Vesicle budding from polymersomes templated by microfluidically prepared double emulsions†
Julian
Thiele‡
*a,
Venkatachalam
Chokkalingam‡
a,
Shaohua
Ma
b,
Daniela A.
Wilson
a and
Wilhelm T. S.
Huck
*a
aRadboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: j.thiele@science.ru.nl; w.huck@science.ru.nl
bUniversity of Cambridge, Department of Chemistry, Melville Laboratory for Polymer Synthesis, Lensfield Road, Cambridge CB2 1EW, UK
First published on 3rd September 2013
Abstract
Many approaches to mimic and understand the dynamics of vesicle budding lack precise control over vesicle membrane properties or require external stimuli to induce budding. We use copolymer-loaded double-emulsion droplets to precisely control size, size distribution, composition and morphology of giant polymersomes. By tuning the copolymer concentration in the polymersome membrane, we identify conditions under which vesicles spontaneously bud from the polymersome surface. Our findings have important implications for the design of copolymer membranes and contribute to the understanding of polymersome formation from double emulsions.
Budding describes the evolution of vesicles from the surface of a membrane;1,2 it is an important phenomenon in intracellular transport between organelles as well as in intercellular transport where vesicles containing signaling molecules bud off from one cell, protect their interior from degradation during transport and fuse with a target cell to release their cargo.3–5 In addition, vesicles play an important role in neurotransmission, immune response and cell aging.6–8 As cells are composed of a crowded mixture of macromolecules surrounded by a membrane of carbohydrates, lipids and proteins,9,10 mimicking the underlying protein machinery of vesicle budding11 in its entire complexity is a challenging task. Giant vesicles based on lipid and copolymer building blocks, liposomes and polymersomes,12,13 respectively, are promising models to mimic a single key aspect of the budding of vesicle membranes and thereby gain a deeper understanding of the physical and chemical mechanisms.14–16 Conventional fabrication techniques such as electroformation and film rehydration rely on the undirected self-assembly of amphiphiles and usually yield polydisperse vesicles that are not suitable to provide reproducible experimental conditions or are too small to mimic the dimension of a cell and accurately describe diffusion and reaction times.17,18 Moreover, vesicle budding does usually not occur unassisted, but requires external triggers such as osmotic shock, temperature change or light.19–21 Howse et al. have obtained polymersomes by hydration of a regular array of micron-sized copolymer patches from which individual polymersomes spontaneously bud.22 The resulting micron-sized polymersomes are monodisperse as the amount of copolymer available for budding is identical in each patch.
