Taking orders from light: progress in photochromic bio-materials
Junji
Zhang
,
Jiaxing
Wang
and
He
Tian
*
Key Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science & Technology, Shanghai 200237, P. R. China. E-mail: tianhe@ecust.edu.cn
First published on 13th September 2013
Abstract
Photochromic materials are a family of compounds which undergo photo-reversible transformations between two different isomers with distinct physical and chemical properties. Most smart photochromic materials have been exploited in research areas such as electro-optical functional materials, while recently their applications have extended to novel bio-materials. Biological systems, such as tissue/cellular imaging, nucleotides, peptides, ion channels, etc., have emerged as a revolutionary research frontier for photochromic materials since both covalent coupling and non-covalent interactions with bio-molecules have been achieved. This review commences with a brief description of exciting progress in this field, and describes strategies for using photochromic functional molecules from bio-sensing and cell imaging to optical manipulation of bio-macromolecules (nucleotides, peptides, ion channels, GFP, etc.). Further development of these photo-switches as well as remaining challenges are also discussed and put in prospect.
 Junji Zhang | Dr. Junji Zhang was born in 1985 and received his PhD from East China University of Science and Technology (ECUST) in 2012 under the supervision of Prof. He Tian. Now he is working at ECUST as a lecturer since 2013. His present research interests are mainly focused on photochromic materials, supramolecular switches and nanopore single-molecule sensing. |
 Jiaxing Wang | Jiaxing Wang was born in 1989 and obtained his BSc degree in Fine Chemicals at East China University of Science and Technology (ECUST) in 2012. He is currently pursuing his PhD degree under the supervision of Prof. He Tian at ECUST. His current research interests are photochromic materials and sensors. |
 He Tian | Prof. He Tian received his PhD in 1989 from ECUST. From 1991 to 1993, he stayed in Siegen University as a postdoc supported by Alexander von Humboldt Foundation. In 1999, he was appointed Cheung Kong Distinguished Professor by the Education Ministry of China. His research interests include the syntheses of novel functional organic dyes and development of interdisciplinary materials science determining the electronic and optical properties of materials. Prof. Tian has published 328 papers in international journals and been awarded 66 Chinese patents, is Co-Editor of Dyes & Pigments and International Advisory Board Member for Polymer Chemistry and Chemical Science. In 2011, he was selected as a member of the Chinese Academy of Science. |
1 Introduction
Light as an energy-efficient, environment-friendly, high-resolution and ultrafast power source, has become one of the widely used driving forces for advanced functional materials in various fields.1 Among all the light-responsive functional materials, photochromic materials, bearing the features of reversibility, modulability and diversity in property regulation, have shown their advantages in material science in the last two decades.2 Photochromism is a term used for a reversible transformation of a specific molecule between its two isomers induced by two different wavelengths of light. The photochromic reaction usually reaches a photo-equilibrium or photostationary state after photo-induction, which demonstrates a mixture state with both photo-isomers in a dynamic equilibrium. During the reversible photoisomerization, several physical and chemical properties, such as absorption spectra, fluorescence emission, π-conjugation, electron conductivity, electrochemical properties, magnetic properties, coordination properties, dipole interaction, refractive index, dielectric constant, geometrical structure, etc., may be tuned by light.3 Due to the flexible tuning ability, scientists are encouraged to apply these kinds of compounds as photo-chemical molecular switches in various functional materials and optoelectronic devices by stimulating them with light. So far, fascinating photochromic compounds, such as dithienylethene, azobenzene, spiropyran, etc. (Fig. 1), have been widely used as key components in molecular data storage and logic gates, photo/electro-functional devices, supramolecular self-assembly, photo-controlled catalysis as well as optical surface/nano-materials.4–8 Works on property studies and applications of several photochromic materials in diverse research fields have been recently reviewed.9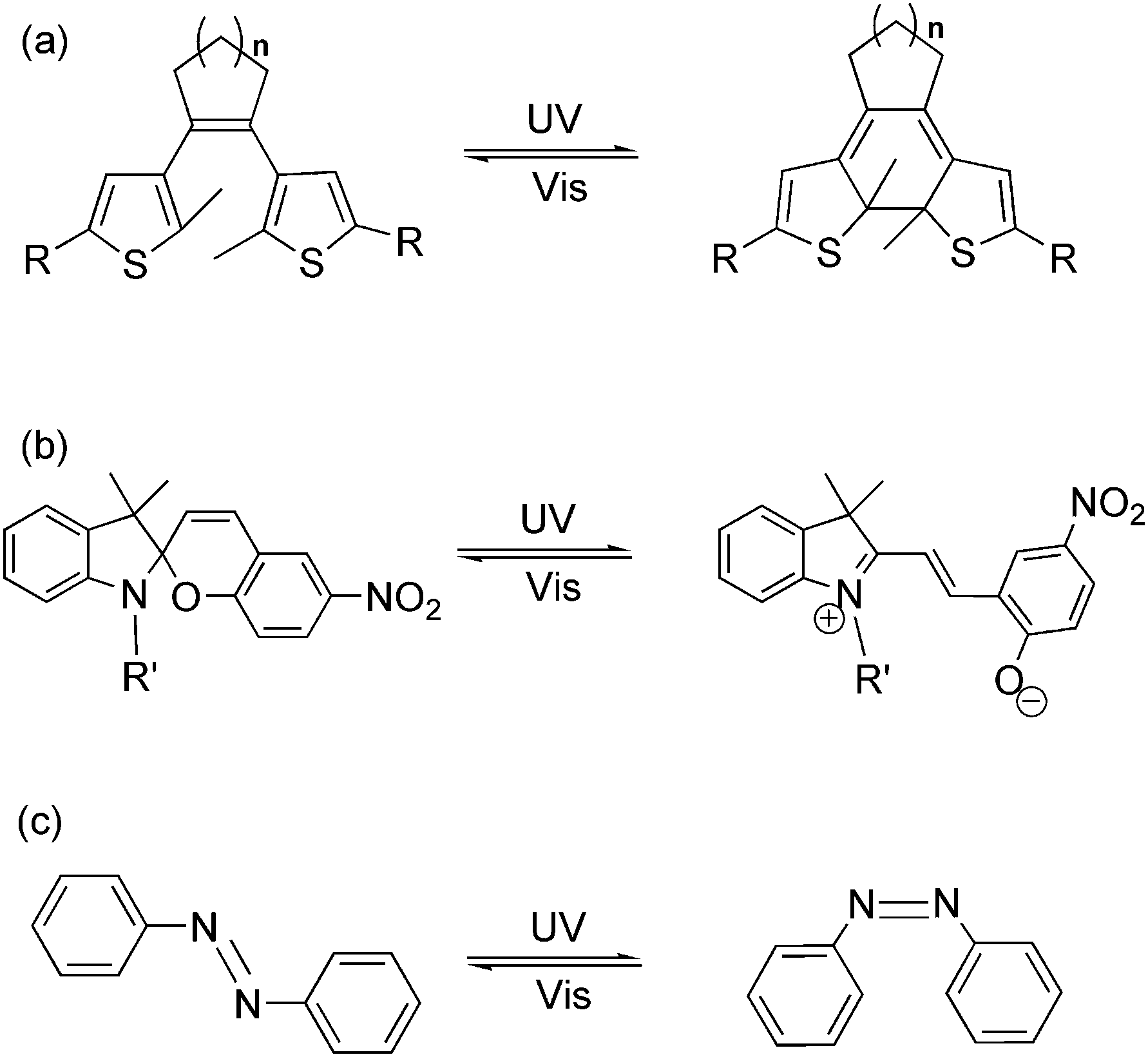 | ||
| Fig. 1 Photochromism of (a) dithienylethene, (b) spiropyran and (c) azobenzene. | ||
Beyond optical/electro-functional materials, the employment of light as the external trigger in regulating bio-chemical, medicinal and physiological systems has aroused wide-spread interest.10,11 Light can provide a controllable high temporal and spatial precision which plays a key role in chemical and biological processes throughout life. Genetic approaches acting in vivo often lack temporal precision, while photochromic molecules can realize greater temporal precision yet suffer from a lack of target specificity.12 A combination or modification of biologically active macromolecules with photochromic compounds endows this hybrid system with both spatial and temporal precision as well as biological specificity, which has proven successful for non-invasive regulation of biological activities and processes in living systems. Generally, two strategies are applied in photo-controllable biological systems: irreversible photo-active or photo-cleavage molecules, and reversible photochromic molecules. Both strategies have their advantages and disadvantages, and the boundary between them is vague.1a In this review, we will focus on notable achievements of photochromic materials in bio-technology and bio-inspired applications over the past five years. This article will not include all accomplishments in this research field but rather points to important developments by selecting a few representative examples from different research areas in photo-regulated bio-technology.
2 Photochromic switches for sensing and cell imaging
As far as we know, photochromic materials, which consist of fluorescence groups and photo-switchable units, have been widely used in sensing and cell imaging. Furthermore, fluorescent photo-switches applied for cell imaging overcome common defects, such as optical interference and autofluorescence.9g It is convenient for us to distinguish the target from the background through turning on and off the fluorophores. Yet, practical applications in vivo are limited by the wavelength of triggering/exciting light which is mostly located in the UV region. Due to low tissue/cell transmission efficiency and cell-damaging issues, ultraviolet light is not the ideal candidate for the triggering force in photo-biological applications. The application of biliproteins, particularly phytochromes, has provided a good solution for this existing challenge,13 while the development of visible or near-infrared light-induced photochromism is another wise choice.2.1 Photochromic sensors
In general, most photo-switches become popular molecular probes in decades. Their spectroscopic properties change once the sensor recognize their targets, and this signal is expected to be visualized or amplified by machine. Although people have paid much attention to sensors, photochromism is rarely mentioned when it comes to sensors for organic molecules and applications in biology.14 Yang and co-workers14a,c,d carried out a lot of researches to detect amino acids and pyrophosphate based on photochromic spiropyran derivatives, which would overcome the obstacles of weak and nonspecific interactions between probes and analytes. The sensing mechanism between spiropyran probes and GSH (glutathione) is shown in Fig. 2. The closed spiropyran (SP) form changes to the open merocyanine (MC) form when GSH is added and induces the open ring reaction via electrostatic interactions. Both 1a and 1b show strong fluorescence signals upon binding with GSH, while only 1a has high selectivity to differentiate GSH from other amino acids such as Glu (glutamic acid) and Cys (cysteine). Furthermore, fluorescence anisotropy and confocal fluorescent microscopy demonstrate that 1a is well transported into human acute T cells and tends to be accumulated. This precedent work provides a potential method for detecting biomolecules via electrostatic interactions and structure complementarities of the host–guest molecules. It should be noted that, although this amino acid sensing behavior does not depend on the photochromicity of spiropyran, the photo-reversible property of this bio-sensor possesses potential in photo-controllable biomolecule capture and transfer15 with high affinity and selectivity.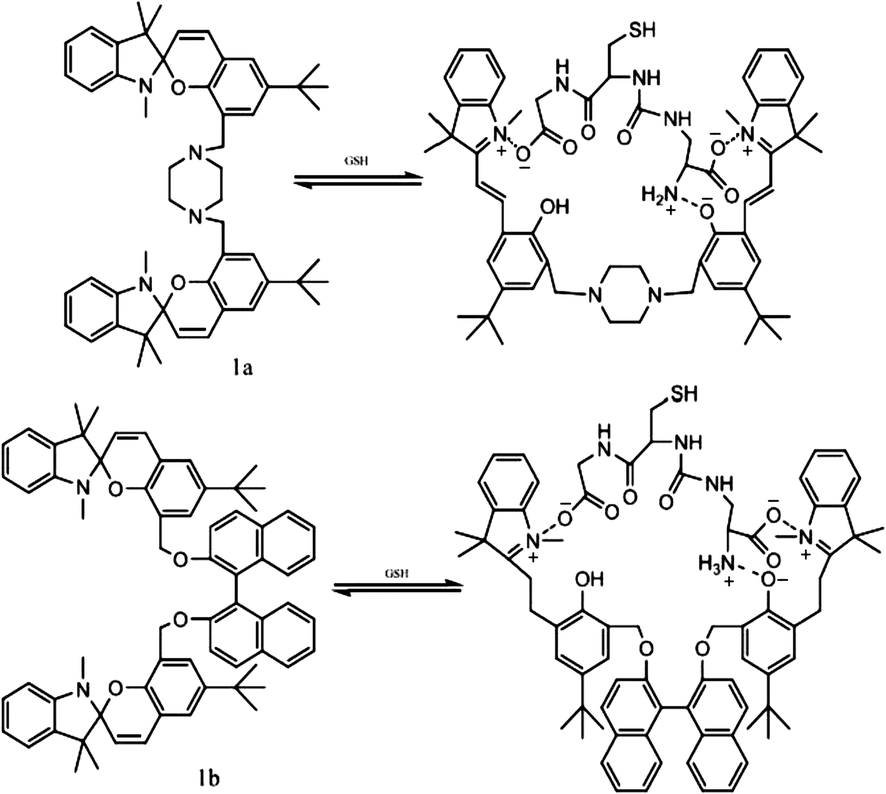 | ||
| Fig. 2 Ring-opening reaction of 1a and 1b in the presence of GSH. | ||
Qu et al.14b synthesized spiropyran-modified MWNTs (multi-walled carbon nanotubes) to regulate the activity of HRP (horseradish peroxidase). The spiro form MWNT-SP exhibits aggregation and the ring-opened merocyanine form MWNT-MC tends to disperse (Fig. 3). MWNT-MC makes a contribution to the catalytic activity enhancement of HRP due to close interactions between the merocyanine and HRP as well as high affinity for hydrophobic substrates on MWNTs because of its high surface-to-volume ratios, while its counterpart MWNT-SP did not enhance the activity of HRP. After addition of 15 mg mL−1 MWNT-MC, the absorption at 652 nm increased by 3.7-fold within 3 min. This feature can be used for label-free colorimetric detection of lysozyme. In the absence of an anti-lysozyme DNA aptamer that binds to lysozyme with high selectivity, HRP activity decreased in the MWNT-MC solution. By adding DNA aptamer to MWNT-MC, the absorption at 652 nm after 3 min decreased by about 57.9%. After adding lysozyme, the absorbance at 652 nm increased which indicates that HRP was bound to MC again. The detection limit was found to be 30 nM.
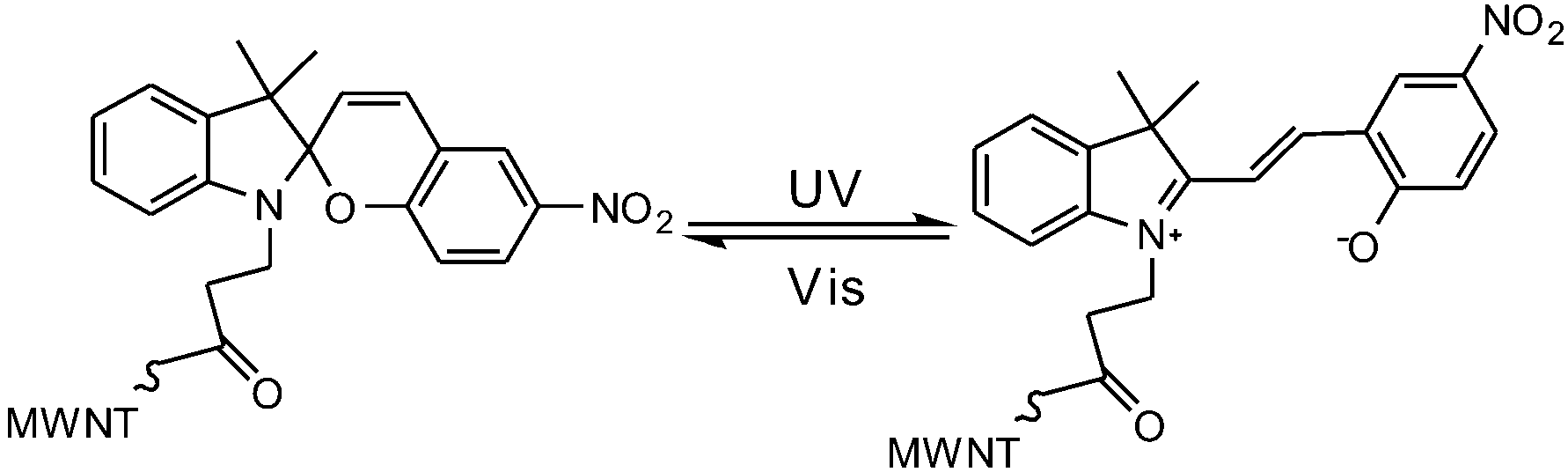 | ||
| Fig. 3 Light regulation of peroxidase activity by spiropyran-modified MWNTs. | ||
Louie et al.16 reported a MRI contrast agent based on spiropyran (Fig. 4). When the SP form turns into the MC form, both the structure and charge of the molecule change, influencing the interaction between water molecules and Gd ions. Consequently, different contrast enhancement is achieved between the two isomers. In the presence of NADH, the initial MC-Gd was reduced to SP-Gd resulting in an almost 100% fluorescence quenching compared to the initial fluorescent MC-Gd state. The complex is thus used as a probe to detect NADH (nicotinamide adenine dinucleotide) in vivo, an important reductive coenzyme in respiration chain. Again, in this work the isomerization of spiropyran is not based on photochromism but on redox-triggered complexation. Yet the introduction of photochromes would shine light on a novel class of “double-check” sensors,17 which could help avoid false positive signals which often occur in traditional sensor applications.
