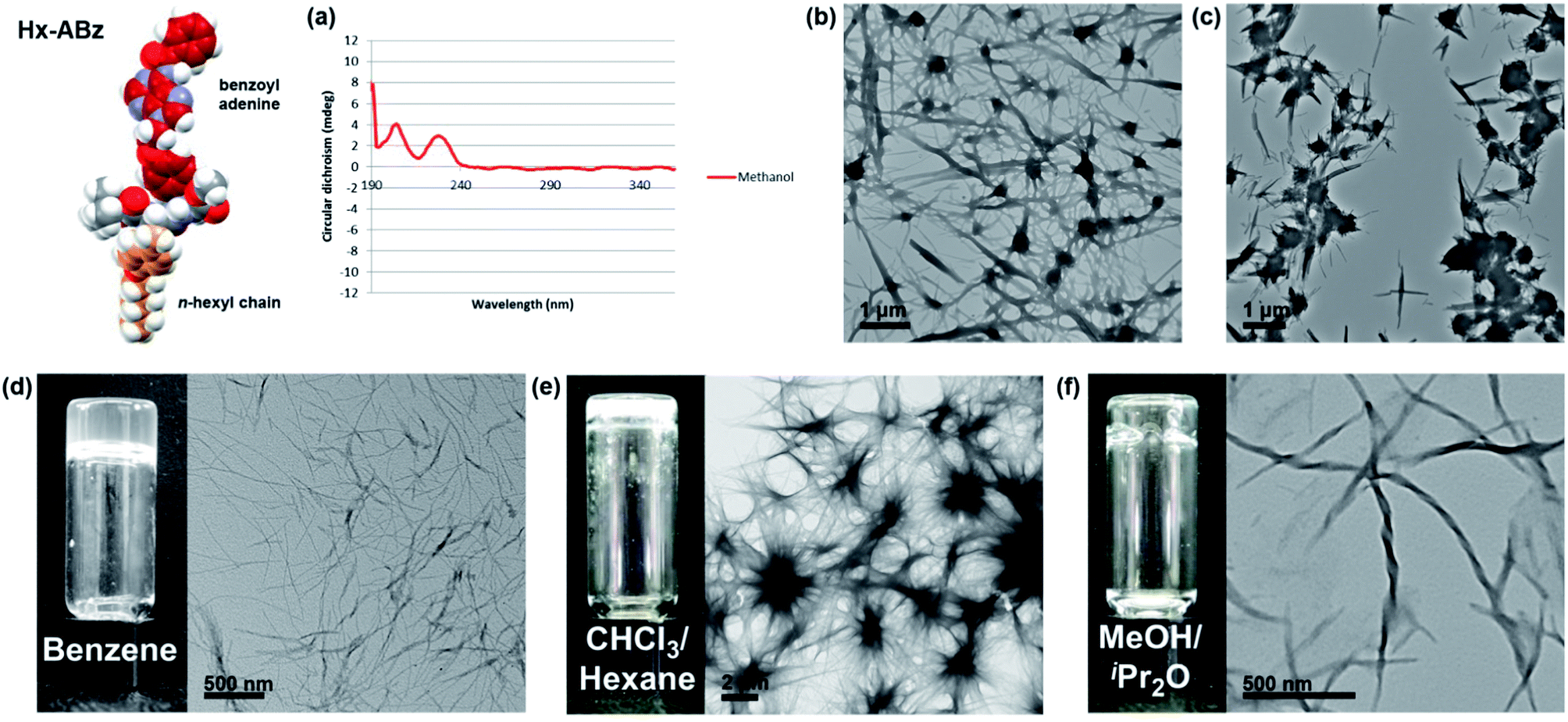 | ||
Fig. 2 Self-assembly of Hx-ABz. (a) CD spectrum in methanol, 298 K. (b) TEM image of precipitate from 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water–methanol. (c) TEM image of precipitate from 9:1 TAEMg buffer:methanol. (d–f) Benzene, chloroform, and methanol gels respectively, with TEM image of fine-structure. Further images can be found in the ESI, Fig. S15–S19.† :1 water–methanol. (c) TEM image of precipitate from 9:1 TAEMg buffer:methanol. (d–f) Benzene, chloroform, and methanol gels respectively, with TEM image of fine-structure. Further images can be found in the ESI, Fig. S15–S19.† | ||
The behaviour of Hx-A was found to be significantly different from Hx-ABz. In methanol, the CD spectrum a positive peak centred at 199 nm and negative peak at 232 nm were seen (Fig. 3b), reminiscent of the β-sheet signature spectrum, although the negative peak is shifted to slightly longer wavelength, which may signify twisting of the sheets.24 Although the differences between the CD spectra of parallel and antiparallel β-sheets are slight, the crossover from negative to positive CD at 195 nm suggests that a parallel β-sheet structure is the more likely orientation.27–29 However, again the signal was low and no peaks in the adenine region (240–290 nm) were seen, consistent the observed light scattering intensity of 8.3 kcps, and indicating limited assembly pertaining only to the peptide region. Dilution of Hx-A in methanol with water or buffer gave a clear solution. In water no CD signal could be measured, neither was there an increase in light scattering intensity (8.8 kcps). However, in buffer a large CD signal was generated, with negative peaks at 208 and 230 nm (i.e. an inversion of the π–π* transition of a twisted β-sheet), a broad positive peak with a maximum at 264 nm, and a third minimum at 284 nm. The extension of the signal into the long wavelength region indicates that the chirality