Inspired by this approach, we present the spontaneous budding of micron-sized vesicles from the surface of monodisperse giant polymersome templates. Similar to the excellent control over lithographically fabricated copolymer patches that template the formation of vesicle buds, the size and membrane properties of our polymersome templates are precisely tuned by employing droplet microfluidics, as sketched in Fig. 1A.23,24 Our approach provides a simple route to study vesicle budding from the surface of polymersomes and is also applicable to high-molecular weight copolymers that do not form vesicle buds by conventional hydration of dried copolymer films.
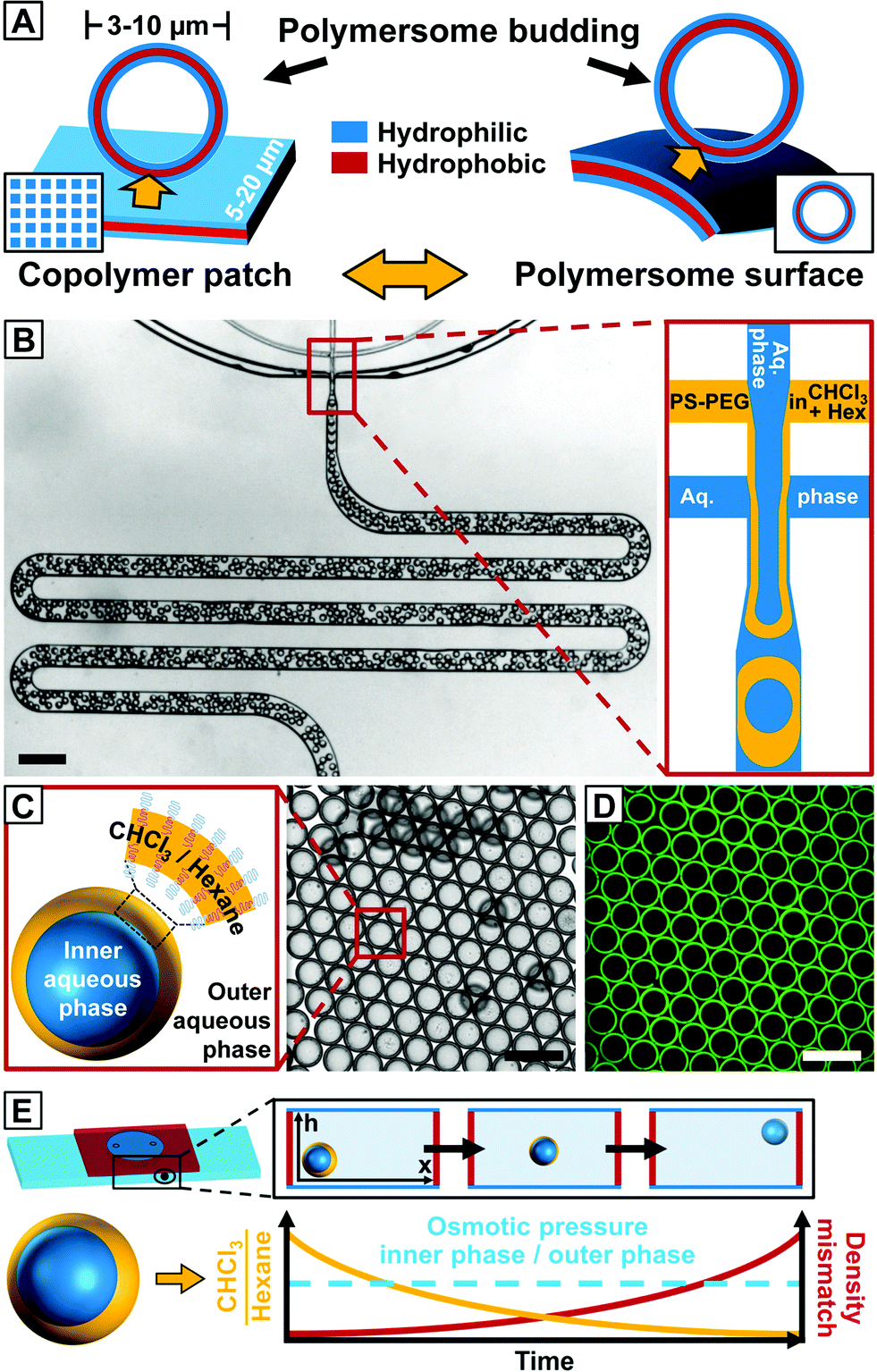 | ||
| Fig. 1 Microfluidic fabrication of copolymer-loaded double emulsions to direct the formation of giant polymersome templates. (A) Schematic representation of vesicle budding from the surface of an array of hydrated copolymer patches according to ref. 22 (left) and the proposed vesicle budding from the surface of a giant polymersome (right). The amount of copolymer available for vesicle budding from the hydrated copolymer membrane is controlled by the size of the copolymer patch and the copolymer concentration in the polymersome membrane, respectively. (B) Fabrication of water-in-organic solvent-in-water (w/o/w) double emulsions stabilized by PS134–PEG45 in a PDMS-based microfluidic device in a one-step emulsification process.26 The scale bar denotes 1 mm. (C) Bright-field microscope image of collected double emulsions. The amphiphilic copolymer assembles at the water–organic-solvent interfaces of the double emulsion. Hexagonal packing indicates monodispersity. The scale bars denote 200 μm. (D) Fluorescence microscope image of the same area as in (B). Nile Blue is well-encapsulated in the hydrophobic middle phase of the emulsion. The scale bar is 200 μm. (E) Upon collection, copolymer-stabilized double emulsion templates float to the top of the microscope chamber slide during solvent evaporation. Chloroform evaporates faster from the droplet template and the ratio of chloroform and hexane (yellow line) inside the double emulsion shell decreases causing a density mismatch (red line) with the surrounding aqueous phase. The osmotic pressure between the inner and outer aqueous phases (dashed blue line) remains constant. | ||
We created double emulsions in poly(dimethylsiloxane) (PDMS)-based microfluidic devices with two flow-focusing cross-junctions, the first being hydrophobic and the second hydrophilic, coated with a durable glass-like layer to resist degradation due to organic solvents.25 This wettability pattern allowed water-in-organic solvent-in-water (w/o/w) double emulsions to be formed, as shown in Fig. 1B (ESI Videos 1 and 2†). We fabricated copolymer-loaded droplets in a one-step emulsification process by removing the dripping instability at the first cross-junction.26 This approach allowed us to control the organic solvent shell thickness and thus the amount of copolymer in the double emulsions over a wider range than in conventional two-step emulsification with one dripping instability at each cross-junction. To account for the diffusion time of the macromolecular surfactant to the w/o interfaces and improve the droplet stability upon product collection outside the device, we included a meander-shaped delay line in the microfluidic design (ESI Video 3†).