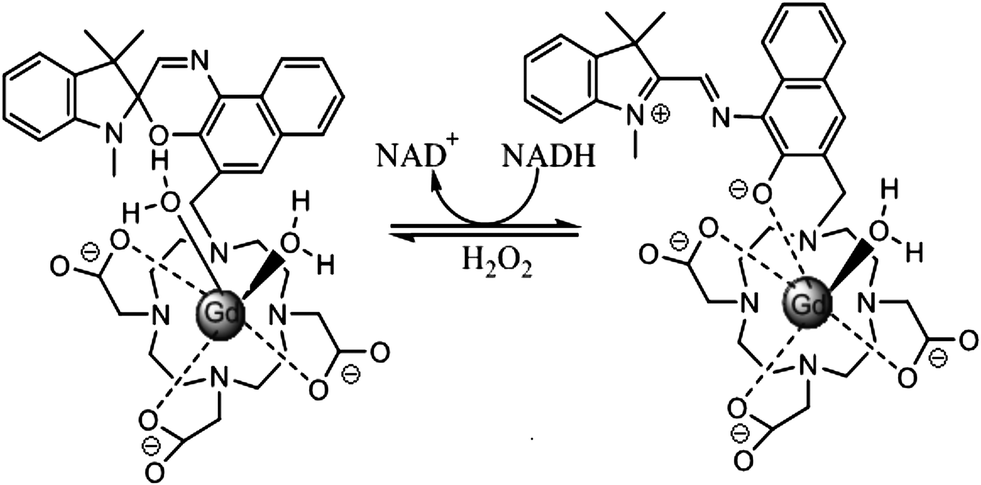 | ||
| Fig. 4 The photo-sensitive complex responds to NADH and hydrogen peroxide. | ||
2.2 Cell imaging with photochromic materials
As a useful tool to study biology dynamic pathways, photo-switchable compounds including spiropyran and azobenzene derivatives have been widely investigated, such as cytotoxicity dynamic pathways and fluorescence imaging in vivo.18Though various spiropyran and azobenzene derivatives are commonly used for cell imaging, their low thermal stability and fatigue hamper their practical utilization in vivo. Thus it is important to introduce more thermal stable and photo-resistant diarylethenes into living cells. Many fluorescent probes based on diarylethenes have already been investigated in recent years.19–23
The pioneer work of diarylethene used as a photo-switchable probe for imaging living cells dates back to the year 2008 (Fig. 5).19 Hydrophilic and hydrophobic chains were introduced into diarylethene molecules which exhibited fluorescence diversity between open and closed isomers. This work exploits a new way to get switchable imaging followed by other turn-off diarylethene probes.
 | ||
| Fig. 5 Amphiphilic diarylethene for cell imaging. | ||
It is well known that many fluorescent diarylethenes have strong fluorescence before UV light irradiation. The fluorescence decreases or even vanishes after isomerization to its closed form due to FRET (Fluorescence Resonance Energy Transfer). In these turn-off fluorescence systems, a major challenge lies in how to apply them for super-resolution fluorescence microscopy which works in the initial dark state. In order to improve higher standard resolution images, development of turn-on fluorescent diarylethenes is of great necessity. Ahn, Kang and Lee20 reported a turn-on fluorescent diarylethene (Fig. 6) for living imaging. In this diarylethene system, the closed-form fluorescence intensity increased remarkably upon UV light irradiation, which showed high sensitivity in TIRFM (total internal reflection fluorescence microscopy) experiments. However, though this diarylethene derivative possesses good thermal stability and reversibility, how to improve the quantum yields upon photochemical ring-opening reactions of turn-on diarylethene still remains to be solved.
 | ||
| Fig. 6 Turn-on fluorescent diarylethene. | ||
Photo-switchable probes are always triggered by UV light. As UV light has low cell penetration ability and will cause damage to living organisms as well as trigger autofluorescence, the keys to solve these problems are using near-infrared light or multi-photon techniques. Li and coworkers reported two-photon photo-switching and two-photon photoluminescence of spiropyran nanoparticles.21 After UV (488 nm) or NIR two-photon (780 nm) irradiation, the SP nanoparticles underwent photo-isomerization (Fig. 7). The white dispersed nanoparticles changed to purple MC nanoparticles and emitted strong red fluorescence. This reversible photo-switching progress can be repeated more than 20 times. The photo-reversible nanoparticles were then injected into Her2 cancer cells. The imaging results proved that anti-Her2 antibody-conjugated photo-switchable nanoparticles had better photochromic and photoluminescence performance upon NIR two-photon irradiation. Results also showed that the fluorescence intensity decreased using single-photon UV light compared with a steady fluorescence intensity using two-photon excitation.
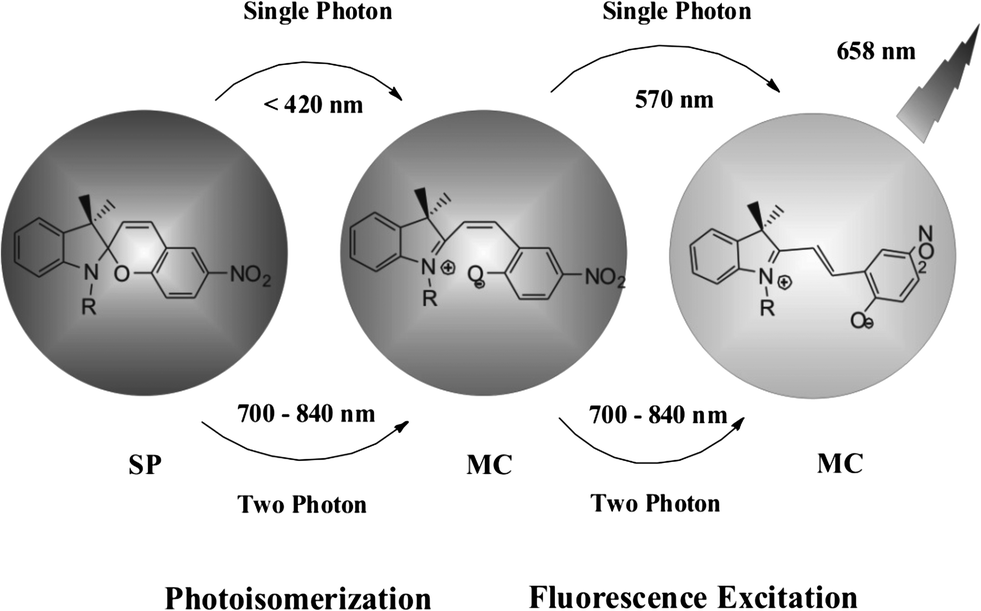 | ||
| Fig. 7 The photo-switching and fluorescence excitation of SP/MC nanoparticles. | ||
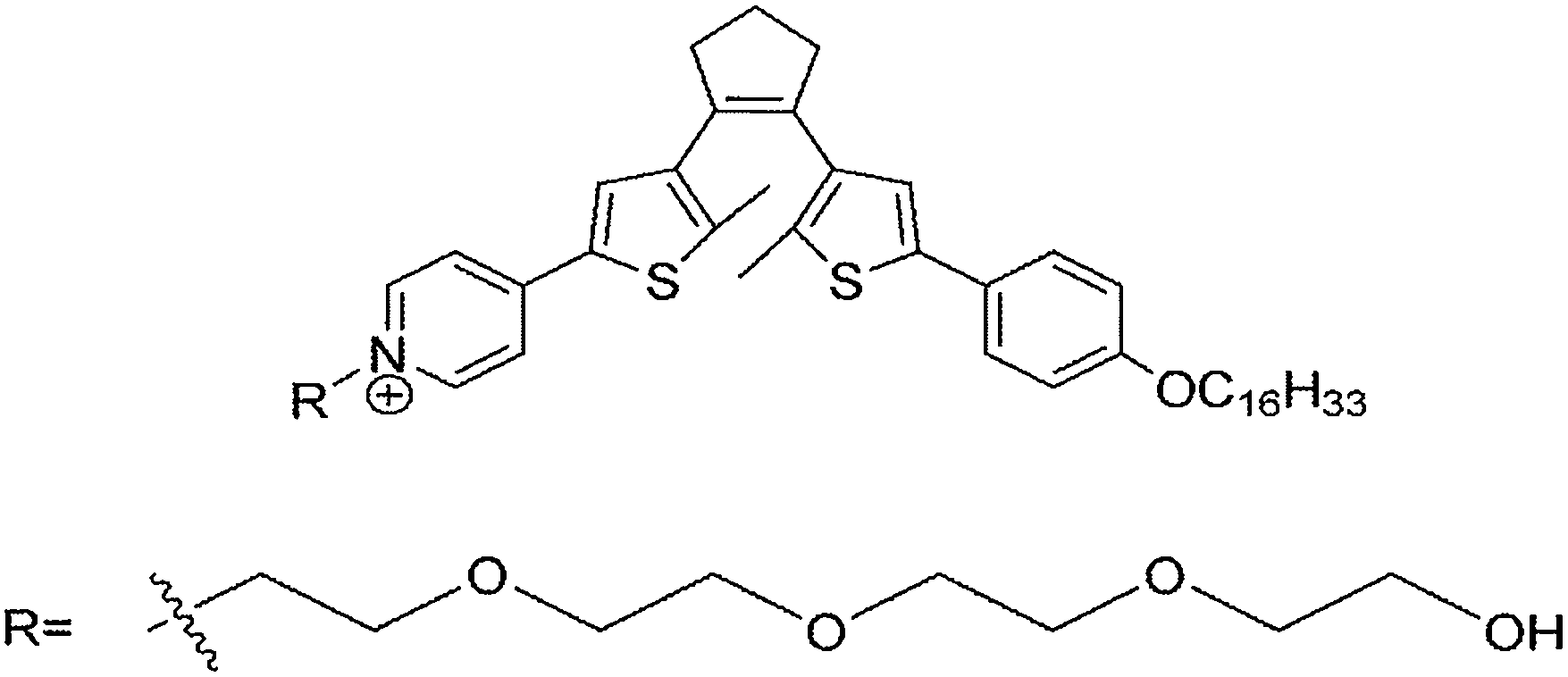
Fluorescence probes containing diarylethenes showed excellent fatigue resistance and thermal stability in imaging living cells. Tian and Yi22 have reported a visible light excited diarylethene–Ir complex for imaging living cells (Fig. 8). The complex Ir(pic)(Py-BTE)2 (pic = 2-picolinic acid) was synthesized to improve water solubility and cell-permeable performance according to previous work.24 Due to the metal-to-ligand charge-transfer (MLCT) transitions, the Ir-complexed diarylethene exhibits visible light photoisomerization which makes it excellent in bio-compatibility and avoiding autofluorescence.
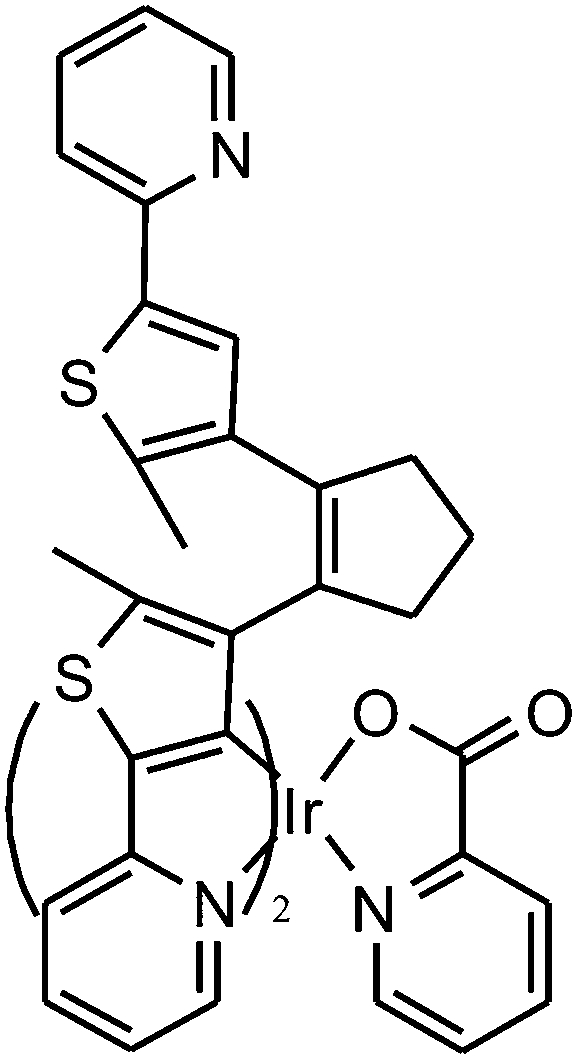 | ||
| Fig. 8 Diarylethene-based complex probe. | ||
In addition, fluorescent nanoparticles with photoluminescence (PL) are another choice to achieve visible light induced photochromism and avoid autofluorescence in biosystems. Based on previous work,25 Branda and Baillie reported diarylethene-decorated upconverting nanoparticles (DTE–UCNPs) with a two-wavelength emission based on different ways of absorption.23 The diarylethene compound is attached to nanoparticles through “click chemistry”. The fluorescence of complex systems, both green and red emission, can be quenched to different extents by photochromism (Fig. 9). Such systems not only possess the ability to reversibly modulate the intensity of the probe emission but also are capable of altering the color of the nanoparticle emission. This smart DTE–UCNP system reduces UV caused photo-degradation and bio-incompatability by using NIR as the absorption light source. Meanwhile the two-wavelength emission property helps avoid undesired interruption during cell imaging and further improves the imaging capabilities of this DTE–UCNP hybridized system.
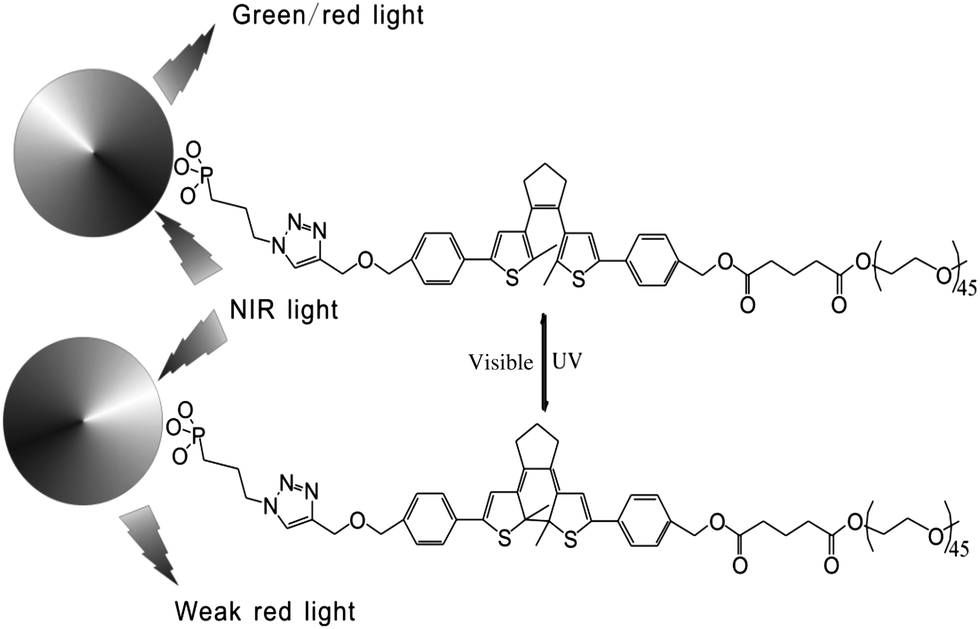 | ||
| Fig. 9 The selective quenching of emission by ring-closing diarylethene. | ||
3 Photochromic switches for drug delivery
Traditional medicines used to cure diseases can get to the target via blood circulation. They usually have common problems such as low aqueous solubility, low selectivity, high toxicity and hardness of degradation. Compared with the disadvantages of conventional medicines, safe and effective drug delivery systems have been widely investigated for decades. Light-responsive drug delivery systems have received much attention due to potential applications in optional sites of the body. Both the drug release rate and precise sites of release can be modulated by light, producing fewer side effects and considerable therapeutic levels of drugs.26 The only critical challenge remaining is also found in other light-controlled molecular switches applied for biocompatible materials. Because of the high level absorbance, shorter wavelength (less than 700 nm) light can hardly penetrate 1 cm into the skin and other tissues. Effective strategies are use of near-infrared light or two near-infrared photons.In light-responsive drug delivery systems, photochromic switches play the key role; they are boosted by light and usually attached to drugs. Although azobenzene dominates in drug delivery rather than other photochromic compounds,27–29 several spiropyrans are also available.30,31
Zhang et al.27 reported an azobenzene light-responsive “plug and play” polyanionic template as a drug delivery system (Fig. 10). Polyacrylic acid (PAA) and azobenzene (Azo) are formed into a polyanionic template, followed by association with “drug-carrying” α-cyclodextrin (α-CD) via trans-azobenzene isomer. α-CD becomes unloaded when the trans form isomerizes to the cis form after UV irradiation, thus completing the drug loading/unloading process. The drug release behaviour of the PnP template has been investigated via studying the release rate of α-CD-RhB and α-CD-Dox. In the dark, the release rates of α-CD-RhB and α-CD-Dox are slow: only 14.7% loaded α-CD-RhB and 14% loaded α-CD-Dox are released from the template within 15 h. However, after UV irradiation, around 80% loaded α-CD-RhB and 82% loaded α-CD-Dox can be released within 15 h, over 500% faster compared to trans isomer modified template. It should be highlighted that polyanionic templates, the surfaces of which are negatively charged, can be repelled by normal cells since normal cells carry negative charges on their membranes. Therefore, this polyanionic template optical drug release system could fulfill the “target elimination” task that specifically targets tumor cells while preventing normal cells from being damaged. Confocal fluorescence images prove that the drug loaded on the polyanionic template cannot be endocytosed by normal tissues during blood circulation. Only after UV irradiation can the drug be released to kill the target cells.
