Magnesium–air batteries: from principle to application
Tianran
Zhang
,
Zhanliang
Tao
and
Jun
Chen
*
Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Synergetic Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China. E-mail: chenabc@nankai.edu.cn
First published on 10th September 2013
Abstract
Metal–air batteries are important power sources for electronics and vehicles because of their remarkable high theoretical energy density and low cost. In this paper, we introduce the fundamental principles and applications of Mg–air batteries. Recent progress in Mg or Mg alloys as anode materials and typical classes of air cathode catalysts for Mg–air batteries are reviewed. In the meantime, different compositions of the electrolyte are also compared. The design of electrode materials both for anodes and cathodes of Mg–air batteries is discussed for further performance improvement. It is noted that in the development of rechargeable Mg–air batteries, bi-functional catalysts with reversible oxygen reduction and evolution reactions are facing challenges and it is worthwhile devoting much effort to this.
 Jun Chen | Jun Chen is the Chair Professor on energy materials chemistry at Nankai University, the Director of the Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education). He received his B.S. and M.S. degrees from Nankai University in 1989 and 1992, respectively, and his Ph.D. from Wollongong University (Australia) in 1999. He held the NEDO fellowship at the National Institute of AIST Kansai Center (Japan) from 1999 to 2002. His research activity focuses on nanomaterials, electrochemistry, batteries, fuel cells and solar cells with efficient energy storage and conversion. |
Introduction
Metal–air batteries have attracted much attention as promising electrochemical energy storage and conversion devices due to their high theoretical energy density and low cost.1–3 Among various types of metal–air batteries, lithium–air and zinc–air batteries have been investigated,4–7 while magnesium (Mg)–air batteries have not been explored as much. The basic structure of a Mg–air battery is shown schematically in Fig. 1, composed of an Mg (or Mg alloy) anode, an air cathode and a saline electrolyte. The reactions involved in Mg–air batteries are as follows:| Anode: Mg → Mg2+ + 2e− | (1) |
| Cathode: O2 + 2H2O + 4e− → 4OH− | (2) |
| Total: 2Mg + O2 + 2H2O → 2Mg(OH)2 | (3) |
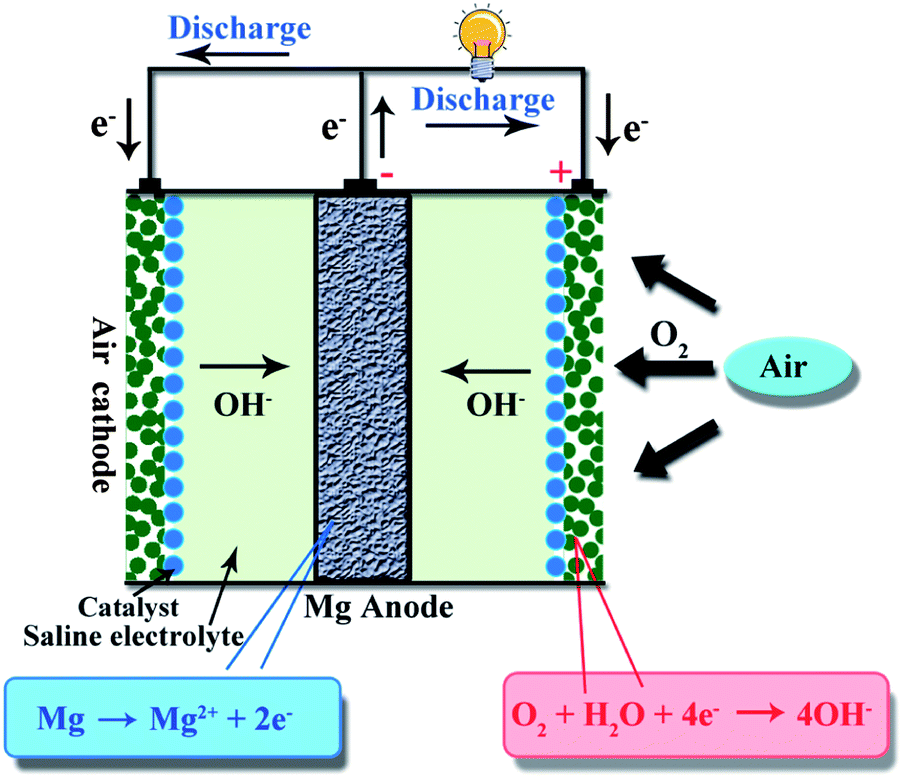 | ||
| Fig. 1 Typical structure and working principle of an Mg–air battery. | ||
During the discharge process, the anode Mg is oxidized to Mg2+, producing two electrons, while at the opposite electrode, O2 passes through the air cathode and is then reduced to OH− by reaction with H2O and electrons.8 The theoretical voltage of the Mg–air battery is 3.1 V and the specific energy density is 6.8 kW h kg−1, as summarized in Table 1. The Mg–air battery is a promising electrochemical energy storage and conversion device since Mg is abundant on the earth, has a high reaction activity, is light weight, has low toxicity and has relatively high safety.9–11 Generally speaking, the present Mg–air battery is a primary battery. Indeed, the Mg–air battery can be re-used mechanically by replacing the spent Mg anode and electrolyte with a fresh Mg anode and electrolyte, making it “refuelable”.12 However, rechargeable Mg–air batteries with reversible oxygen reduction and evolution reactions are facing challenges and it is worthwhile devoting much effort to this.
| Selected batteries | Theoretical cell voltage (V vs. SHE) | Theoretical specific energy densitya (kW h kg−1) |
|---|---|---|
| a The theoretical specific energy density (εM = −nFE/∑M, where F is the Faraday constant, E is the reaction potential and M is the molar mass of reactants) is calculated excluding O2 for metal–air batteries. | ||
| Li-ion (0.5C6Li + Li0.5CoO2 ↔ 3C + LiCoO2) | 3.8 | 0.387 |
| Li–air (2Li + O2 → Li2O) | 2.91 | 13.0 |
| Li–sulfur (2Li + S ↔ Li2S) | 2.2 | 2.6 (ref. 4) |
| Zn–air (2Zn + O2 → 2ZnO) | 1.65 | 1.3 |
| Mg–air (Mg + 0.5O2 + H2O → Mg(OH)2) | 3.1 | 6.8 |
Though Mg–air batteries have a relative high voltage and energy density, there are still scientific problems limiting their widespread application. The main issue of Mg–air batteries is the high polarization and low coulombic efficiency. For example, the working voltage is normally below 1.2 V and the practical specific energy is lower than ten percent of the theoretical one. This issue is caused by two aspects: the corrosion of the Mg anode arising from the reaction of Mg and the electrolyte, and the sluggish kinetics of the oxygen reduction reaction in the air cathode.
To overcome the existing problems of Mg–air batteries, more investigation and deep understanding are obviously needed. Herein, we review Mg–air batteries from basic principles to applications. In particular, the corrosion mechanism of Mg, the recent progress on Mg anode materials, the characterization of the electrolyte, the air electrode structure and the catalyst design for the oxygen reduction reaction are summarized to provide a more in-depth understanding of Mg–air batteries. This should shed light on further research and design of the electrodes, electrolytes, and structures for both primary and rechargeable Mg–air batteries. This review is organised in the following sequence: the Mg anode, electrolyte, air cathode, and applications are introduced, followed by conclusions.