We used poly(styrene)-block-poly(ethylene glycol) (PS134–PEG45, Mw = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) as an amphiphile. In previous work, copolymers with hydrophobic and hydrophilic blocks of equal molecular weight as well as large hydrophilic blocks have been employed to form double emulsions.27,28 However, these copolymers either do not naturally form vesicles or they lower the interfacial tension, thus preventing stable droplet formation. While PS134–PEG45 has a significantly higher molecular weight than copolymers employed in conventional polymersome fabrication techniques such as electroformation, we found that a long hydrophobic block enhances the stability of the double emulsion templates and obviates the need for an additional homopolymer that has been used in previous work to stabilize the droplet templates and prevent coalescence.27–29
000) as an amphiphile. In previous work, copolymers with hydrophobic and hydrophilic blocks of equal molecular weight as well as large hydrophilic blocks have been employed to form double emulsions.27,28 However, these copolymers either do not naturally form vesicles or they lower the interfacial tension, thus preventing stable droplet formation. While PS134–PEG45 has a significantly higher molecular weight than copolymers employed in conventional polymersome fabrication techniques such as electroformation, we found that a long hydrophobic block enhances the stability of the double emulsion templates and obviates the need for an additional homopolymer that has been used in previous work to stabilize the droplet templates and prevent coalescence.27–29
Polymersome templates were formed from 8% (w/w) PS–PEG-loaded double emulsions, which is well above previously reported copolymer concentrations to form giant polymersomes from copolymer-loaded double emulsions.30 This concentration ensures a high interfacial density of copolymer in the double emulsions (thus increasing droplet stability during double-emulsion-to-polymersome transition) and provides sufficient excess of copolymer in the later polymersome membrane to form vesicle buds. We injected an aqueous solution of sodium dodecyl sulfate (SDS) as well as PS–PEG in chloroform–hexane into our microfluidic device, as sketched in Fig. 1B. Although SDS can be incorporated into vesicles and dissolve their membrane,31 we require SDS to stabilize the inner aqueous-phase droplet upon emulsification inside the microfluidic device and find that aqueous solutions of our giant polymersomes are stable for at least several weeks despite the use of SDS. By using a mixture of organic solvents, the solubility of PS–PEG as well as the density and evaporation rate of the organic phase and thus the transition from a double emulsion to a polymersome, which we employed to study vesicle budding, was precisely controlled. The optimal solvent ratio of chloroform, which is a good solvent for PS and PEG, and hexane, which is a bad solvent for PEG, was identified in bulk experiments.32 To prevent osmosis-driven shrinkage and collapse of the droplet templates, we balanced the osmolarity of the inner and outer aqueous phases by adding glucose. The double emulsions showed excellent monodispersity, as indicated by their hexagonal packing, presented in Fig. 1C and D. Owing to a higher vapor pressure and better solubility in water, chloroform evaporated faster than hexane from the droplet shell, and the emulsion became density-mismatched with the surrounding aqueous media floating to the top of the collection chamber, as sketched in Fig. 1E. However, the volume of the inner aqueous phase that determined the size of the later polymersome template remained constant.
Upon collection of the double emulsions, chloroform and hexane evaporated, the shell width decreased, as shown in Fig. 2A1 and A2, and the copolymer monolayers at the inner-middle and the middle-outer interface assembled into a polymersome membrane. Instead of a unilamellar bilayer that is usually assumed to be formed involving double-emulsion templates,28,33 the thickness of the as-formed polymersome membranes indicated the self-assembly of two copolymer monolayers encapsulating excess copolymer between them (see below). From the undisturbed shape of these polymersomes, we assume that the osmotic pressure remains constant inside the vesicles, as a change in osmotic pressure would cause the PS–PEG membrane to buckle,28 as we observed before, when collecting polymersomes on simple glass slides in air. As hexane was nearly evaporated, the polymersome membrane swelled in the surrounding aqueous media, as indicated by the surface wrinkling, Fig. 2A3 and A4, and smaller vesicles spontaneously budded off from the polymersome surface, Fig. 2A5 and A6 (ESI Video 4†). Compared to conventional vesicle fabrication methods, our monodisperse double emulsions yielded monodisperse polymersomes, as shown in Fig. 2C, which thus did not require further processing such as high-pressure extrusion to narrow their size distribution.17,18 We performed confocal microscopy, as shown in the image sequence in Fig. 2D. Encapsulation of Nile Blue in a thin hydrophobic layer around the aqueous core indicated the formation of a polymersome membrane from which the smaller vesicle buds evolved, as highlighted in the last image of the sequence. Vesicle buds are out-of-focus in the image sequence and appear as bright fluorescent spheres.