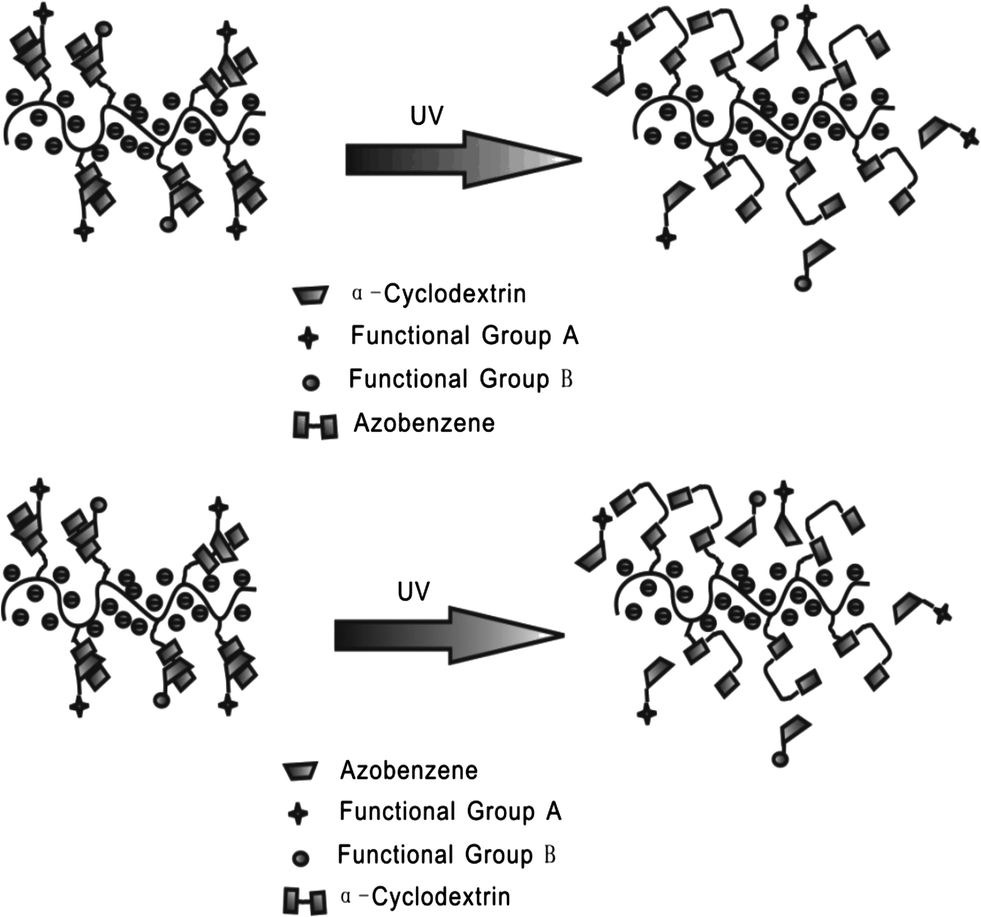 | ||
| Fig. 10 Photo-active polyanionic template as a drug delivery system. | ||
Apart from polymeric drug delivery systems, Zhang et al.28 also developed layer-by-layer (LbL) microcapsules as optical drug carriers. It has been already demonstrated that LbL microcapsule-shaped drugs deliver superiority to cure cancer cells.32 Since noncovalent interactions between the azo groups and α-CD can be easily controlled, the authors constructed a supramolecular hollow microcapsule system with PAA-C12-Azo, CMD-g-α-CD and α-CD modified drug as shown in Fig. 11. Driven by UV irradiation, hollow microcapsules disassociated and released more than 60% of drug comparing to only 5% released in the dark. This type of drug delivery system also inhibits the antineoplastic drug endocytosis of normal cells via packaging the drug in microcapsules. These novel photo-switchable microcapsules show great potential for clinical applications.
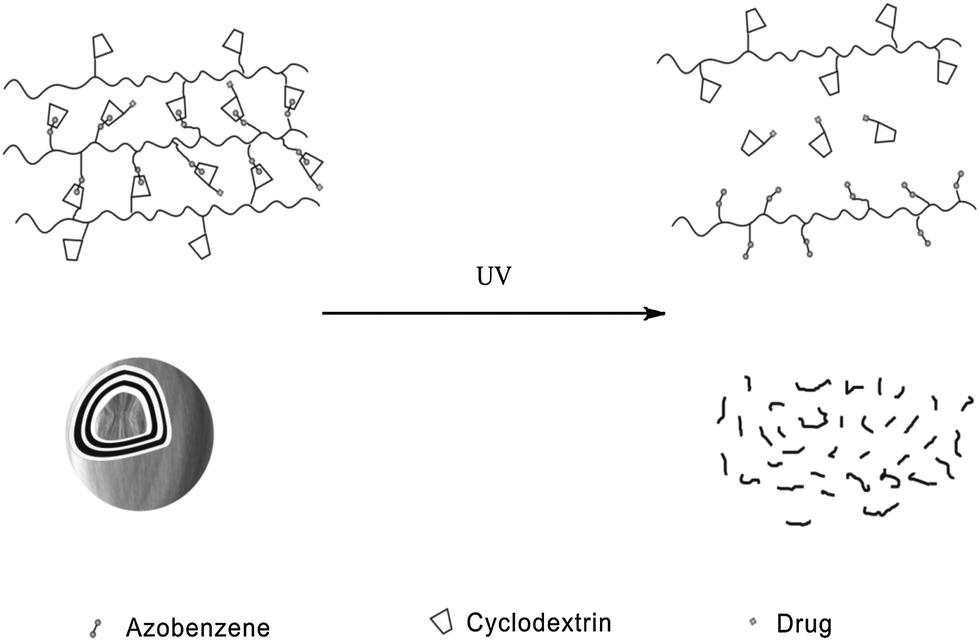 | ||
| Fig. 11 Azobenzene modified layer-by-layer (LbL) microcapsules as drug delivery systems. | ||
Meanwhile, photo-responsive hydrogels are becoming a promising class of bio-materials because of their multi-functional role in photo-patterning33 as well as drug delivery.34 Tan and Zhang29 applied hydrogel gel–sol conversion for drug delivery. The hydrogels are composed of two DNA–polymer conjugates and an azo-incorporated DNA linker (ADL) as shown in Fig. 12. In the presence of ADL, two DNA polymer chains self-assemble with each other immediately and form a yellow gel. Then the two chains are spread out followed by gel-to-sol conversion after UV irradiation. Moreover, the capability to release small molecules, large NPs and bioactive enzymes (e.g. fluorescein, doxorubicin, horseradish peroxidase, and gold nanoparticles) is also realized by excitation at 350 nm. Both the release rate and release amount of NPs are affected by the concentration of hydrogel: 50 μM hydrogel turned out to have the fastest release rate (6.88 ± 0.70 × 10−4 nmol min−1), while 100 μM hydrogel has the highest release amount (66.9%). More importantly, this system built up a model to reclaim drugs triggered by visible light, which highlights its great potential in future applications in drug and gene delivery and tissue engineering.
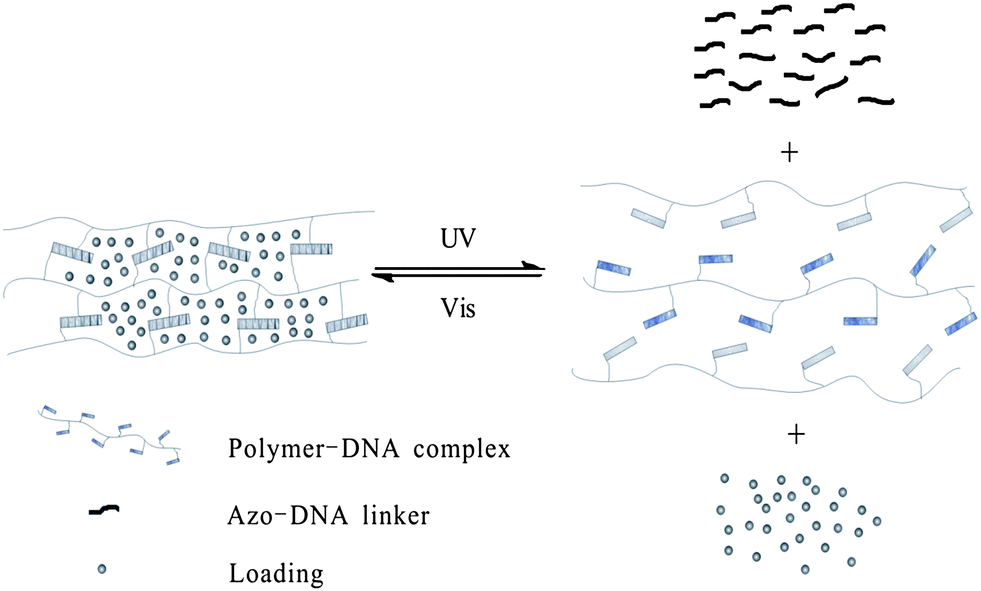 | ||
| Fig. 12 Photo-responsive hydrogels as drug delivery systems. | ||
Kohane and coworkers30 developed a spiropyran-based nanoparticulate drug delivery system (Fig. 13). Besides the release rate factor mentioned above, the size of loading nanoparticles is another crucial determinant of penetration into target tissue. Triggered by UV/visible light, both the size and the release rate can be easily modulated. The nanoparticle size shrank from 150 to 40 nm upon UV irradiation and was restored by visible light in the dynamic light scattering experiment. In the absence of UV, only 7.2% drugs and dyes were released within 1 h and full release was completed within 48–72 h; while the release rate can be significantly enhanced to 29.3% within the same time scale upon UV irradiation. Meanwhile, the concentration of released dye was much higher after UV irradiation compared to that of the initial state. What's more, this system showed enhanced diffusion ability in the cornea. The color conversion of the cornea indicated that the loaded Cy5 dye was released in 1 min when exposed to UV light. The mechanism of this optically controlled drug release system is as follows: the SP nanoparticle is composed of a hydrophilic PEG shell, lipid and SP-C9. At first, SP perturbs the alkyl chain packing inside the hydrophobic cores of the nanoparticle, leading to a loose structure. When SP converts to MC, zwitterionic MC tends to move outward to the hydrophilic microenvironments, allowing the alkyl chains of the DSPE (1,2-distearoyl-sn-glycero-3-phosphoethanolamine) and lecithin to accumulate inside the hydrophobic cores. As a result, the size of the nanoparticle shrinks.
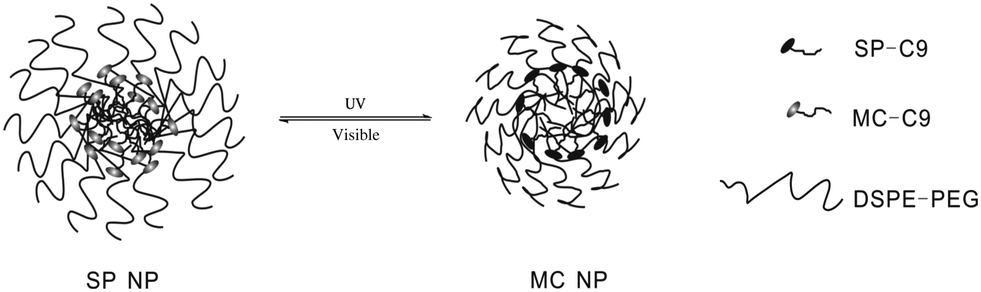 | ||
| Fig. 13 The photo-switching of an SP nanoparticle. | ||
4 Photo-modulated nucleotides and peptides
4.1 Photo-switchable nucleotides
DNA is one of the most exciting biomolecules in life science because of its key role as the carrier of genetic information.35 Moreover, DNA as a main character in multi-disciplinary research such as biosensing, nanotechnology and molecular computing, is a thriving subject nowadays.36 Using light as the driving force in DNA chemistry thus becomes one of the major interests in this new area. The photo-controlled nucleic acids have been constructed in two manners: (i) non-covalent interaction between photochromic molecules and specific base pairs in DNA strands; (ii) covalent tethering of photochromic molecules to the oligonucleotide structure.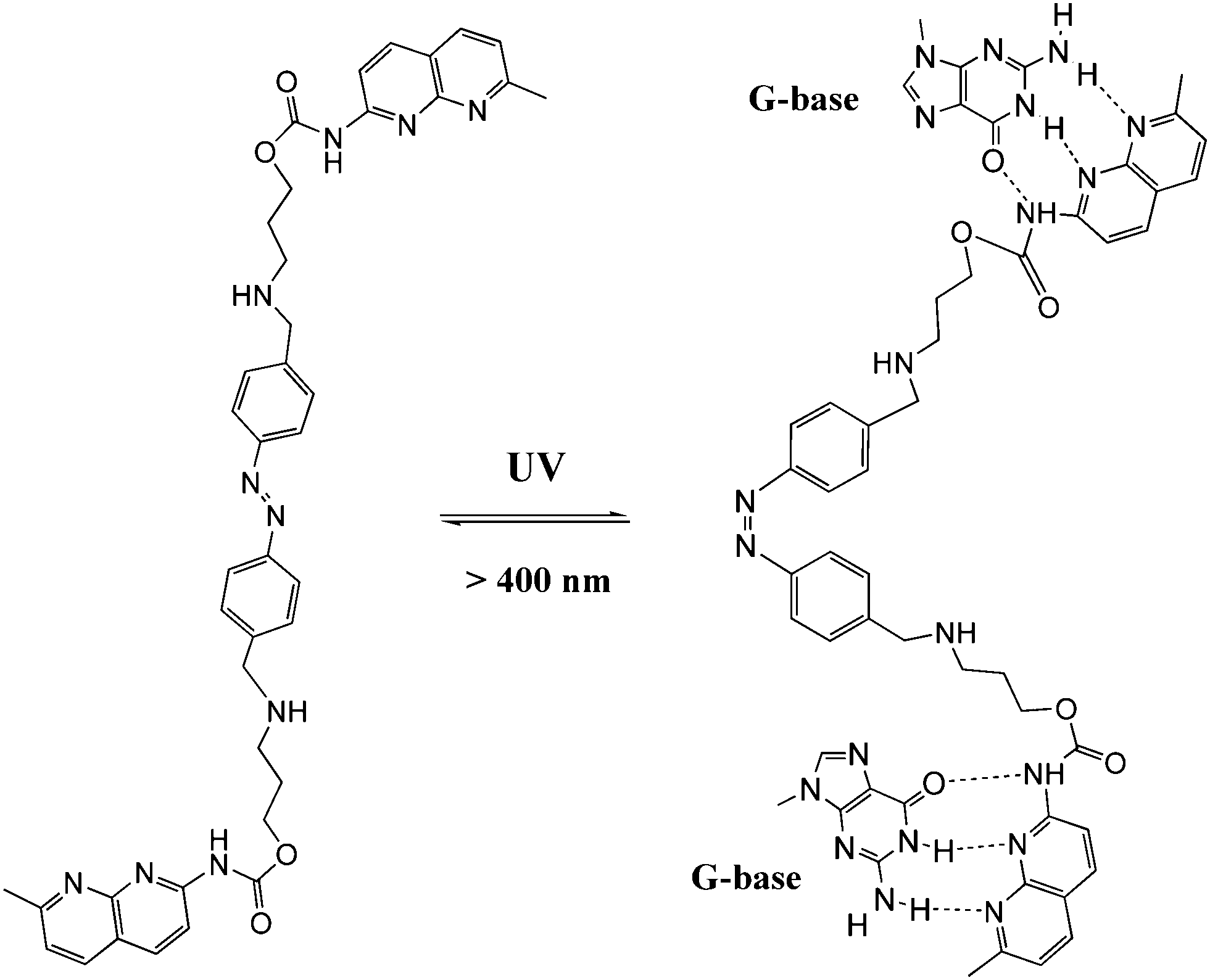 | ||
| Fig. 14 Azobenzene-based DNA molecular glue. | ||
Zhou and colleagues38 further modulated the structure of a G-quadruplex with a piperidinium modified azobenzene photo-switch in metal ion (e.g. K+/Na+) free conditions (Fig. 15). Unlike the hydrogen-binding azobenzene glue above, in this work the major force between the photo-active molecule and DNA is electrostatic interaction. The stretched trans form stabilized the G-quadruplex, while the cis form de-folded the G-quadruplex into a single strand form induced by visible light with a photo-conversion rate of 84.6% (determined by 1H NMR). CD (circular dichroism) spectroscopy and thermal denaturation were used to monitor the photo-controlled stretching/folding motion of the G-quadruplex. The binding constant of trans-azobenzene dipiperidinium with the telomere DNA was determined by UV/Vis-absorption titration experiment to be around (3.9 ± 0.5) × 106 M−1. Exonuclease I catalyzed hydrolysis, which is based on the folding/unfolding conversion of G-quadruplex, was further used as an assay to evaluate the photo-modulation of the G-quadruplex. In the K+ stabilized G-quadruplex solution, exonuclease I could not degrade DNA since the G-quadruplex was in its folding mode. The same phenomenon was observed when trans-azobenzene dipiperidinium was present in the system, while the exonuclease I catalyzed hydrolysis was switched on after irradiation with 350 nm UV light, demonstrating the unfolding of the G-quadruplex due to the formation of the cis-azobenzene isomer. These two non-covalent photo-switchable systems directly converted light into mechanical work, thereby offering the potential to turn off/on gene expression39 and enable the development of DNA nano-device applications.