of the assembly extends to the nucleobase. A significant increase in light scattering (29.6 kcps) was also noted in buffer. The amplification of both CD and light scattering signal in buffer is all the more remarkable given the tenfold dilution of the samples. While examination of the water solution by AFM revealed only a handful of irregular aggregates, the buffer solution sample (Fig. 3c) displayed elongated micellar structures packed into a dense layer with a uniform height of 0.6 nm and minimum spacing of 17 nm. Some larger globular structures could be observed in dense regions, but close examination showed these to be decorated with the same patterns. Interestingly, in less densely deposited areas, the fibres formed laterally striated ribbons, with widths of 0.1–1.5 μm, likely arising from self-sorting of rod-like micelles, or cooperative growth of adjacent rods. At the edges, individual fibres could be seen. Similar features could be observed by TEM (Fig. 3d). We are currently investigating the molecular origin of these size-sorted sheets, however a probable hierarchy of self-assembly already emerges from the results discussed above (Fig. 3a). Firstly, in methanol β-sheet structures (probably parallel) are formed; the weak CD and light scattering signals indicates these are likely to be only one NPA wide and of limited length, and the form of the CD indicates that the side-chains are disordered. Secondly, upon addition of buffer, the hydrophobic hexyl chains aggregate, bringing the β-sheets together into larger assemblies (hence the higher CD and light scattering intensities), and distorting the peptidic hydrogen bonding motifs, as visualised by the change in shape of the CD spectrum, leaving the nucleobases exposed. The hydrophobic collapse must also induce some curvature into the structure, resulting in the rod-like structures seen in AFM and TEM. These rods then align upon deposition, potentially through adenine–adenine interactions.