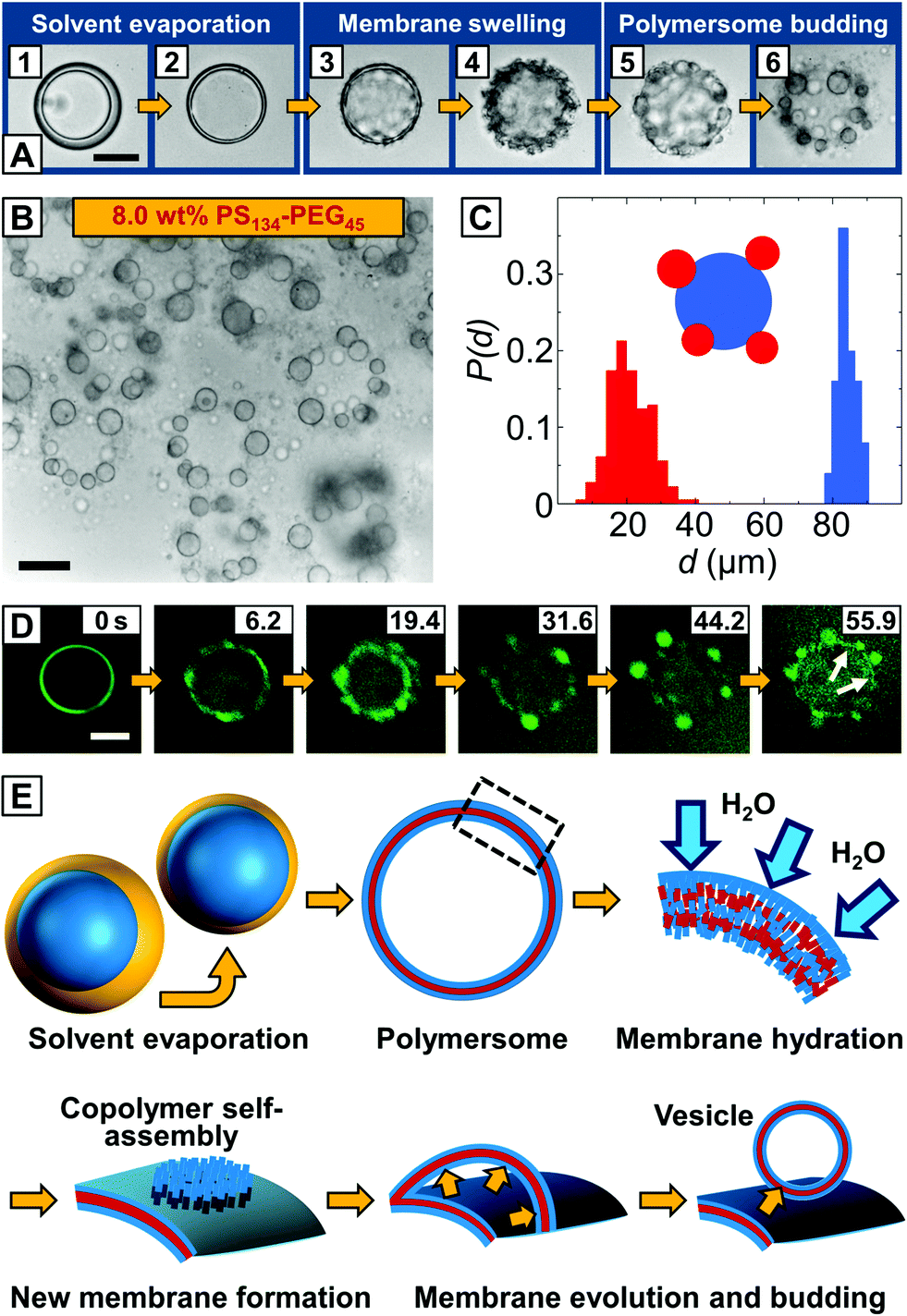 | ||
| Fig. 2 Budding of vesicles from the surface of giant PS–PEG polymersomes. (A) Bright-field microscopy image sequence of a double emulsion droplet transitioning into a polymersome from which multiple vesicles bud off. The scale bar for all images is 50 μm. (B) Bright-field microscope image of monodisperse mother vesicles decorated with small daughter vesicles. Scale bar denotes 50 μm. (C) Size distributions of polymersomes and vesicle buds. Monodisperse polymersomes with controlled size are templated by double emulsions with uniform size. (D) Confocal microscopy image sequence of a double emulsion-to-polymersome transition. Fluorescent Nile Blue is added to the organic solvent phase and imaged using green false color for higher contrast. The aqueous core of the double-emulsion template is surrounded by a thin vesicle membrane (white arrows), from which smaller polymersomes bud off. The scale bar is 50 μm. (E) The proposed mechanism of vesicle budding from the surface of a polymersome fabricated from copolymer-stabilized double emulsions. For simplicity, only the uppermost copolymer layer of the non-unilamellar polymersome is sketched. The polymersome membrane is hydrated by the surrounding aqueous phase, whereas excess copolymer in the polymersome membrane self-assembles into a new membrane that evaginates from the polymersome by the inflow of the external aqueous phase, followed by membrane closure yielding a new vesicle. | ||
Based on these observations, we envisage budding from the surface of our polymersomes as the evagination of a copolymer membrane from the hydrated polymersome surface, followed by membrane closure and vesicle formation, as sketched in Fig. 2E. In the first step, monolayers of PS–PEG at the inner-middle and the middle-outer droplet interface, respectively, close in upon solvent evaporation. As solvent evaporation is accompanied by a drop in solvent quality, the PS block phase-separates and a vesicle membrane forms.30 We do not observe the release of excess copolymer from the as-formed polymersome membrane in a dewetting transition of the organic solvent phase, as previously observed in double emulsion-to-polymersome transitions,27–30 and conclude that excess PS–PEG remains trapped inside the copolymer membrane. Followed by hydration and swelling of the polymersome membrane by the surrounding aqueous phase, excess copolymer molecules inside the swollen membrane become mobile and self-assemble into a new copolymer bilayer by lateral diffusion, as previously described for lipid bilayers.34 Although budding occurs spontaneously and is undirected, our vesicle buds exhibit well-defined sizes, as shown in Fig. 2C. Starting from a homogeneous distribution of the copolymer within the polymersome membrane, each bud can grow from an equal amount of copolymer until the surrounding polymersome membrane is depleted from excess copolymer or the newly formed copolymer membrane attains a critical size which is determined by its edge and bending energy. It then partitions off from the polymersome surface, as shown in Fig. 2A5, and closes up to minimize its surface energy whereas it encapsulates the outer aqueous phase and a new vesicle forms. As we observe a dark layer surrounding the vesicle buds in the bright-field microscope images in Fig. 2A6 and B as well as non-uniform fluorescence in the confocal microscope images in Fig. 2D, we assume that part of the excess copolymer in the polymersome template does not contribute to the vesicle budding, but diffuses into the membrane of the newly formed vesicle buds, thus considered not to be unilamellar.