Mg anode
The Mg anode plays a critical role in Mg–air batteries. During discharge, Mg in the anode is dissolved to produce Mg2+, producing two electrons (eqn (1)). The standard electrode potential of the reaction is −2.37 V and this electrochemical reaction can produce 2.2 A h g−1 of capacity. However, for Mg–air batteries, a high level of polarization is displayed. One issue is the side-reaction in the Mg anode, that is to say, the corrosion of Mg. Therefore, in order to improve the performance of Mg–air batteries, it is necessary to discuss the corrosion mechanism of the Mg anode. In the Pourbaix diagram of a Mg–water system at 25 °C,13,14 as shown in Fig. 2a,15 Mg is thermodynamically stable below −2.37 V with a large pH range. Unfortunately, the area is below the region of water stability. Mg2+ is the stable substance in a pH range from 0 to 11, above which Mg(OH)2 is the stable one. Consequently, in neutral and acidic aqueous environments, Mg spontaneously converts to Mg ions. These further react with water through an electrochemical reaction to produce magnesium hydroxide and hydrogen gas, often involving micro-galvanic coupling between cathodic areas (where the electrochemical reduction of water happens) and anodic areas (where the electrochemical oxidation of Mg occurs).16 This process is the corrosion of Mg with the following three parts:| Mg → Mg2+ + 2e− (anodic reaction) | (4) |
| 2H2O + 2e− → H2 + 2OH− (cathodic reaction) | (5) |
| Mg2+ + 2OH− → Mg(OH)2 (product formation) | (6) |
| Mg + 2H2O → Mg(OH)2 + H2 | (7) |
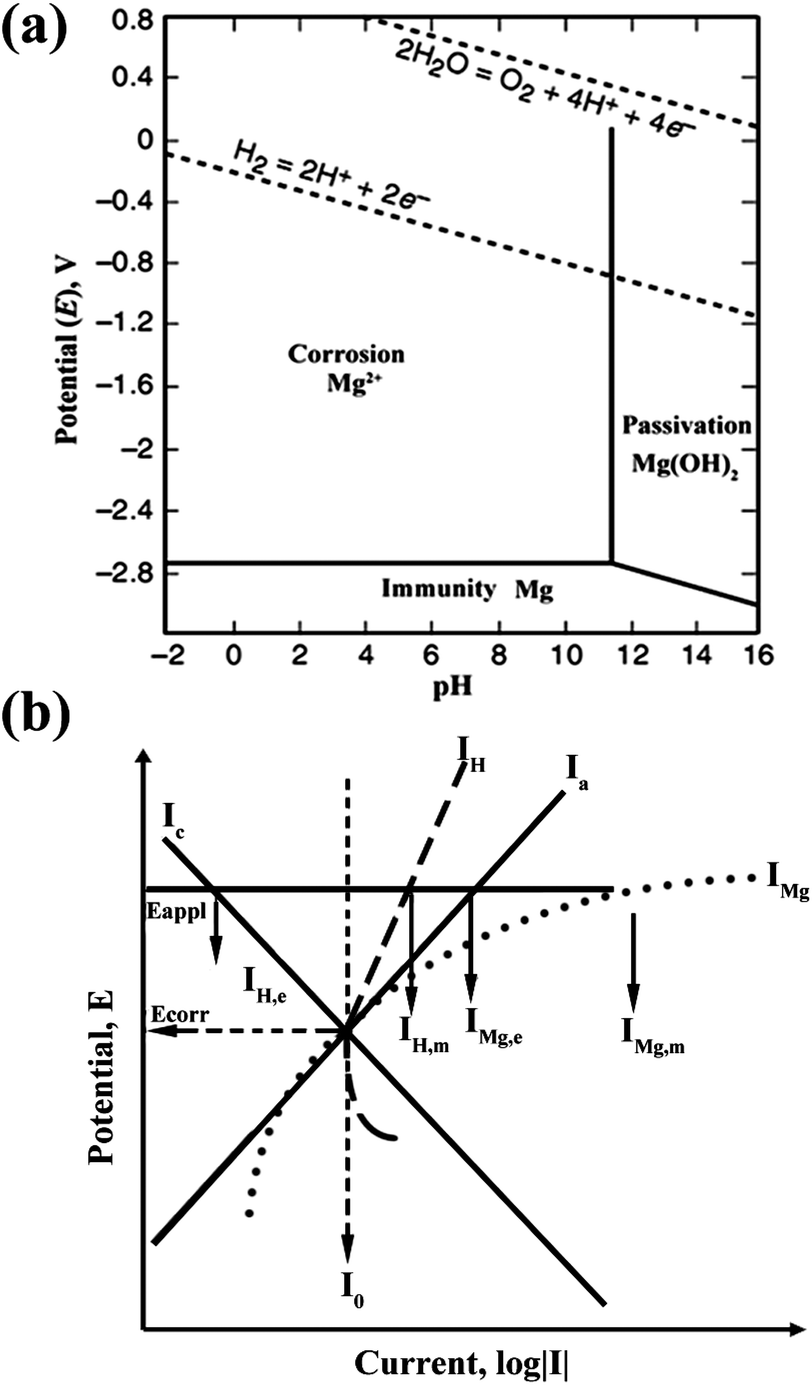 | ||
| Fig. 2 (a) Pourbaix diagram for the magnesium–water system at 25 °C. Adapted from ref. 15. (b) Negative difference effect (NDE) of magnesium. Reproduced from ref. 16 by permission. Copyright 2003, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Besides the HER, other factors also give rise to the corrosion of Mg. The negative difference effect (NDE) is an important one. Generally, the corrosion reaction is either an anodic or cathodic process. For most metals such as iron, zinc or steel, when the applied potential increases, the anodic current rises, accompanied by the cathodic current decreasing simultaneously (Ia and Ic curves in Fig. 2b). However, Mg displays quite different behaviour. It is clearly shown in Fig. 2b that the anodic current IMg (corresponding to eqn (4)) and the cathodic current IH (corresponding to eqn (5)) both rise with an increase of the applied potential. This strange performance of Mg is called the negative difference effect (NDE). As the NDE increases the hydrogen evolution current, it accelerates the corrosion of Mg. The reasons for the NDE have been investigated for decades and different possible mechanisms are proposed. However, none of the mechanisms can completely interpret the NDE.16,17
Another factor of Mg corrosion is galvanic corrosion, which is caused by the impurities in Mg plate such as metallic iron, nickel, or copper. These elements construct micro-cells with Mg and expedite the cathodic reaction in eqn (5), resulting in further corrosion of the Mg plate.8 It is found that the galvanic corrosion is directly associated with the impurity content.16 In particular, once the impurity content exceeds the “tolerance limit”, the corrosion rate is greatly increased while with a lower impurity content, the corrosion rate remains low. For example, the tolerance limits of Fe, Ni, and Co are about 0.2 wt%,16 indicating that trace amounts of these elements are harmful to Mg.
According to the discussions above, the HER and NDE as well as impurities are the main factors for the corrosion of Mg. A suitable Mg anode material requires a lower HER reaction rate, a smaller NDE and no impurities. Meanwhile, the by-product Mg(OH)2 (eqn (7)) should also be easily removed from the anode in order to gain refreshed active sites. Therefore, searching for new Mg-based anode materials with high reaction activity and slow corrosion rate is important for the development of Mg–air batteries. So far, efforts have focused on two directions as summarized in Table 2. One is alloying Mg with other metals to inhibit the HER and the other is to improve the properties of Mg itself.
| Type | Main constituents | Morphology | Properties | Ref. | |
|---|---|---|---|---|---|
| a AZ31 (96 wt% Mg, 3 wt% Al and 1 wt% Zn), AZ61 (93 wt% Mg, 6 wt% Al and 1 wt% Zn), AZ91 (90 wt% Mg, 9 wt% Al and 1 wt% Zn). b AM50 (∼94 wt% Mg, 5 wt% Al, 0.6 wt% Mn), AM60 (∼93 wt% Mg, 6 wt% Al, 0.6 wt% Mn), MA8M06 (∼97 wt% Mg, 1.3 wt% Mn, 0.12 wt% Zn, 0.12 wt% Al, 0.2 wt% Fe). | |||||
| Simple Mg | Commercial Mg | Mg | — | High corrosion rate, large negative difference effect | 1 |
| Nano/mesoscale Mg | Mg | Nanospheres | Better corrosion resistance and high current density as well as higher rate discharge ability than commercial Mg | 19 | |
| Nanoplates | |||||
| Nanorods | |||||
| Urchin-like | |||||
| Mg-alloy | AZ31, AZ61, AZ91a | Mg/Al/Zn | — | Better corrosion resistance, better strength and tarnish resistance and higher working voltage than commercial Mg | 12 and 18 |
| AM50, AM60, MA8M06b | Mg/Al/Mn | — | Better corrosion resistance, smaller crystalline grains and higher working voltage than commercial Mg | 20 | |
| Mg–Li alloys | Mg/Li | — | Better corrosion resistance, higher energy density and higher working voltage than commercial Mg | 18 and 21 | |
Alloying Mg with other metals such as Al, Mn, or Zn, which prevent the hydrogen evolution reaction, has attracted a lot of attention. With the fast development of metallurgical technology, Mg alloys have been widely explored for Mg–seawater batteries.22,23 The effect of alloyed metals on the corrosion rate of Mg alloys has been deeply investigated.15 One of the most interesting series of Mg alloys is Mg–Al alloys. In the phase diagram of Mg–Al alloys,24 when the concentration of Al is lower than 10 wt%, the Mg phase is obtained after solidification with a hexagonal closely-packed hcp structure. The Al introduced to the Mg will not only enhance the physical strength but also prevent the HER. Therefore, Mg–Al alloys suppress the self-corrosion of the anode.
