 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ambient mass spectrometry technologies for the detection of falsified drugs
María J.
Culzoni
abc,
Prabha
Dwivedi
a,
Michael D.
Green
d,
Paul N.
Newton
efg and
Facundo M.
Fernández
*a
aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: facundo.fernandez@chemistry.gatech.edu; Tel: +1 404 385 4432
bLaboratorio de Desarrollo Analítico y Quimiometría (LADAQ), Cátedra de Química Analítica I, Facultad de Bioquímica y Ciencias Biológicas, Universidad Nacional del Litoral, Ciudad Universitaria, Santa Fe S3000ZAA, Argentina
cConsejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Avda. Rivadavia 1917, Buenos Aires C1033AAJ, Argentina
dDivision of Parasitic Diseases and Malaria, Center for Global Health, Centers for Disease Control and Prevention, Atlanta, GA, USA
eLao-Oxford-Mahosot Hospital-Oxford-Wellcome Trust Research Unit, Microbiology Laboratory, Mahosot Hospital, Vientiane, Lao PDR
fCentre for Tropical Medicine, Churchill Hospital, Nuffield Department of Medicine, University of Oxford, Oxford, England, UK
gWorldWide Antimalarial Resistance Network, Churchill Hospital, University of Oxford, Oxford, UK
First published on 15th October 2013
Abstract
Increased globalization of the pharmaceutical market has facilitated the unobstructed and fast spread of poor-quality medicines. Poor-quality medicines include spurious/falsely-labeled/falsified/counterfeit drugs (those that are deliberately and fraudulently mislabeled with respect to content and/or origin), substandard drugs (legitimate drugs that do not meet their quality specifications), and degraded medicines (good quality pharmaceuticals that suffered from deterioration caused by improper storage or distribution). Consumption of poor-quality pharmaceuticals is likely to increase morbidity and mortality. Moreover, poor-quality drugs can also contribute to the development of resistance to anti-infective medicines and decrease the quality of health care received by patients. To assess the true prevalence of poor quality drugs, tiered technology approaches enabling the testing of drug samples collected at points of sale are required, thus ensuring public health standards. High throughput and high resolution ambient mass spectrometry techniques allow investigation of pharmaceuticals with minimal or no sample preparation, thus possessing capabilities to survey a large number of drug samples for their authenticity.
1. Scope of the review: background on falsified drugs
This review discusses advances in the field of ambient sampling/ionization mass spectrometry (ambient MS) with a specific focus on the detection of spurious/falsely-labeled/falsified/counterfeit (SFFC) medicines, and provides several examples of poor quality drugs that have been analyzed using these approaches since 2006 (Table 1).| Technique | Acronym | Figure | Analyte | Ref. |
|---|---|---|---|---|
| Plasma and solid–liquid extraction-based techniques | ||||
| Direct analysis in real time | DART | 1 | Artesunate | 20,22 and 58 |
| “Yaa Chud” | 54 | |||
| Etoricoxib | 55 | |||
| Alprazolam | 11 | |||
| Glycin, noortropyl, anapril, mexidol, biseptol | 56 | |||
| Tadalafil | 57 | |||
| Desorption atmospheric pressure chemical ionization | DAPCI | 2 | Amoxicillin | 63 |
| Desorption atmospheric pressure photoionization | DAPPI | 3 | Anabolic steroids | 82 |
| Desorption electrospray ionization | DESI | 1 | Artesunate | 20–22,34,58,69 and 73 |
| Oseltamivir | 67 | |||
| Laser desorption/ablation techniques | ||||
| Infrared laser assisted desorption electrospray ionization | IR-LADESI | 4 | Dihydroartemisinin, piperaquine, chloroquine, quinine, artesunate | 87 |
| Infrared laser ablation metastable-induced chemical ionization | IR-LAMICI | 5 | Artesunate, acetaminophen | 93 |
| Multimode techniques | ||||
| Desorption electrospray/metastable-induced ionization | DEMI | 6 | Artesunate | 94 |
With the number of reports on cases involving drug counterfeiting increasing globally,1 the risk to public health is at an all-time high. The most widely used definition of counterfeit medicines that has been used by the World Health Organization (WHO) states that: “A counterfeit medicine is one which is deliberately and fraudulently mislabeled with respect to identity and/or source. Counterfeiting can apply to both branded and generic products and counterfeit products may include products with the correct ingredients, wrong ingredients, without active ingredients, with insufficient quantity of active ingredient or with fake packaging”.2 Counterfeit drugs are the products of criminals. Frequently, incorrect active pharmaceutical ingredients (APIs) are present in counterfeits with the purpose of mimicking both the organoleptic characteristics or symptom-relief effects of authentic medicines, or perhaps due to the use of waste powders left over from the manufacturing of other pharmaceuticals.3
It has been argued that the use of the term ‘counterfeit medicines’ and that some proposed definitions wrongly include legitimate generics and will therefore obstruct the trade in generic medicines by invoking intellectual property (IP) issues.4 To avoid any IP connotations the term falsified is used here instead of counterfeit, except when this term is used in the primary publication.5
Thorough characterization of the chemical composition of fake medicines is of utmost importance since their consumption can trigger severe side effects, or even death. Falsified medicines containing wrong APIs or insufficient quantity of the correct APIs might also be responsible for the emergence of drug resistance, although the connection between the two has been difficult to establish. Substandard medicines, another type of poor quality medicine, arise as a result of lack of expertise, poor manufacturing practices, insufficient infrastructure, or in some cases criminal negligence.6 Degraded medicines are good quality pharmaceuticals that suffered from deterioration caused by improper storage or distribution, an issue most commonly encountered in anti-infectives that are sold in tropical areas.7–9