By contrast, we did not observe vesicle formation from dried films of the same copolymer concentration in conventional bulk rehydration experiments. While re-dissolution of dried PS134–PEG45 films is energetically hindered due to the glassy nature of the long PS-block,35 residues of solvent in our droplet templates plasticize the non-unilamellar polymersome membrane allowing for lateral diffusion of PS–PEG molecules and thus their re-assembly towards vesicle buds. Due to the poor solubility of SDS in chloroform and hexane, which plasticize the polymersome membrane, we did not detail the influence of SDS as a second amphiphile on bud formation and emphasized on the copolymer concentration that determines vesicle budding. However, future work will focus on the influence of secondary amphiphiles on vesicle budding, as discussed by Lipowsky and Hyodo, for instance.36,37
Although the copolymer membrane was hydrated by both the inner and outer aqueous phases, budding occurred preferentially on the outer surface. For zero spontaneous curvature, vesicle buds have the same bending and edge energy on both sides of the membrane of the polymersome templates.34 Our observation can be rationalized with a numerical difference of copolymer molecules at the inner and outer side of the membrane which breaks this symmetry and induces a preference for budding on the outside of the copolymer membrane, as observed for all vesicle buds in Fig. 2A–D. Occasionally, the vesicle buds can be detached from the surface which was promoted by shearing the polymersome solution.
We did not observe merging of neighboring vesicle buds on the polymersome surface. Vesicle membranes evolved from spaced-out locations on the polymersome surface, preventing early merging of the newly formed membranes, whereas at a later stage of the budding process, the microscopic vesicle buds had gained sufficient stability due to the nature of the copolymer as well as additional copolymer inside the vesicle bud membrane.27–29,38,39
To elucidate the control over the size of vesicle buds by the amount of copolymer provided in the membrane of our polymersome templates, we formed polymersomes from double emulsions loaded with PS–PEG from 0.5 to 16% (w/w). The copolymer concentration had a significant influence on the mechanism of double emulsion-to-polymersome transition. While a polymersome membrane simply formed by adhesion of two copolymer monolayers due to solvent evaporation at 8% (w/w) PS–PEG, as discussed above, double emulsion-to-polymersome transition proceeded via dewetting of the organic solvents from the inner double-emulsion droplet at lower copolymer concentrations (0.5%, 1.0%, 2.5% and 5.0% w/w) to give rise to a polymersome membrane. In all cases, as the dewetting process is strongly dependent on the relative surface energy of the inner, middle and outer phases of the double emulsion,30 small changes in the copolymer concentration strongly influenced the double emulsion-to-polymersome transition, and the dewetting process in particular. The vesicle membrane of the as-formed giant polymersomes, exemplarily shown for 2.5% (w/w) in Fig. 3A, is only slightly larger than a unilamellar bilayer of PS134–PEG45, and we expect small amounts of excess copolymer, which do not assemble at the w/o interfaces, to be trapped inside the vesicle membrane. However, the amount of copolymer inside the polymersome membrane did not support the formation of vesicle buds, as indicated by the smooth surface of the freeze-dried polymersome in the cryo-scanning electron microscope (cryo-SEM) image in Fig. 3A. Comparing the respective bright-field microscope images, vesicle buds shown in Fig. 2A6 and B exhibit a significantly thicker membrane, again indicating their non-unilamellarity. As we doubled the copolymer concentration to 5.0% (w/w), the greater part of excess copolymer accumulated in an organic solvent droplet, as observed before in double emulsion-to-polymersome transitions.27,28 Upon solvent evaporation, an aggregate of PS–PEG remained attached to the polymersome surface which re-swelled in the surrounding aqueous phase, as shown in Fig. 3B, lower row. Again, the amount of excess copolymer that remained in the polymersome membrane did not support the formation of vesicle buds.