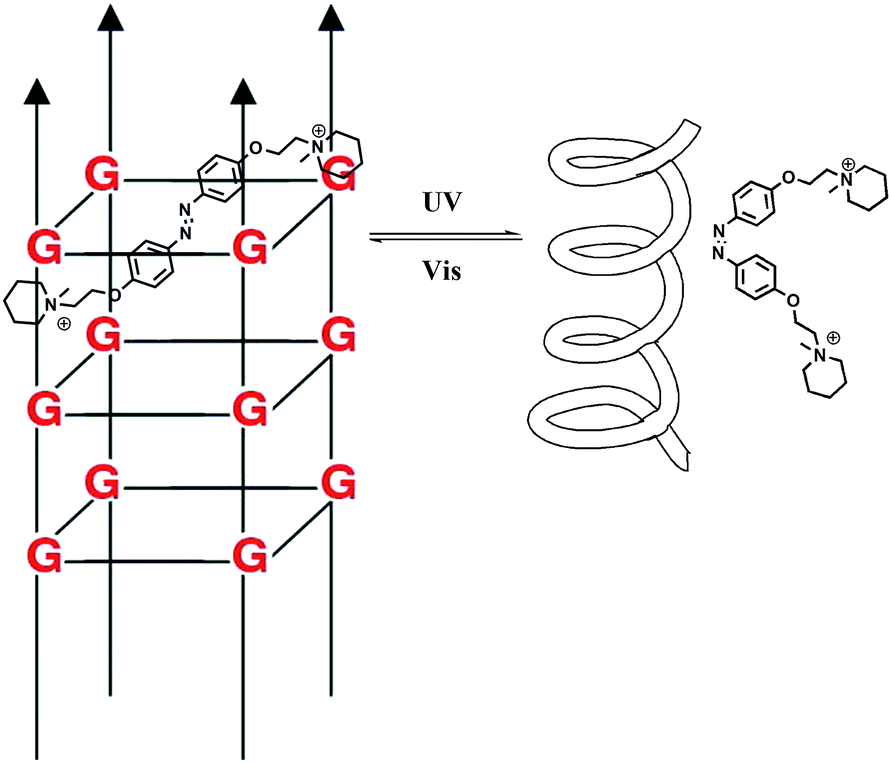 | ||
| Fig. 15 Azobenzene photo-induced folding and stretching of G-quadruplex. | ||
Andréasson et al.40 reported dramatically different DNA-binding performances between two isomers of a spiropyran photo-switch. The intercalator open merocyanine form could be shifted to the nonbinding closed form with irradiation using visible light, and vice versa with UV light (with a photostationary distribution of 54/46 spiropyran/merocyanine and 100% back conversion with visible light, calculated from NMR data). The zwitterionic merocyanine intercalator would resonate between two isomeric structures: zwitterionic and neutral. While the zwitterionic merocyanine tends to stay in the more polar water environment, the latter neutral isomer favors the interaction with hydrophobic nucleobases causing a pronounced solvatochromism (92 nm blue shift), which makes it an excellent candidate for DNA probing. Absorption spectra and flow-oriented LD (linear dichroism) were used to probe the whole switching process, and the binding constant (ca. 2 × 10−4 M−1) as well as binding affinity sequences (GC < CT < AT) were also determined. Further construction of a DNA AND logic system was achieved based on this spiropyran–DNA hybrid with an additional proton stimulus, which has potential applications in logically controlled anticancer drug release (Fig. 16).41 Spiropyran, as an anti-aptamer small molecule, also showed dramatically different binding properties with specific RNA aptamers during photo-switching between its open and closed isomers.42 This developed photochemical riboswitch can be used to spatially and temporally regulate the gene function in a reversible fashion as well as in future biological data-processing circuits and storage devices.
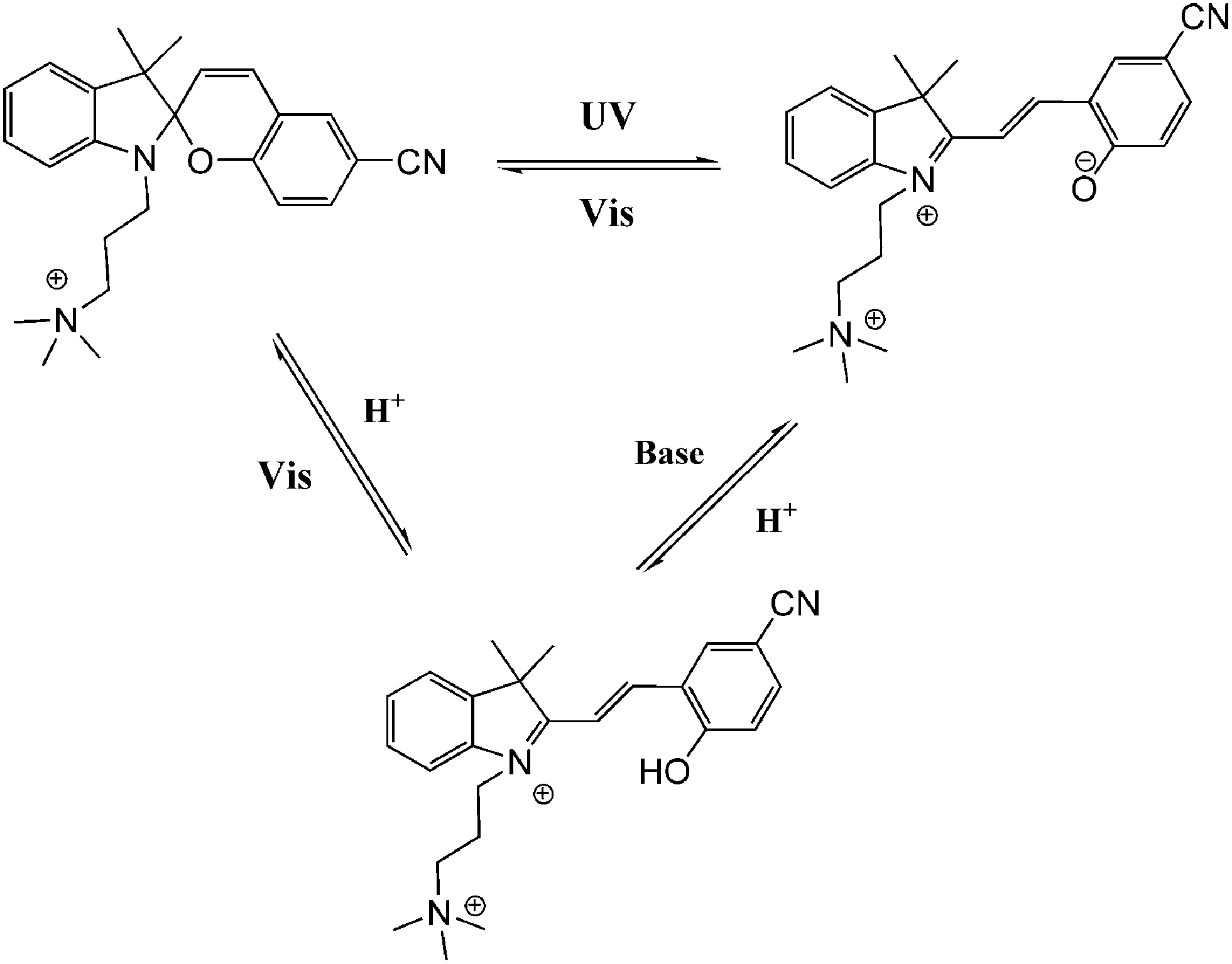 | ||
| Fig. 16 Multi-state switches between DNA-binding spiropyran. | ||
Dithienylethenes (DTE), which form another major family of photochromic materials, were also active in non-covalent DNA binding. Feringa and colleagues synthesized an amine-terminated dithienylethene A (Fig. 17, left) and intercalated them into a DNA duplex via electrostatic interaction between the amine group of DTE and the phosphate group of DNA.43 Due to its minor geometric change after UV-Vis irradiation, the hybridization of the target DNA did not change as azobenzene and spiropyran did. However, an evident chiroptical response of the DNA–DTE hybrid was shown after alternating UV/Vis irradiation, indicating that a chiral photo-switching DTE–DNA complex was formed. Recently, another DNA-intercalating dithienylethene B (Fig. 17, right) showing high enantioselectivity upon open–closed isomerization was also reported by Andréasson et al.44 Compared to azobenzene and spiropyran, the photoisomerization rates of dithienylethenes in the two examples above are both above 90% with full back conversion upon visible light irradiation, endowing these DTE functionalized bio-materials with high photo-conversion efficiency as well as distinctive photo-reversibility.
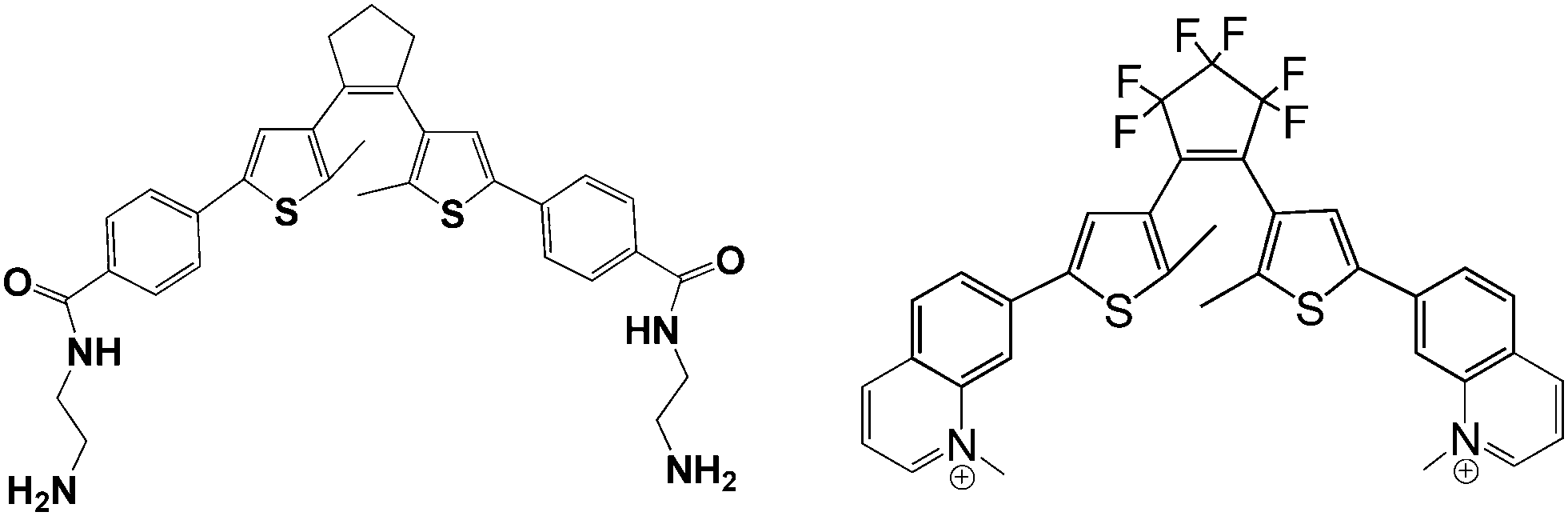 | ||
| Fig. 17 Diarylethenes A (left) and B (right). | ||
Tan and colleagues46 designed a DNA hairpin structure with azobenzene molecules tethered in the DNA backbone through the stem moiety (Fig. 18). A fluorescein (FAM) and a Dabcyl group were attached at the 5′ and 3′ ends, respectively, of this DNA hairpin as a fluorophore/quencher (F/Q) pair. Before irradiation with UV light, azobenzene was in its trans form, which stabilized the hairpin structure and resulted in a low fluorescence due to the efficient FRET effects. Upon UV irradiation, trans-azobenzene was transformed into its cis form, which dissociated the hairpin structure to a single strand. The distance between the F/Q pair was significantly lengthened, leading to a large decrease of FRET effects along with a reciprocal much higher fluorescence signal. Therefore, the photo-reversible motion of this DNA molecular motor could be tracked with alternating fluorescence variation. Compared with previous studies on photo-controllable DNA hybridization, such a photo-regulated DNA nanomotor has the advantages of easy manipulation and high reproducibility because of the single-molecule feature requiring no additional DNA strands as fuel. Based on this strategy, further research on enhancing the photo-isomerization efficiency of DNA nanomotors on local plasmon resonance-induced near field47 and azobenzene incorporated DNA aptamers have also been reported sequentially.48
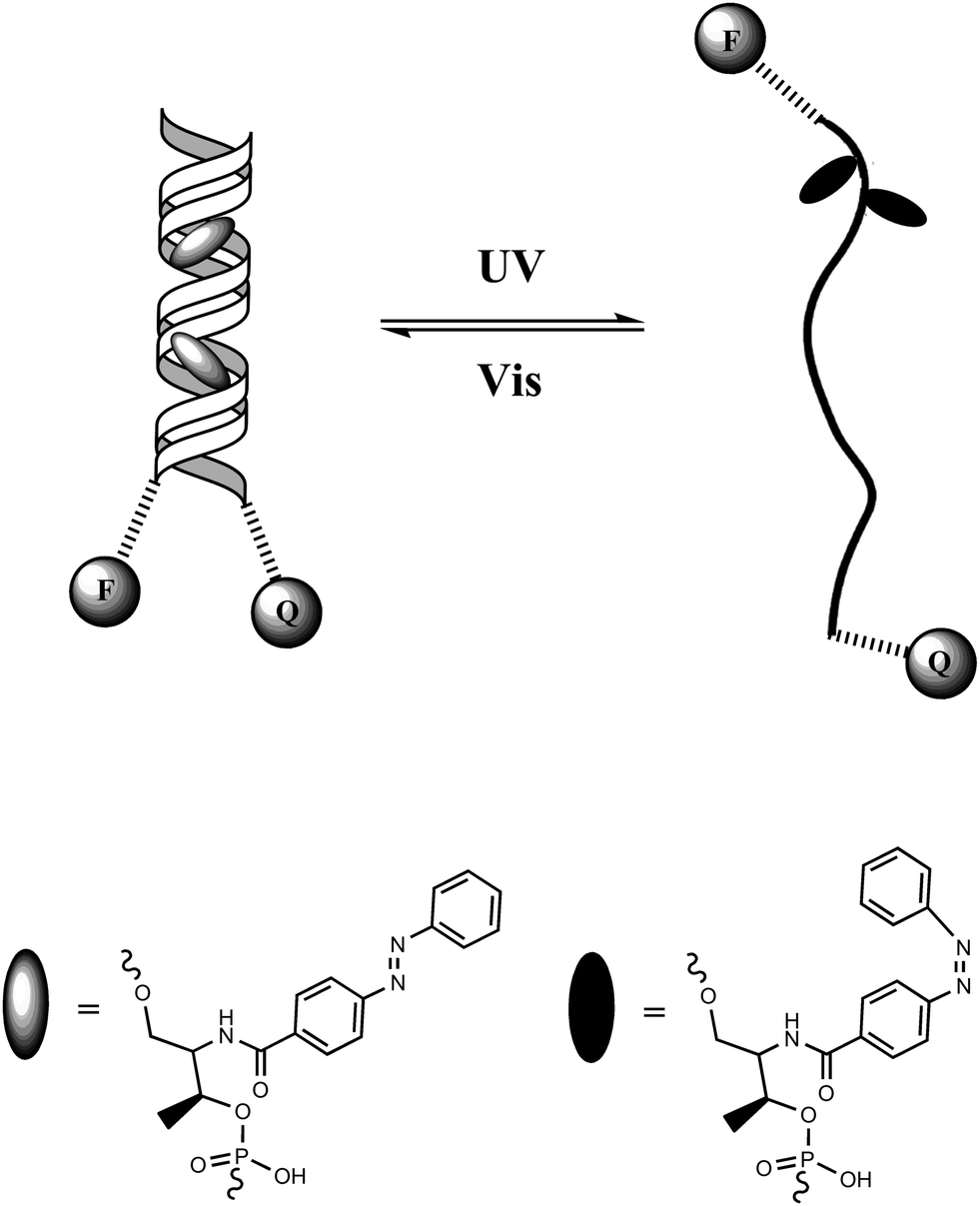 | ||
| Fig. 18 Azobenzene optical DNA motor. | ||
DNAzymes are DNA molecules which have the ability to perform a chemical reaction as catalysts. Asanuma et al.constructed a photo-responsive DNA machine based on a DNAzyme,49 which could perform the photo-regulation of RNA digestion by enzymes. As shown in Fig. 19, when azobenzene is in its trans from, a very stable hairpin structure with a trans-azobenzene stabilized stem duplex is formed. In this state, the RNA-cleavage activity is suppressed due to the topologically constrained higher order structure of the catalytic loop, which cannot offer the correct conformation for cleavage. After triggering with UV light, the cis form of azobenzene cannot stabilize the stem duplex and thus opens the hairpin structure, leading to an estimated 38-fold acceleration of DNAzyme catalyzed RNA cleavage. Importantly, the RNA digestion rate is greatly influenced by the topology, which provides a strategy for the general design of optically controllable RNA digestion DNAzyme structures. Furthermore, this design of modified DNAzyme can be extended to exonuclease, which facilitates its applications in vivo for photo-regulating gene expression as well as for in vitro photo-switchable nano-robots.