 | ||
| Fig. 3 Self-assembly of Hx-A. (a) Overview of hierarchical self-assembly to yield aligned nanorods. (b) CD spectra in methanol and aqueous systems, plus interaction with T10 in 9:1 TAEMg:methanol, 298 K. (c) AFM images illustrating hierarchy of fibre aggregation in 9:1 TAEMg:methanol (d) TEM images of the same features. (e) TEM and AFM images of structures formed by interaction of T10 with Hx-A. (f) Hx-A hydrogel (with 5% DMSO), accompanied by TEM image of fine-structure. Further images can be found in the ESI, Fig. S20–S25.† | ||
If the model described above is correct, then the nucleobases at the solvent interface should be free to interact with DNA, altering either the structure of the DNA or the NPA. The nucleobase pairing was tested by adding deca(deoxythymidine phosphate) (T10) DNA to methanolic Hx-A at the same time as the buffer and then heating to 60 °C for half an hour, followed by cooling to room temperature. The resultant CD of the NPA (Fig. 3b) was slightly reduced in magnitude in the peptide region with negative peaks remaining at 206 and 231 nm, but changed in form above 240 nm, remaining negative until 262 nm, and exhibiting a positive peak at 273 nm with no negative tail (reminiscent of the CD spectrum of A-form DNA30). This indicates that A–T interactions impose a DNA-type chirality environment on the adenines, while maintaining the peptide assembly in its overall structure. Additionally, an increase in light scattering intensity was also recorded (47.1 kcps), suggesting that larger constructs were produced. Examination by AFM and TEM (Fig. 3e) revealed rods with a broad distribution of lengths (80 nm–1.7 μm) which could not be seen in samples containing only T10. Similar structures were observed in just one small section of the TEM upon which Hx-A (TAEMg) had deposited (Fig. S21, ESI†), and in the background of the AFM images of Hx-A/T10 in some places a linearly patterned surface could be observed (Fig. S22, ESI†), suggesting that influence of DNA upon the self-assembly of Hx-A causes a shift in equilibrium between different constructs, rather than simply forming an NPA–DNA duplex. The differences in CD, light scattering, and morphology confirm that in self-assembled Hx-A systems the adenines are accessible to nucleic acid molecules. Hydrogels could be formed from Hx-A by diluting a DMSO solution twenty-fold with water with stirring, followed by heating to close to boiling, giving a translucent white hydrogel upon cooling and standing (Fig. 3f). A minimum gelator concentration of 5 mg mL−1 (0.5 wt%) was required, which is significantly lower than that for reported nucleopeptides (2 wt%).24 TEM imaging of the Hx-A/DMSO hydrogel revealed a highly branched network