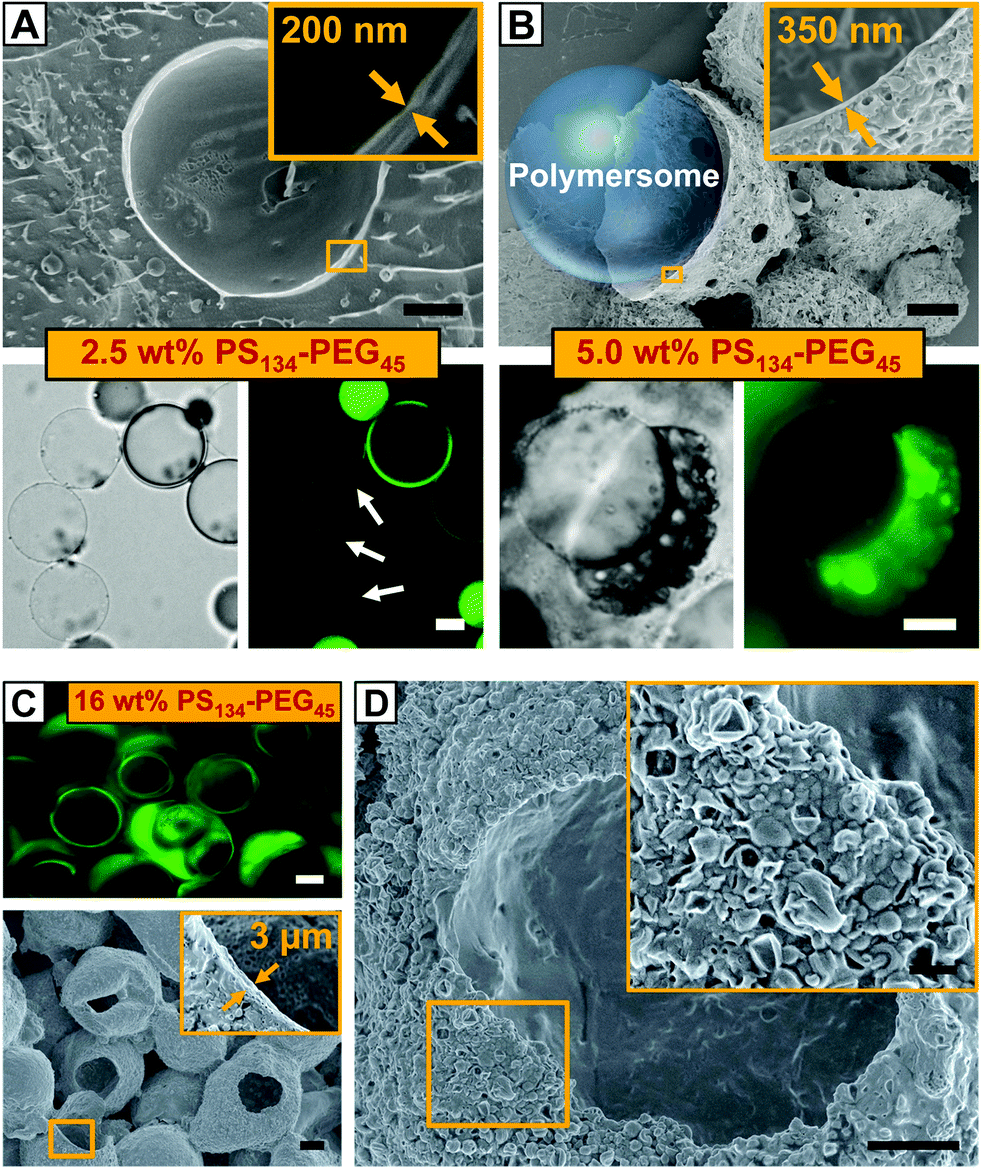 | ||
| Fig. 3 (A and B) Polymersomes formed from w/o/w double emulsions loaded with 2.5% (w/w) PS–PEG and 5.0% (w/w) PS–PEG, respectively. (A) Cryo-SEM image of a single polymersome with a homogeneous vesicle membrane, upper row, bright-field and corresponding fluorescence microscope images, lower row. White arrows point at polymersomes and organic solvent droplets appear as bright spots. Two double-emulsion droplets have not transformed into polymersomes, yet. (B) Dry-SEM image of agglomerates of excess copolymer, upper row, corresponding bright-field and fluorescence microscope images of an intact polymersome, lower row. The polymersomes have ruptured during SEM sample preparation in vacuo. A schematic of one polymersome is inserted in the SEM image. The insets in the SEM images in (A) and (B) illustrate the thickness of the vesicle membrane. The scale bars denote 10 μm for the upper row and 20 μm for the lower row. Green false color is used in fluorescence microscope images for enhanced contrast. (C and D) Copolymer capsules formed from w/o/w double emulsions loaded with 16% (w/w) PS–PEG. (C) A 2D laser-scanning confocal microscope slice showing microcapsules loaded with Nile Blue, upper row, the SEM image of dried capsules with a ruptured shell, lower row. The inset depicts the average shell thickness. The scale bars denote 20 μm. (D) SEM image of a dried copolymer capsule. The surface is not smooth but covered with nanometer-sized, dried polymersomes, as highlighted in the inset. The scale bar is 10 μm and 3 μm for the inset. | ||
The thickness of the polymersome membrane upon solvent evaporation increased with the copolymer concentration from approximately 250 nm at 2.5% (w/w) PS–PEG to 350 nm at 5.0% (w/w), as shown in the insets of Fig. 3A and B. However, in the case of 16% (w/w) copolymer concentration, both fluorescence microscopy and SEM of dried samples revealed that we yielded copolymer capsules instead of a polymersome membrane, as exemplified in Fig. 3C. Upon solvent evaporation, the double emulsion templates did not undergo any double emulsion-to-polymersome transition. Instead, PS–PEG immediately formed a dense copolymer shell with a thickness of approximately 3 μm. Although we did not immediately observe vesicle budding from these capsules on the micro-scale, SEM imaging revealed that a large number of polymersomes probably budded from the capsule's surface, as shown in the magnified view of Fig. 3D. However, these vesicles were nanometer-sized and thus significantly smaller than the microscopic vesicle buds formed at 8% (w/w) PS–PEG which were 20 μm in diameter on average. We assume that organic-solvent residues cannot sufficiently plasticize the dense capsule, thus limiting free diffusion along the rigid capsule and preventing the formation of larger copolymer membranes from which vesicle buds could evolve. However, by choosing copolymers which form flexible, mobile polymersome membranes, we expect to extend the copolymer concentration range that allows for spontaneous vesicle budding.