Meanwhile, the addition of a small amount of zinc to Mg–Al alloys to produce Mg–Al–Zn alloys such as AZ31, is widely employed for Mg–air batteries. The discharge testing of AZ31 in neutral 3.5 wt% NaCl solution with 5 mA cm−2 current density shows that the operating voltage is 1.125 V and the specific discharge capacity is 1125 mA h g−1.18 In comparison with AZ31, the same type of AZ61 shows better performance in Mg–air batteries with a 7 wt% NaCl electrolyte, where the cell voltage is about 1.28 V.12
Moreover, Mg–Al alloys containing manganese are also an important option. The existence of manganese reduces the particle size of the alloys. The performances of AM50, AM60, and MA8M06 as anode materials of Mg–air batteries have been investigated, among which MA8M06 is the best one with a higher voltage and more positive corrosion potential even than the AZ series alloys.20
Recently, alloying Mg with Li has been considered for potential battery anodes due to the more negative standard potential and high Faradic capacity, as well as the high specific energy of Mg–Li alloys.18,21 Taking Mg–14Li–1Al–0.1Ce as an example, a higher electrochemical activity and lower self-corrosion rate are observed than for Mg and AZ31. Fig. 3 shows the discharge behaviour of Mg–air batteries with Mg–14Li–1Al–0.1Ce, pure Mg, and AZ-31 as the anodes at a current density of 2.5 mA cm−2. The specific capacity of the Mg–air cell with Mg–14Li–1Al–0.1Ce anode in 3.5 wt% NaCl solution is 2076 mA h g−1 and its operating voltage is 1.272 V, higher than that with pure Mg and AZ-31 anodes.18 In summary, Mg alloys show high performance as anode of Mg–air batteries. However, Mg alloys still suffer from corrosion, leading to lower working voltages in comparison with the theoretical ones. Thus, it is still necessary to further reduce the corrosion rate and improve the reaction activity of the anode.
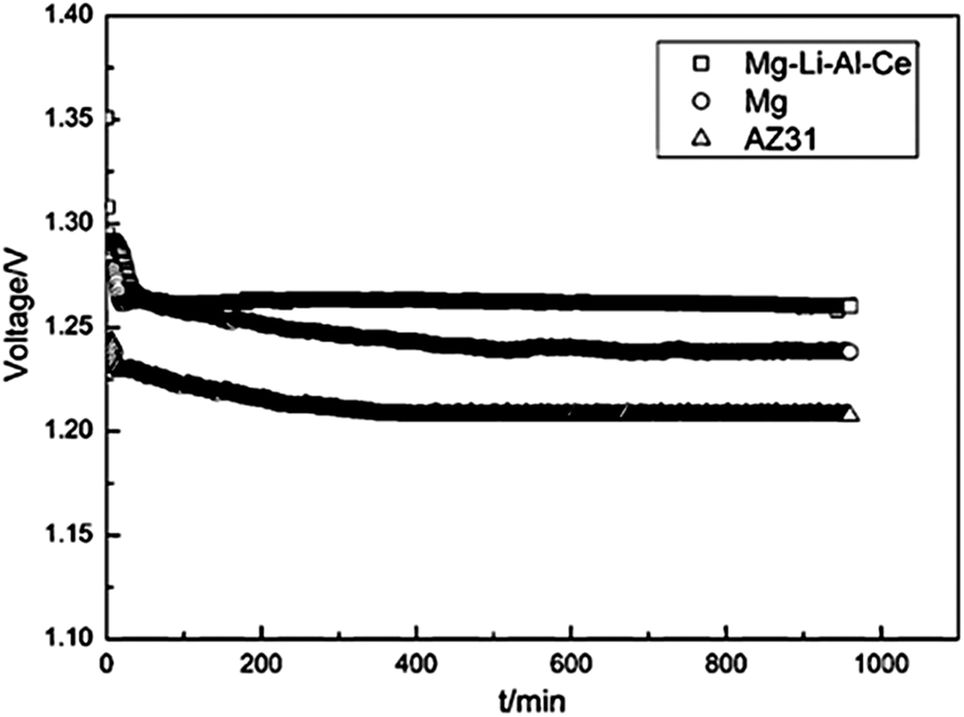 | ||
| Fig. 3 Discharge behaviour of Mg–air batteries with different anode materials at a current density of 0.5 mA cm−2. Reproduced from ref. 18 by permission. Copyright 2011, Elsevier. | ||
Another strategy to improve the performance of the Mg anode is to modify the intrinsic properties of Mg itself. In this case, the purity and morphology are important factors. As mentioned above, commercial Mg plate contains harmful impurities such as Fe, Co or Ni. Reducing the amount of impurities in the Mg plate is helpful for improving the performance of Mg–air batteries. High-purity Mg (99.99%) shows better performance as an anode with higher corrosion resistance (meaning lower corrosion rate) than even some Mg alloys.20
In addition to just suppressing the corrosion, increasing the activity of the anode is also important. It is accepted that nanostructures exhibit novel physical and chemical properties that are not present in the corresponding macroscale structures.25,26 Furthermore, nanomaterials have already been investigated in batteries and fuel cells.27–29 In Mg–air batteries, with controllable morphology, Li et al. explored the electrochemical activity of micro/nanostructured Mg anodes in Mg–air cells.19 Samples with microspheres, microplates, nanorods and sea-urchin-like nanostructures prepared by the vapour-transport method have shown better performance in comparison with that of commercial Mg powders in the Mg(NO3)2 + NaNO2 electrolyte (Fig. 4). Among the micro/nanostructures, sea-urchin-like Mg displays the highest energy density and high-rate performance. The results demonstrate that porous and network nanostructures increase the number of active sites and accelerate the sedimentation of the by-product (Mg(OH)2) in a certain electrolyte, and hence improve the performance of the Mg–air battery. One thing that should be mentioned is that a high specific area of the Mg anode does enhance the activity of the cell reaction, but at the same time it also increases the corrosion rate of Mg. Therefore, just tuning the Mg anode is not enough and a suitable electrolyte is highly desired, which is discussed later.
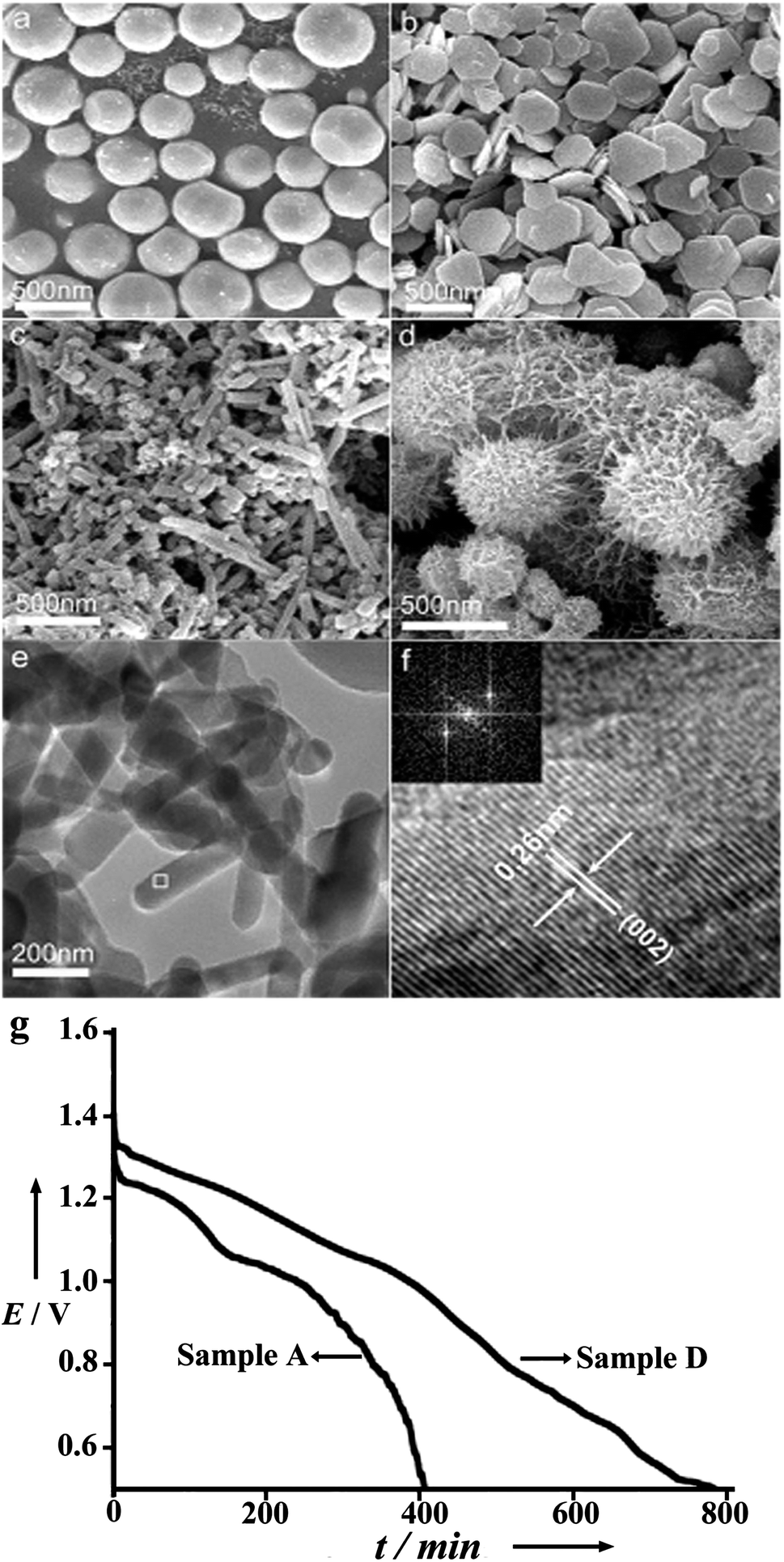 | ||
| Fig. 4 Typical scanning electron microscopy (SEM) images of as-prepared Mg: (a) microspheres, (b) microplates, (c) nanorods, (d) sea-urchin-like nanostructures. (e) Representative transmission electron microscopy (TEM) image of nanorods and (f) high-resolution transmission electron microscopy (HRTEM) image of the white square marked in (e) with the inset showing the corresponding fast Fourier transform (FFT) pattern. (g) Representative discharge curves of the Mg–air batteries made from Mg microspheres and sea-urchin-like nanostructures at a constant current of 0.5 mA and a temperature of 25 °C. Reproduced from ref. 19 by permission. Copyright 2006, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
According to the previous efforts on Mg anode materials, it is concluded that for developing Mg anodes with high-performance, a small size of Mg particles, especially with nanostructures, promotes the reaction activity. The high specific surface of Mg particles results in more Mg atoms being exposed to the electrolyte and the amount of Mg(OH)2 by-product is reduced quickly. In the meantime, Mg alloying with elements such as Al, Mn or Li suppresses the HER and reduces the self-corrosion of Mg. Therefore, hollow or porous nanostructures of Mg or Mg alloys should be promising candidates for Mg–air batteries. Moreover, with the rapid development of computer technology, first-principles calculations have been widely used for designing materials, especially for batteries.30,31 For Mg-based materials, efforts have been devoted to energy storage and the electronic structures of Mg alloys.8,32,33 It is expected that using calculations as a tool, functional Mg anode materials are to be designed.