of interlinked and frayed fibres (8–40 nm in width), which was confirmed by AFM (Fig. S25, ESI†). A hydrogel displaying nucleobases on its constituent fibres would be a promising material for nucleotide technologies: base-recognition could be used to enforce sequence-selective electrophoretic mobility. For example, it may be possible to append a T-tract to oligonucleotides, which will selectively restrict electrophoretic mobility, acting analogously to the affinity columns31 such as those used in conjunction with the ubiquitous His-tag32 in protein purification. The highly defined surface deposition patterns (Fig. 3c) on mica substrates can also be exploited as a tool in DNA nanotechnology to align other DNA nanostructures or gold nanoparticles with appended polyT strands.
Although its nucleobase is protected, the self-assembly of PEG-ABz was interesting to study because its amphiphilicity is opposite to that of Hx-A – the nucleobase has become hydrophobic whereas the other side chain is now hydrophilic. In methanol PEG-ABz displayed a CD curve (Fig. 4b) almost identical to that of Hx-ABz, meaning that the secondary structure is influenced by the protection of the nucleobase. Again the light scattering intensity was low (1.6 kcps), showing that PEG-ABz is not extensively ordered in methanol. Addition of water to PEG-ABz in methanol resulted in a clear solution, which gave a very large CD signal (Fig. 4b) with a positive peak at 199 nm, and a negative peak at 222 nm, and a change of sign at 192 nm, classic signifiers of a (likely parallel) β-sheet morphology.27 A broad positive peak between 240 and 280 nm, followed by a negative peak at 292 nm indicated that the chiral environment pertained to the nucleobase as well as the peptide backbone. Light scattering was very intense (257.4 kcps), consistent with large-scale self-assembly. Analysis of the system by TEM (Fig. 4c) displayed a library of related shapes. In places, elongated and curved structures were seen (ca. 20 × 300 nm); it could be seen that aggregation of these led to patterned lamellae (up to 1 μm wide), and that the lamellae could grow extensively in one direction to produce ribbons ten of microns in length. AFM produced similar results (Fig. 4d), determining that the height of the lamellae or ribbons was 5 nm, and that they displayed longitudinal striations with a peak frequency of approximately 25 nm. When TAEMg buffer was used in place of pure water, the CD spectrum was changed only slightly (Fig. 4b), while the light scattering intensity was reduced by half (124.2 kcps). AFM and TEM analysis (Fig. 4e and f) showed only the lamellae form, indicating that the buffer impeded the formation of elongated ribbons. Cryo-EM images of the assemblies formed in water (Fig. S30†) demonstrated that lamellar structures are present in solution, rather than being an artefact of drying. Addition of T10 to these samples had no significant effect upon their CD spectra, confirming that the protected nucleobase does not partake in base pairing interactions. Combining the experimental results for PEG-ABz suggests a new hierarchical assembly pattern (Fig. 4a). In methanol a limited level of self-assembly occurs (CD and light scattering intensity), in which the peptide association has a structure related to β-sheet and the nucleobase is disordered. Upon addition of aqueous solvents, the peptide rearranges to a classical parallel β-sheet and the nucleobase becomes ordered (CD and light scattering). It is likely that the hydrophobically protected nucleobase ordering involves expulsion of solvent, leaving the soluble PEG chains at the water interface. Since AFM and TEM imaging indicates that the assemblies associate differentially in different directions, it is probably that the smallest species in aqueous solutions are anisotropic lamellae rather than rods with a circular cross-section. In pure water it appears that longitudinal growth dominates over lateral association, producing elongated ribbons whereas in buffer aggregation of the lamellae in a side-by-side fashion, yielding plates. Since the striations along the ribbons or plates show variations in frequency, it is likely that they arise due to packing defects during edgeways merging of lamellae of different widths.
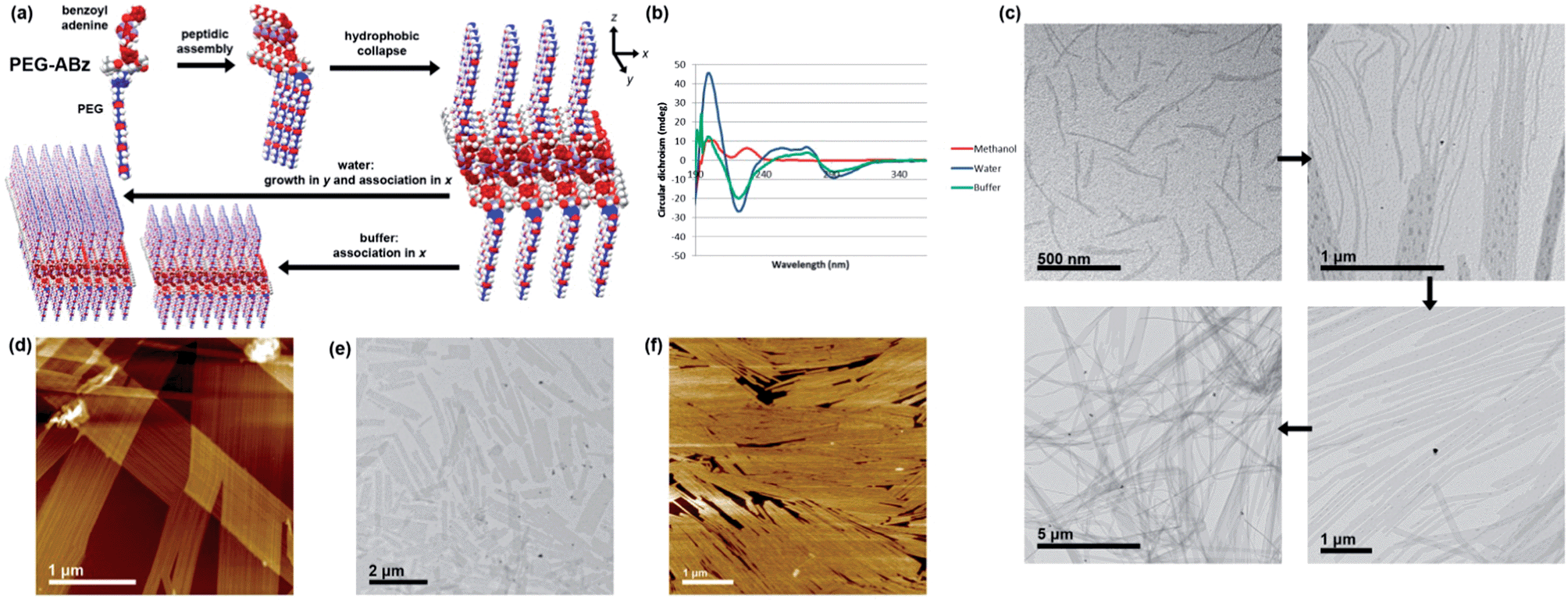 | ||
| Fig. 4 Self-assembly of PEG-ABz. (a) Overview of hierarchical self-assembly. (b) CD spectra in methanol and aqueous systems, 298 K. (c) TEM images illustrating hierarchy of rod-like micelle aggregation into lamellae and ribbons in 9:1 water–methanol (d) AFM image of the same features. (e) TEM images of shorter lamellae formed by in 9:1 TAEMg:methanol (f) AFM images of shorter lamellae formed by in 9:1 TAEMg:methanol. Further images can be found in the ESI, Fig. S26–S30.† | ||
As with Hx-A, in methanol PEG-A displayed a CD spectrum suggestive of a parallel β-sheet secondary structure, albeit weak (Fig. 5b), consistent with light scattering (6.2 kcps). Given that PEG-A is fully water soluble (adenine deprotection changes the solubility properties of NPAs drastically), it was not surprising that addition of either water or buffer did not result in any significant self-assembly. No CD signal could be measured in either of these systems, although some light scattering intensity was observed in TAEMg (41.9 kcps). Examination by AFM or TEM showed only amorphous aggregates. Inclusion of T10 in the mixture had no effect; base-pairing in water requires shielding of the bases from solvent, and it is likely that such shielding does not occur with PEG-A in the absence of a secondary structure. This result suggests that base-pairing interactions with NPAs rely first upon the self-assembly of the NPAs to create a polyvalent partner, and that of the three programmed self-assembly motifs, the hydrophobic component is a prerequisite for the other two.
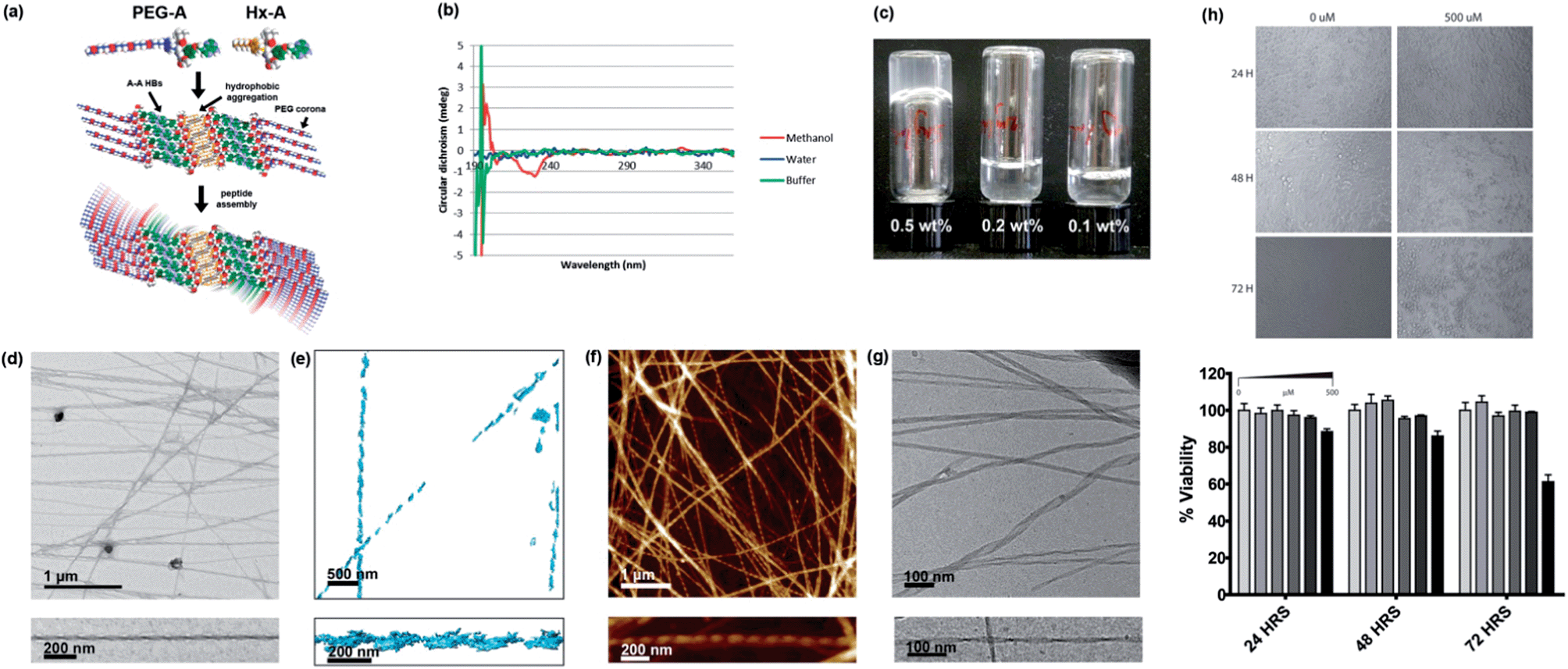 | ||
| Fig. 5 Self-assembly of PEG-A, on its own and in combination with Hx-A. (a) Proposed self-assembly scheme of 1:1 PEG-A:Hx-A. (b) CD spectra of PEG-A in methanol and aqueous systems, 298 K. (c) Gelation of water using 1:1 Hx-A:PEG-A. (d) TEM image of helical fibres in the gel, with magnification of a single example. (e) 3D tomographic reconstruction of fibres from TEM images. (f) AFM image of helical fibres in the gel, with magnification of a single example. (g) Cryo-EM image of helical gel fibres, with magnification of a single example. Further images can be found in the supporting information, Fig. S31–S34.† (h) MTS HeLa cell viability assay results at varying concentration of NPA mixture and timepoints. | ||
Interestingly, while PEG-A is fully soluble and does not associate in aqueous solution, it can form hydrogels by mixing a 1:1 ratio of PEG-A and the otherwise insoluble Hx-A in water at 5 mg mL−1 (0.5 wt%) followed by heating to give a clear solution and standing overnight (Fig. 5c). TEM (Fig. 5d) showed that the Hx-A/PEG-A gel consisted of thicker (27 ± 5 nm), more rigid, and less branched ribbons, when compared to the Hx-A/DMSO hydrogel. These ribbons displayed helical twisting with a pitch of ca. 120 nm. Tomographic reconstruction of a set of TEM images (Fig. 5e) collected with a stage tilt of ±55° resulted in a 3D model of two individual ribbons, each displaying a left-handed helical twist. AFM imaging gave similar images (Fig. 5f), again visualising a left-handed chirality to the helix in all the ribbons in which the direction could be clearly discerned. Due to the interesting hierarchical structure, the gel was also submitted to analysis by cryo-EM (Fig. 5g), providing confirmation that the observed helical ribbons are those present in the aqueous phase. The finer detail in cryo-EM images revealed that the width varied from 10 to 35 nm, and could distinguish that the thicker ribbons displayed a coiled helical morphology whereas the finer ones took on the appearance of twisted ribbons with a helical pitch of ca. 110 nm, matching well with the AFM and TEM data. The height of the ribbons was more consistent at 8.1 nm. In some places the ribbons joined together, indicating that attractive ribbon–ribbon interactions exist.
Although light absorption and scattering were problematic at gelating concentrations, hindering analysis, the CD spectrum of the gel (Fig. S35†) resembled that of Hx-A in TAEMg. Hence, we propose a structure (Fig. 5a) consisting of a core of β-sheets of Hx-A assembled into a bilayer through hydrophobic aggregation of the alkyl chains, surrounded by β-sheets of PEG-A assembled via A–A interactions33 mediated through hydrogen bonding on both the Hoogsteen and Watson–Crick faces of the nucleobases. The architecture leaves the PEG groups, the most hydrophilic in this system, exposed to the aqueous environment. If a fully-stretched conformation were adopted, such a structure would have a height of 9.8 nm; the value measured in solution by cryo-EM is consistent with moderate compaction which would be expected to fulfil the demands of close-packing. A degree of twisting must then exist in the β-sheet packing to produce the helical fibres.
Hydrogels which display good shape persistence at low gelator concentrations are attractive candidates for biofabrication technologies,34 particularly when they display bioactive moieties. The combination of biomolecular interactions, rigid rod formation, and fouling-resistant PEG chains make this gel a highly interesting material. Furthermore, as with previously reported nucleopeptides, it may be possible to use the affinity of the gel for nucleic acids to assist in delivery of therapeutic DNAs or RNAs to cells. As a preliminary test to check for cytotoxicity, an MTS assay35 using HeLa cells was performed (Fig. 5h). No noticeable cell death was observed at concentrations up to 200 μM over 72 hours. Even at 500 μM, an extremely high dose if the system were expected to be bioactive, significant cytotoxicity was seen not seen until 72 hours, indicating that the Hx-A/PEG-A system is generally well tolerated by cells. We are currently investigating the potential of NPA hydrogels for improved transfection-free cellular delivery of nucleic acids and 3D DNA nanostructures.
Conclusions
We have developed a new class of dipeptide-based materials – nucleobase peptide amphiphiles (NPAs) based on dityrosine. Our NPAs have been shown to be capable both of solution-phase hierarchical assembly to give helical or curved rod-like micelles, leading to extended nanosheets, and gelation of a range solvents, including water, and interaction with DNA. In these molecules, three levels of orthogonal supramolecular association are possible: formation of peptide-like secondary structure motifs; hydrophobic/hydrophilic amphiphile self-assembly; and complementary nucleobase interactions. The combination of the peptide assembly and the amphiphilicity result in the formation of large-scale scaffolds decorated with addressable nucleobases for further modulation of the self-assembly, or to influence the spatial distribution of nucleic acids. We are working to elucidate the internal structures in fine detail of the large-scale assemblies, incorporate all four nucleobases, expand the library of amphiphilicity modulating groups, and to bring forward applied biological studies on NPAs.Acknowledgements
The authors would like to thank NSERC, the Canada Research Chairs program and the Qatar National Research Fund for financial support. C. J. Serpell would like to thank the Banting Postdoctoral Fellowships program. H. Sleiman is Cottrell scholar of the Research Corporation.Notes and references
- N. R. Zaccai, B. Chi, A. R. Thomson, A. L. Boyle, G. J. Bartlett, M. Bruning, N. Linden, R. B. Sessions, P. J. Booth, R. L. Brady and D. N. Woolfson, Nat. Chem. Biol., 2011, 7, 935–941 CrossRef CAS PubMed.
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042–1046 CrossRef CAS PubMed.
- N. Koga, R. Tatsumi-Koga, G. Liu, R. Xiao, T. B. Acton, G. T. Montelione and D. Baker, Nature, 2012, 491, 222–227 CrossRef CAS PubMed.
- H. Gradišar, S. Božič, T. Doles, D. Vengust, I. Hafner-Bratkovič, A. Mertelj, B. Webb, A. Šali, S. Klavžar and R. Jerala, Nat. Chem. Biol., 2013, 9, 362–366 CrossRef PubMed.
- K. Morris and L. Serpell, Chem. Soc. Rev., 2010, 39, 3445–3453 RSC.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- V. Percec, A. E. Dulcey, V. S. K. Balagurusamy, Y. Miura, J. Smidrkal, M. Peterca, S. Nummelin, U. Edlund, S. D. Hudson, P. A. Heiney, H. Duan, S. N. Magonov and S. A. Vinogradov, Nature, 2004, 430, 764–768 CrossRef CAS PubMed.
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem. – Eur. J., 2008, 14, 6870–6881 CrossRef CAS PubMed.
- X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed., 2007, 46, 2431–2434 CrossRef CAS PubMed.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1–18 CrossRef CAS PubMed.
- C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC.
- F. Versluis, H. R. Marsden and A. Kros, Chem. Soc. Rev., 2010, 39, 3434–3444 RSC.
- S. Koutsopoulos, L. Kaiser, H. M. Eriksson and S. Zhang, Chem. Soc. Rev., 2012, 41, 1721–1728 RSC.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed.
- T. Kar, S. Debnath, D. Das, A. Shome and P. Das, Langmuir, 2009, 25, 8639–8648 CrossRef CAS PubMed.
- T. Kar, S. K. Mandal and P. K. Das, Chem. – Eur. J., 2011, 17, 14952–14961 CrossRef CAS PubMed.
- C. E. Stanley, N. Clarke, K. M. Anderson, J. A. Elder, J. T. Lenthall and J. W. Steed, Chem. Commun., 2006, 3199–3201 RSC.
- D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18–33 RSC.
- O. Yamashita, Y. Kato, T. Yamane and T. Ashida, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 2657–2663 CrossRef.
- R. Afrasiabi and H.-B. Kraatz, Chem. – Eur. J., 2013, 19, 1769–1777 CrossRef CAS PubMed.
- X. Li, Y. Kuang, H.-C. Lin, Y. Gao, J. Shi and B. Xu, Angew. Chem., Int. Ed., 2011, 50, 9365–9369 CrossRef CAS PubMed.
- X. Li, K. Yi, J. Shi, Y. Gao, H.-C. Lin and B. Xu, J. Am. Chem. Soc., 2011, 133, 17513–17518 CrossRef CAS PubMed.
- X. Li, Y. Kuang and B. Xu, Soft Matter, 2012, 8, 2801–2806 RSC.
- M. M. Kelly, E. S. Pysh, G. M. Bonora and C. Toniolo, J. Am. Chem. Soc., 1977, 99, 3264–3266 CrossRef CAS.
- J. Applequist, Biopolymers, 1982, 21, 779–795 CrossRef CAS.
- M. C. Manning, M. Illangasekare and R. W. Woody, Biophys. Chem., 1988, 31, 77–86 CrossRef CAS.
- E. Charney and H. H. Chen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 1546–1549 CrossRef CAS.
- L. A. Phillips, R. H. L. Pang, J. J. Park, V. W. Hollis and F. Famuyiwa, Prep. Biochem., 1980, 10, 11–26 CrossRef CAS PubMed.
- E. Hochuli, W. Bannwarth, H. Dobeli, R. Gentz and D. Stuber, Nat. Biotechnol., 1988, 6, 1321–1325 CrossRef CAS.
- N. Safaee, A. M. Noronha, D. Rodionov, G. Kozlov, C. J. Wilds, G. M. Sheldrick and K. Gehring, Angew. Chem., Int. Ed., 2013, 52, 10370–10373 CrossRef CAS PubMed.
- J. Malda, J. Visser, F. P. Melchels, T. Jüngst, W. E. Hennink, W. J. A. Dhert, J. Groll and D. W. Hutmacher, Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- A. H. Cory, T. C. Owen, J. A. Barltrop and J. G. Cory, Cancer Commun., 1991, 3, 207–212 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, synthetic protocols and characterisation of NPAs, and further microscopy images. See DOI: 10.1039/c3mh00154g |
| This journal is © The Royal Society of Chemistry 2014 |