Conclusions
By precisely tuning the copolymer concentration of PS–PEG-stabilized w/o/w double-emulsions, we have shown the unassisted formation of microscopic vesicle buds from the surface of giant polymersomes. A prerequisite for budding is residual organic solvent in the polymersome membrane: it plasticizes the polymersome membrane and facilitates the copolymer mobility that is crucial for vesicle budding. Our experiments further elucidate the mechanism of double emulsion-templated polymersome formation beyond the influence of solvent composition and interfacial tension30,33 and can help to mimic the budding of vesicles from crowded biomembranes.40,41Experimental section
Fabrication of microfluidic devices
Microfluidic devices were fabricated using soft lithography in PDMS.42 The channel width at the first and the second cross-junction was 60 μm and 120 μm, respectively. A PDMS replica of this channel design was bonded to a glass slide using oxygen plasma treatment. PDMS-based microfluidic devices need to resist degradation due to organic solvents and require spatially controlled wettability to form water-in-organic solvent-in-water (w/o/w) double emulsions.43,44 The devices were coated with a photo-reactive sol–gel. The sol–gel is intrinsically hydrophobic, but can be rendered hydrophilic by grafting hydrophilic patches of poly(acrylic acid) onto the sol–gel using spatially patterned UV light, as further detailed in the ESI.†Formation of PS–PEG-stabilized double emulsions
All solutions were injected using gastight syringes (Hamilton 1000 series) mounted onto syringe pumps (Cetoni® neMESYS, 14.5 gear) connected to the microfluidic device via PTFE tubing (Novodirect GmbH, inner diameter = 0.53 mm, outer diameter = 1.03 mm). We formed copolymer-stabilized double emulsions by injecting an aqueous solution of glucose (50 mM) with sodium dodecyl sulfate (0.1% w/w) as the inner and outer phases and chloroform–hexane (80:20 v/v) with 0.5% to 16% (w/w) PS–PEG as the double emulsion's shell phase into the microfluidic device. A typical set of flow rates of the inner, middle and outer phases was 150, 330, and 750 μL h−1, respectively. The outlet tubing of the microfluidic device was fed into a microscope chamber slide separated by a silicone isolator (SecureSeal™, diameter = 20 mm, height = 0.5 mm), where copolymer-stabilized double emulsions were collected for further characterization. This reduced the rate at which the organic solvents evaporated and allowed us to monitor the formation of giant polymersome templates as well as vesicle budding using optical microscopy. If the double emulsion was simply left in air, the organic solvents would evaporate too quickly and destabilize the emulsion upon double emulsion-to-polymersome transition. In addition, a sudden change in concentration due to evaporation could affect the osmotic pressure between the inner and outer phases of the as-formed polymersomes and cause their membrane to buckle.28
Acknowledgements
J. T. is a Feodor–Lynen fellow of the Alexander von Humboldt Foundation. D. A. W. and W. T. S. H. received financial support from the European Research Council under the European Union’s Seventh Framework Program FP7/2007-2012/ ERC-StG 307679 “StomaMotors” and ERC Advanced Grant 246812 “Intercom”.References
- H. T. McMahon and J. L. Gallop, Nature, 2005, 438, 590 CrossRef CAS PubMed.
- J. S. Bonifacino and B. S. Glick, Cell, 2004, 116, 153 CrossRef CAS.
- R. Schekman and L. Orci, Science, 1996, 271, 1526 CAS.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37 CrossRef CAS PubMed.
- G. L. Kolling and K. R. Matthews, Appl. Environ. Microbiol., 1999, 65, 1843 CAS.
- J. Rohrbough and K. Broadie, Nat. Rev. Neurosci., 2006, 5, 139 Search PubMed.
- C. Théry, M. Ostrowski and E. Segura, Nat. Rev. Immunol., 2009, 9, 581 CrossRef PubMed.
- G. J. Bosman, F. L. Willekens and J. M. Werre, Cell. Physiol. Biochem., 2005, 16, 1 CrossRef CAS PubMed.
- S. B. Zimmerman and A. P. Minton, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 27 CrossRef CAS PubMed.
- F. Spira, N. S. Mueller, G. Beck, P. Von Olshausen, J. Beig and R. Wedlich-Söldner, Nat. Cell Biol., 2012, 14, 640–648 CrossRef CAS PubMed.