With overwhelming literature evidence, it is now well established that drug counterfeiting is a major threat to public health worldwide.1 This fraudulent activity not only affects consumers, but also health care providers, drug manufacturers, and governments. Falsified medicines may be spread not only by illegitimate channels, but also inadvertently by legitimate channels such as through governments, private hospitals and pharmacies. Falsified medicines may be visually indistinguishable from the authentic pharmaceutical product, and licensed distributors, pharmacists, health care providers or patients are unable to visually distinguish them.10 For example, a recent investigation by the US Institute of Forensic Sciences into suspicious alprazolam tablets determined that their visual appearance mimicked the authentic product almost exactly, but that melatonin was present instead of the correct active ingredient.11
Although falsified drugs occur worldwide, their incidence is greatest in regions where regulatory and enforcement systems for medicine quality are weakest, such as developing countries of Africa, Asia and Latin America.12 According to the latest estimates reported by the International Medical Products Anti-Counterfeiting Taskforce (IMPACT) set up by the WHO and partners to fight against counterfeit medicines, it is estimated that more than 30% of the medicines on sale in parts of Africa, Asia and Latin America can be counterfeit. It is believed that counterfeits make up more than 20% of the market in the former Soviet Republics, up to 1% of sales in developed countries, and approximately 10% in developing countries, on average.13
According to the WHO, products that are subjected to counterfeiting include medicines for a wide set of conditions, ranging from the treatment of life-threatening diseases to inexpensive versions of painkillers and antihistamines. Amongst the most targeted medicines for counterfeiting are those which are expensive, innovative, or in high demand, as well as generic drugs.14 Regarding online pharmacies, over 50% of the medicines sold over the Internet have been found to be counterfeit.12 An alert posted by the US Food and Drug Administration in 2010 regarding the online sale of a “generic” version of Tamiflu, which contained penicillin cloxacillin instead of the correct API oseltamivir, is an example of such fraudulent activities prevalent over the Internet space. Consumption of such fake Tamiflu had the potential to induce severe allergic reactions in those allergic to penicillin.15
Medicine fraudsters prey on regions with insufficient regulatory and legal oversight compounded with poor reporting practices. The Pharmaceutical Security Institute (PSI), a not-for-profit innovative pharmaceutical organization, states that medicines in every therapeutic category are targeted by criminal organizations with medicines in the genito-urinary and anti-infective categories being targeted the most. PSI data also show an increasing year-to-year trend in several other therapeutic categories: hormones (+76%), cytostatic (+48%), central nervous system (+29%), musculo-skeletal (+27%), and respiratory (+22%). Artesunate-based antimalarials have been one of the pharmaceuticals with the highest frequency of reported falsification. The high incidence of malaria in many countries of SE Asia and Africa has made this class of drugs a profitable niche for counterfeiters since 1998,8,16–19 with some estimates suggesting that up to 33% to 53% of artesunate monotherapy tablets were falsified.20 Artemisinin derivatives are essential for efficacious treatment of falciparum malaria. Therefore, the dissemination of falsified antimalarials promotes avoidable death, contributes to the growing problem of drug resistance, reduces confidence in the drug, and damages health systems and the pharmaceutical industry.20 The potential for artemisinin resistance is of major concern due to the manufacture of medicines containing sub-therapeutic amounts of artesunate which are probably added in order to defeat the colorimetric methods currently used to detect artesunate.20–22
The falsification of a wide range of other essential medicines is well-documented in the scientific literature.8,14,23,24 Some recently reported cases include falsified Avastin used in the treatment of cancer, which did not contain the active ingredient, bevacizumab, and may have resulted in patients not receiving needed therapy;25 Truvada and Viread, which are pharmaceuticals used in the treatment of HIV/AIDS, and were found to be diverted authentic products in falsified packaging;26 Zidolam-N, containing lamivudine, zidovudine and nevirapine, also employed against HIV/AIDS, which affected nearly 3000 patients that were under antiretroviral therapy;27 and an anti-diabetic traditional medicine, which contained six times the normal dose of glibenclamide, and led to the death of two people, while the other nine were hospitalized.12 This evidence strongly suggests that the proliferation of falsified drugs is a very significant, but neglected, public health problem. This persistent problem has been accelerated by the globalization of the pharmaceutical industry, which may favor the access of criminals to technologies required to manufacture medicines mimicking genuines.9,24
Laboratory methodologies allowing for rapid, cost effective pharmaceutical analysis are nowadays a required part of toolbox needed to combat the ever-growing number of both crude and sophisticated falsified medicines.20 We hypothesize that the implementation of powerful, high throughput and reliable technologies to screen for falsified medicines will play a key role in providing a realistic picture of the prevalence of poor quality medicines in the global market, identifying new fakes and improving public health through contributing to the fight against such criminal activities.