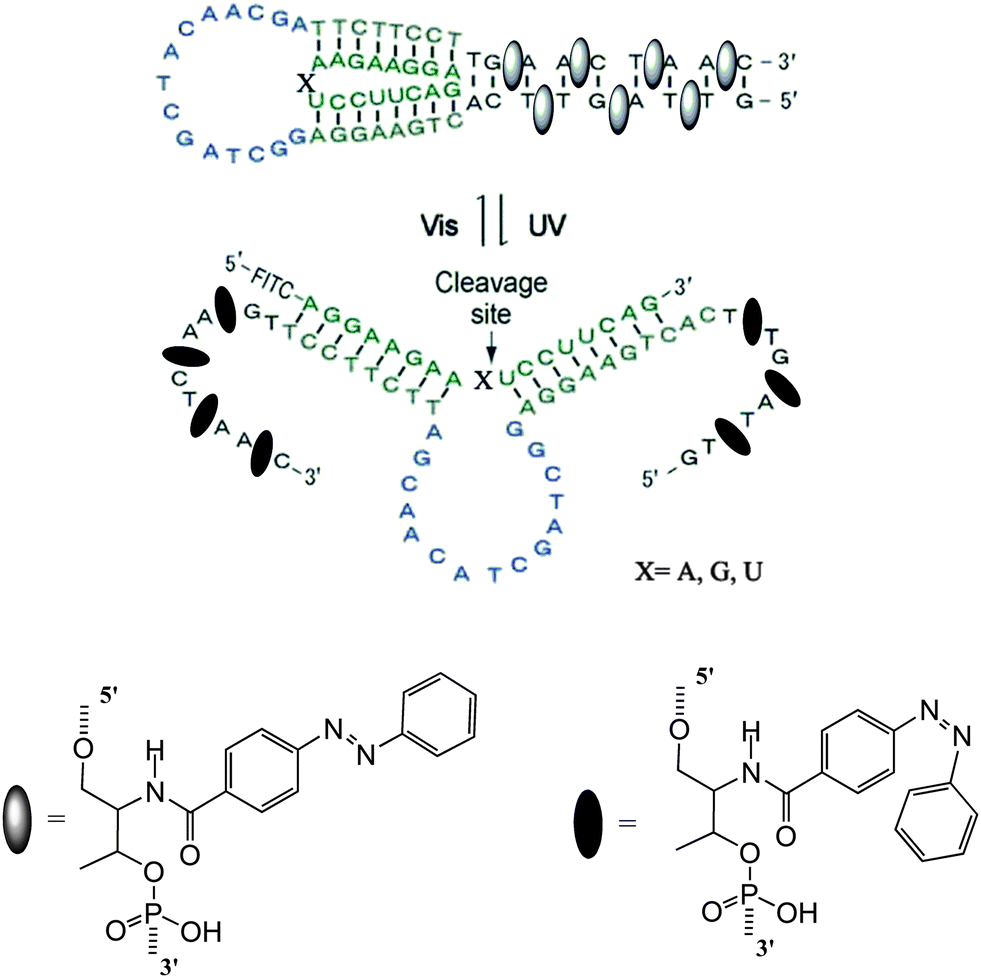 | ||
| Fig. 19 Light-driven azobenzene-tethered DNA nano-machine. | ||
In nature, nucleic acids and proteins perform their specific tasks based on functional self-assembly. Recently, artificially self-assembled nano-devices have been also reported to mimic bio-behaviors and bio-functions. Tan et al.50 constructed a photo-reversible DNA–enzyme catalytic cascade by incorporating azobenzene groups in DNA strands. Two self-assembled DNA cascades are illustrated in Fig. 20. In Fig. 20a, an azobenzene-tethered DNA strand with GOx can hybridize with another wild type DNA strand modified with HRP when the azobenzene moieties are in trans form. Thereby, sequential catalytic reactions of glucose oxidation and hydrogen peroxide reduction are carried out by this self-assembled DNA–enzyme duplex. The cis-azobenzene excited by UV light dissociates the duplex cascade, thus separating the two enzymes causing a maximum 25-fold decrease in catalytic activity. Therefore, the sequential catalytic reactions are hampered. Similar results are shown in Fig. 20b. In this situation, in contrast, the DNA–enzyme cascade is stabilized by cis-azobenzene. A HRP mimic G-quadruplex DNAzyme is formed and incorporates GOx on the other end to present a GOx/DNAzyme catalytic cascade in the presence of hemin. While irradiating with visible light, trans-azobenzene is formed which tends to self-assemble into a duplex structure. As a result, the HRP mimic G-quadruplex DNAzyme is lost together with the dissociation of the GOx/DNAzyme catalytic cascade (maximum 12-fold decrease in catalytic activity). This smart approach can be applied to design various types of photo-regulated protein enzyme or DNAzyme self-assembly cascades. With the increase in number of DNAzymes/protein enzymes and the incorporated functional molecules, more complex multi-responsive bio-catalytic cascades will be constructed for future bio-mimic, biomedical and pharmaceutical applications.
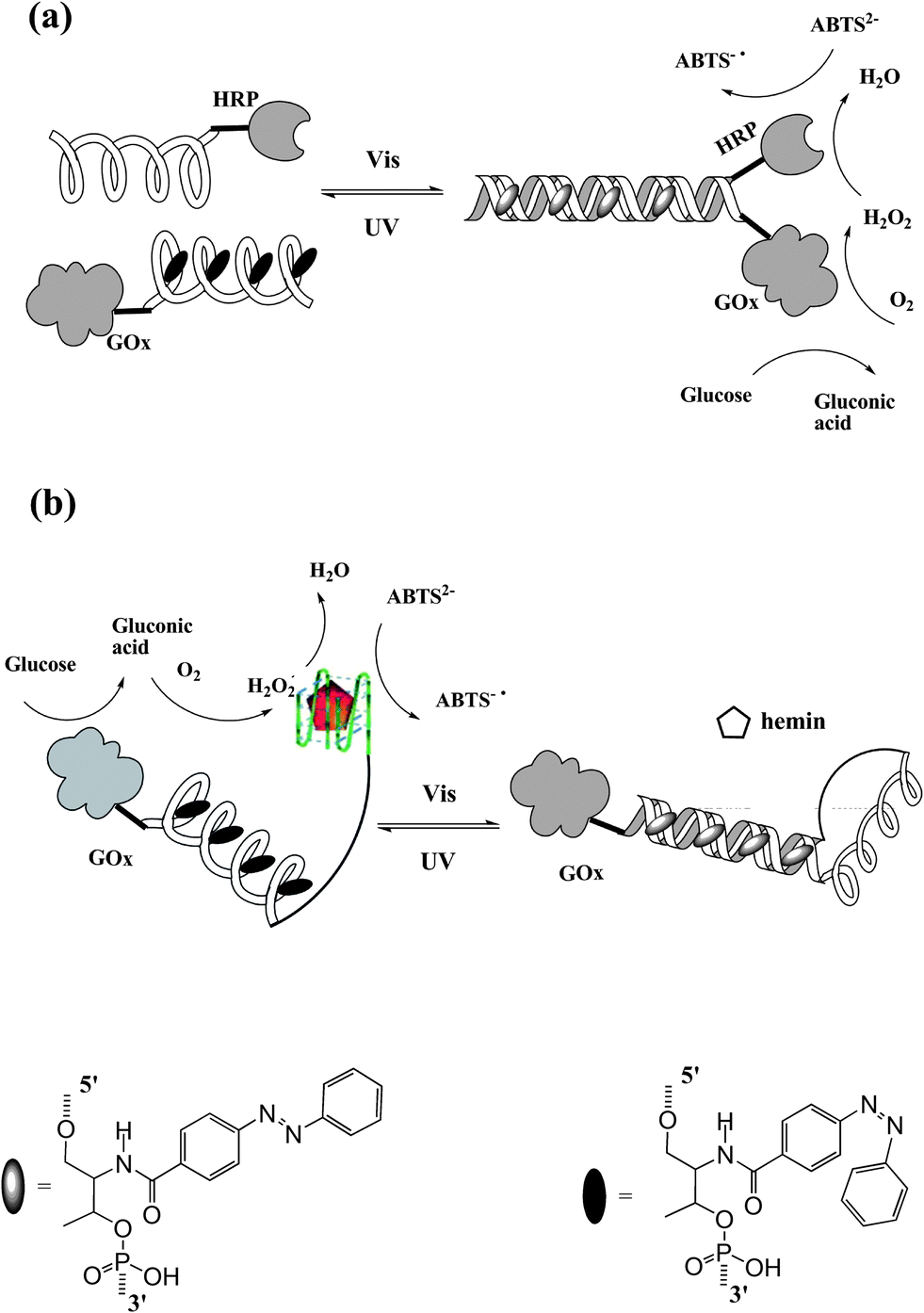 | ||
| Fig. 20 Working mechanism of photo-regulated DNA–enzyme cascades. (a) Light-triggered azobenzene-incorporated DNA duplex controlling GOx/HRP enzyme cascade. (b) DNA duplex/G-quadruplex switch for optically controlled GOx/HRP mimic DNAzyme cascade. | ||
Besides DNA and RNA,51 photo-switchable molecules have been attached to other kinds of nucleic acids to perform their smart reversible magic. PNAs (peptide nucleic acids) are synthetic nucleic acid analogs which exhibit nucleobases on a repeating N-(2-aminoethyl)glycine polyamide backbone.52 Owing to their hybridization with DNA and RNA through standard Watson–Crick and Hoogsteen base pairing,53 PNAs are often used in modulating diverse biochemical processes. Stafforst and Hilvert54 incorporated an azobenzene derivative into specific sites of PNA oligomers and studied their photo-controllable behaviors over the triplex binding of PNA with a DNA strand (Fig. 21). The transformation between cis- and trans-azobenzene upon irradiation caused different states of PNA–DNA triplex complex with a site-dependent magnitude of cis–trans discrimination. More importantly, the distinct transcription of optically controlled behaviors of different isomer tethered PNA–DNA triplexes was also exploited. Such photo-switchable PNA–DNA triplex compounds are showing considerable potential in biotechnology, diagnostics and medicinal applications.
 | ||
| Fig. 21 Azobenzene modified building block for incorporation into PNAs. | ||
Diarylethenes, a major family of photochromic materials, did not achieve their potential in DNA chemistry as they did in other bio-technologies until Jäschke et al.55a synthesized a series of base-analogue functionalized diarylethene derivatives. Diarylethenes are flexible in the design of acrylic substituents in their core structure. Therefore, a DNA analogue, 7-deazaadenine, was introduced as one of the two aryl moieties which made this novel diarylethene molecule more accessible than other photochromes to oligonucleotides. Just recently, Jäschke and Cahová went one step forward to incorporate diarylethene as a new nucleobase constituent in the DNA strand through DNA polymerization and bio-compatible Suzuki coupling, presenting a first example of diarylethene “nucleobased” oligonucleotides.55b Noticeably, the carbon atoms involved in the photo-cyclization of diarylethene are exactly the same as those that take part in the UV-induced cyclodimerization photo-damage reaction of DNA (Fig. 22), therefore this facilitates its high photo-conversion efficiency, electrocyclic rearrangement in DNA strands. Upon alternating irradiation with UV/Vis light, chemical bonds, hybridization, bond order as well as geometry were reversibly switched between two distinctive states. Compared to traditional dithienylethenes, this “nucleic acid” based diarylethene system not only possesses advantages such as high photo-conversion efficiency (over 90%), thermo-stability and excellent photo-reversibility (less than 0.5% of closed isomer after visible light photo-decyclization), but also improves their bio-compatibility which may promote novel applications of the diarylethene family to innovations in biology, microscopy and nanotechnology.
 | ||
| Fig. 22 Photochromism of diarylethene “nucleobased” oligonucleotides. | ||
Photo-responsive oligonucleotide applications in other phase or inter-phase studies, such as DNA nanostructure (origami) photo-engineering,56 photo-controllable DNA motors or hybridization on surfaces,57 are also becoming more and more popular nowadays. Due to space limitations, these exciting works will not be discussed in detail herein.
4.2 Photo-switchable peptides
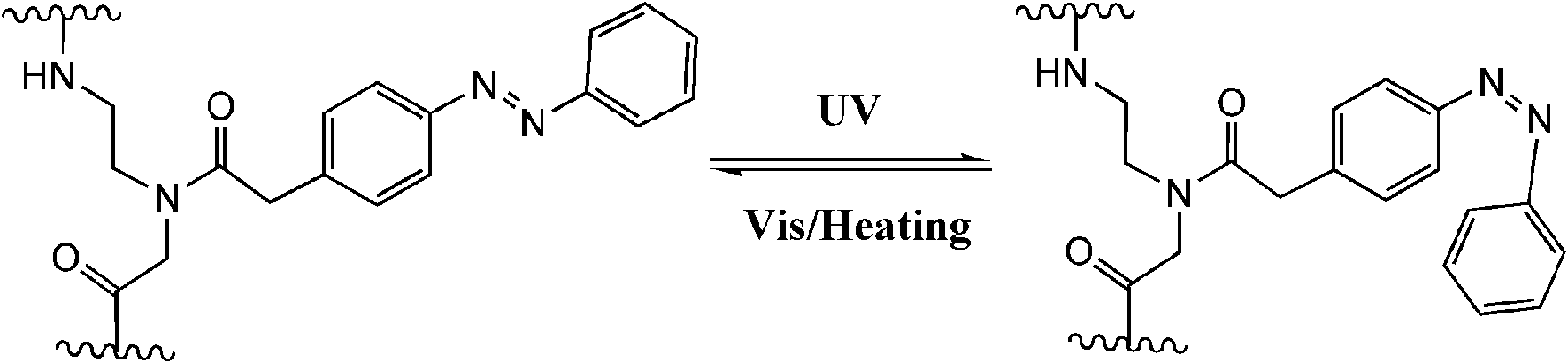
Peptides, consist of short chains of amino acids linked by amide bonds, possess an important functional role in biology, serving as hormones, ligands (coenzymes and cofactors) or linkers to other bio-macromolecular assemblies (DNA, RNA, etc.). Consequently, manipulating the activity of a specific peptide/protein inside living organisms offers exciting prospects for studying protein function in vivo. In the past decades, photo-regulating functional peptides by incorporating azobenzene in both the backbone and side-chains of peptides have been explored by the groups of Moroder58 and Allemann,59 which have been recently reviewed.12aPhoto-responsive cross-linkers have also been used in protein helix modulation. A study on photo-controlling the AP-1 transcription factor (Jun and Fos) heterodimer interaction was demonstrated by Woolley and colleagues.60 An azobenzene derivative cross-linker was introduced into the dominant negative AFosW peptides. As shown in Fig. 23, the UV triggered isomerization of azobenzene cross-linker from trans to cis enhanced the helicity of XAFosW/cJun, thereby favoring the coiled-coil formation and leading to the 10-fold enhancement of the inhibition of DNA binding. The photo-controlled DNA binding inhibition behavior was further investigated in vivo after injection into living cells. In the cell experiment, the author used the luciferase reporter under the control of an AP-1 promoter together with a β-galactosidase reporter under the control of a constitutive rous sarcoma virus (RSV) promoter. Ratios of luciferase activity to β-galactosidase activity were used as an indication of specific inhibition of AP-1 activity. The in vivo photo-isomerization results in a 40% decrease in AP-1 activity after UV irradiation, also confirmed that the cis form was a more powerful inhibitor than the trans form, consistent with the results in vitro. Yet, two main obstacles still remain in this work. First, the excitation wavelength for trans to cis photo-switching is 365 nm which will cause photo-damage to cells. This problem may be solved by the recent development of visible-light triggered azobenzene isomerization.61 Another problem is the thermo relaxation nature of azobenzene, that is, even in the dark the cis-azobenzene will isomerize back to trans-azobenzene after heating. This certainly shortens the half-life of cis-azobenzene during measurements. In this work, photo-relaxation in vivo with 460 nm was not observed due to the fast thermal relaxation at 37 °C. To tackle this problem, new thermo-resistant photochromes are to be explored.
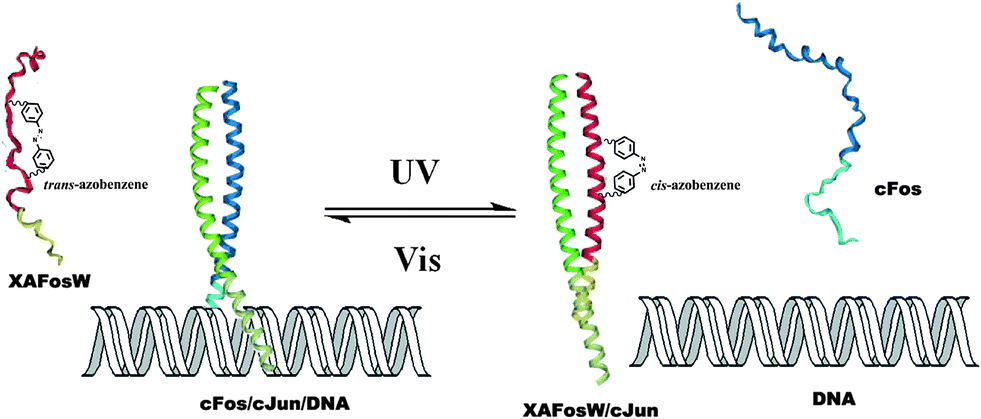 | ||
| Fig. 23 Trans-azobenzene linked XAFosW turned to its cis form and thus enhanced its helicity and coiled-coil formation with cJun after irradiation with UV light. This photo-induced formation change leads to the inhibition of DNA binding. | ||
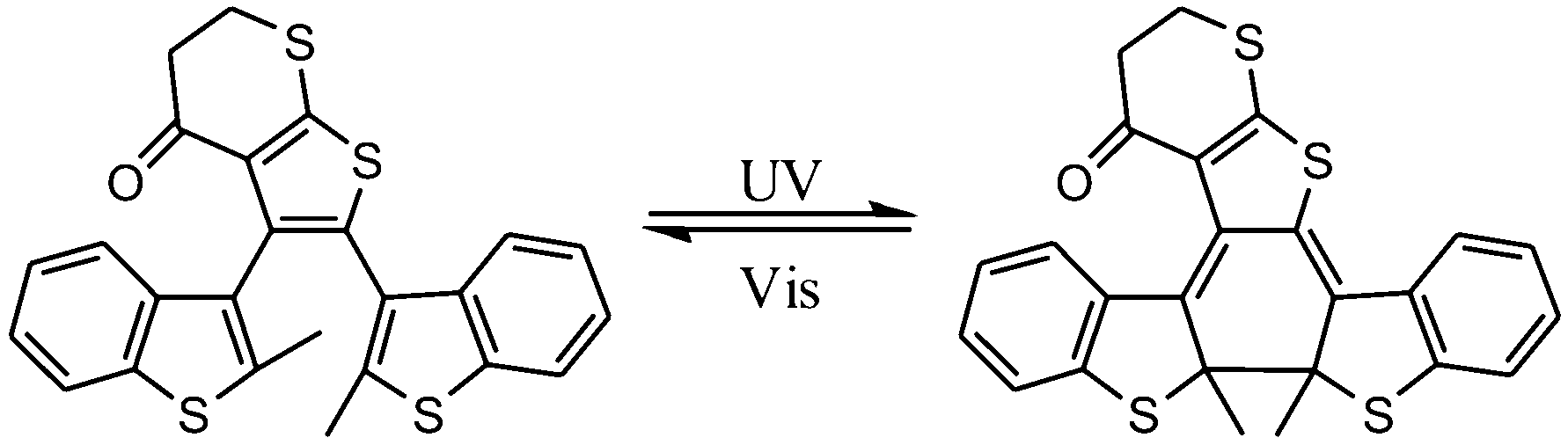
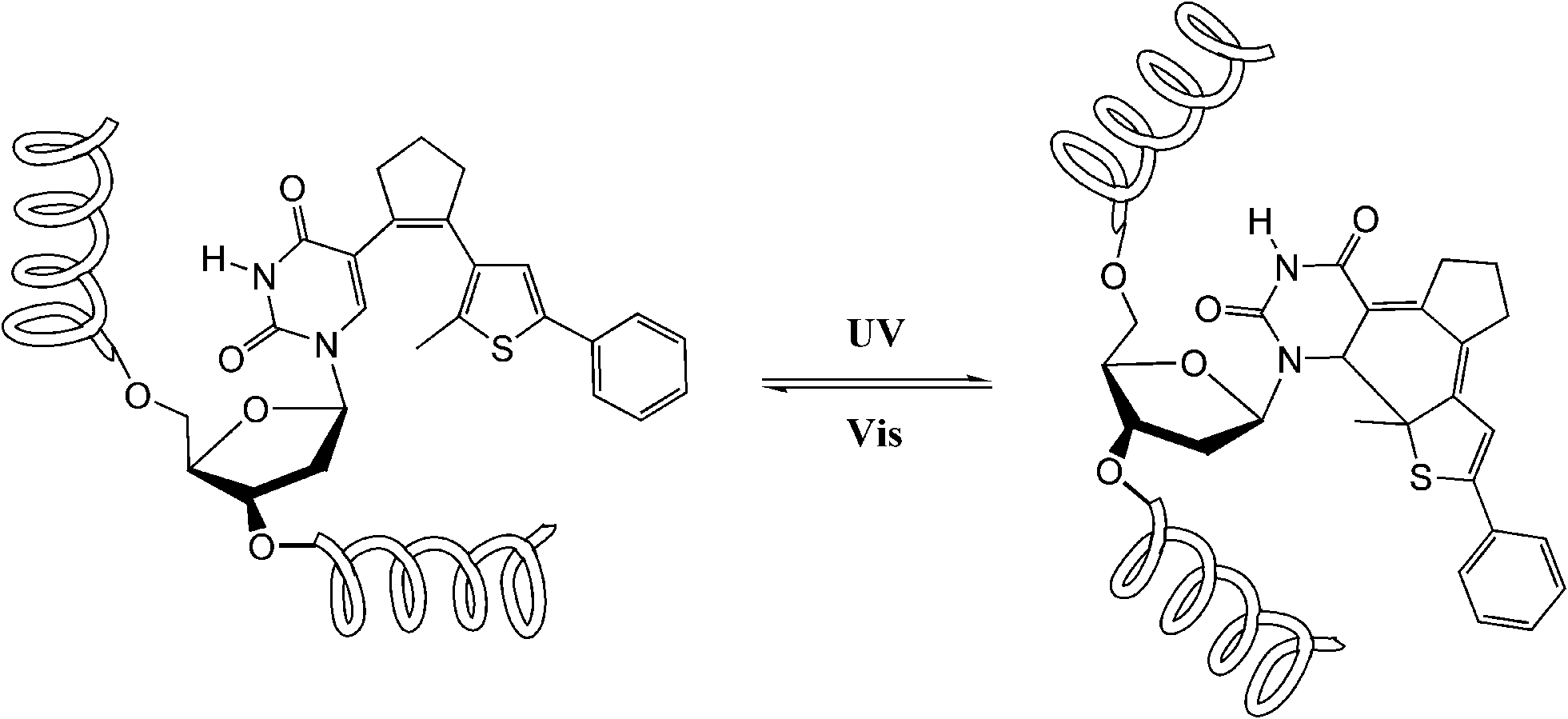
Dithienylethene, as mentioned before, is famous for its high thermo-stability. Thus, it was selected as a candidate photo-responsive and thermo-resistant cross-linker by Fujimoto and colleagues to reversibly regulate the peptide helices.62 When the dithienylethene cross-linker is in its open form, the peptides fold into stable α-helices. Upon UV irradiation, the closed isomer of dithienylethene forms and destabilizes the helical structures of peptides (Fig. 24). The photo-conversion efficiency of DTE-tethered peptides is over 90% with excellent photo-reversibility. This photo-modulation motif was further introduced into the peptide–DNA binding study detected by QCM analysis. Upon interaction with the DNA, the open form linked peptide, which possessed a more stable α-helical structure, exhibited a higher binding affinity than its closed photo-isomer counterpart which dissociated the α-helical structure. Upon alternating irradiation with UV/Vis light, the [θ222] of CD spectra varies around 30–40%, indicating a reversible photoisomerization of DTE, which regulates the helical structures of the DNA-binding peptides.
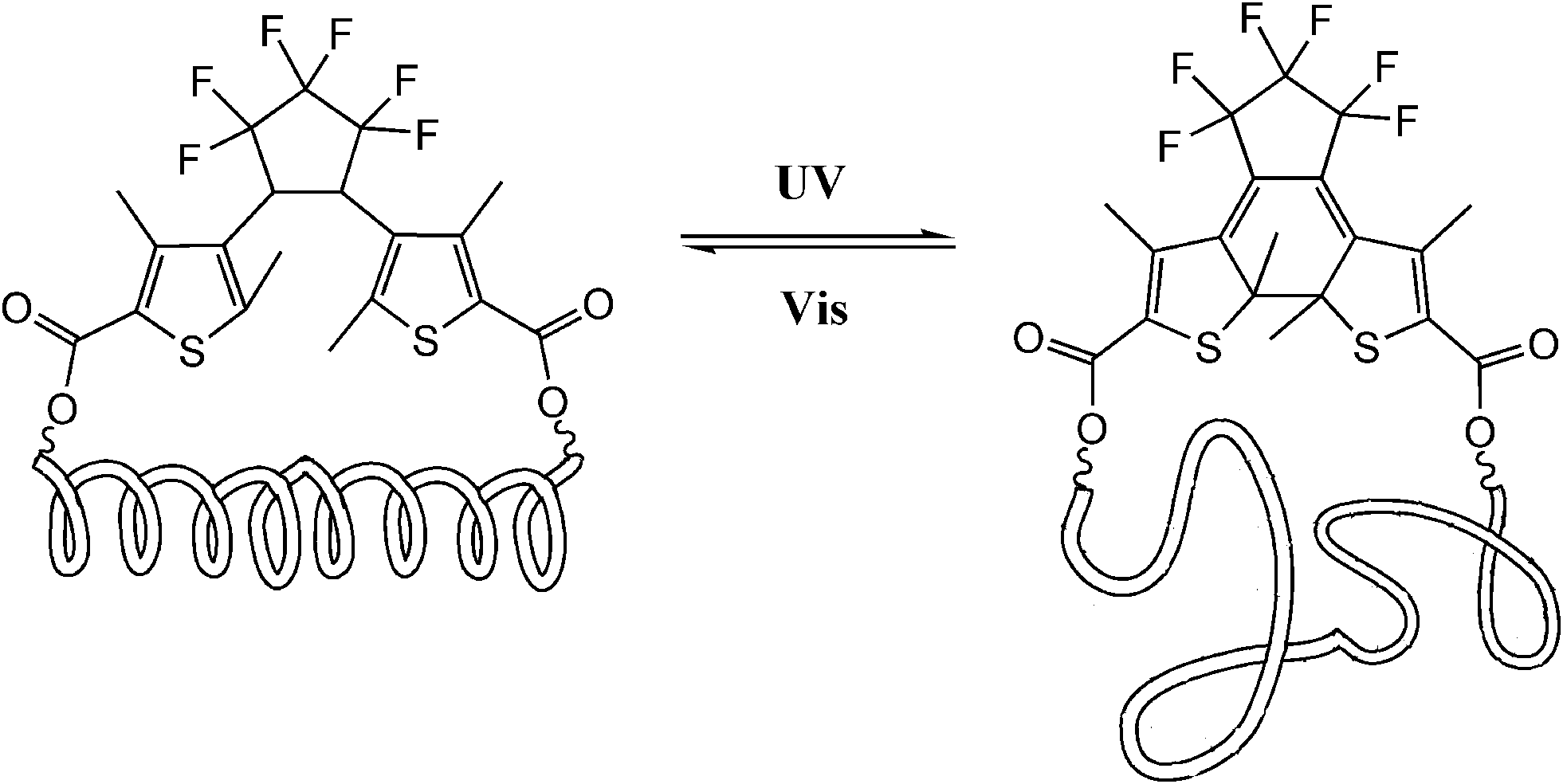 | ||
| Fig. 24 Photo-regulation of α-helix by a diarylethene derivative. | ||
5 Photo-regulated biological channels
The transport of ions across the cell membrane is a prerequisite for many biological processes. Biological nanochannels, including ion channels and water channels, play a key role in physiology and pathology. For example, potassium ion channels contribute to the regulation of the action potential duration in cardiac muscle. So malfunction of potassium channels may cause life-threatening arrhythmias. Therefore, regulating biological nanochannels with waste-free external signals, such as light, will have great value in neurobiology and therapeutics. Though bio-inspired study of the design and development of photo-switchable artificial biomimetic channels has received a great deal of attention in the past decade,63 we will only focus on the photo-modulated biological nanochannels in this review.Trauner and Kramer64 synthesized a series of acrylamide–azobenzene–quaternary ammonium (AAQ) derivatives, and studied the photo-regulation behavior of AAQs on the potassium channel as PCL (photochromic ligands). AAQs were originally designed by the authors as PTLs (photo-switchable tethered ligands), which were hypothesized to be covalently functionalized at the external tetraethylammonium (TEA) binding site. Yet, further research results showed that AAQs predominantly acted as non-covalent PCLs at the internal TEA binding site instead. As shown in Fig. 25, upon alternating irradiation with 380 nm and 500 nm light, the ion channel appears to be blocked and activated, respectively, revealing 30-fold block potency upon photoisomerization. Then the authors studied the detailed function of different structures of AAQ PCLs with potassium ion channels. Though the photochromic efficiencies of these different azobenzene derivatives are in the same range (∼80%), the ion channel blocking potency varied from zero to >95%. According to the results above, principles have been established to guide the design of additional functional photochromic ligands for other voltage-gated ion channels, such as sodium and calcium channels.
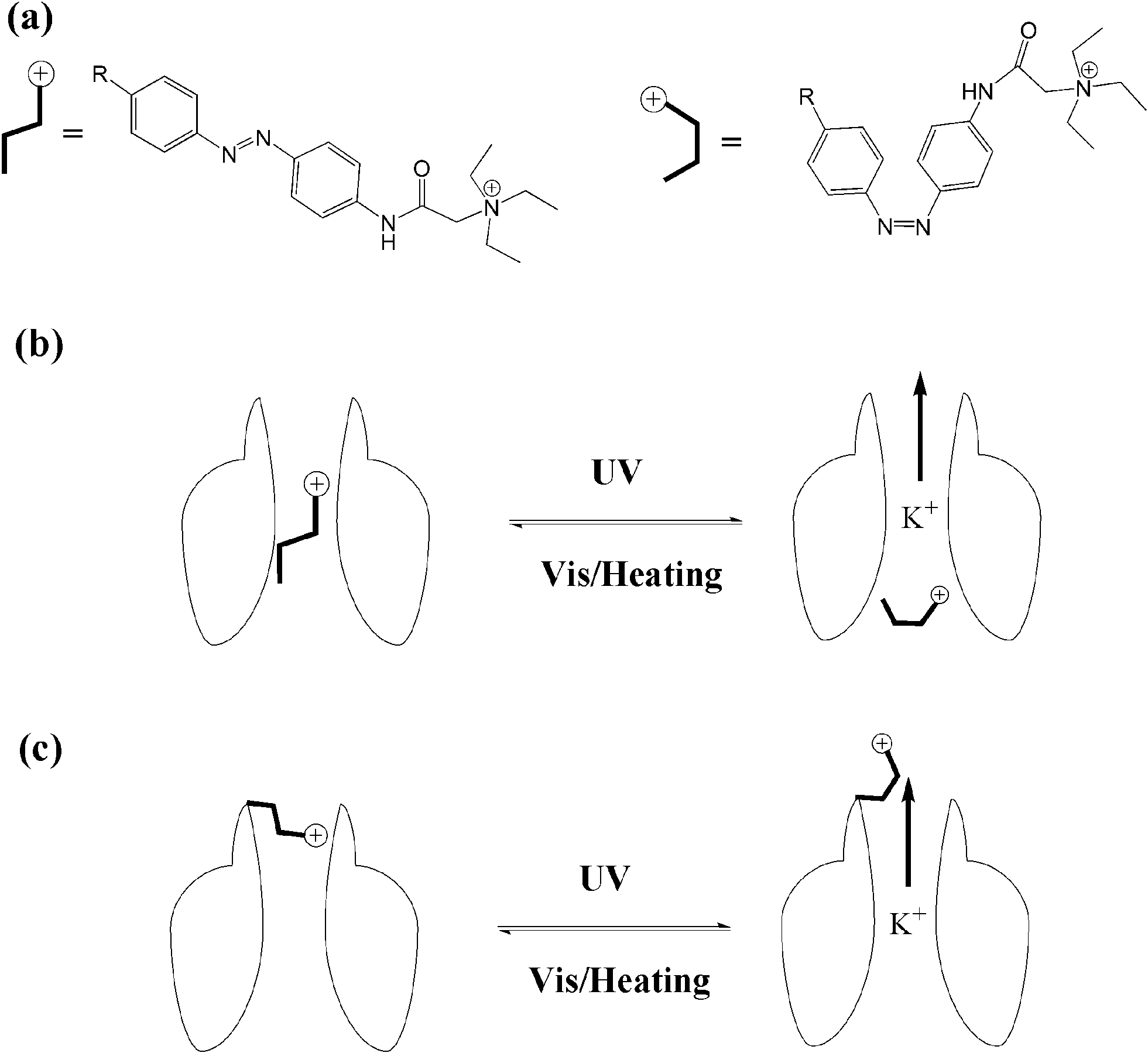 | ||
| Fig. 25 (a) Photo-isomerization of azobenzene blockers. (b) PCL photo-reversible blocking of ion channels. (c) PTL photo-reversible blocking of ion channels. | ||
Trauner and Kramer65 further tethered maleimide–azobenzene–acylcholine (MAACh) and maleimide–azobenzene–homocholine (MAHoCh) ligands to mutated nicotinic acetylcholine receptors (nAChRs), introducing a genetically engineered, light-controlled nAChR (LinAChR) ion channel. nAChRs are pentameric ligand-gated cation channels expressed throughout the mammalian nervous system and at the neuromuscular junction.66 In this study, two azobenzene derivatives, MAACh and MAHoCh (Fig. 26), were used as PTL agonist and antagonist to photo-reversibly activate and inhibit the engineered nAChR channels at specific mutated sites, respectively. The acylcholine modified MAACh ligand was found to activate the engineered nAChR channels photo-reversibly, while the homocholine tethered MAHoCh inhibited the engineered ion channels. The ion channel blocking percentages varied corresponding to different mutation sites for both azobenzene PTLs; while for MAHoCh the concentration of ACh is another factor to be considered. Moreover, the thermo-relaxation properties of photo-switchable ligands were investigated, showing that ligand binding increased the thermo-stability of cis-MAACh while that of cis-MAHoCh stayed the same. This research will be of great use for future studies of photo-regulated heteromeric nAChRs in physiology and pathology based in the brain and periphery.