- J. E. Rothman, Protein Sci., 1996, 5, 185 CrossRef CAS PubMed.
- A. Jesorka and O. Orwar, Annu. Rev. Anal. Chem., 2008, 1, 801 CrossRef CAS PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323 CrossRef CAS PubMed.
- M. S. Long, C. D. Jones, M. R. Helfrich, L. K. Mangeney-Slavin and C. D. Keating, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5920 CrossRef CAS PubMed.
- M. M. Hanczyc and J. W. Szostak, Curr. Opin. Chem. Biol., 2004, 8, 660 CrossRef CAS PubMed.
- F. Meng, G. H. M. Engbers and J. Feijen, J. Controlled Release, 2005, 101, 187 CrossRef CAS PubMed.
- S. Lecuyer, W. D. Ristenpart, O. Vincent and H. A. Stone, Appl. Phys. Lett., 2008, 92, 104105 CrossRef.
- J. C.-M. Lee, H. Bermudez, B. M. Discher, M. A. Sheehan, Y.-Y. Won, F. S. Bates and D. E. Discher, Biotechnol. Bioeng., 2001, 73, 135 CrossRef CAS PubMed.
- R. Rodríguez-García, M. Mell, I. López-Montero, J. Netzel, T. Hellweg and F. Monroy, Soft Matter, 2011, 7, 1532 RSC.
- C. Sanson, J.-F. Le Meins, C. Schatz, A. Soum and S. Lecommandoux, Soft Matter, 2010, 6, 1722 RSC.
- G. P. Robbins, M. Jimbo, J. Swift, M. J. Therien, D. A. Hammer and I. J. Dmochowski, J. Am. Chem. Soc., 2009, 131, 3872 CrossRef CAS PubMed.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507 CrossRef CAS PubMed.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846 CrossRef CAS PubMed.
- A. Perro, C. Nicolet, J. Angly, S. Lecommandoux, J.-F. Le Meins and A. Colin, Langmuir, 2011, 27, 9034 CrossRef CAS PubMed.
- A. R. Abate, J. Thiele, M. Weinhart and D. A. Weitz, Lab Chip, 2010, 10, 1774 RSC.
- A. R. Abate, J. Thiele and D. A. Weitz, Lab Chip, 2011, 11, 253 RSC.
- J. Thiele, A. R. Abate, H. C. Shum, S. Bachtler, S. Förster and D. A. Weitz, Small, 2010, 6, 1723 CrossRef CAS PubMed.
- H. C. Shum, J.-W. Kim and D. A. Weitz, J. Am. Chem. Soc., 2008, 130, 9543 CrossRef CAS PubMed.
- A. T. Nikova, V. D. Gordon, G. Cristobal, M. R. Talingting, D. C. Bell, C. Evans, M. Joanicot, J. A. Zasadzinski and D. A. Weitz, Macromolecules, 2004, 37, 2215 CrossRef CAS.
- R. C. Hayward, A. S. Utada, N. Dan and D. A. Weitz, Langmuir, 2006, 22, 4457 CrossRef CAS PubMed.
- A. Tan, A. Ziegler, B. Steinbauer and J. Seelig, Biophys. J., 2002, 83, 1547 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley & Sons, New York, U. S., 4th edn, 1999 Search PubMed.
- H. C. Shum, E. Santanach-Carreras, J.-W. Kim, A. Ehrlicher, J. Bibette and D. A. Weitz, J. Am. Chem. Soc., 2011, 133, 4420 CrossRef CAS PubMed.
- F. Jülicher and R. Lipowsky, Phys. Rev. Lett., 1993, 70, 2964 CrossRef.
- S. Hocine, D. Cui, M.-N. Rager, A. Di Cicco, J.-M. Liu, J. Wdzieczak-Bakala, A. Brûlet and M.-H. Li, Langmuir, 2013, 29, 1356 CrossRef CAS PubMed.
- R. Lipowsky, J. Phys. II France, 1992, 2, 1825 CrossRef CAS.
- S. Yamamoto and S. Hyodo, J. Chem. Phys., 2003, 118, 7937 CrossRef CAS.
- T. P. Smart, C. Fernyhough, A. J. Ryan and G. Battaglia, Macromol. Rapid Commun., 2008, 29, 1855 CrossRef CAS.
- Y.-L. Lin, H.-Y. Chang, Y.-J. Sheng and H.-K. Tsao, Soft Matter, 2013, 9, 4802 RSC.
- H. Terasawa, K. Nishimura, H. Suzuki, T. Matsuura and T. Yomo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5942 CrossRef CAS PubMed.
- B. Städler, A. D. Price, R. Chandrawati, L. Hosta-Rigau, A. N. Zelikin and F. Caruso, Nanoscale, 2009, 1, 68 RSC.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153 CrossRef CAS.
- A. R. Abate, D. Lee, T. Do, C. Holtze and D. A. Weitz, Lab Chip, 2008, 8, 516 RSC.
- M. H. Schneider, H. Willaime, Y. Tran, F. Rezgui and P. Tabeling, Anal. Chem., 2010, 82, 8848 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mh00043e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |