Additive-assisted supramolecular manipulation of polymer:fullerene blend phase morphologies and its influence on photophysical processes†
E.
Buchaca-Domingo
*a,
A. J.
Ferguson
*b,
F. C.
Jamieson
c,
T.
McCarthy-Ward
c,
S.
Shoaee
c,
J. R.
Tumbleston
d,
O. G.
Reid
b,
L.
Yu
e,
M.-B.
Madec
cf,
M.
Pfannmöller
g,
F.
Hermerschmidt
a,
R. R.
Schröder
gh,
S. E.
Watkins
i,
N.
Kopidakis
b,
G.
Portale
j,
A.
Amassian
e,
M.
Heeney
c,
H.
Ade
d,
G.
Rumbles
bk,
J. R.
Durrant
c and
N.
Stingelin
a
aDepartment of Materials and Centre for Plastic Electronics, Imperial College London, London, UK. E-mail: e.buchaca-domingo@imperial.ac.uk
bChemical and Materials Science Center, National Renewable Energy Laboratory, Golden, Colorado, USA. E-mail: andrew.ferguson@nrel.gov
cDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London, UK
dDepartment of Physics, North Carolina State University, Raleigh, North Carolina, USA
eDivision of Physical Sciences and Engineering, Solar and Photovoltaic Engineering Research Center, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
fSolvay Interox, Warrington, UK
gCellNetworks, BioQuant, Heidelberg University, Heidelberg, Germany
hInnovationLab GmbH, Heidelberg, Germany
iCSIRO Materials Science and Engineering, Clayton, VIC, Australia
jDUBBLE CRG BM26@ESRF, Netherlands Organization for Scientific Research, Horowitz, Grenoble, France
kDepartment of Chemistry and Biochemistry, University of Colorado, Boulder, USA
First published on 22nd November 2013
Abstract
It is well known that even small variations in the solid-state microstructure of polymer:fullerene bulk heterojunctions can drastically change their organic solar cell device performance. We employ pBTTT:PC61BM as a model system and manipulate co-crystal formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (by weight) blends with the assistance of fatty acid methyl esters as additives. This allows us to evaluate the role of the intermixed phase in such binary blends through manipulation of their phase morphology—from fully intercalated to partially and predominantly non-intercalated systems—and its effect on the exciton- and carrier- dynamics and the efficiency of charge collection, with relevance for future device design and manufacturing.
:1 (by weight) blends with the assistance of fatty acid methyl esters as additives. This allows us to evaluate the role of the intermixed phase in such binary blends through manipulation of their phase morphology—from fully intercalated to partially and predominantly non-intercalated systems—and its effect on the exciton- and carrier- dynamics and the efficiency of charge collection, with relevance for future device design and manufacturing.
Despite the rapid and significant progress in polymer:fullerene blends for use as the light-harvesting active layer in organic photovoltaic (OPV) devices, there is still a lack of complete understanding of the actual phase morphology achieved in the active layer (i.e. the number of phases present and the consequent complexity of the resulting microstructure) and its correlation to device performance. Recently, significant efforts have been devoted to elucidate the role intermixed phases play. One of the best studied systems in this context is the bulk heterojunction (BHJ) composed of poly(3-hexylthiophene) (P3HT) and [6,6]-phenyl C61-butyric acid methyl ester (PC61BM), which has been investigated since the early 1990s,1 and for which multiple phases have been identified. For instance, following the initial observation of polymer:fullerene miscibility and the measurement of a bimodal composition by Watts et al., groups including Collins et al., Treat et al. and Yin et al. found that PCBM aggregates and/or molecular species are miscible and mobile in disordered P3HT.2 Pfannmöller et al. used energy-filtered transmission electron microscopy (EFTEM) to detect such a third intermixed (‘composite’) phase,3 which exists in addition to the previously proposed BHJ microstructure of relatively phase-pure domains of the polymer and the fullerene. Further characterisation with X-ray diffraction and neutron reflectometry performed by Ro et al. showed that the mixing of P3HT and PCBM is influenced by many factors, such as the volumetric degree of ordering in the polymer and whether or not the PCBM is (pre-)aggregated.4 Other authors have calculated the weight fraction of PCBM able to interdiffuse in regiorandom P3HT to be between 20 wt% (for PC61BM) to 41 wt% (for PC71BM).5 It appears also that the high miscibility of fullerene derivatives with amorphous polymers or the disordered phase of semi-crystalline polymers is a rather universal behavior for binary polymer:fullerene systems;6 however the ability to control the extent of mixing has not been demonstrated, and it is still poorly understood what the optimum phase morphology is for efficient devices.
Clearly, if we want to reach the maximum performance within polymer:fullerene BHJ solar cells, we need to gain a more in-depth knowledge and control of donor:acceptor blends in order to correlate their optical and electronic properties with their solid-state microstructure and phase morphology. Thereby, it is critical to identify the required fraction of intermixed phases that provides the best compromise between relevant optoelectronic processes for maximizing their photovoltaic performance. Here, we present a versatile way to manipulate, and thus easily study, such functional two-component, multi-phase blend architectures. We propose a strategy which exploits the fact that several conjugated polymers are capable of hosting certain fullerene derivatives such as PC61BM in “cavities” within their molecular arrangements to form what is termed a co-crystal,7 representing an ordered intermixed phase in contrast to the intermixed amorphous solid-solutions formed by, for instance, the molecularly disordered fractions in P3HT:PC61BM binaries.
One example of such a co-crystal formation is the supramolecular arrangement obtained in blends of the thiophene-based polymer, poly(2,5-bis(3-alkyl-thiophene-2-yl)thieno[3,2-b]thiophene)s (pBTTT)8–11 and PC61BM, whose structure, photophysical properties and device performance have been well studied.12–15 We have, for instance, previously shown that, films of this blend of 1:1 (by weight) composition comprise primarily the co-crystal, with minimal pure phases, resulting in rapid electron–hole recombination and consequently poor photocurrent generation, although there is still a measurable long-lived mobile polaron population.16
Co-crystal formation makes the binary pBTTT:PC61BM an ideal model system for controlling and characterizing the amount/type of phases that are present in this system by manipulation of the extent of fullerene intercalation in the polymer; i.e. by controlling the amount of this ordered intermixed phase. We therewith aim to answer the two following questions: (i) an intermixed phase (i.e. PCBM molecules intimately mixed with the polymer chains; in the present case-in-point, the co-crystal) seems to be present in many OPV devices: what is its importance and what role does it play in the photophysics? (ii) Can we design and fabricate an ideal phase morphology that maximizes the yield of long-lived free charges?
With these questions in mind we describe here a methodology to modify the phase morphology of 1:1 pBTTT:PC61BM blends using commercially available and economically viable additives. Through introduction of such additives, we induce either a fully intercalated, partially intercalated, or a predominantly non-intercalated system. We discuss how these changes in the phase morphology affect the exciton and carrier dynamics, and deduce the corresponding efficiency of charge collection.
We selected for our purpose asymmetrical compounds as processing aids—methyl esters of two fatty acids: dodecanoic acid methyl ester (Me12) and heptanoic acid methyl ester (Me7)—with the idea that their polar heads would favor PC61BM while their alkyl ‘tail’ would favor the side-chains of pBTTT. Fig. 1a shows the chemical structures of pBTTT, PC61BM and these additives. We expected the additives to direct the supramolecular assembly of the two OPV components. Both additives, i.e. Me12 and Me7, were, however, not intended to act as plasticizers such as processing additives like 1,8-diiodooctane (DIO), 1,8-octanedithiol (ODT) and 1,8-dichlorooctane (DCO), which are symmetrical and frequently applied to modify the morphology of the active layer to realize an improvement of device performance.17 Indeed, Cates et al. did not find improvement in device performance when using DIO in 1:1 pBTTT:PC71BM devices14 indicating that these species cannot be used to control the phase morphology – which is the objective of this work.
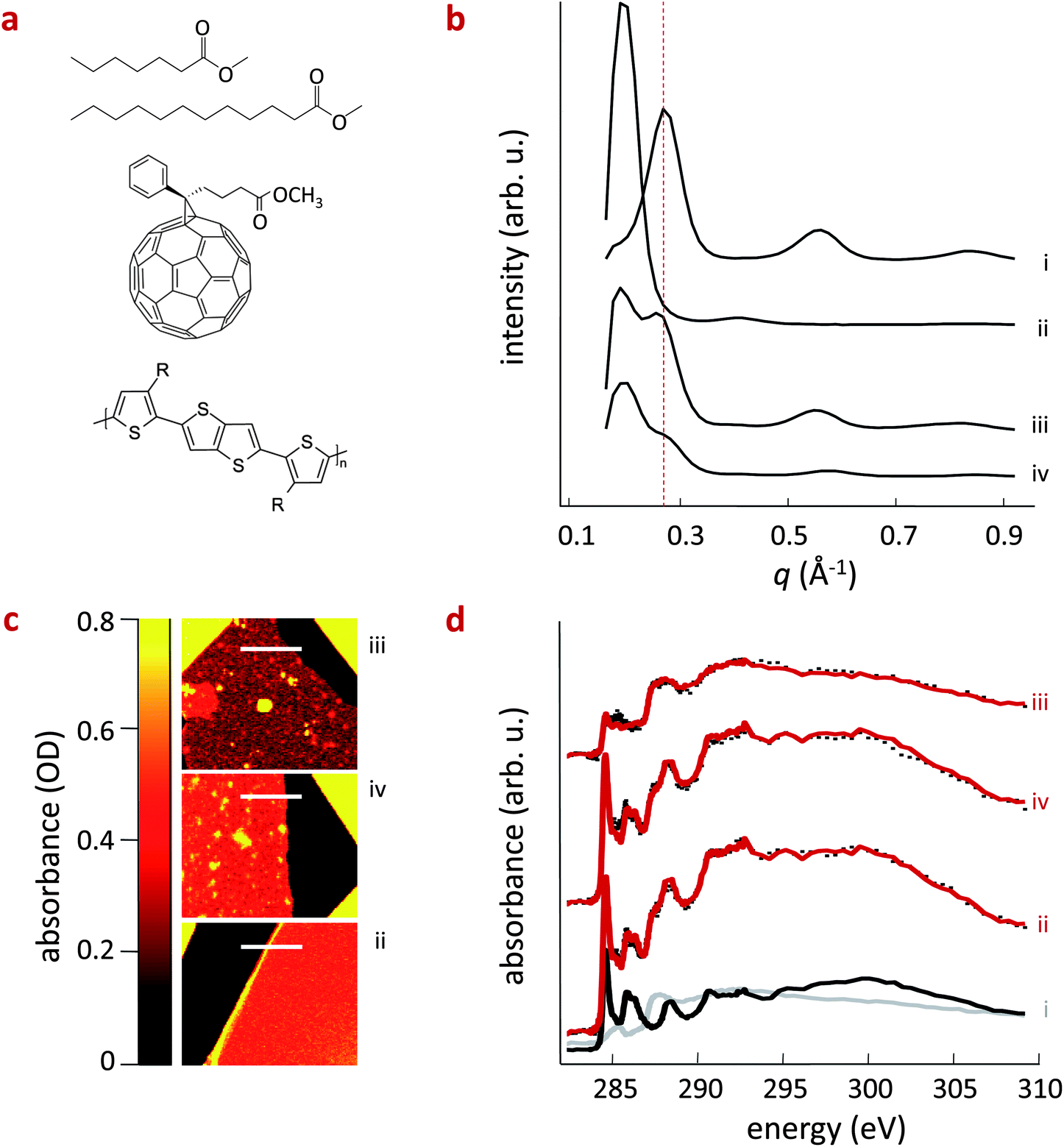 | ||
| Fig. 1 (a) From top to bottom, chemical structures of Me7, Me12, PC61BM and pBTTT where R = (CH2)15CH3. (b) Grazing-angle incidence X-ray diffractograms of neat pBTTT (i), pBTTT:PC61BM, forming co-crystals (ii), pBTTT:Me12:PC61BM (iii) and pBTTT:Me7:PC61BM (iv). The dashed line indicates the position of the (100) diffraction associated with the lamellar packing distance of the neat polymer at q ∼ 0.28 Å−1. (c) STXM images (left panels) and absorbance spectra (right panels) of pBTTT:PC61BM (ii), pBTTT:Me7:PC61BM (iv) and pBTTT:Me12:PC61BM (iii). The spectra of neat pBTTT ((i), grey line) and PC61BM (black line) are included for direct comparison. The absorbance spectra were employed to estimate the composition of each film at the position marked with a white line in the STXM images. We calculate a PC61BM content of approximately 42 wt% and 43 wt% for (ii) and (iv), respectively, indicating the likely presence of an intermixed phase (co-crystal) in such films, while only about 19 wt% of PC61BM was detected in (iii), lowering the availability of PC61BM for co-crystal formation. | ||
When using 1:1 (by weight) pBTTT:PC61BM blends and adding 10 molar equivalents of the corresponding additive per monomer unit of the pBTTT, a first observation can be readily made: by varying the length of the alkyl ‘tails’ (C7H15vs. C12H25) of the additive the amount of fullerene that is intercalated in the polymer seems to be altered. Note here that we found the use of additives with different tail lengths to be more efficient than varying the additive fraction. In fact, the longer alkyl additive seems to be able to expel considerable fractions of the fullerene, leading to a noticeable amount of relatively phase-pure pBTTT regions. This can be deduced from the grazing-angle incidence wide-angle X-ray scattering (GIWAXS) data shown in Fig. 1b, where we compare the neat polymer (i), with the pBTTT:PC61BM binary (ii) and the corresponding ternary systems. In the pBTTT:PC61BM blend (ii), the first (100) polymer diffraction order at q ∼ 0.28 Å−1 is shifted to higher distances (q ∼ 0.2 Å−1) due to co-crystal formation.11–14 However, when Me12 (iii) and Me7 (iv) are introduced into the system, we observe two (100) diffractions, one at q ∼ 0.2 Å−1 characteristic for the co-crystal, and one at q ∼ 0.28 Å−1 that can be attributed to the neat polymer. This is a clear indication that for both ternaries the formation of an intercalated phase is at least partly prevented, with the Me12 additive seeming to have a more pronounced effect, leading to a higher fraction of phase-pure pBTTT domains, as evident from the stronger diffraction observed at q ∼ 0.28 Å−1 for this ternary compared to the system comprising Me7.
These findings are supported and quantified by scanning transmission X-ray microscopy (STXM; Fig. 1c/d), which also allows us to determine the composition of the different blends. Fig. 1d displays absorbance measurements near the carbon 1s absorption edge of pBTTT:PC61BM blend films (along the white lines indicated in the different panels of Fig. 1c), processed with Me12 (iii) and Me7 (iv) and without additives (ii). The spectra are fit using a linear combination of the neat pBTTT and PC61BM reference spectra following previously established procedures.5,6c,18 We find that both the pBTTT:PC61BM binary (ii) and the pBTTT:Me7:PC61BM ternary (iv) comprise a similar average PCBM content in the pBTTT matrix: about 42 wt% and 43 wt%, respectively. These data reinforce the idea that a considerable fraction of intermixed phase is still present in the blends comprising the short-chain additive Me7 (iv), as also indicated by the pronounced diffraction at q ∼ 0.2 Å−1 in GIWAXS. In contrast, only about 19 wt% of PC61BM is detected in blends comprising Me12 (iii), supporting the picture that longer additives such as Me12 are able to efficiently macro-phase separate the polymer and the fullerene and, thus, deplete a larger amount of PC61BM molecules from the mixed phase (here, the co-crystal), leading to a phase morphology dominated by the co-existence of relatively pure phases of both components (i.e. pBTTT and PC61BM).
Optical microscopy highlights the consequence the various phase morphologies has on the film-forming characteristics of these blends (see ESI Fig. S1/a†): the presence of additives increases the surface roughness of the films, which exhibit large fullerene domains (verified with STXM) when the long-chain additive Me12 is used, likely due to the phase separation of the polymer and fullerene, leading to a stronger aggregation of the PC61BM component in pBTTT:Me12:PC61BM ternaries. In addition, we found that longer tailed additives, such as C13H27 (called here Me14), are able to expel even higher amounts of PC61BM as shown in ESI Fig. S1a/b.† Note that the features observed in pBTTT:Me7:PC61BM microscope images are due to surface roughness and not large-scale phase separation. This was confirmed with STXM data at different photon energies supporting that those features seen for pBTTT:Me7:PC61BM are not PC61BM aggregates (see ESI Fig. S1/c†). At the same time, Resonant Soft X-ray Scattering (R-SoXS)19 shows that films comprising Me12 and Me7 have nano-morphologies with very similar spatial frequency distributions that are characterized by a median characteristic length scale of ∼25 nm (see ESI Fig. S2†). Moreover, R-SoXS data show different intensities for (ii) and (iv) providing further evidence that pBTTT:Me7:PC61BM consists of a 3-phase morphology producing contrast between the co-crystal (i.e. fully intercalated) phase as well as the (relatively) phase-pure polymer and PC61BM regions. In contrast, the pBTTT:PC61BM binary forms a 1-phase system quenching any possible contrast between the polymer and the fullerene over the measurement length scale of R-SoXS (10 nm to 1 μm). Note also that the separation between polymer chains for the fully intercalated blend is ∼3 nm (from WAXS measurements), which is below the smallest lengths scale probed with R-SoXS. Thus, only volume fraction and not the length scale of the nano-morphology was manipulated.
The latter observation is in agreement with data obtained by time-correlated single-photon counting (TCSPC), where the photoluminescence (PL) decays (Fig. 2a) are close to the instrument response limit for both the pBTTT:PC61BM binary (ii) and pBTTT:Me7:PC61BM ternary (iv), i.e. systems for which GIWAXS indicates the presence of a considerable amount of intercalated material. Apparently, the fine intermixing of the donor and acceptor molecules, as found in a co-crystal, results in very rapid exciton quenching.16b In contrast, the PL decay measured for pBTTT:Me12:PC61BM (iii), whilst still strongly quenched relative to neat pBTTT [note we show here the decay of pBTTT:Me12 (v) which is similar to the neat polymer], is significantly slower than those observed for the pBTTT:PC61BM (ii) and pBTTT:Me7:PC61BM (iv) blends, pointing to the fact that excitons are longer-lived in these samples. Since ternaries comprising Me12 (iii) appear to possess a microstructure comprising more defined, relatively phase-pure pBTTT as indicated by our GIWAXS and STXM data, the extended exciton lifetime is likely to result from the necessity for the exciton to reach an interface between the neat pBTTT and PC61BM domains. Note though that the steady-state photoluminescence quenching (PLQ) for all systems comprising PC61BM was high (Fig. 2b); the addition of fatty acid methyl esters did decrease the PLQ, with the pBTTT:Me12:PC61BM ternaries featuring the least quenching in agreement that Me12 is better able to prevent intercalation of the fullerene into the pBTTT than its Me7 counterpart.
 | ||
| Fig. 2 Photophysical response of films of pBTTT:PC61BM (ii), pBTTT:Me12:PC61BM (iii), pBTTT:Me7:PC61BM (iv) and as reference, pBTTT (i) and pBTTT:Me12 (v) structures. (a) Photoluminescence decays: faster decays are observed for samples containing a noticeable amount of co-crystal such as (ii) and (iv); (b) steady-state photoluminescence (PL) quenching measured for the various systems. The longer alkyl methyl ester (Me12) results in less quenching compared to ternaries comprising Me7. (c) Schematic illustrations of the phase morphologies we deduce from the structural data presented in Fig. 1 and results in (a) and (b): pBTTT:PC61BM (ii) forms a 1-phase morphology; in pBTTT:Me12:PC61BM ternaries (iii), introduction of the additive noticeably expels the fullerene from the polymer leading to a considerable fraction of phase-pure pBTTT and PC61BM regions; while for pBTTT:Me7:PC61BM (iv) still a considerable amount of co-crystal is present beside neat polymer and fullerene minority phases. A schematic for the neat polymer (i) is also shown. | ||
Based on the GIWAXS, STXM, R-SoXS, TCSPC and PLQ data we conclude that the microstructure of pBTTT:PC61BM systems can be manipulated by use of additives and they can be utilized to control the amount/type of phases present depending on their precise nature (see schematics presented in Fig. 2c). The pBTTT:PC61BM binary (ii), at the composition selected (1:1 by weight) is well known to form a co-crystal (i.e. a 1-phase system) in agreement with our data. When adding Me12 to the system, a considerable amount of the fullerene is expelled into macrophase-separated domains, lowering the average PCBM concentration of the nano-morphology. Hence, a noticeable fraction of neat pBTTT and, likely, neat PC61BM phases are present in these thin-film architectures, with the phase separation being directed by the Me12 (iii). Me7 seems less efficient to induce phase separation; as a consequence a 3-phase morphology is induced in pBTTT:Me7:PC61BM ternaries (iv) that is dominated by the highly intermixed phase comprising the co-crystal, with the pure pBTTT and PC61BM regions being minority phases.
Having established a microstructural picture of the various binaries and ternaries discussed here, we probed their electronic landscape, and in particular, the photoinduced charge carrier generation and decay in such systems. To this end, we performed Transient Absorption Spectroscopy (TAS) and Time-Resolved Microwave Conductivity (TRMC) measurements. TRMC is a contactless, pump-probe technique that is sensitive to mobile carriers and is a complementary technique to TAS. Charge generation and decay are monitored through the time-resolved changes in absorbed microwave power by the sample, which are related to the transient photoconductance, ΔG(t).20 Note though that with both techniques also relevant information on the phase morphology can be deduced; for instance, Jamieson et al. recently reported TAS data of pBTTT:PC61BM blends of varying composition, where significant generation of long-lived charges was only observed for PC61BM loadings ≥50 wt%,16a corresponding to a composition where all available intercalation sites are filled and relatively phase-pure PC61BM domains are formed; while with TRMC, Rance et al. showed for pBTTT:fullerene blends, that upon formation of pure domains of PC71BM or bis-PC61BM mobile electrons also begin to contribute to the high-frequency mobility and enhanced mobile carrier lifetimes are observed.16b
We first discuss the TAS data obtained on the various pBTTT:PC61BM blends, measured by monitoring the absorbance of pBTTT polarons at 980 nm (Fig. 3a). The observed transients all exhibit similar power-law decays on the 20–400 ns timescale, characteristic of non-geminate recombination of separated (free) polarons.21 The magnitude of these signals is therefore indicative of the yields of dissociated polarons. The largest photoinduced absorption (ΔOD) is observed for the pBTTT:Me7:PC61BM (iv) ternary, indicating that this 3-phase system which comprises a considerable amount of intermixed phase in the form of the co-crystal beside some predominantly pure regions of pBTTT and possibly PC61BM, exhibits the highest separated polaron yield (Table 1).
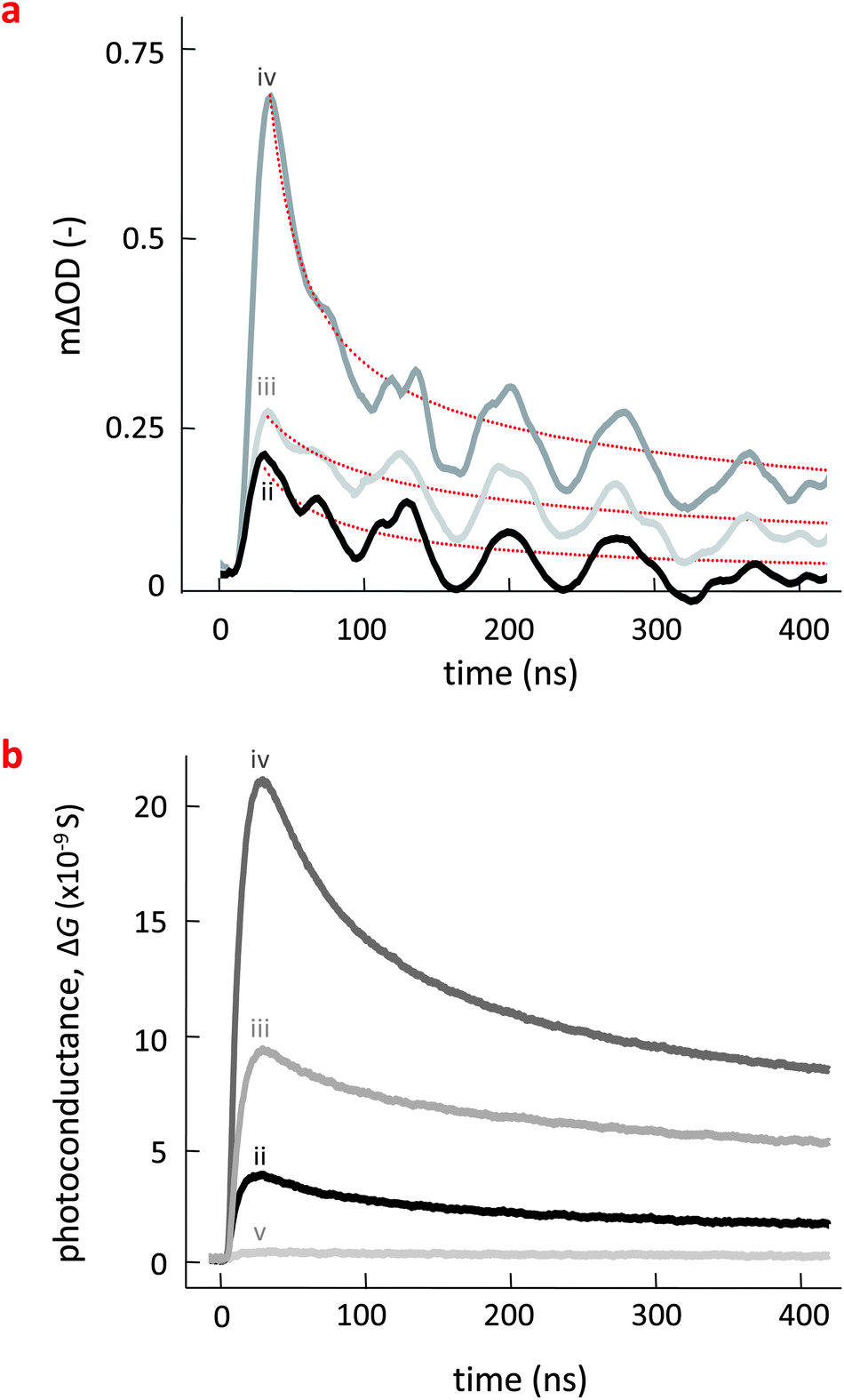 | ||
| Fig. 3 (a) Transient absorption decay for pBTTT:PC61BM (ii), pBTTT:Me12:PC61BM (iii), pBTTT:Me7:PC61BM (iv) showing the photoinduced absorption of long lived pBTTT polarons following pulsed laser excitation. Highest yield was obtained for ternary comprising the Me7 additive (iv) forming a three-phase morphology dominated by the intermixed co-crystal phase. (b) Photoconductance transients, measured by Time-Resolved Microwave Conductivity (TRMC), which follow the same trend in their dynamics and the development of the peak signals as the TAS data: maximum photoconductance is obtained for the ternary system containing Me7 (iv). Data for pBTTT:Me12 (v) binary is also shown. | ||
| pBTTT | pBTTT:Me7 | pBTTT:Me12 | pBTTT:PC61BM | pBTTT:Me7:PC61BM | pBTTT:Me12:PC61BM | |
|---|---|---|---|---|---|---|
| a Calculated from the extrapolation of the peak photoconductance to low absorbed photon flux using eqn (3) (Experimental section). b Measured using time-correlated single-photon counting. c Calculated from the exciton lifetimes for the corresponding pBTTT:Me7 and pBTTT:Me12 sample with and without PC61BM. d Amplitude of transient absorption signals measured at ∼44 ns, probing the polymer cations in all cases. e Calculated from the amplitude of the transient absorption signals (mΔOD) of each system relative to the pBTTT:Me7:PC61BM, assuming that the charge generation yield (ϕCG,TAS) and exciton quenching yield (ϕEQ) are equivalent for pBTTT:Me7:PC61BM—this assumption means that these values represent upper limits estimated using TAS measurements. f Calculated from ϕΣμ assuming that. g Only mobile holes in the polymer contribute to the photoconductance with a high-frequency mobility of 0.015 cm2 V−1 s−1.22 h Calculated as a lower limit the for sum of mobilities, assuming that the exciton quenching yield (ϕEQ) for that sample represents an upper limit for the transient absorption spectroscopy charge generation yield (ϕCG,TAS) for the pBTTT:Me7:PC61BM sample. i Calculated using the exciton quenching yield (ϕEQ) and the transient spectroscopy charge generation yield (ϕCG,TAS) as upper and lower limits, respectively, for the actual charge generation yield measured by TRMC. | ||||||
| ϕΣμa (× 10−3 cm2 V−1 s−1) | 0.38 | 0.40 | 0.43 | 4.99 | 25.82 | 11.31 |
| τ PL (ns) | 1.08 | 0.96 | 0.80 | 0.054 | 0.048 | 0.202 |
| ϕ EQ (%) | — | — | — | 95.1 | 95.0 | 75.4 |
| mΔODd | — | — | — | 0.151 | 0.693 | 0.231 |
| ϕ CG,TAS (%) | — | — | — | 20.7 | 95.0 | 36.1 |
| ϕ CG,TRMC (%) | 2.53 | 2.68 | 2.85 | 33.3 | — | — |
| Σμ (× 10−3 cm2 V−1 s−1) | 15.0g | 15.0g | 15.0g | 15.0g | >27.2h | 15.0–31.3i |
A similar trend is observed in the corresponding photoconductance transients obtained in TRMC (Fig. 3b). The decay dynamics are, for instance, comparable to those observed in TAS, indicating that they are indeed dominated by non-geminate recombination, as discussed above. Also, the highest photoconductance signal is again obtained for the pBTTT:Me7:PC61BM system (iv), followed by the pBTTT:Me12:PC61BM ternary (iii). The lowest signal is observed for the pBTTT:PC61BM binary (ii). [Note that the photoconductance of pBTTT:Me12 (v) is also plotted here to demonstrate that these additives do not change significantly the mobilities of holes inside neat pBTTT. An essentially identical behavior was observed for pBTTT:Me7].
We furthermore estimated the charge-carrier yield combining our TRMC data with those obtained from TAS and photoluminescence decay measurements (Table 1). For this purpose we utilized the fact that the measured peak photoconductance signal is related to the product of the yield for free carrier generation and the sum of the mobilities of free charge carriers (ϕΣμ), as shown in eqn (2) in the Experimental section. By extrapolating the peak photoconductance signal to the linear regime at low light intensities (see ESI Fig. S3†), using eqn (3) in the Experimental section, we can estimate the peak ϕΣμ values for neat pBTTT, and all blends with and without additives. For the neat pBTTT (as well as binaries of the polymer with the additives, i.e. pBTTT:Me7 and pBTTT:Me12), we moreover assume that photoinduced electrons are trapped and that the photoconductance is therefore dominated by mobile holes, which possess a high-frequency mobility of ∼0.01 cm2 V−1 s−1.22 This results in a free-carrier yield in the neat polymer of ∼2.5% which increases slightly (but remains very low) for pBTTT:Me7 (2.7%) and pBTTT:Me12 (2.9%).
The situation changes when PC61BM is added to the polymer or polymer:additive system: for all blend films studied photoluminescence quenching data (steady-state and time-resolved) indicates that the efficiency of PBTTT exciton quenching is ≥75%. As such, the increase in separated polaron yields observed in the TRMC and TAS data with methyl ester addition cannot be assigned to enhanced exciton quenching with respect to the pBTTT:PC61BM binary (indeed Me12 addition slightly reduces the efficiency of exciton quenching). We have previously reported that the long-lived polaron yields observed for the 1:1 (by weight) pBTTT:PC61BM binary in the absence of additives is partially limited by electron–hole recombination in the highly intermixed co-crystal in the film.16a As such the increase in the long-lived separated polaron yields with methyl ester addition can be assigned to increased phase segregation, as indicated from the structural data above, with the presence of relatively pure pBTTT and PC61BM phases assisting the co-crystal in spatial separation of photogenerated electrons and holes.
In the following we attempt to quantify the separated polaron yields and determine the contribution of mobile electrons to the measured photoconductance signals for the blends with PC61BM (Table 1). For the pBTTT:PC61BM binary, which is fully intercalated, we assume that the electrons are unable to resonate at the frequency of the microwave probe beam, consistent with the observations made for blends of pBTTT with PC71BM, another fullerene derivative, prior to the formation of relatively phase-pure acceptor domains at higher PC71BM loadings.16b This results in a predicted free-carrier yield of ∼33%, indicating that intimate mixing of the polymer and fullerene derivative in this blend results in reasonably efficient dissociation of excitons into long-lived free carriers. However, the relative exciton lifetimes in neat pBTTT and pBTTT:PC61BM, as measured using TCSPC, indicate an exciton quenching yield of ∼95% in the blend. The discrepancy between the yields predicted from the PL lifetimes and the photoconductance signal observation suggests that there is an additional process that results in exciton quenching but does not give rise to mobile carriers, presumably due to rapid electron–hole recombination below the response time of our TRMC system. [We note that in previous work the binary pBTTT:fullerene films were subjected to a high temperature anneal at 180 °C that drives extensive formation of the fully intercalated co-crystal and leads to higher yield for free carrier generation measured by TRMC than obtained here.16b The high temperature anneal is not performed here since the scope of our study is to control the morphology by using appropriate additives.]
For the pBTTT:Me7:PC61BM blends, we use the exciton lifetime measured by TCSPC to estimate the maximum possible free carrier yield, also giving a value of 95%. Since the data for the pBTTT:PC61BM binary suggests that intercalation results in a large discrepancy between the exciton quenching and free carrier yield, this value of 95% represents an upper limit for the free carrier yield in pBTTT:Me7:PC61BM; which means that the lower limit for the mobility sum, Σμ, is ∼0.027 cm2 V−1 s−1. This calculation implies that mobile electrons begin to contribute to the measured photoconductance signal in pBTTT:Me7:PC61BM ternaries (contrary to pBTTT:PC61BM systems), likely because some phase-pure PC61BM domains are formed as a result of the expulsion of the fullerene from the pBTTT domains by the Me7 additive (as observed in GIWAXS), confirming our picture of a three-phase morphology in this ternary system.
For pBTTT:Me12:PC61BM samples, we can also use our TCSPC data in combination with the relative transient polaron absorption to estimate upper and lower limits for the free carrier yield. This approach results in values for ϕ of 36 and 75%, which places bounds on Σμ of 0.015 and 0.031 cm2 V−1 s−1. The presence of a reduced amount of intimately mixed pBTTT and PC61BM in this ternary suggests that once again the free carrier yield is likely to tend toward to lower value, which suggests that mobile electrons can also be detected for this ternary, consistent with our structural analysis, which indicates the presence of considerable fractions of relatively phase-pure PC61BM domains.
The question that remains is how these changes in exciton and charge-carrier generation and dynamics affects device performance. To this end, we fabricated OPV devices coating the active layer with a wire-bar coater, the performance of which was highly reproducible (see Fig. 4a and ESI, Table S1†). For the pBTTT:PC61BM binary (ii) we observe poor OPV performance, with low open-circuit voltage (Voc), short-circuit current (Jsc), and fill factor (FF), consistent with previous reports12–14 and in agreement with extensive PC61BM intercalation and the absence of neat domains of the fullerene derivative, which results in rapid electron–hole recombination preventing extraction of charges from the device and thus low photocurrent density.14,16a Devices fabricated using the pBTTT:Me12:PC61BM ternary (iii) as the active layer show a slight improvement in device performance, mainly due to improved Jsc and FF. In accord with the TAS and TRMC data, the optimum performance is achieved for pBTTT:Me7:PC61BM (iv) active layer. Our data, hence, support the picture that an optimum phase morphology is present in systems where charge generation occurs in a finely intermixed polymer–fullerene phase followed by spatial separation of electrons and holes at the interface of this mixed phase and relatively phase-pure domains of the polymer or the fullerene.16a The presence of a relatively large fraction of such an intermixed phase coexisting with pure domains appears favorable. Indeed, the ternaries comprising the Me12 additive, leading to a more pronounced phase-segregation of the pBTTT and PC61BM, seem to be somewhat limited by exciton diffusion. Moreover, our work shows that quite a considerable amount of intermixed phase needs to be present for efficient device performance. So far common belief in the field has generally been that large amounts of phase-pure domains will lead to optimum OPV efficiency. It is also rather unambiguous from our data that fully intermixed systems are detrimental for OPV device performance: the binary pBTTT:PC61BM blend, as well as the ternary pBTTT:Me7:PC61BM blend (iv) after annealing at 150 °C (anniv)—leading also to a fully intercalated blend [see Electron Spectroscopic Images (ESI) in Fig. 4b and temperature-dependent WAXS data in the ESI S4†]—feature the lowest device efficiencies.
 | ||
| Fig. 4 (a) Current density–voltage (J–V) curves for organic photovoltaic (OPV) devices fabricated using active layers comprised of pBTTT:PC61BM (ii), pBTTT:Me12:PC61BM (iii) and pBTTT:Me7:PC61BM (iv). For the 1:1 binary (pBTTT:PC61BM) a comparable device performance as reported by Cates et al. was measured.14 The best device performance was obtained for pBTTT:Me7:PC61BM (iv), which is comprised of three phases. When annealing this specific ternary (denoted here as (anniv)) at 150 °C, which drives fullerene intercalation and, hence, formation of a 1-phase system (see ESI Fig. S4†), a J–V curve similar to the pBTTT:PC61BM binary is obtained (red line). (b) Electron Spectroscopic Images (ESI) of pBTTT:Me7:PC61BM films before and after annealing at 150 °C ((iv) and (anniv), respectively), illustrating that some phase separation is observed in as-cast structures (left and middle images), while essentially no phase contrast is obtained in annealed samples (right image), indicative of a finely intermixed phase. | ||
Finally, we note that Bartelt et al. have recently come to very similar conclusions having investigated a three-phase morphology that included an amorphous, mixed regions, the composition of which was manipulated by annealing.6a The presence of the mixed region was critical and required a specific threshold PCBM concentration to be reached for high performance. Intercalation, as used here, is a natural way of keeping the PCBM concentration in the mixed regions sufficiently high.
Conclusions
We demonstrated that the extent of intercalation of PC61BM into the crystalline domains of pBTTT can be controlled through the use of long fatty acid methyl ester as additives. We showed that the shorter chain additive, Me7, hinders to a certain extent intercalation of PC61BM into the polymer structure, resulting in the formation of a 3-phase system, consisting of predominantly intercalated co-crystals that co-exist with relatively pure pBTTT and PC61BM minority phases. [Note: we do not exclude the presence of partially intercalated co-crystals]. In contrast, the longer chain additive, Me12, appears to be more effective in preventing formation of co-crystalline structures and leads to a more pronounced macro-phase separation of pBTTT and PC61BM. We used TAS and TRMC spectroscopic techniques to show that in the presence of PC61BM, exciton quenching and free carrier generation are enhanced in all blends. In the absence of any additives, i.e. in the case of the fully-intercalated 1:1 pBTTT:PC61BM binary, the efficient exciton quenching does, however, not correlate with quantitative free carrier formation. An optimum free carrier yield is obtained for the partially-intercalated pBTTT:Me7:PC61BM ternary comprising a considerable fraction of co-crystal, which seems to also provide the ideal microstructure for long-range carrier transport. Our results highlight the importance of having a certain amount of relatively pure domains of either the polymer or the fullerene or both.23 These can act as energetic sinks for the generation of charges, relative to amorphous or mixed domains,16a and/or provide an entropic (carrier density gradient) driving force due to formation of the carriers in the mixed phase and lack of electron density in the relatively phase-pure PC61BM phase.16b In the strongly phase-separated pBTTT:Me12:PC61BM blend, free carrier formation seems to be limited by the ability of the pBTTT excitons to reach the interface with PC61BM, resulting in a reduced OPV device performance, despite the presence of some neat PC61BM domains capable of transporting electrons. Reassuringly, the spectroscopic data are consistent with the microstructures predicted by structural techniques and schematically drawn in Fig. 2c. For instance, the three-phase morphology achieved with the Me7 additive will enhance charge formation due to a large donor–acceptor interface within the co-crystal (the intermixed phase), while also providing a network of neat PC61BM to maximize the long-lived carrier density and facilitate carrier extraction in a device.
In summary, we, for the first time, provide donor:acceptor blends that are structurally well-defined whereby we can manipulate the intermixed phases present and, thus, the phase morphology of such systems. Hence, our work is contributing to the on-going discussion stimulated by, for instance, Treat et al. and Collins et al.2b,c and will assist in establishing guidelines to fabricate high-performance reproducible devices by selecting the individual components for their both electronic properties as well their structural compatibility. Despite these observations that the microstructure can be easily controlled by use of suitable additives a number of questions remain: (i) how much of this intermixed phase is actually present for the ternary system with Me7? (ii) What are the dimensions of the formed domains? (iii) How does the phase morphology affect the energy levels of the individual phases? (iv) How does the microstructure affect the photocarrier dynamics and carrier transport; and (v) which is the mechanism behind these additives? These are questions that we are intending to address in future.
Experimental section
pBTTT was synthesized as reported previously (Mw ≈ 66 kg mol−1, Mn ≈ 34 kg mol−1) and measured by GPC against polystyrene standards.24 PC61BM was purchased from Solenne and used as received. Heptanoic acid methyl ester (Me7) and dodecanoic acid methyl ester (Me12) additives were purchased from Fluka and Aldrich, respectively. Solutions were prepared weighing a 1:1 (by mass) mixture of pBTTT and PC61BM, and then adding 10 molar equivalents of the respective additive per monomer unit of the polymer (11 μL of Me7 and 16.4 μL of Me12). The total concentration of the pBTTT:PCBM mixture was 20 mg mL−1 in 1,2-ortho-dichlorobenzene (1,2-oDCB, Aldrich) and all solutions were left stirring for more than 4 hours at 100 °C to fully dissolve the active material. Grazing-angle incidence wide-angle X-ray scattering (GIWAXS) measurements were carried out at D-line at the Cornell High Energy Synchrotron Source (CHESS) at Cornell University. A 0.5 × 0.1 mm beam size with a wavelength of 1.15 Å and wide band pass (1.47%) was generated from double-bounce multilayer monochromator. The incidence angle was 0.15° with respect to the substrate plane, as established by performing X-ray reflectivity using an ion chamber. A 50 × 50 mm charged coupling device (CCD) area detector (Medoptics) with pixel size of 46.9 μm was placed at a distance of 103.6 mm from the sample stage. A 1.5 mm-wide tantalum rod was used to block the intense scattering at low angles of incidence. Films were deposited on glass by wire-bar coating from hot solution (∼85–90 °C) at RT or 35 °C. Variable temperature WAXS measurement was performed at BM26B-DUBBLE Dutch-Belgian beamline of the European Synchrotron Radiation Facility (ESRF) equipped with a Linkam THMS600 temperature-controlled stage. Film was prepared by drop-casting a hot 1,2-oDCB solution onto substrate kept at RT. The obtained film was powdered and then placed between Kapton sheets properly pressed. Scanning transmission X-ray microscopy (STXM) and resonant soft X-ray scattering (R-SoXS) were conducted at Beamline 5.3.2.2 (ref. 25) and 11.01.2,19b of the Advanced Light Source at Lawrence Berkeley National Laboratory, respectively. For these measurements, films were deposited onto polystyrene sulfonate (PSS)-coated glass substrates by wire-bar coating hot solutions (∼85–90 °C) at RT or 35 °C and then floated onto TEM grids. Photoluminescence decays were recorded, for films prepared by drop-casting hot 1,2-oDCB solutions onto substrates kept at either RT or 35 °C, after excitation at 550 nm with a 10 MHz train of pulses (pulse-width: ∼6 ps fwhm; instrument response function: ∼160 ps fwhm) from a high-power fiber laser (Fianium WhiteLase Supercontinuum SC400-2), for emission at 725 nm, with a cooled photon counting photomultiplier tube (Hamamatsu H6279), using the time-correlated single-photon counting technique.26 The PL decays were analyzed using an established non-linear least squares iterative reconvolution procedure,27 where the finite width of the instrument response function was effectively deconvoluted from the measured data to give an overall temporal response of ∼20 ps. Data were fit to a sum of two or three exponentials and the quality of fit judged using stringent statistical procedures.26Photoluminiscence quenching was measured using steady state spectrofluorimeter (Horiba Jobin Yvon, Spex FLuoromax 1), all films were deposited on glass by wire-bar coating from hot solution (∼85–90 °C) at RT or 35 °C and excited at their absorption maxima. Transient absorption (TA) decays were measured by exciting the sample film, under a nitrogen (and oxygen) atmosphere, pumped with a Nd:YAG laser (Lambda Photometrics). The excitation wavelength used was 560 nm, with a pump intensity of 0.4–50 μJ cm−2 and a repetition frequency of 20 Hz. A photodiode (Thorlabs ITC502) was used as the probe. The probe light passing through the sample film was detected with a silicon photodiode (Hamamatsu Photonics, S1722-01). The signal from the photodiode was pre-amplified and sent to the main amplification system (Costronics Electronics). The amplified signal was collected with a digital oscilloscope (Tektronics, TDS220), which was synchronised with a trigger signal of the pump laser pulse from a photodiode (Thorlabs Inc., DET210). To reduce stray light, scattered light and sample emission, appropriate optical cut-off filters were placed before and after the sample. Films were deposited on glass by wire-bar coating from hot solution (∼85–90 °C) at RT or 35 °C. Time-Resolved Microwave Photoconductivity. Films were prepared by drop-casting hot 1,2-oDCB solutions onto substrates kept at either RT or 35 °C. Photocarrier dynamics were studied using time-resolved microwave photoconductivity (TRMC), a contactless, pump-probe technique where both the initial generation of mobile carriers and their eventual decay back to equilibrium are monitored through the time-resolved changes in absorbed microwave power by the sample.20a,28 TRMC measurements were performed at NREL using a system that has been described fully elsewhere.28b,29 The sample was placed in an X-band microwave cavity terminated with a grating reflective to microwaves but transparent to the optical excitation that was used to generate carriers within the film. All drop cast films were excited with 5 ns laser pulses at 550 nm from an optical parametric oscillator (Continuum Panther) pumped by a Q-switched Nd:YAG laser (Continuum Powerlite). The transient change in photoconductance, ΔG(t), was measured via changes in the microwave power, ΔP(t), due to absorption of microwaves by the generated carriers, and is given by: | (1) |
 | (2) |
The sub-linear dependence of the peak photoconductance, ΔGEOP, can be extrapolated to the linear response limit using:
 | (3) |
Acknowledgements
The authors are very grateful to Lee J. Richter and Dean M. DeLongchamp and Paul Smith for highly fruitful and stimulating discussions regarding this manuscript, as well as Richard Sweeney and Pabitra Shakya Tuladhar for their help. This work was supported by UK's Engineering and Physical Sciences Research Council (EP/J500021/1 and EP/G060738/1) and a KAUST Global Collaborative Research Academic Excellence Alliance (AEA) grant. NS is in addition supported by a European Research Council (ERC) Starting Independent Research Fellowship under the grant agreement no. 279587. The TRMC system described here was funded by the Solar Photochemistry Program, Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy (DOE), Grant DE-AC36-08GO28308. The experimental implementation of the TRMC technique was supported by the Laboratory Directed Research and Development (LDRD) Program at the National Renewable Energy Laboratory under task number 06RF1201. STXM and R-SoXS characterization and analysis by JT and HA were supported by Materials Chemistry Program, Materials Sciences and Engineering Division, Office of Basic Energy Sciences, DOE, Grant DE-FG02-98ER45737; data were acquired at Beamline 5.3.2.2 (ref. 25) and 11.01.2 (ref. 19b) of the ALS, which is supported by DOE (DE-AC02-05CH1123). Thanks is also given to David Kilcoyne for instrument support. Finally, the authors extend their thanks to ESRF (Dubble Beamline) for their assistance with the temperature-dependent WAXS experiment.Notes and references
- (a) J. F. Chang, J. Clark, N. Zhao, H. Sirringhaus, D. W. Breiby, J. W. Andreasen, M. M. Nielsen, M. Giles, M. Heeney and I. McCulloch, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 115318 CrossRef; (b) R. Zhang, B. Li, M. C. Iovu, M. Jeffries-El, G. Sauvé, J. Cooper, S. Jia, S. Tristram-Nagle, D. M. Smilgies, D. N. Lambeth, R. D. McCullough and T. Kowalewski, J. Am. Chem. Soc., 2006, 128, 3480 CrossRef CAS PubMed; (c) M. Theander, O. Inganäs, W. Mammo, T. Olinga, M. Svensson and M. R. Andersson, J. Phys. Chem. B, 1999, 103, 7771 CrossRef CAS; (d) K. E. Aasmundtveit, E. J. Samuelsen, W. Mammo, M. Svensson, M. R. Andersson, L. A. A. Pettersson and O. Inganäs, Macromolecules, 2000, 33, 5481 CrossRef CAS; (e) I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281 CrossRef CAS.
- (a) B. Watts, W. J. Belcher, L. Thomsen, H. Ade and P. C. Dastoor, Macromolecules, 2009, 42, 8392 CrossRef CAS; (b) B. A. Collins, E. Gann, L. Guignard, X. He, C. R. McNeill and H. Ade, J. Phys. Chem. Lett., 2010, 1, 3160 CrossRef CAS; (c) N. D. Treat, M. A. Brady, G. Smith, M. F. Toney, E. J. Kramer, C. J. Hawker and M. L. Chabinyc, Adv. Energy Mater., 2011, 1, 1 CrossRef; (d) W. Yin and M. Dadmun, ACS Nano, 2011, 5, 4756 CrossRef CAS PubMed.
- M. Pfannmöller, H. Flügge, G. Benner, I. Wacker, C. Sommer, M. Hanselmann, S. Schmale, H. Schmidt, F. A. Hamprecht, T. Rabe, W. Kowalsky and R. R. Schröder, Nano Lett., 2011, 11, 3099 CrossRef PubMed.
- H. W. Ro, B. Akgun, B. T. O'Connor, M. Hammond, R. J. Kline, C. R. Snyder, S. K. Satija, A. L. Ayzner, M. F. Toney, C. L. Soles and D. M. DeLongchamp, Macromolecules, 2012, 45, 6587 CrossRef CAS.
- X. He, B. A. Collins, B. Watts, H. Ade and C. R. McNeill, Small, 2012, 8, 1920 CrossRef CAS PubMed.
- (a) J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Fréchet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364 CrossRef CAS; (b) J. R. Tumbleston, A. C. Stuart, E. Gann, W. You and H. Ade, Adv. Funct. Mater., 2013, 23, 3463 CrossRef CAS; (c) B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65 CrossRef CAS; (d) B. A. Collins, Z. Li, C. R. McNeill and H. Ade, Macromolecules, 2011, 44, 9747 CrossRef CAS; (e) N. D. Treat, A. Varotto, C. J. Takacs, N. Batara, M. Al-Hashimi, M. J. Heeney, A. J. Heeger, F. Wudl, C. J. Hawker and M. L. Chabinyc, J. Am. Chem. Soc., 2012, 134, 15869 CrossRef CAS PubMed.
- N. C. Miller, E. Cho, R. Gysel, C. Risko, V. Coropceanu, C. E. Miller, S. Sweetnam, A. Sellinger, M. Heeney, I. McCulloch, J. L. Brédas, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2012, 2, 1208 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328 CrossRef CAS PubMed.
- M. L. Chabinyc, M. F. Toney, R. J. Kline, I. McCulloch and M. Heeney, J. Am. Chem. Soc., 2007, 129, 3226 CrossRef CAS PubMed.
- M. Baklar, P. H. Wobkenberg, D. Sparrowe, M. Goncalves, I. McCulloch, M. Heeney, T. Anthopoulos and N. Stingelin, J. Mater. Chem., 2010, 20, 1927 RSC.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488 CrossRef CAS PubMed.
- J. E. Parmer, A. C. Mayer, B. E. Hardin, S. R. Scully, M. D. McGehee, M. Heeney and I. McCulloch, Appl. Phys. Lett., 2008, 92, 113309 CrossRef.
- A. C. Mayer, M. F. Toney, S. R. Scully, J. Rivnay, C. J. Brabec, M. Scharber, M. Koppe, M. Heeney, I. McCulloch and M. D. McGehee, Adv. Funct. Mater., 2009, 19, 1173 CrossRef CAS.
- N. C. Cates, R. Gysel, Z. Beiley, C. E. Miller, M. F. Toney, M. Heeney, I. McCulloch and M. D. McGehee, Nano Lett., 2009, 9, 4153 CrossRef CAS PubMed.
- N. C. Miller, E. Cho, M. J. N. Junk, R. Gysel, C. Risko, D. Kim, S. Sweetnam, C. E. Miller, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, A. Amassian, D. Acevedo-Feliz, C. Knox, M. R. Hansen, D. Dudenko, B. F. Chmelka, M. F. Toney, J. L. Brédas and M. D. McGehee, Adv. Mater., 2012, 24, 6071 CrossRef CAS PubMed.
- (a) F. C. Jamieson, E. Buchaca- Domingo, T. McCarthy-Ward, M. Heeney, N. Stingelin and J. R. Durrant, Chem. Sci., 2012, 3, 485 RSC; (b) W. L. Rance, A. J. Ferguson, T. McCarthy-Ward, M. Heeney, D. S. Ginley, D. C. Olson, G. Rumbles and N. Kopidakis, ACS Nano, 2011, 5, 5635 CrossRef CAS PubMed; (c) T. J. Savenije, W. J. Grzegorczyk, M. Heeney, S. Tierney, I. McCulloch and L. D. A. Siebbeles, J. Phys. Chem. C, 2010, 114, 15116 CrossRef CAS.
- (a) J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619 CrossRef CAS PubMed; (b) I. W. Hwang, S. Cho, J. Y. Kim, K. Lee, N. E. Coates, D. Moses and A. J. Heeger, J. Appl. Phys., 2008, 104, 033706 CrossRef; (c) Y. Gu, C. Wang and T. P. Russell, Adv. Energy Mater., 2012, 2, 683 CrossRef CAS; (d) N. Shin, L. J. Richter, A. A. Herzing, R. J. Kline and D. M. DeLongchamp, Adv. Energy Mater., 2013, 3, 938 CrossRef CAS.
- (a) B. A. Collins and H. Ade, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 119 CrossRef CAS PubMed; (b) A. P. Hitchcock, T. Tyliszezak, I. Koprinarov, H. Stover, W. H. Li, Y. M. Heng, K. Murti, P. Gerroir, J. R. Dutcher, K. Dalnoki-Veress and H. W. Ade, AIP Conf. Proc., 2000, 507, 231 CrossRef CAS.
- (a) T. Araki, H. Ade, J. M. Stubbs, D. C. Sundberg, G. E. Mitchell, J. B. Kortright and A. L. D. Kilcoyne, Appl. Phys. Lett., 2006, 89, 124106 CrossRef; (b) E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Rev. Sci. Instrum., 2012, 83, 045110 CrossRef CAS PubMed.
- (a) J. E. Kroeze, T. J. Savenije, M. J. W. Vermeulen and J. M. Warman, J. Phys. Chem. B, 2003, 107, 7696 CrossRef CAS; (b) M. P. De Haas and J. M. Warman, Chem. Phys., 1982, 73, 35 CrossRef CAS.
- S. Cook, H. Ohkita, Y. Kim, J. J. Benson-Smith, D. D. C. Bradley and J. R. Durrant, Chem. Phys. Lett., 2007, 445, 276 CrossRef CAS PubMed.
- O. G. Reid and G. Rumbles, J. Phys. Chem. Lett., 2013, 4, 2348 CrossRef CAS.
- D. Veldman, O. Ipek, C. Meskers, J. Sweelssen, M. Koetse, S. Veenstra, J. Kroon, S. v. Bavel, J. Loos and R. Janssen, J. Am. Chem. Soc., 2008, 130, 7721 CrossRef CAS PubMed.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. Chabinyc, R. Kline, M. McGehee and M. Toney, Nat. Mater., 2006, 5, 328 CrossRef CAS PubMed.
- A. L. D. Kilcoyne, T. Tyliszczak, W. F. Steele, S. Fakra, P. Hitchcock, K. Franck, E. Anderson, B. Harteneck, E. G. Rightor, G. E. Mitchell, A. P. Hitchcock, L. Yang, T. Warwick and H. Ade, J. Synchrotron Radiat., 2003, 10, 125 CrossRef CAS PubMed.
- D. Phillips and D. V. O'Connor, Time-Correlated Single-Photon, Academic Press, London, 1984 Search PubMed.
- A. Grinvald and I. Z. Steinberg, Anal. Biochem., 1974, 59, 583 CrossRef CAS.
- (a) G. Dicker, M. P. de Haas, L. D. A. Siebbeles and J. M. Warman, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 045203 CrossRef; (b) A. J. Ferguson, N. Kopidakis, S. E. Shaheen and G. Rumbles, J. Phys. Chem. C, 2008, 112, 9865 CrossRef CAS.
- J. Piris, N. Kopidakis, D. C. Olson, S. E. Shaheen, D. S. Ginley and G. Rumbles, Adv. Funct. Mater., 2007, 17, 3849 CrossRef CAS.
- T. J. Savenije, M. P. de Haas and J. M. Warman, Z. Physiol. Chem., 1999, 212, 201 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mh00125c |
| This journal is © The Royal Society of Chemistry 2014 |