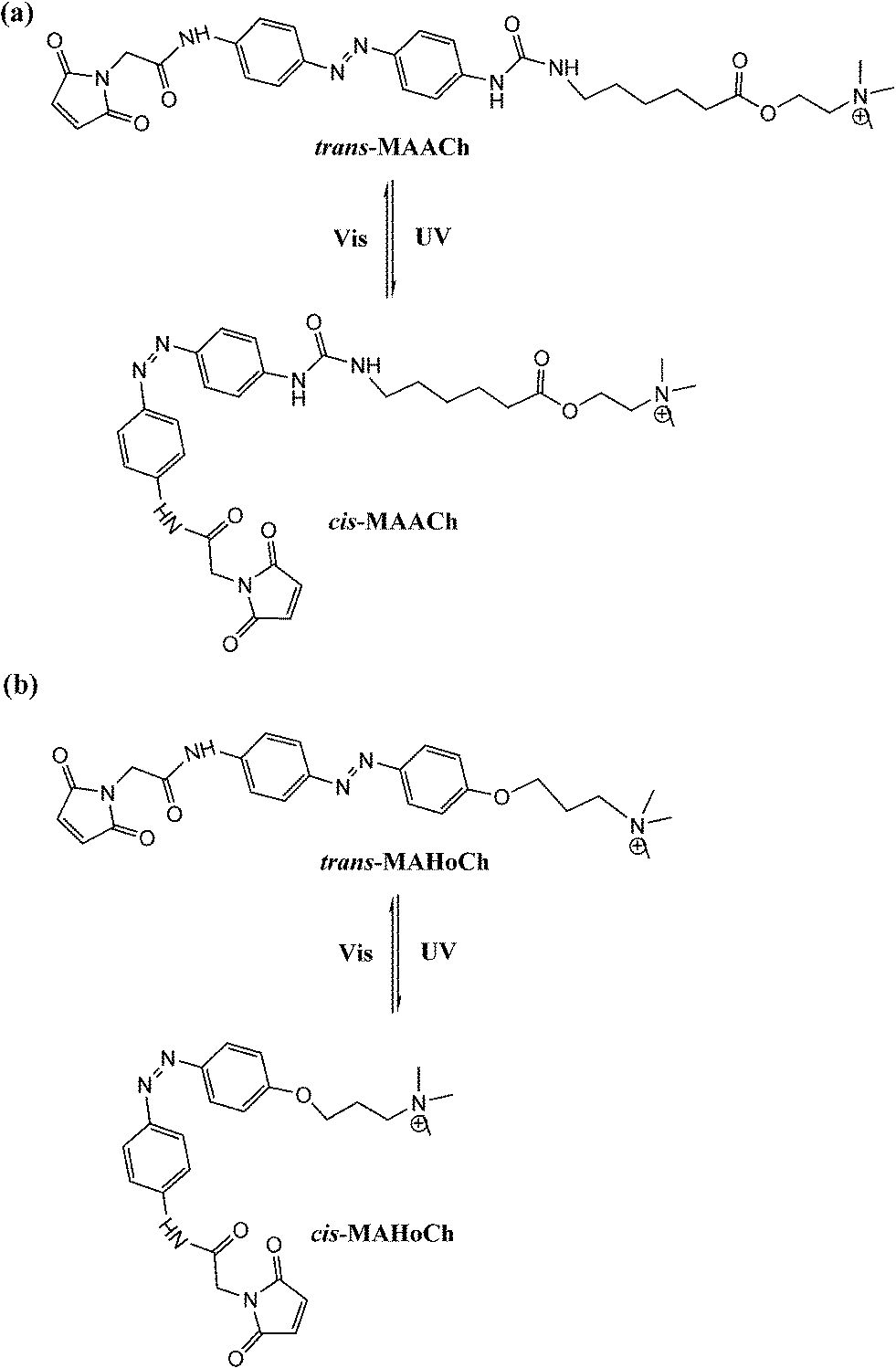 | ||
| Fig. 26 Photoswitchable ligands for optical control of nAChRs: (a) MAACh, (b) MAHoCh. | ||
Trauner and Sigel67 developed an azobenzene based propofol derivative as a photo-switchable potentiator rather than agonist/antagonist or blocker in GABAA receptors (Fig. 27). GABAA receptors are another pentameric ligand-gated ion channel which are activated by γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the mammalian brain.68 GABA-induced chloride ion currents were potentiated when the propofol azobenzene was in its trans form, and reversed by excitation with UV light, approximately 9-fold stimulation changes were observed. Further investigation of the photo-reversible anesthetic activity of this azobenzene derivative was carried out in albino Xneopus laevis tadpoles. Thus, a potential light-induced anesthetic was developed based on azobenzene propofol analogs which would potentially play an important role in future pharmacology of anesthesia. More systemic work addressing the application of azo-propofol analogs in other living systems, such as brain slices or retinas lacking innate photoreceptors, is worthy of exploration.
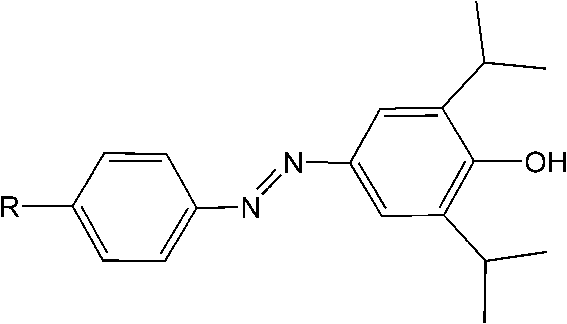 | ||
| Fig. 27 Azobenzene-based photoreactive propofol derivative. | ||
α-Hemolysin (α-HL) forms a heptameric ligand-gated aqueous channel with moderate anion selectivity on the membrane of target cells. The transport of molecules and ions from the target cells through α-HL leads to the ultimate rupture of the cell membrane.69 Realizing nature's functions of α-HL, it became an ideal nanoscale material for probing molecular transport and recognition processes, e.g. the analysis of nucleic acids and the development of DNA sequencing methods.70 Further functionalizing natural channels by using synthetic chemistry would provide biological nanopores with novel properties and applications. Tian, Long et al.71 implemented a photochromic host–guest supramolecular system to reversibly regulate the open/close state of the α-HL nanopore (Fig. 28). The sulfonato-calix[4]arene (SC4) host molecule binds to the lysine residues of α-HL via host–guest interaction (Ka = 753 M−1), causing the collapse of the stem base and consequently leading to the closed state of α-HL. After adding azobenzene-viologen guest molecule, the SC4 host molecule is displaced from the amino acid residue and binds with viologen via a much stronger host–guest interaction (Ka ∼ 105 M−1). This releases the crucial amino acid residues which constitute the β-barrel of α-HL at the glycine-rich stem base, thus reviving the open state of α-HL. More importantly, a photo-regulated open–closed state switch was also achieved in this work. As the trans-azobenzene viologen completes the release of SC4 from the lysine residues at the stem base region and opens the α-HL, its cis counterpart shows inhibition behavior of α-HL pore opening. It should be highlighted that the inhibition frequency of cis-azobenzene viologen (28%) is similar to the photoisomerization efficiency in solution (33%). This result places emphasis on the efficacy of the nanopore biosensor to study the photochromic host–guest system at the single-molecule level.
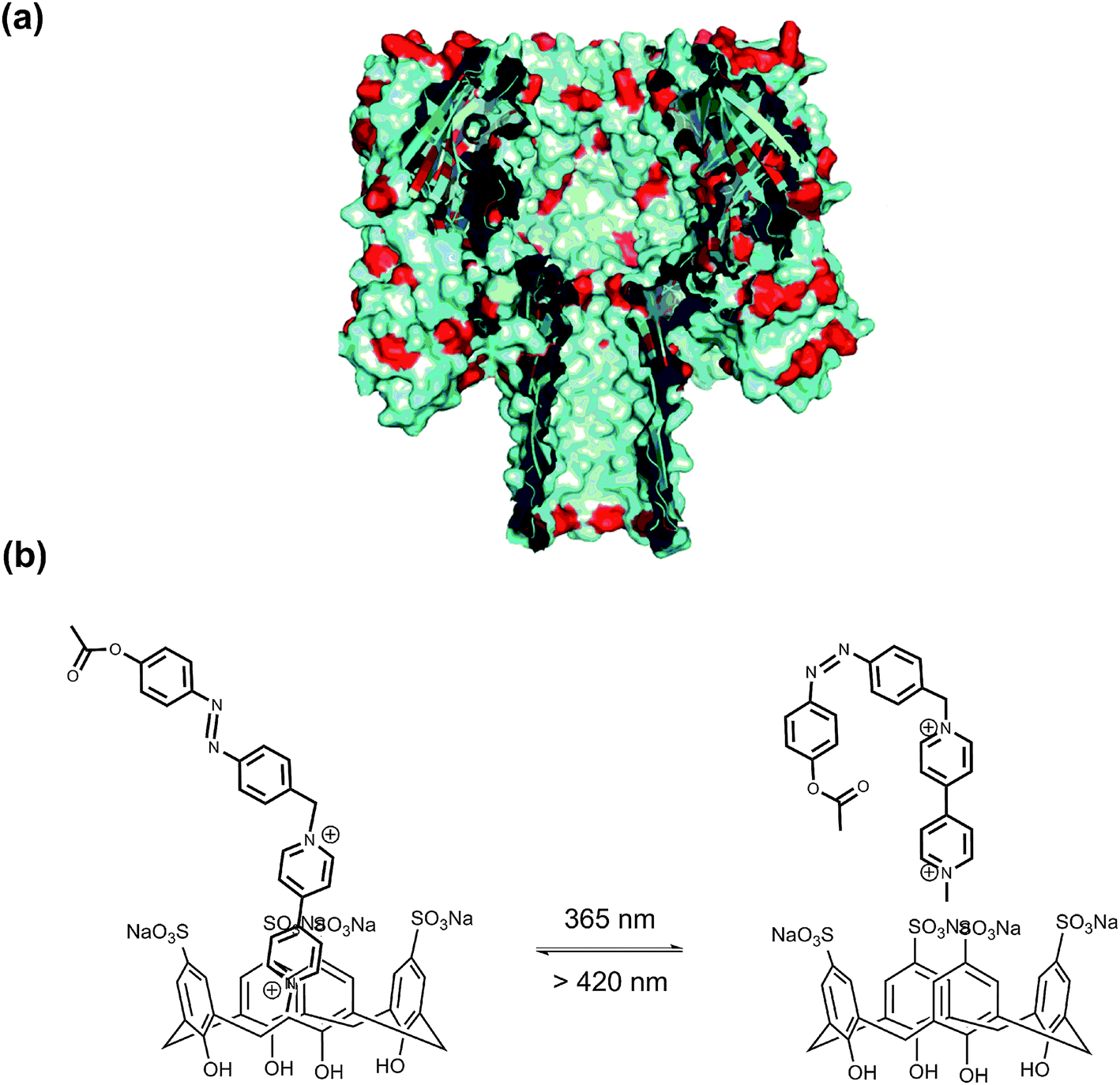 | ||
| Fig. 28 (a) Cross section of α-HL. (b) The photoisomeric reaction between V2+-trans-Az:SC4 and V2+-cis-Az:SC4 upon irradiation with alternating UV and visible light. | ||
6 Photochromic green fluorescent protein (GFP)
Compared to traditional photochromes, other photo-reversible structures, e.g. photochromic fluorescent proteins (PFP), have attract more and more attention from chemists because of their great value for in vivo cell/tissue imaging and noninvasive analysis of protein localization and dynamics in living cells.72 Dronpa, a mutant from Pectiniidae family, is the most popular PFP. Dronpa can photo-switch between its strong green fluorescent state and dark off state with alternating UV/blue light over 100 times with minor loss of emission. The chromophore of Dronpa is formed autocatalytically from residues of Cys,62 Tyr63 and Gly,64 with a rationalized four-state scheme entailing the isomerization of the chromophore between the cis anionic bright state and the trans neutral dark state (Fig. 29).73 PFP is generally used as an excellent switchable fluorophore in super-resolution microscopy techniques such as PALM/STORM. Recently, PFPs have also been utilized in data storage and all-optical writing with sub-diffraction size and spacing on account of their reversible photo-switching ability and super-resolution performances.74 To further expand their usage in biotechnology, chemical and molecular biological modifications/mutations of new PFP have been carried out to construct new structures with novel functionalities,75 enhance their photochromism,76 encompass optogenetic control,77etc. Since there are more reviews specific to PFPs,78 we will not go into the details of fruitful studies in this area.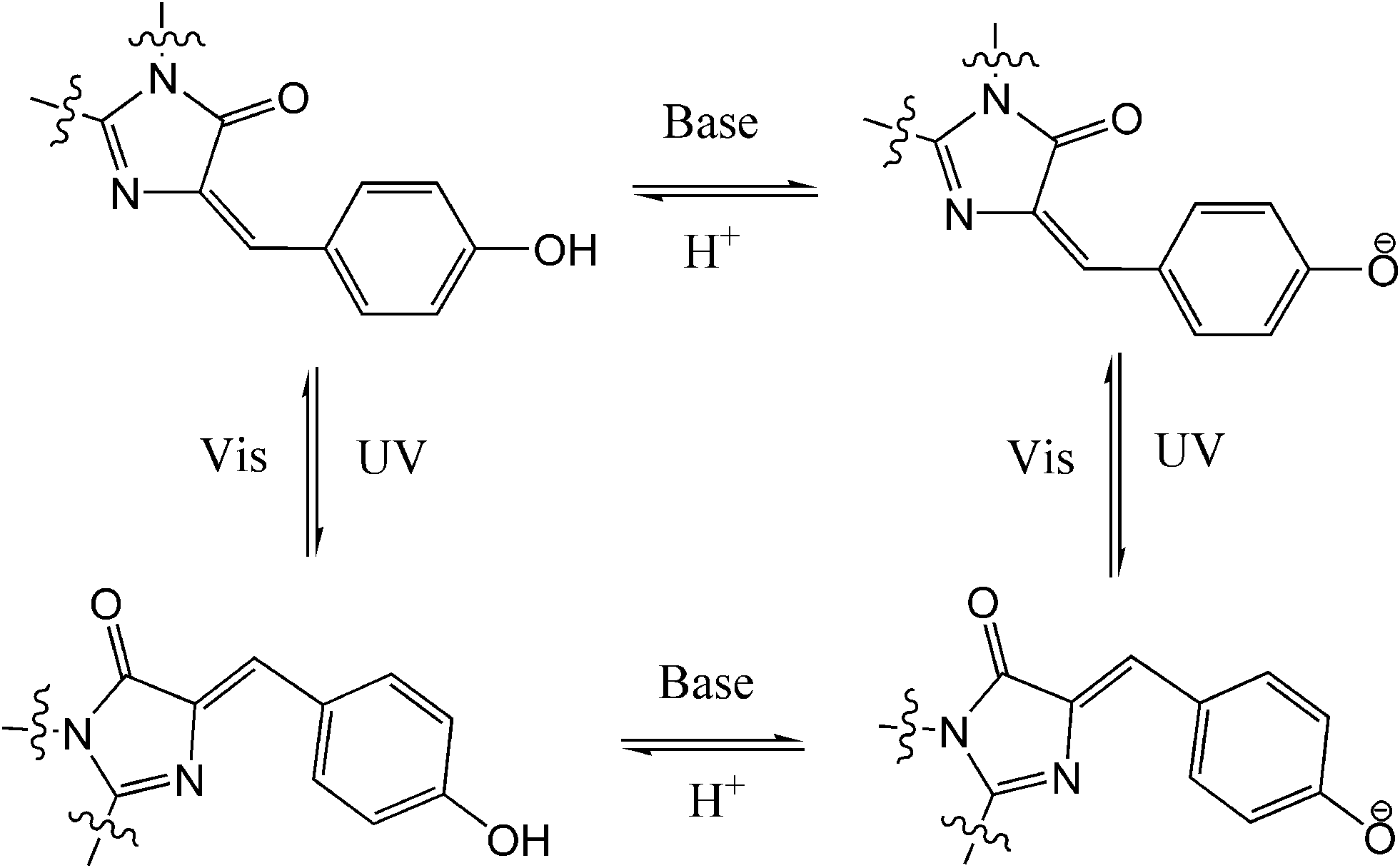 | ||
| Fig. 29 Optically and pH induced four-state interchange of Dronpa. | ||
7 Conclusion and outlook
Photochromic materials are proven to be very promising with potential in various research fields including chemistry, physics, materials science and especially bio-technology. Nowadays, most efforts have been devoted to the construction of functional bio-materials with photo-reversible fragments. Azobenzenes are the most widely used photo-switches due to their significant steric and geometrical transformations upon light excitation. Since many bio-molecules are charged entities (e.g. nucleotides, peptides), spiropyran and its zwitterionic merocyanine counterpart may perform a key role in bio-molecule photo-modulation via electrostatic interactions. Diarylethenes, though they undergo negligible steric and geometrical changes, display high thermo-stability and photon fatigue as well as optical/electronic coupling properties (e.g. energy transfer, donor–acceptor, conductivity, ion complexation). These smart performances suggest diarylethene derivatives as a rising star in photo-controlled bio-material design and applications.Although great achievements have been made in photochromic bio-materials, several key issues still remain to be resolved. One of the major challenges which lies ahead is the excitation wavelength range for photo-isomerization in bio-systems. Using visible or infrared light as the photo-stimulus provides great opportunities in photochromic bio-materials, since UV light has limitations in cell/tissue penetration together with damage-causing effects as mentioned before. Yet, photochromism is commonly triggered under UV light. Although recently several solutions, such as upconversion nanoparticles (UCNPs),23,25,79 metal to ligand charge transfer (MLCT)9b,22,80 and multi-photon excitation processes,21,81 have been proposed, they are still in their infancy. Another major challenge for bio-compatible photochromic materials is bio-toxicity, which is of great concern in medicinal applications and in vivo sensing. Covalently linking photochromic molecules to bio-molecules will “dilute” the toxicity to some extent, yet we consider developing natural photo-switchable molecules, such as green fluorescence protein (GFP) should be the preference in resolving the toxicity concerns. Other problems, e.g. the quantum efficiency and ON/OFF ratio, should also be considered in future studies and developments. Taking into account the present achievements and remaining challenges, it may be imagined that photochromic materials will present a prosperous future in smart bio-materials research and will provoke many further biological and medicinal applications.
Acknowledgements
This work has been supported by NSFC/China, National Basic Research 973 Program (2013CB733700). Thanks are extended to all co-workers working on photochromic materials in our lab.References
-
(a) C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446 CrossRef CAS PubMed
; (b) Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849 CrossRef CAS
-
(a) M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed
-
(a) H. Tian and S. J. Yang, Chem. Soc. Rev., 2004, 33, 85 RSC
-
(a) H. Li, J. X. Wang, H. Lin, L. Xu, W. Xu, R. M. Wang, Y. L. Song and D. B. Zhu, Adv. Mater., 2010, 22, 1237 CrossRef CAS PubMed
-
(a) E. Orgiu, N. Crivillers, M. Herder, L. Grubert, M. Patzel, J. Frisch, E. Pavlica, D. T. Duong, G. Bratina, A. Salleo, N. Koch, S. Hecht and P. Samori, Nat. Chem., 2012, 4, 675 CrossRef CAS PubMed
-
(a) S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596 CrossRef CAS PubMed
-
(a) B. M. Neilson and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 12693 CrossRef CAS PubMed
-
(a) J. Zhang, M. Riskin, R. Freeman, R. Tel-Vered, D. Balogh, H. Tian and I. Willner, ACS Nano, 2011, 5, 5936 CrossRef CAS PubMed
-
(a) B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544 CrossRef CAS PubMed
-
(a) D. Vomasta, C. Högner, N. R. Branda and B. König, Angew. Chem., Int. Ed., 2008, 47, 7644 CrossRef CAS PubMed
-
(a) H.-M. Lee, M. A. Priestman and D. S. Lawrence, J. Am. Chem. Soc., 2010, 132, 1446 CrossRef CAS PubMed
-
(a) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422 RSC
-
(a) N. C. Rockwell and J. C. Lagarias, ChemPhysChem, 2010, 11, 1172 CrossRef CAS PubMed
-
(a) Y. H. Li, Y. Duan, J. S. Li, J. Zheng, H. Yu and R. H. Yang, Anal. Chem., 2012, 84, 4732 CrossRef CAS PubMed
-
(a) J. Sunamoto, K. Iwamoto, Y. Mohri and T. Kominato, J. Am. Chem. Soc., 1982, 104, 5502 CrossRef CAS
- C. Q. Tu, R. Nagao and A. Y. Louie, Angew. Chem., Int. Ed., 2009, 48, 6547 CrossRef CAS PubMed
- J. Jin, X. Li, J. Zhang, P. Zhao and H. Tian, Isr. J. Chem., 2013, 53, 288 CrossRef CAS
-
(a) J. R. Nilsson, S. Li, B. Önfelt and J. Andréasson, Chem. Commun., 2011, 47, 11020 RSC
- Y. Zou, T. Yi, S. Xiao, F. Li, C. Li, X. Gao, J. Wu, M. Yu and C. Huang, J. Am. Chem. Soc., 2008, 130, 15750 CrossRef CAS PubMed
- S. C. Pang, H. Hyun, S. Lee, D. Jang, M. J. Lee, S. H. Kang and K. H. Ahn, Chem. Commun., 2012, 48, 3745 RSC
- M.-Q. Zhu, G.-F. Zhang, C. Li, M. P. Aldred, E. Chang, R. A. Drezek and A. D. Q. Li, J. Am. Chem. Soc., 2011, 133, 365 CrossRef CAS PubMed
- W. J. Tan, J. Zhou, F. Y. Li, T. Yi and H. Tian, Chem.–Asian J., 2011, 6, 1263 CrossRef CAS PubMed
- J. C. Boyer, C. J. Carling, S. Y. Chua, D. Wilson, B. Johnsen, D. Baillie and N. R. Branda, Chem.–Eur. J., 2012, 18, 3122 CrossRef CAS PubMed
- W. J. Tan, Q. Zhang, J. J. Zhang and H. Tian, Org. Lett., 2009, 11, 161 CrossRef CAS PubMed
- J.-C. Boyer, C.-J. Carling, B. D. Gates and N. R. Branda, J. Am. Chem. Soc., 2010, 132, 15766 CrossRef CAS PubMed
- C. Alvarez-Lorenzo, L. Bromberg and A. Concheiro, Photochem. Photobiol., 2009, 85, 848 CrossRef CAS PubMed
- W. Xiao, W. H. Chen, X. D. Xu, C. Li, J. Zhang, R. X. Zhuo and X. Z. Zhang, Adv. Mater., 2011, 23, 3526 CrossRef CAS PubMed
- W. Xiao, W. H. Chen, J. Zhang, C. Li, R. X. Zhuo and X. Z. Zhang, J. Phys. Chem. B, 2011, 115, 13796 CrossRef CAS PubMed
- H. Z. Kang, H. P. Liu, X. L. Zhang, J. L. Yan, Z. Zhu, L. Peng, H. H. Yang, Y. Kim and W. H. Tan, Langmuir, 2011, 27, 399 CrossRef CAS PubMed
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848 CrossRef CAS PubMed
- I. Yildiz, S. Impellizzeri, E. Deniz, B. McCaughan, J. F. Callan and F. M. Raymo, J. Am. Chem. Soc., 2011, 133, 871 CrossRef CAS PubMed
- Y. Yan, C. J. Ochs, G. K. Such, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2010, 22, 5398 CrossRef CAS PubMed
- S. Matsumoto, S. Yamaguchi, S. Ueno, H. Komatsu, M. Ikeda, K. Ishizuka, Y. Iko, K. V. Tabata, H. Aoki, S. Ito, H. Noji and I. Hamachi, Chem.–Eur. J., 2008, 14, 3977 CrossRef CAS PubMed
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088 CrossRef CAS PubMed
- C. Teller and I. Willner, Curr. Opin. Biotechnol., 2010, 21, 376 CrossRef CAS PubMed
-
(a) Z. G. Wang, J. Elbaz and I. Willner, Nano Lett., 2011, 11, 304 CrossRef CAS PubMed
- C. Dohno, S. Uno and K. Nakatani, J. Am. Chem. Soc., 2007, 129, 11898 CrossRef CAS PubMed
- X. Wang, J. Huang, Y. Zhou, S. Yan, X. Weng, X. Wu, M. Deng and X. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5305 CrossRef CAS PubMed
- X. Xing, X. Wang, L. Xu, Y. Tai, L. Dai, X. Zheng, W. Mao, X. Xu and X. Zhou, Org. Biomol. Chem., 2011, 9, 6639 CAS
- J. Andersson, S. Li, P. Lincoln and J. Andréasson, J. Am. Chem. Soc., 2008, 130, 11836 CrossRef CAS PubMed
- M. Hammarson, J. Andersson, S. Li, P. Lincoln and J. Andréasson, Chem. Commun., 2010, 46, 7130 RSC
- D. D. Young and A. Deiter, ChemBioChem, 2008, 9, 1225 CrossRef CAS PubMed
- A. Mammana, G. T. Carroll, J. Areephong and B. L. Feringa, J. Phys. Chem. B, 2011, 115, 11581 CrossRef CAS PubMed
- T. C. S. Pace, V. Müller, S. Li, P. Lincoln and J. Andréasson, Angew. Chem., Int. Ed., 2013, 52, 4393 CrossRef CAS PubMed
- H. Asanuma, T. Ito, T. Yoshida, X. Liang and M. Komiyama, Angew. Chem., Int. Ed., 1999, 38, 2393 CrossRef CAS
- H. Kang, H. Liu, J. A. Phillips, Z. Cao, Y. Kim, Y. Chen, Z. Yang, J. Li and W. Tan, Nano Lett., 2009, 9, 2690 CrossRef CAS PubMed
- Q. Yuan, Y. Zhang, Y. Chen, R. Wang, C. Du, E. Yasun and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9331 CrossRef CAS PubMed
- J. A. Phillips, H. Liu, M. B. O'Donoghue, X. Xiong, R. Wang, M. You, K. Sefah and W. Tan, Bioconjugate Chem., 2011, 22, 282 CrossRef CAS PubMed
- M. Zhou, X. Liang, T. Mochizuki and H. Asanuma, Angew. Chem., Int. Ed., 2010, 49, 2167 CrossRef CAS PubMed
- M. You, R.-W. Wang, X. Zhang, Y. Chen, K. Wang, L. Peng and W. Tan, ACS Nano, 2011, 5, 10090 CrossRef CAS PubMed
- H. Ito, X. Liang, H. Nishioka and H. Asanuma, Org. Biomol. Chem., 2010, 8, 5519 CAS
-
(a) M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Nordén and P. E. Nielsen, Nature, 1993, 365, 566 CrossRef CAS PubMed
- T. Bentin, H. J. Larsen and P. E. Nielsen, Biochemistry, 2003, 42, 13987 CrossRef CAS PubMed
- T. Stafforst and D. Hilvert, Angew. Chem., Int. Ed., 2010, 49, 9998 CrossRef CAS PubMed
-
(a) M. Singer and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8372 CrossRef CAS PubMed
-
(a) F. Tanaka, T. Mochizuki, X. Liang, H. Asanuma, S. Tanaka, K. Suzuki, S. Kitamura, A. Nishikawa, K. Ui-Tei and M. Hagiya, Nano Lett., 2010, 10, 3560 CrossRef CAS PubMed
-
(a) M. McCullagh, I. Franco, M. A. Ratner and G. C. Schatz, J. Am. Chem. Soc., 2011, 133, 3452 CrossRef CAS PubMed
-
(a) T. E. Schrader, W. J. Schreier, T. Cordes, F. O. Koller, G. Babitzki, R. Denschlag, C. Renner, M. Loweneck, S. L. Dong, L. Moroder, P. Tavan and W. Zinth, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15729 CrossRef CAS PubMed
-
(a) S. Kneissl, E. J. Loveridge, C. Williams, M. P. Crump and R. K. Allemann, ChemBioChem, 2008, 9, 3046 CrossRef CAS PubMed
- F. Zhang, K. A. Timm, K. M. Arndt and G. A. Woolley, Angew. Chem., Int. Ed., 2010, 49, 3943 CrossRef CAS PubMed
-
(a) O. Sadovski, A. A. Beharry, F. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484 CrossRef CAS PubMed
- K. Fujimoto, M. Kajino, I. Sakaguchi and M. Inouye, Chem.–Eur. J., 2012, 18, 9834 CrossRef CAS PubMed
-
(a) L. Wen, X. Hou, Y. Tian, F.-Q. Nie, Y. Song, J. Zhai and L. Jiang, Adv. Mater., 2010, 22, 1021 CrossRef CAS PubMed
- M. R. Banghart, A. Mourot, D. L. Fortin, J. Z. Yao, R. H. Kramer and D. Trauner, Angew. Chem., Int. Ed., 2009, 48, 9097 CrossRef CAS PubMed
- I. Tochitsky, M. R. Banghart, A. Mourot, J. Z. Yao, B. Gaub, R. H. Kramer and D. Trauner, Nat. Chem., 2012, 4, 105 CrossRef CAS PubMed
- E. X. Albuquerque, E. F. R. Pereira, M. Alkondon and S. W. Rogers, Physiol. Rev., 2009, 89, 73 CrossRef CAS PubMed
- M. Stein, S. J. Middendorp, V. Carta, E. Pejo, D. E. Raines, S. A. Forman, E. Sigel and D. Trauner, Angew. Chem., Int. Ed., 2012, 51, 10500 CrossRef CAS PubMed
- R. L. Macdonald and R. W. Olsen, Annu. Rev. Neurosci., 1994, 17, 569 CrossRef CAS PubMed
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859 CrossRef CAS
-
(a) J. Kasianowicz, E. Brandin, D. Branton and D. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770 CrossRef CAS
- Y.-L. Ying, J. Zhang, F.-N. Meng, C. Cao, X. Yao, I. Willner, H. Tian and Y.-T. Long, Sci. Rep., 2013, 3, 1662, DOI:10.1038/srep01662
- D. M. Chudakov, S. Lukyanov and K. A. Lukyanov, Trends Biotechnol., 2005, 23, 605 CrossRef CAS PubMed
-
(a) S. Habuchi, R. Ando, P. Dedecker, W. Verheijen, H. Mizuno, A. Miyawaki and J. Hofkens, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9511 CrossRef CAS PubMed
- T. Grotjohann, I. Testa, M. Leutenegger, H. Bock, N. T. Urban, F. Lavoie-Gardinal, K. I. Willig, C. Eggeling, S. Jakobs and S. W. Hell, Nature, 2011, 478, 204 CrossRef CAS PubMed
- X. Liu, J. Li, C. Hu, Q. Zhou, W. Zhang, M. Hu, J. Zhou and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 4805 CrossRef CAS PubMed
- R. Bizzarri, M. Serresi, F. Cardarelli, S. Abbruzzetti, B. Campanini, C. Ciappiani and F. Beltram, J. Am. Chem. Soc., 2010, 132, 85 CrossRef CAS PubMed
- X. X. Zhou, H. K. Chung, A. J. Lam and M. Z. Lin, Science, 2012, 338, 810 CrossRef CAS PubMed
-
(a) V. Sample, R. H. Newman and J. Zhang, Chem. Soc. Rev., 2009, 38, 2852 RSC
- Z. G. Zhou, H. Hu, H. Yang, T. Yi, K. W. Huang, M. X. Yu, F. Y. Li and C. H. Huang, Chem. Commun., 2008, 4786 RSC
-
(a) P. H.-M. Lee, C.-C. Ko, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 6058 CrossRef CAS PubMed
- Y. Feng, Y. Yan, S. Wang, W. Zhu, S. Qian and H. Tian, J. Mater. Chem., 2006, 16, 3685 RSC
| This journal is © The Royal Society of Chemistry 2014 |