Electrolyte
In general, the electrolyte of an Mg–air battery is a neutral saline aqueous solution that contacts with the electrodes, greatly influencing the electrode reactions. Since the high polarization and low coulombic efficiency of the Mg–air battery is not only related to the electrodes but also to the electrolyte, choosing a suitable electrolyte is crucial to the performance of Mg–air batteries.For the anode reaction, the electrolyte has a large influence on the corrosion of Mg. Table 3 summarizes the corrosion potential of “bare” Mg in various aqueous solutions. It is clear that Mg has a higher resistance in alkaline solution than in acidic or neutral solution. Recent research also shows that by tuning the pH value above 10, an electrolyte consisting of a near-saturated aqueous solution of LiCl, MgCl2 or mixture of the two salts can suppress the hydrogen evolution reaction and promote the corrosion resistance of Mg.34 The reason for the high corrosion resistance of Mg in the alkaline solution is due to the partial formation of Mg(OH)2 film on the surface of the Mg or Mg alloys. This could protect the anode active material from corrosion. However, too much Mg(OH)2 film on the electrode prevents further reaction of the anode, leading to a delayed response to an increase in the load.1 Thus, a neutral electrolyte is usually used in Mg–air batteries.
| Electrolyte | E r (V vs. NHE) |
|---|---|
| NaCl | −1.72 |
| Na2SO4 | −1.75 |
| HCl | −1.68 |
| HNO3 | −1.49 |
| NaOH | −1.47 |
| NH3 | −1.43 |
In addition to the influence of pH, the anions in the aqueous solution also play an important role. A large number of electrolytes containing different anions have been screened with the purpose of understanding the influence of the anions.36 Upon comparison with NaCl, KHCO3, NH4NO3, NaNO3, NaNO3 + HNO3, Mg(NO3)2·6H2O + NaNO2, NaNO2, Na2SO4, MgCl2, and MgBr2, the Mg(NO3)2·6H2O + NaNO2 electrolyte can result in low, uniform corrosion and low anodic polarization of Mg at useful current densities as well as fast coagulation of the by-products (Mg(OH)2).19 It has been found that Cl− and SO42− are more likely to attack Mg and thus lead to high corrosion rates, while NO3− or NO2− ions, though they also attack magnesium, do not do so to the same extent as chlorides.17 Therefore, Mg(NO3)2·6H2O + NaNO2 is more suitable than other salts.
Another factor related to the electrolyte is the concentration of salt in the electrolyte. The corrosion rate of pure Mg in seawater is about 0.25 mm per year while that in 3 M MgCl2 solution is 1200 times larger.35 Therefore, a low concentration of salt is usually necessary. Also, adjusting the metal/electrolyte interface is a relevant method to promote resistance to corrosion. Khoo et al.37 used phosphonium chloride ionic liquid and water as an electrolyte and tested the discharge performance of an Mg–air battery. The results demonstrated that the amorphous gel-like interface formed on Mg led to a level of passivation when the cell was at open circuit. This stabilized the metal/electrolyte interface and enhanced the performance of the Mg–air battery.
In addition to searching for new electrolytes, some hydrogen evolution inhibitors such as stannates, quaternary ammonium salts, dithiobiuret and their mixtures can be added into the electrolyte to suppress the HER. A mixture of stannate and quaternary ammonium salt can improve the efficiency of the AZ31 alloys, leading to a higher voltage potential.8 Therefore, it is worth engaging in research focused on developing inhibitors for the HER.
Air cathode
The performance of Mg–air battery is tightly associated with the air cathode. The structure of a typical air cathode is composed by four layers: a waterproof breathable layer, a gas diffusion layer, a catalyst layer and a current collector layer, as shown in Fig. 5. The waterproof layer is usually a water-repellent porous substance (e.g. paraffin wax or Teflon), which is used to separate the electrolyte and air, and at the same time, is permeable only to O2 and blocks CO2 and H2O. The gas diffusion layer has a highly porosity and high electronic conductivity, and is usually made from acetylene black containing hydrophobic materials such as polytetrafluoroethylene (PTFE). The catalyst layer is composed of active catalysts for the oxygen reduction reaction (ORR), which are dispersed in the surface of the gas diffusion layer near the electrolyte. The commonly used catalysts are noble metals (Ag). In addition to the anode corrosion, the low coulombic efficiency of the Mg–air battery is attributed to the overpotential of the air cathode caused by the sluggish kinetics of the ORR. Therefore, it is essential to improve the performance of the air cathode.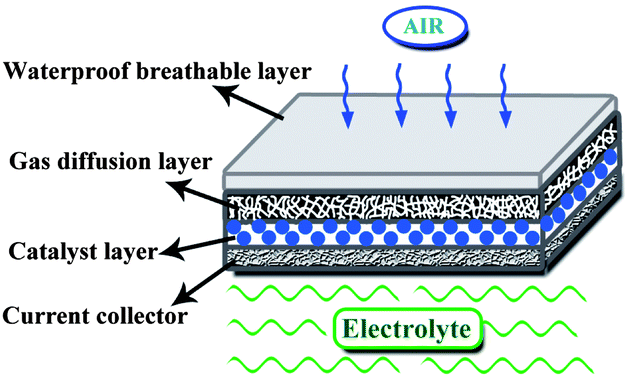 | ||
| Fig. 5 Structure of a typical air cathode with four layers. | ||
In a neutral electrolyte, oxygen is reduced to OH− at the interface of the gas–solid–liquid ternary phases in the air cathode. As the ORR occurs in the three-phase interface, it involves complex physical and chemical reactions. In general, the possible reaction pathways involved in the ORR in a neutral solution can be expressed as:38
| O2 + 2H2O + 4e− → 4OH− | (8) |
| O2 + H2O + 2e− → HO2− + OH− | (9) |
| HO2− + H2O + 2e− → 3OH− | (10) |
| 2HO2− → 2OH− + O2 | (11) |
The standard electrode potential of the oxygen reduction reaction (eqn (8)) is 0.44 V. However, a large overpotential and high polarization are usually observed for the reaction, leading to a bad performance of the Mg–air battery. This is due to the sluggish kinetics of the ORR. Therefore, to lower the overpotential, highly efficient catalysts are required. As the direct four-electron process (eqn (8)) is the most efficient, a suitable catalyst should have direct four-electron catalytic ability. Additionally, the catalyst should be abundant on the earth and have a low cost.
Many types of catalysts have been investigated for the ORR, either in acidic or alkaline solutions. However, research focused on developing catalysts for neutral solutions is limited. According to previous literature,39 the performances of catalysts in neutral solutions are comparable to that in weak alkaline solution. Therefore, the catalysts suitable for weak alkaline solutions are also suitable for neutral solutions. Herein, we only briefly review the catalysts used in neutral and alkaline solutions while those for acidic solutions have been discussed in other comprehensive reviews.3,38
The catalysts used for metal–air batteries are summarized in Table 4. Among these, platinum shows superior ORR catalytic activity. The effects of particle size and crystal plane have been considered, showing that a catalyst consisting of carbon supported 2–4 nm Pt particles with the (111) surface is the state-of-the-art ORR catalyst.40,41 However, the high price and the scarcity of Pt results in a high cost of the metal–air battery, meaning that the search for low Pt and non-Pt catalysts receives a lot of attention.38
| Type | Representative examples | Remarks | Ref. | |
|---|---|---|---|---|
| a r-GO is reduced graphene oxide. b N-GA is N-doped graphene aerogel. c CoPcF16 is fluorinated cobalt phthalocyanine. | ||||
| Noble metals | Pt-based | Pt | High catalytic activity; low overpotential and large limited current density; four-electron reaction mechanism; commercially used catalyst; high price and scarcity; not quite stable | 40 |
| Pt alloys | Higher activity than pure Pt; four-electron reaction mechanism; lower price; better stability | 45–51 | ||
| Non-Pt | Pd, Au, Ag,… | Lower activity than Pt; four electron reaction mechanism; high price and scarcity | 52–54 | |
| Non-Pt alloys | Higher activity than pure noble metals and comparable to Pt; four-electron reaction mechanism; lower price | 55–57 | ||
| Carbon-based materials | Carbons | Porous carbon, nanotubes, graphene | Much higher overpotential and smaller limited current density than metals; two-electron reaction mechanism; better stability; usually used as substrates | |
| Modified carbons | N-doped graphene, N-doped nanotubes, P-doped graphene,… | Much improved catalytic activity over pure carbons and close to Pt; quasi-four-electron reaction mechanism; better durability; promising substitute for noble metal; difficult to produce | 58–63 | |
| Transition metal oxides | Pure oxides | MnOx, CaMnO3, CoMn2O4,… | Lower electrocatalytic activity than Pt; serial two-electron plus two-electron mechanism; stable; low cost; bad electronic conductivity | 39 and 64–66 |
| Complexes | Co3O4/r-GO,a MnCoO/r-GO,a Fe3O4/N-GAs,b… | High activity and comparable to Pt; improved electron conductivity over metal oxides; four-electron reaction mechanism; stable; promising substitute for noble metal | 67–69 | |
| N-containing metallic complex | FeTMPP/C, CoTMPP/C, CoPcF16@Ag/C,…c | High activity and comparable to Pt/C; better durability than Pt; low cost; four-electron reaction mechanism | 70 and 71 | |
For low-Pt catalysts, alloying Pt with other cheap transition metals has attracted much attention, due to better activity than pure Pt. With the help of spectroscopy and theoretical calculation,42 the reason for the enhanced activity of Pt-alloys is well understood and a trend for their ORR activity is proposed, as shown in Fig. 6a. The tight binding of the intermediates such as OH* and O* on the surface is the main reason for the activity attenuation of Pt-based catalysts.43 The weakening the O* and OH* adsorption, which are good descriptors for ORR activity, is a principle reason for the higher activity of Pt alloys. According to Fig. 6a, a better catalyst should involve O atom binding strengths 0–0.4 eV weaker than the Pt (111) surface with the optimum value at about 0.2 eV. Based on these findings, the Pt3Y alloy is outstanding with the highest activity.42 Moreover, the O adsorption energy is also related to the Pt d-band-center, where lowering the d-band-center can result in faster Pt–O electro-reduction and increase the ORR kinetics.44 For example, X-ray absorption near edge structure (XANES) measurements of PtCo/C indicate a lower variation of the Pt 5d band vacancy from lower to higher potentials due to the presence of Co. This increases the Pt electronegativity and decreases the Pt–OH coverage, resulting in a higher catalytic activity than Pt/C.45 Besides, tuning the morphology and structure of Pt alloys also enhances the activity and stability toward the ORR. A “spongy” structure with numerous voids is obtained via chemical dealloying of intermetallic nanoparticles of Cu3Pt, resulting in enhanced specific and mass activities and higher stability relative to Pt/C.46 Recently, a new class of Pt–Co nanocatalysts composed of ordered Pt3Co intermetallic cores with a 2–3 atomic-layer-thick Pt shell has been developed with high activity and stability, which is attributed to the Pt-rich shell and especially to the stable intermetallic Pt3Co core arrangement.47 This provides a new direction for catalyst performance optimization. Furthermore, some new and low cost preparation methods of Pt alloys have been developed. Using Pt3Fe nanoparticles as an example, a surfactant-free NP–KCl matrix method (NP stands for nanoparticle) has been developed for the synthesis of nanoparticles with controlled sizes and structures. Insoluble KCl serves as a matrix that avoids particle agglomeration and controls the coalescence of nanoparticles during thermal annealing.48 One thing that should be mentioned is that the transition metals (Fe, Co, Ni) in Pt-alloys, which may be dissolved during the ORR, have little effect on the Mg anode. In the Mg anode, the impurities are Fe, Ni and Co metals, while the cathodic dissolved transition metal is mainly in the form of ion states. As interpreted above, Fe, Ni and Co metals facilitate the hydrogen evolution reaction due to the construction of micro-cells with Mg, but the ion states (Fe2+, Co2+ and Ni2+) can't form such micro-cells. Therefore, these transition metals in Pt-alloys will not induce the corrosion of the Mg anode.
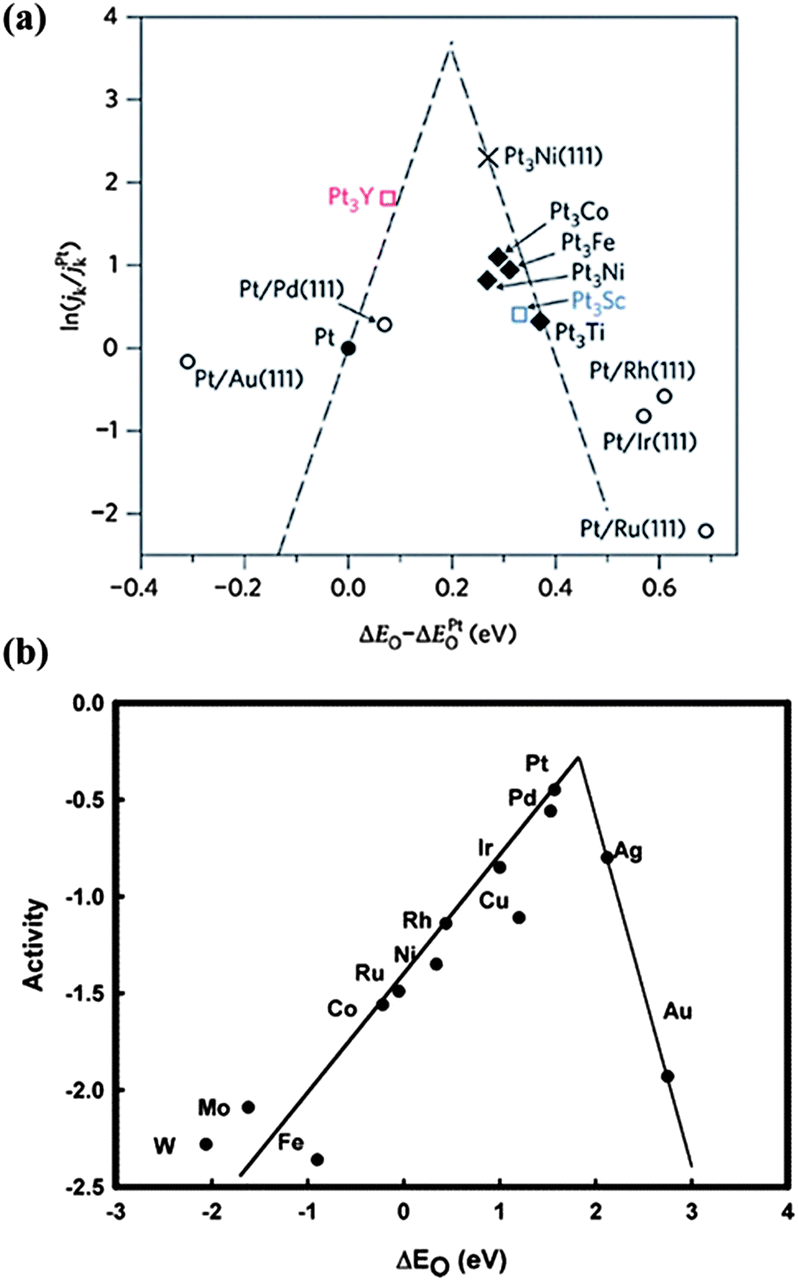 | ||
| Fig. 6 Trends in oxygen reduction activity plotted as a function of the O binding energy of (a) ‘Pt skins’, reproduced from ref. 42 by permission (copyright 2009, Macmillan Publishers Limited) and (b) metals, reproduced from ref. 72 by permission of the American Chemical Society. | ||
In addition to Pt-based materials, other noble metals such as Pd, Cu and Ag or their alloys achieve much attention as ORR catalysts with the activity trend shown in Fig. 6b. Though non-Pt noble metals such as Ag are not comparable with Pt,72 their much lower cost makes them candidates for the ORR. A carbon-supported Ag catalyst exhibits a high ORR activity with a predominately four-electron reaction mechanism in 0.1 M NaOH solution. Upon increasing the loading amount of Ag, a more positive initial potential is obtained, indicating better catalytic properties.73 Furthermore, non-Pt alloys exhibit higher activities. For example, with Ag-rich AgPd nanoalloys, the ORR mass activities per total amount of metal are 1.6, 2.7 and 3.2 times that for pure Pd for AgPd2, Ag9Pd and Ag4Pd alloys, respectively.74
To further control the cost of Mg–air batteries, much research is devoted to developing non-noble metal catalysts such as carbon-based materials and transition metal oxides. Carbon materials are ubiquitous in air electrodes in Mg–air batteries, not only as catalysts and conductive agents but also as a gas diffusion layer. For catalysts, carbon involves a two-electron reaction with the production of H2O2. This reaction has sluggish kinetics, so carbon materials alone are not good ORR catalysts. However, by doping with heteroatoms (P or N), the ORR activities of the carbons improve a lot, especially for high surface areas such as mesoporous carbons58,59 and nanotubes,61 as well as graphene.62,63,75 For instance, vertically aligned nitrogen-containing carbon nanotubes (VA-NCNTs) display much better electrocatalytic activity and long-term operation stability than platinum in alkali with a four-electron reaction process, as shown in Fig. 7. The presence of nitrogen, which imparts a high positive charge on adjacent carbon atoms, plays an important role for activity enhancement.61 A similar result is also obtained in the case of N-containing graphene.62 These results indicate that N-doped high surface area carbon materials are promising substitutes for Pt. The effects of different types of nitrogen are definite. Graphitic N determines the limiting current density, while pyridinic N improves the onset potential.63 However, one thing that should be mentioned is that as the production of graphene and nanotubes is still costly, much effort should be further devoted to a more facile and lower cost synthesis of graphene and nanotubes.
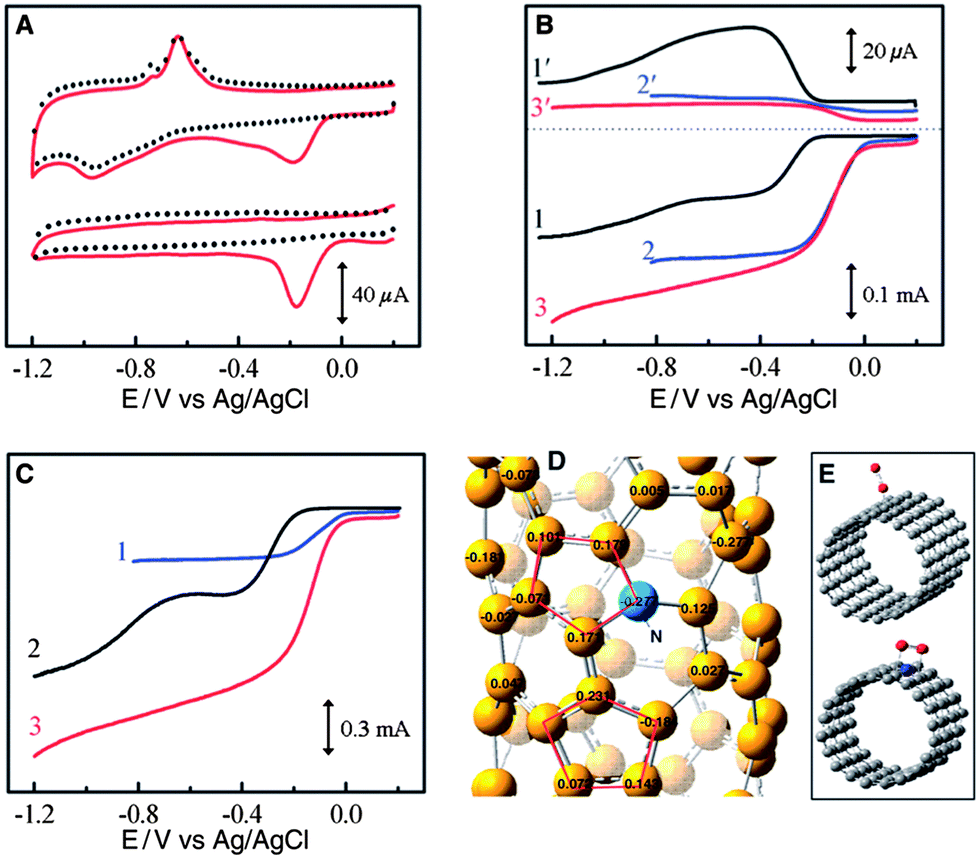 | ||
| Fig. 7 (a) CVs for oxygen reduction at the unpurified (upper) and electrochemically purified (bottom) VA-NCNT/GC electrodes in argon-protected (dotted curves) or air-saturated 0.1 M KOH (solid red curves) at a scan rate of 100 mV s−1. Rotating Ring-Disk Electrodes (RRDE) plots and the corresponding amperometric responses for the oxygen reduction in air saturated 0.1 M KOH at (b) NA-CCNT/GC (curves 1 and 1′), Pt–C/GC (curves 2 and 2′), and NA-NCNT/GC (curves 3 and 3′) electrodes and at (c) Pt–C/GC (curve 1), VA-CCNT/GC (curve 2), and VA-NCNT (curve 3) electrodes. (d) Calculated charge density distribution for NCNTs. (e) Schematic representations of possible adsorption modes of an oxygen molecule at CCNTs (top) and NCNTs (bottom). VA-NCNT, NA-CCNT and GC stand for vertically aligned nitrogen-containing carbon nanotubes, nitrogen-free nonaligned carbon nanotubes and glass carbon, respectively. Reproduced from ref. 61 by permission of AAAS. | ||
Transition metal oxides are another important family of non-noble metal ORR catalysts.38 The ORR activities of different kinds of transition metal oxides have been thoroughly investigated from their crystallographic structures, morphologies and doping effects. Taking MnO2 as an example, the ORR properties become worse for different MnO2 phases as follows: α- > β- > γ-MnO2, and nanostructures are superior to microstructures.64 Meanwhile, doping MnO2 with Cu or Ni improves the catalytic activity.76 In order to accelerate the development of highly active transition metal oxide catalysts, a design principle has been identified. For perovskite, eg-filling with a value of 1 exhibits the maximum activity due to stabilization of the O22−/OH− exchange, which is the rate-limiting step. However, an important hindrance for transition metal oxides is the low electronic conductivity. To improve this, constructing transition metal oxides with highly conductive materials such as graphene or porous carbons is a useful strategy. Although transition metal oxides or carbons alone have low catalytic activity, their hybrids show an unexpected, surprisingly high ORR activity due to synergetic chemical coupling effects.67,69,77 Similar to Co3O4/N-doped graphene, they exhibit a similar catalytic activity but superior stability to Pt in alkaline solutions, as shown in Fig. 8. The ORR activity of this hybrid is further improved by tuning the morphology of the carbons.67 Moreover, this hybrid also displays a high oxygen evolution reaction (OER) activity.68
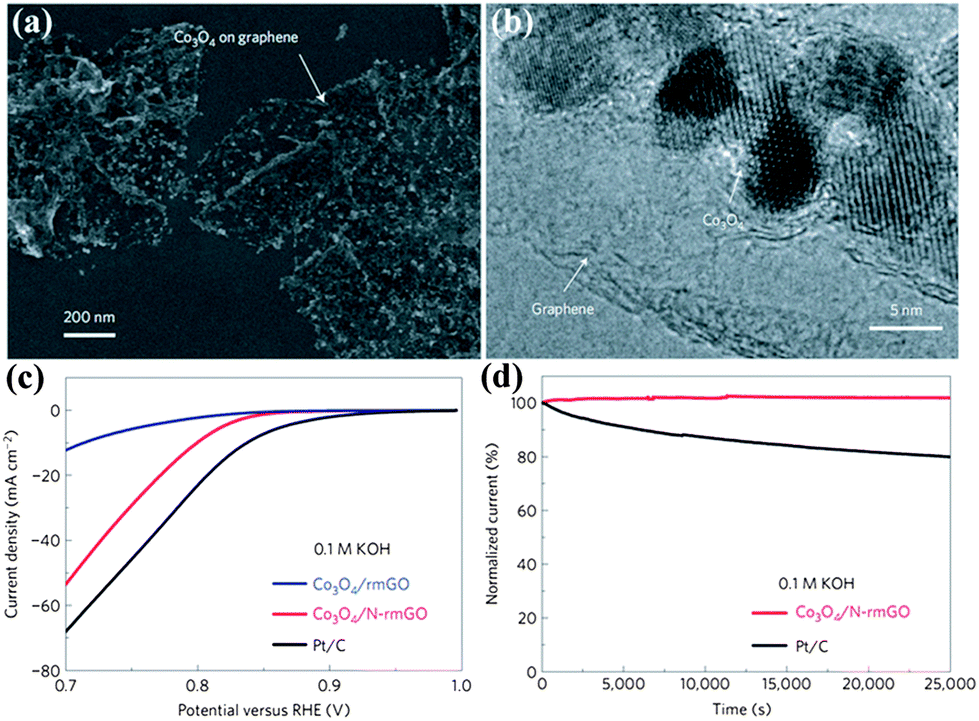 | ||
| Fig. 8 (a) SEM images and (b) TEM images of Co3O4/N-rmGO hybrid. (c) Oxygen reduction polarization curves and (d) chronoamperometric responses (percentage of current retained versus operation time) of Co3O4/rmGO, Co3O4/N-rmGO and Pt/C. rmGO stands for reduced mildly oxidized graphene oxide. Reproduced from ref. 68 by permission. Copyright 2011, Macmillan Publishers Limited. | ||
It should be noted that N-containing metal macrocyclic compounds are also important catalysts for the ORR. Cobalt and iron tetramethoxyphenylporphyrin (CoTMPP and FeTMPP) catalysts loaded on carbons or with preheating show high ORR activity in alkaline solutions.78–80 CoTMPP is more stable and catalyzes oxygen reduction through a two-electron process, and is more active than FeTMPP. In addition to tetramethoxyphenylporphyrins, other metal macrocyclic compounds have been produced recently. For example, a new series of trinuclear metal N4 complexes [MN4]n on carbon black have been successfully prepared, revealing much better electrochemical activity and long-term stability than the commercially available Pt/C catalyst in an alkaline electrolyte.70
There are certainly many other catalysts such as nitrides for the ORR.81 One can see that catalysts such as N-doped graphenes are already comparable to Pt. Further decreasing the cost and increasing the durability of the catalysts, especially in a neutral solution, is the required direction for developing cathode catalysts in Mg–air batteries.
Applications
The application of Mg–air batteries has a long history. As early as the 1960s, General Electric (GE), an American company, obtained a neutral NaCl solution Mg–air fuel cell. Nowadays, an important application of Mg–air batteries is as backup systems for electric and solar power, which can be employed in hospitals and schools for emergencies. In the period in which the batteries are not used, they are stored in the dry state for a long time. When they are to be used, the electrolyte is added to the batteries. Such easy operation makes their use convenient. Recently, the Canadian Greenvolt Power Company successfully developed a serial Mg/saltwater/air fuel cell (MASWFC),82 which had a much higher energy density than a lead–acid battery. This system could provide electronic energy not only for TVs and phone cells but also for vehicles. Another Canadian company, MagPower™ Systems, also developed a Mg–air battery combining magnesium, oxygen and a saltwater electrolyte.83 In this system, hydrogen inhibitors were added to prevent hydrogen generation, showing an efficiency rate of 90% and an operating range of −20 °C to 55 °C. Potential markets for this Mg–air battery include remote military and telecommunication sites. In the meantime, this system can also be used as a backup system for electric and solar power devices.Another important application of Mg–air batteries is for undersea instruments. Such a system employs Mg alloys as the anode, seawater as the electrolyte and the oxygen dissolved in seawater as the cathode. In 1996, Norway and Italy cooperatively developed a Mg–air fuel cell, which was used in the automatic control systems for offshore oilfield exploration.8 The fuel cell was composed of two large piles with an energy density of 650 kW h and a life of 15 years. Undersea cells are also utilized in lighthouses, floats and undersea monitoring equipment. Meanwhile, the Mg–air battery is an option for military application. The Navy considered the potential of a hybridized Mg–air fuel cell and nickel–zinc battery as energy providers for detectors, which could provide 25 kW of pulse power over two weeks.84
Conclusions
In this review, we highlight recent progress in Mg–air batteries from fundamental principles to the available applications. Mg–air batteries contains three parts: a Mg anode, an air cathode and a saline neutral aqueous electrolyte. The reactions involved in the batteries are Mg electrochemical oxidation to Mg ions in the anode and the oxygen reduction reaction in the cathode. Mg plates are common materials for the Mg anode and the drawback is the high level of corrosion. Mg alloys and Mg nanoparticles can improve the performance of the Mg anode. The air cathode contains four layers and the sluggish kinetics of the ORR hinders the performance of the cathode. Therefore, catalysts as well as multilayer structures play a key role in the air cathode. Noble metals such as Pt and Ag are the usual catalysts in the air cathode. To lower the cost, different types of catalyst such as N-doped carbonaceous, metal oxides and metal oxide–carbonaceous mixtures are used as substitutes for noble metals. The neutral electrolyte consists of salts such as NaCl, NaNO2 and KHCO3, among which a mixed electrolyte containing Mg(NO3)2·6H2O and NaNO2 shows a high performance. The applications of Mg–air batteries currently include undersea equipment, military and backup systems.High polarization and low coulombic efficiency are the main problems of Mg–air batteries. Methods to overcome these issues mainly include searching for electrode materials with high activities and low corrosion rates. For the anode, further efforts will be devoted to searching for high hydrogen overpotential elements for alloys, increasing the number of active sites and utilization of Mg through structure design, and optimizing the preparation process. For the cathode, the directions to be taken are lowering the cost of the catalysts by searching for new catalysts, improving the activity and durability of the catalysts through hybrid technology, and increasing the contact between oxygen, electrolyte and catalyst by tuning the multilayer structure. At the same time, the exploration of highly stable electrolytes is also important.
It should be noted that currently Mg–air batteries are primary batteries. The production of rechargeable Mg–air batteries is still challenging. Compared with primary Mg–air batteries, rechargeable Mg–air batteries are quite different, and reversible oxygen reduction reactions and oxygen evolution reactions are taking place. A non-aqueous system, which is similar to that for a rechargeable Li–air battery, should be a good choice. Possible directions for developing rechargeable Mg–air batteries would focus on the following points: (1) the first and most important mission is to find a fully rechargeable Mg-base material. As we know, magnesium oxide is not quite rechargeable, and magnesium peroxide may be a candidate due to higher activity. (2) Searching for a suitable electrolyte that is stable and does not react with the discharged products. In this case, it is important to investigate the electrolyte of rechargeable Mg-ion batteries. (3) Exploring electro-catalysts that are highly active to both the oxygen reduction reaction and oxygen evolution reaction, which reduce the polarization of the cell and accelerate the decomposition of the discharged products. Though many difficulties are faced, it is still worth applying much effort to develop rechargeable Mg–air batteries.
Acknowledgements
This work was supported by the Programs of National 973 (2011CB935900), NSFC (21231005), 111 Project (B12015), and Tianjin High-Tech (13JCQNJC06400).References
- D. Linden and T. B. Reddy, Handbook of Batteries, McGraw-Hill, New York, 2002 Search PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- P. Sapkota and H. Kim, J. Ind. Eng. Chem., 2009, 15, 445–450 CrossRef CAS PubMed.
- J. S. Lee, S. Tai Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- W. Yang, S. Yang, G. Sun and Q. Xin, Chin. J. Power Sources, 2005, 29, 182–186 CAS.
- B. Peng, J. Liang, Z. Tao and J. Chen, J. Mater. Chem., 2009, 19, 2877–2883 RSC.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- X. Wang, Y. Hou, Y. Zhu, Y. Wu and R. Holze, Sci. Rep., 2013, 3, 1014 Search PubMed.
- R. P. Hamlen, E. C. Jerabek, J. C. Ruzzo and E. G. Siwek, J. Electrochem. Soc., 1969, 116, 1588–1592 CrossRef CAS PubMed.
- M. Pourbaix, Atlas D'equilibres Electrochimiques, Gauthier Villars, Paris, 1963 Search PubMed.
- G. G. Perrault, J. Electroanal. Chem. Interfacial Electrochem., 1974, 51, 107–119 CrossRef CAS.
- B. A. Shaw, Corrosion Resistance of Magnesium Alloys, ASM International, 2003 Search PubMed.
- G. Song and A. Atrens, Adv. Eng. Mater., 2003, 5, 837–858 CrossRef CAS.
- G. L. Song and A. Atrens, Adv. Eng. Mater., 1999, 1, 11–33 CrossRef CAS.
- Y. Ma, N. Li, D. Li, M. Zhang and X. Huang, J. Power Sources, 2011, 196, 2346–2350 CrossRef CAS PubMed.
- W. Li, C. Li, C. Zhou, H. Ma and J. Chen, Angew. Chem., Int. Ed., 2006, 45, 6009–6012 CrossRef CAS PubMed.
- Y. D. Milusheva, R. I. Boukoureshtlieva, S. M. Hristov and A. R. Kaisheva, Bulg. Chem. Commun., 2011, 43, 42–47 CAS.
- Y. Lv, M. Liu, Y. Xu, D. Cao and J. Feng, J. Power Sources, 2013, 225, 124–128 CrossRef CAS PubMed.
- R. Balasubramanian, A. Veluchamy, N. Venkatakrishnan and R. Gangadharan, J. Power Sources, 1995, 56, 197–199 CrossRef CAS.
- M. G. Medeiros and E. G. Dow, J. Power Sources, 1999, 80, 78–82 CrossRef CAS.
- J. L. Murray, ASM Handbook Volume 3: Alloy Phase Diagrams, ASM International, 1998 Search PubMed.
- J. Chen and F. Y. Cheng, Acc. Chem. Res., 2009, 42, 713–723 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- Y. Qiao and C. M. Li, J. Mater. Chem., 2011, 21, 4027–4036 RSC.
- Y. S. Meng and M. E. Arroyo-de Dompablo, Energy Environ. Sci., 2009, 2, 589–609 CAS.
- G. Ceder, G. Hautier, A. Jain and S. P. Ong, MRS Bull., 2011, 36, 185–191 CAS.
- D. Shin and C. Wolverton, Scr. Mater., 2010, 63, 680–685 CrossRef CAS PubMed.
- R. W. P. Wagemans, J. H. van Lenthe, P. E. de Jongh, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2005, 127, 16675–16680 CrossRef CAS PubMed.
- B. W. Jensen, M. Gaadingwe, D. R. Macfarlane and M. Forsyth, Electrochim. Acta, 2008, 53, 5881–5884 CrossRef PubMed.
- H. P. Godard, W. P. Jepson, M. R. Bothwell and R. L. Lane, The Corrosion of Light Metals, John Wiley & Sons, 1967 Search PubMed.
- S. Sathyanarayana and N. Munichandraiah, J. Appl. Electrochem., 1981, 11, 33–39 CrossRef CAS.
- T. Khoo, P. C. Howlett, M. Tsagouria, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2011, 58, 583–588 CrossRef CAS PubMed.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- F. Y. Cheng, J. Shen, B. Peng, Y. D. Pan, Z. L. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- J. Perez, E. R. Gonzalez and E. A. Ticianelli, Electrochim. Acta, 1998, 44, 1329–1339 CrossRef CAS.
- L. Genies, R. Faure and R. Durand, Electrochim. Acta, 1998, 44, 1317–1327 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Gronbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 CAS.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, J. Chem. Phys., 2004, 120, 10240–10246 CrossRef CAS PubMed.
- F. H. B. Lima, J. R. C. Salgado, E. R. Gonzalez and E. A. Ticianelli, J. Electrochem. Soc., 2007, 154, A369–A375 CrossRef CAS PubMed.
- D. Wang, Y. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nano Lett., 2012, 12, 5230–5238 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- H. Chen, D. Wang, Y. Yu, K. A. Newton, D. A. Muller, H. Abruña and F. J. DiSalvo, J. Am. Chem. Soc., 2012, 134, 18453–18459 CrossRef CAS PubMed.
- F. H. B. Lima and E. A. Ticianelli, Electrochim. Acta, 2004, 49, 4091–4099 CrossRef CAS PubMed.
- M. Oezaslan, F. Hasche and P. Strasser, J. Electrochem. Soc., 2012, 159, B444–B454 CrossRef CAS PubMed.
- L. H. Jiang, A. Hsu, D. Chu and R. R. Chen, J. Electroanal. Chem., 2009, 629, 87–93 CrossRef CAS PubMed.
- Y. F. Yang, Y. H. Zhou and C. S. Cha, Electrochim. Acta, 1995, 40, 2579–2586 CrossRef CAS.
- L. Jiang, A. Hsu, D. Chu and R. Chen, Electrochim. Acta, 2010, 55, 4506–4511 CrossRef CAS PubMed.
- T. J. Schmidt, V. Stamenkovic, M. Arenz, N. M. Markovic and P. N. Ross, Electrochim. Acta, 2002, 47, 3765–3776 CrossRef CAS.
- B. Li and J. Prakash, Electrochem. Commun., 2009, 11, 1162–1165 CrossRef CAS PubMed.
- M. C. Oliveira, R. Rego, L. S. Fernandes and P. B. Tavares, J. Power Sources, 2011, 196, 6092–6098 CrossRef CAS PubMed.
- D. A. Slanac, W. G. Hardin, K. P. Johnston and K. J. Stevenson, J. Am. Chem. Soc., 2012, 134, 9812–9819 CrossRef CAS PubMed.
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- W. Yang, T. P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- D. S. Geng, Y. Chen, Y. G. Chen, Y. L. Li, R. Y. Li, X. L. Sun, S. Y. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- F. Y. Cheng, Y. Su, J. Liang, Z. L. Tao and J. Chen, Chem. Mater., 2010, 22, 898–905 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. S. Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- R. L. Liu, C. von Malotki, L. Arnold, N. Koshino, H. Higashimura, M. Baumgarten and K. Mullen, J. Am. Chem. Soc., 2011, 133, 10372–10375 CrossRef CAS PubMed.
- J. S. Guo, H. X. Li, H. He, D. Chu and R. R. Chen, J. Phys. Chem. C, 2011, 115, 8494–8502 CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. S. Guo, A. Hsu, D. Chu and R. R. Chen, J. Phys. Chem. C, 2010, 114, 4324–4330 CAS.
- D. A. Slanac, W. G. Hardin, K. P. Johnston and K. J. Stevenson, J. Am. Chem. Soc., 2012, 134, 9812–9819 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- T. N. Lambert, D. J. Davis, W. Lu, S. J. Limmer, P. G. Kotula, A. Thuli, M. Hungate, G. Ruan, Z. Jin and J. M. Tour, Chem. Commun., 2012, 48, 7931–7933 RSC.
- I. Roche and K. Scott, J. Appl. Electrochem., 2009, 39, 197–204 CrossRef CAS.
- Q. Zhou, C. M. Li, J. Li, X. Cui and D. Gervasio, J. Phys. Chem. C, 2007, 111, 11216–11222 CAS.
- M. Maja, C. Orecchia, M. Strano, P. Tosco and M. Vanni, Electrochim. Acta, 2000, 46, 423–432 CrossRef CAS.
- S. L. Gojković, S. Gupta and R. F. Savinell, J. Electrochem. Soc., 1998, 145, 3493–3499 CrossRef PubMed.
- H. Wu and W. Chen, J. Am. Chem. Soc., 2011, 133, 15236–15239 CrossRef CAS PubMed.
- GreenVolt confirms a 150% power increase in its new magnesium–air–saltwater-fuel-cell -TM- without an increase in size & weight, The Free Library, Business Wire 2001, http://www.thefreelibrary.com/GreenVolt+Confirms+A+150%25+Power+Increase+In+Its+New…-a078823900.
- MAGPOWER, http://www.magpower.pt/X/home.cgi.
- Hybridized magnesium air fuel cell with Ni–Zn battery or electrochemical capacitor as the ideal energy source for USV sensor payloads, Department of Defense, Navy, N04–080, 2004, http://sbirsource.com/sbir/awards/6590-hybridized-magnesium-air-fuel-cell-with-ni-zn-battery-or-electrochemical-capacitor-as-the-ideal-energy-source-for-usv-sensor-payloads.
| This journal is © The Royal Society of Chemistry 2014 |