2. Ambient ionization techniques applied to the detection of falsified drugs
The chemical analysis of medicines of unknown quality can be approached by analytical methods ranging from simple thin layer chromatography (TLC) and colorimetric tests to more sophisticated fingerprinting approaches such as near-infrared spectroscopy (NIR), Raman spectroscopy, X-ray fluorescence, nuclear magnetic resonance spectroscopy (NMR), and/or gas and liquid chromatography coupled to mass spectrometry (GC-MS and LC-MS, respectively) via electrospray ionization (ESI) and atmospheric-pressure chemical ionization (APCI).8,9,14,23 GC-MS and LC-MS enable not only the quantitation of the API amount, but also the identification of unknown sample ingredients, and are the most commonly used techniques by laboratories in pharmaceutical companies and regulatory agencies in developed countries. In addition, extensive research has been conducted in recent years to develop complementary techniques to improve the limited sample throughput of LC and GC-MS which results from extensive sample preparation requirements prior to analysis, and from the length of the chromatographic runs themselves.23 With billions of dollars of pharmaceuticals in the market and increasing fraud, analytical technologies that can provide faster and reliable answers in a short period of time are highly desirable.MS is one of the most popular technologies for chemical analysis due to its high selectivity, accuracy and quantitative capabilities provided the investigated analytes are effectively converted to gas phase ions. Ambient ionization methods for MS comprise a large and growing family of sampling/ionization techniques that allow the creation of ions under ambient conditions with minimal or no sample preparation prior to analysis.28 Their high throughput capabilities make them particularly suitable for the quick and reliable investigation of large numbers of solid or liquid pharmaceutical samples of various complexities.
Ambient ion sources operate in the open air, outside of the mass spectrometer vacuum environment, and the analysis probes the sample directly, without pretreatment.28 Ambient ionization techniques share common features such as the possibility of being interfaced with most types of mass spectrometers fitted with atmospheric pressure interfaces, and the ability to generate ions softly.29,30 These techniques involve various desorption (laser, plasma, thermal, liquid jet) and ionization (chemical ionization, electrospray, photoionization) methods either alone or combined with each other.31
Amongst the more than 30 different types of ambient ionization methods reported in the last 9 years,31–33 desorption electrospray ionization (DESI) and direct analysis in real time (DART) have been the two most widely adopted for detection and characterization of poor-quality pharmaceutical products. Their introduction in 2004 (DESI) and 2005 (DART) resulted in widespread interest in ambient MS approaches. Besides allowing investigation of pharmaceuticals without sample pretreatment in a few seconds, their coupling to high resolution mass spectrometers allows the structural identification of unknown compounds through accurate mass and tandem MS experiments.9 Though HPLC and spectrophotometric tests are traditionally used for analysis of known chemicals in pharmaceutical samples, these approaches are not as well suited for the forensic analysis of unknowns. Identifying wrong APIs in falsified drugs, for example, is of major interest, since their presence could be more harmful than the complete absence of any API and also for sample fingerprinting purposes. In this regard, ambient ionization techniques have significantly expanded the pharmaceutical analysis toolbox by enabling screening, quantitation, and identification of unknowns directly from the sample with high throughput.34
Apart from APIs, inactive excipients play a major role in any pharmaceutical formulation during the drug development phase. Inactive ingredients are added to drug formulations as fillers, binders, diluents, coloring agents, coatings, lubricants, disintegrants and/or preservatives with the aim of masking their unpleasant taste, making them easier to swallow, controlling the API release, increasing their resistance to adverse environmental conditions, and/or improving their appearance.35 Spectral signatures of these inactive ingredients obtained through mass spectrometry based analytical platforms serve as forensic fingerprints that are specific to a particular drug, and thus play a crucial role in the quality characterization of a sample when compared against genuine sample signatures. On the down side, these inactive ingredients in certain cases might interfere with identification and quantitation of chemical constituents, and may produce false positive or false negative results. Particularly, many genuine as well as poor-quality tablets are coated with thin polymer-based films that can interfere with API ionization and detection because of charge competition and/or ionization suppression during the ionization process. Physical removal of tablet coating prior to ambient MS analysis is found to minimize API signal interference from such coatings. In addition, splitting of the tablet to expose the tablet's core to the ionizing plume can mitigate signal masking by surface coatings as well as any other surface contaminant. During ambient MS analysis, it may be difficult to minimize API signal suppression due to signals from certain chemical components of a formulation. In such cases, method development using genuine samples is required, and statistically relevant deviation in the forensic fingerprint from that of a genuine should be considered as a sign of falsification, and should be subjected to further investigations for confirmation. However, quality characterization of a sample is always performed against a reference standard, and the presence along with the effects of these inactive ingredients on analytical response(s) of APIs are known prior to the sample analysis. Unknown sample characterization on the other hand would require sample interrogation by multiple technologies including separations prior to MS analysis for chemical identification, validation, and confirmation.
2.1. Plasma and solid–liquid extraction-based techniques
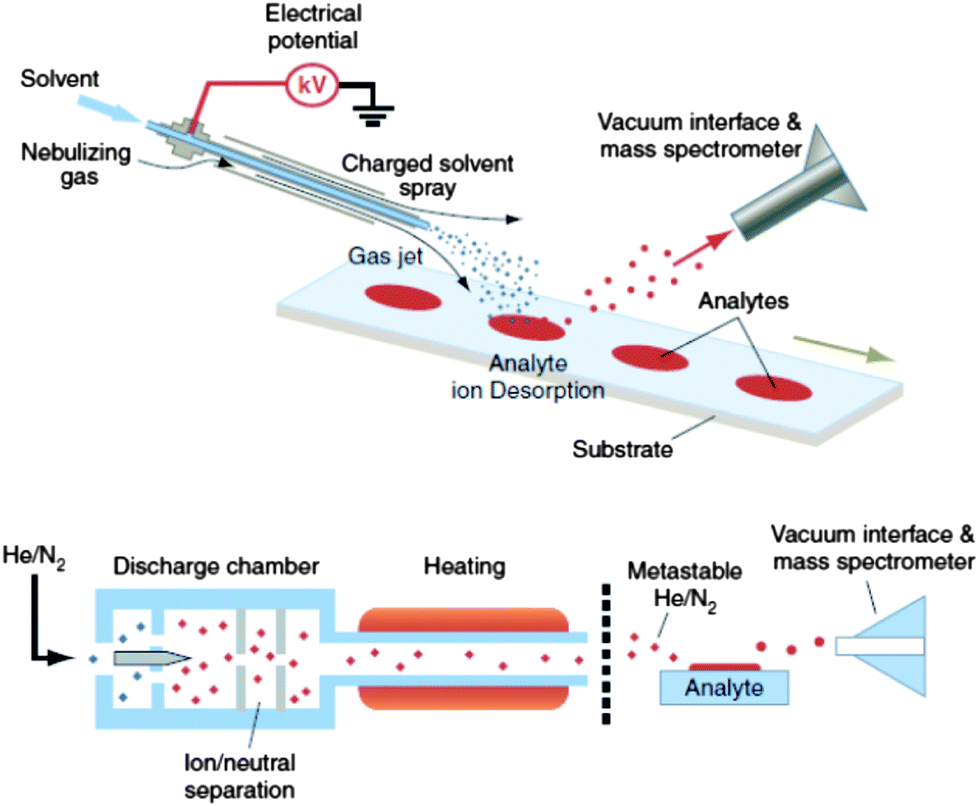 | ||
| Fig. 1 DESI (upper) and DART (lower) analyses for ambient high-throughput mass spectrometric analysis of unprepared samples (skin, bricks, urine spots, clothing, tissue, etc.). Reprinted with permission from ref. 28. Copyright © 2006, American Association for the Advancement of Science. | ||
The details of DART ionization are described elsewhere.30,46,47 DART uses a negatively biased point-to-plane atmospheric pressure glow discharge to generate metastable species within the discharge supporting gas, typically He or N2. In positive ionization mode, protonated water clusters (the reactive reagents) are produced by Penning ionization of atmospheric water molecules through collision with metastables. After analyte thermal desorption by the heated gas stream in the region between the DART ion source outlet and the mass spectrometer inlet, proton transfer proceeds from the reactive reagents to the analytes. Collisions at the atmospheric pressure inlet interface lead to fully desolvated protonated analytes, which are transported toward the mass spectrometer inlet mainly by the gas flow lines.48 It is believed that analyte desorption is mainly a thermal process,49 the thermal conductivity of the gas used for desorption being a critical parameter affecting the sensitivity of the measurements. However, it has also been suggested that desorption of analytes might be facilitated by charged clusters via a chemical sputtering mechanism.46,50,51 In the negative ion generation mode, metastable atoms react with atmospheric water to produce negatively charged oxygen-water clusters as reactant ions that consequently interact with analyte neutrals in the gas phase to produce negatively charged species.46 Samples are introduced between the plasma plume and the mass spectrometer inlet for rapid characterization of sample chemical composition in a single step.
Since DART is a gas phase ionization technique, formation of complex cluster ions, metal adduct ions, and multiply charged ions that are usually observed through solution phase ionization techniques such as DESI (Section 2.1.3) are rarely observed.52 Relative disadvantages of DART when compared to other ambient MS techniques include the dilution impinged in the desorbed analyte by the discharge gas and the unavoidable ion fragmentation observed at high discharge gas temperatures.53 DART has mainly been used for providing a rapid but qualitative screening of pharmaceutical sample composition, but its quantitative capabilities still remain relatively under-explored.52
DART applications. Falsified and genuine artesunate antimalarial tablets collected from SE Asia have been investigated by DART MS in both negative and positive ion detection modes in order to establish not only the presence or absence of the API, but also the potential presence of other unstated ingredients.22 Artesunate was found to preferentially form positive ions through adduction with ammonium ions generated in the pool of reactant species. To eliminate false negatives and to enhance the ionization efficiency of artesunate through formation of the artesunate [M + NH4]+ ions, the population of ammonium reactants was increased by introduction of ammonia vapor as a dopant in the ionization region. In this study, 83% of the samples analyzed by DART were found to contain no detectable artesunate. Instead, early generation antimalarials such as pyrimethamine/sulfadoxine, chloramphenicol, and chloroquine; acetaminophen, metamizole, and metronidazole were found in some tablets. Even more concerning was the detection of a sample that contained a lower than stated quantity of artesunate along with acetaminophen, which was later confirmed by HPLC to contain only 20% of the artesunate amount expected in the tablet. Such formulations with sub-therapeutic quantities of API pose a grave threat to disease control as they can pass simple colorimetric field screening tests and thereafter be consumed by patients in ineffective amounts. Such “medicines” would facilitate the development and propagation of drug resistant parasitic strains and eventually render medications and disease control efforts ineffective.
As part of an international epidemiological collaboration and criminal investigation focused on obtaining evidence as to the source of falsified artesunate in SE Asia, Newton et al. analyzed a total of 391 antimalarial samples.20 Chemical content characterization of the samples by DART MS and DESI MS demonstrated that 50% of the analyzed samples were not genuine and thus of concern to public health. Samples were found to have subtherapeutic quantities of artesunate or a wide diversity of wrong active ingredients such as dipyrone, a variety of antibiotics, and safrole – a banned raw material used in manufacturing ecstasy. Hence, the finding of this wrong API suggested links between manufacture of falsified antimalarials and illicit drugs.
DART MS has also been applied to the identification of APIs in tablets sold without their original packaging. For example, when patients with fever seek treatment from private providers in certain parts of SE Asia, they commonly opt to purchase a little unlabeled plastic bag containing up to 8 colorful tablets and capsules. These bags are called “Yaa chud” in the Thailand/Myanmar border region, or “tnam psom” in Cambodia, meaning “combination medicine” or “drug cocktails”, and are sold without prescription or medical assessment. The content of these cocktails is usually unknown to the patient and they are used as the first-line malaria treatment. Newton and co-workers characterized 50 packets of “Yaa chud” purchased from 44 shops located in the Thailand–Myanmar border region.54 Identification of each pill was first visually attempted by a pharmacist, followed by chemical analysis by high resolution DART MS and atomic spectroscopy to discover that only 14% of the samples had the potential to cure malaria. The most frequently detected active ingredients were acetaminophen (22.0%), chlorpheniramine (13.4%), chloroquine (12.6%), tetracycline/doxycycline (11.4%), and quinine (5.1%). The potential of unregulated local practices impeding the fight against malaria cannot be ignored and such ineffective antimalarial drugs are likely to increase malaria morbidity, mortality and health costs, and engender the emergence and spread of antimalarial drug resistance. Moreover, about 82% of the bags contained medicines that in spite of being sold to pregnant women are contraindicated in pregnancy. Other examples on the application of DART MS to the screening for falsified drugs have been reported by Helmy et al. (etoricoxib),55 Samms et al. (alprazolam),11 Chernetsova et al. (glycin, nootropyl, anaprilin, mexidol, and biseptol),56 and Moffat et al. (tadalafil).57
In forensic investigations of falsified pharmaceuticals requiring identification and identity validation of unknown components, the use of multiple complementary analytical methods is often necessary. To illustrate this multi-tiered approach, several ambient MS techniques including DART MS and DESI MS (see Section 2.1.3), and 2D DOSY 1H NMR were performed to integrally characterize artesunate antimalarial tablets collected from Burma, Cambodia and the Lao People's Democratic Republic (Laos). Of 16 samples only 6 were found to have the correct API while the rest of the formulations contained either wrong APIs such as acetaminophen or dipyrone, or contained only pharmaceutical excipients.58 Analytical results from multiple techniques provided a more confident picture on the chemical composition and homogeneity of such samples than the one that would be obtained from the use of a single technique.
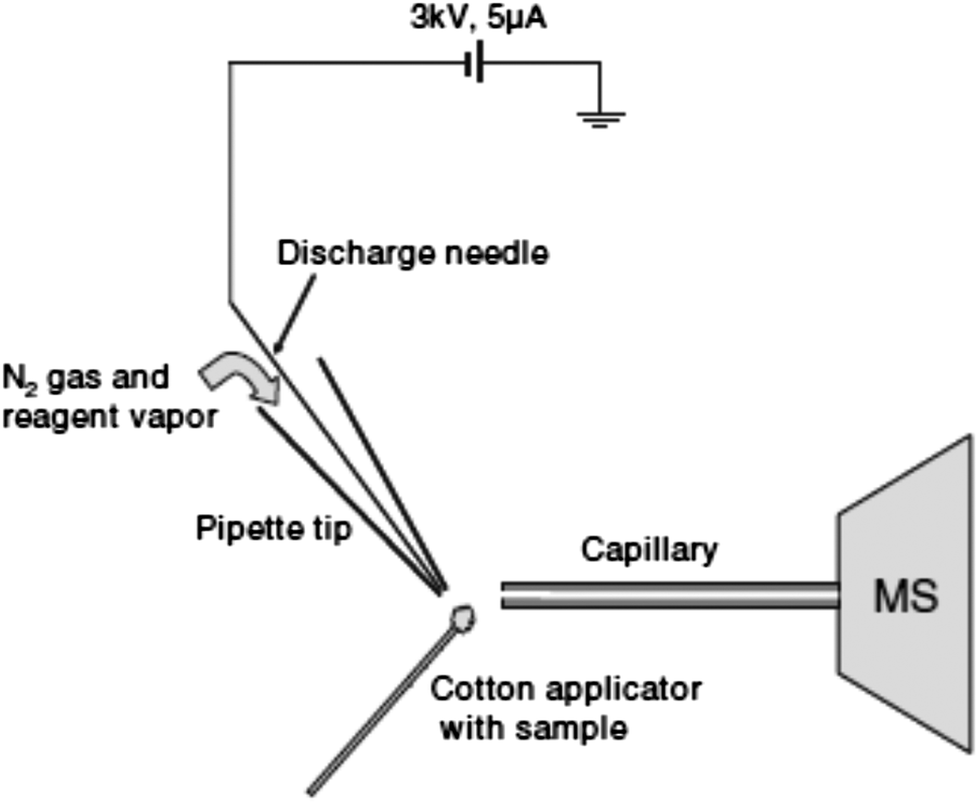 | ||
| Fig. 2 Schematic of the desorption atmospheric pressure chemical ionization experiment. Reprinted with permission from ref. 96. Copyright © 2006, John Wiley & Sons, Ltd. | ||
DAPCI has been applied to rapid analysis of common drugs, common antibiotics and complex pharmaceuticals.61–63 Zang et al. used DAPCI MS for drug quality control and chemical origin differentiation applications. Amoxicillin capsules were found to contain starch instead of amoxicillin. Furthermore, the method was used to rapidly screen amoxicillin preparations, and to discriminate between expired and unexpired drugs. This technique also proved to be a useful alternative to track the origin of pharmaceutical products and to differentiate their overall quality, through chemical fingerprinting.63
In DESI (Fig. 1), a pneumatically assisted continuous electrospray jet is directed towards the surface of a solid sample, forming a micron thickness thin solvent film on the sample where molecules are extracted and dissolved. The solvent flow from the spray dynamically dislodges the surface film resulting in the generation of analyte-containing charged secondary droplets which are sampled downstream of the mass spectrometer inlet.64 Gas phase ions from secondary droplets are formed by mechanisms similar to those in ESI.68 The signal is influenced by several parameters such as spray tip-to-surface distance, sample-to-spectrometer orifice distance, spray incident angle, collection angle, and sample properties such as hardness and sample shape.69 DESI is mostly used as a surface characterization technique because the jet of charged solvent used to ionize the analyte dissolves a small area on the surface, for instance, of a pharmaceutical tablet. If this action is prolonged over time, and high repetition rate scans are acquired, enough mass will be removed from the surface to allow the analysis of compounds located deeper into the sample.9
Addition of selective chemical reagents into the DESI spray solution in order to induce selective ion–molecule reactions at the interface between the charged microdroplets and the condensed phase analytes is referred to as reactive DESI.21,70–72 This approach enables the analysis of materials over a large mass range with the same sample throughput as conventional DESI, the added advantage of improved selectivity, and likely sensitivity enhancement due to increased ion evaporation rates and/or reduced fragmentation brought upon by the choice of the chemical reagent.21 This alternative allows the high throughput screening of target species present in complex matrices avoiding chromatographic separation by taking advantage of host–guest chemistries and molecular recognition phenomena.67
DESI applications. The previously described DART analysis of genuine and falsified tablets collected in SE Asia (Section 2.1.1) was also validated by coupling DESI to ion trap MS.22 Both DESI MS and DART MS produced spectra with good signal-to-noise ratios in approximately five seconds, providing two-orders of magnitude improvement in sample throughput over LC-MS methods.
Ricci and co-workers used micro-attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopic imaging, complemented and validated by both reagentless and reactive DESI MS in order to characterize counterfeit antimalarial tablets collected in SE Asia.73 These two techniques were complementary to each other in their application to tablet quality control. ATR-FTIR allowed drug distribution imaging in a non-destructive way, while DESI MS provided the m/z of the spatially localized species. This approach demonstrated the presence of dipyrone and acetaminophen in some of the fake tablets. Although artesunate levels in genuine tablets are high enough to allow qualitative analysis by DESI MS, substandard or falsified tablets containing lower artesunate concentrations may not produce a detectable signal. To increase sensitivity and reduce ion fragmentation, a reactive DESI method was developed to detect artesunate at very low concentrations in samples previously analyzed by HPLC. Dodecylamine was added to the DESI spray, allowing the detection of artesunate as the proton-bound non-covalent artesunate complex with the amine. The identity of this adduct species was then confirmed by DESI MS/MS.73 With this method, a sensitivity gain of up to 170-fold was obtained in comparison to reagentless DESI, depending on both the type of amine added to the spray and its concentration. Reactive DESI was further exploited for the direct quantitative analysis of APIs in antimalarials purchased from shops and pharmacies in SE Asia. In this quantitative approach,69 the internal standard (IS) d4-artesunic acid was synthesized, and incorporated into a solid tablet in a controlled and well-dispersed way, with the purpose of removing the dependences of the analyte signal on the geometry variables of the DESI setup. With dodecylamine as the reagent for reactive DESI, the analyte-to-IS signal ratio was found to depend on tablet hardness. A 6% precision in the amount of artesunate measured was obtained. The precision observed was inferior to that commonly achieved with HPLC methods (2%), but the high throughput capability of the method easily offset the somewhat reduced precision. It was found that up to 94% quantification accuracy could be achieved by reactive DESI.
Another reactive DESI method for direct API quantitation purposes was reported and applied to the investigation of antiviral Tamiflu® samples purchased over the Internet.67 The method was based on the formation of stable gas-phase complexes between protonated oseltamivir and crown ethers added to the DESI spray solvent. By allowing competition for complex formation to take place between two different crown ethers, rapid and sensitive quantitation without requiring addition of an internal standard was achieved.
DESI ambient mass spectrometry imaging (MSI) is a versatile powerful tool that has been used to probe the spatial resolution of analytes on the surface of intact pharmaceutical samples with high throughput.74,75 DESI MS was performed in the imaging mode76,77 as a tool to contrast the homogeneity in API distribution between genuine and falsified artesunate antimalarial tablets.58 DESI MSI not only facilitates detection of APIs and excipients, but also detection of any minor impurities present in the sample which would remain undetected should the sample be crushed, dissolved and diluted. Homogeneity in API distribution is highly important in certain antimalarial therapies, in which tablets have to be split to adjust for body weight medication dosage. DESI MS images of genuine artesunate tablets not only showed the presence of the correct API homogeneously distributed on the tablet, and its absence in the fakes, but also the homogeneous distribution of acetaminophen in the counterfeits, pointing towards sophisticated procedures in manufacturing.
Considering that DESI MS analysis at trace levels is likely to suffer from background chemical interferences arising from the ESI-like ionization process, a platform coupling DESI to differential mobility (DM) ion separation, and interfaced to a TOF mass spectrometer (DESI-DM-MS) was developed.34 In this approach, only ions of interest generated by DESI are transmitted into the mass spectrometer for detection, allowing discrimination and simplification of the mass spectrum by filtering undesired chemical noise. This strategy was implemented to analyze counterfeit antimalarial tablets, and improved both the sensitivity, i.e. enhanced signal-to-noise ratios by 70–190%, and the selectivity when compared to standalone DESI MS.
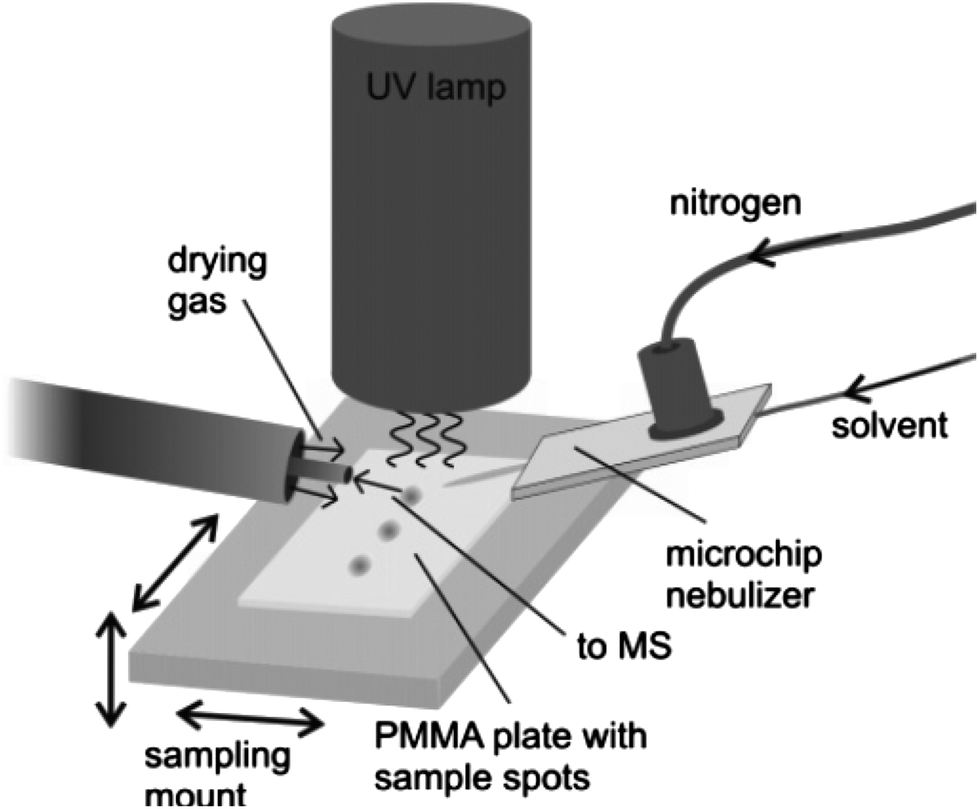 | ||
| Fig. 3 Schematic view of the DAPPI setup. Reprinted with permission from ref. 80. Copyright © 2007, American Chemical Society. | ||
Kauppila et al. have applied DAPPI to the direct analysis of illegal anabolic steroids, which are frequently sold in the black market and online pharmacies.82,83 For analysis, an aliquot of the sample soaked into a kitchen paper was placed on a glass microscope slide that was positioned under the DAPPI source and in front of the mass spectrometer inlet. Direct analysis of the samples by DAPPI MS and MS2 revealed the presence of certain compounds that were not detected by traditionally employed gas chromatographic procedures. In many cases, the chemicals detected by DAPPI MS were different than those stated by the manufacturer, and the steroids detected in seemingly identical samples were also different.
2.2. Laser desorption/ablation techniques
This subfamily of laser-based ambient two-step techniques involves coupling of laser desorption/ablation processes to electrospray or plasma secondary ionization. In general terms, the plume that is generated when the surface material is ablated or desorbed by UV or IR lasers is further merged with an electrospray ion plume from an electrospray source or with a reactive plasma beam from a plasma source. Since desorption and ionization events are spatially and temporally separated, these processes can be independently optimized for selective and sensitive measurement of semi-volatile as well as non-volatile analytes of interest from solids and liquids.29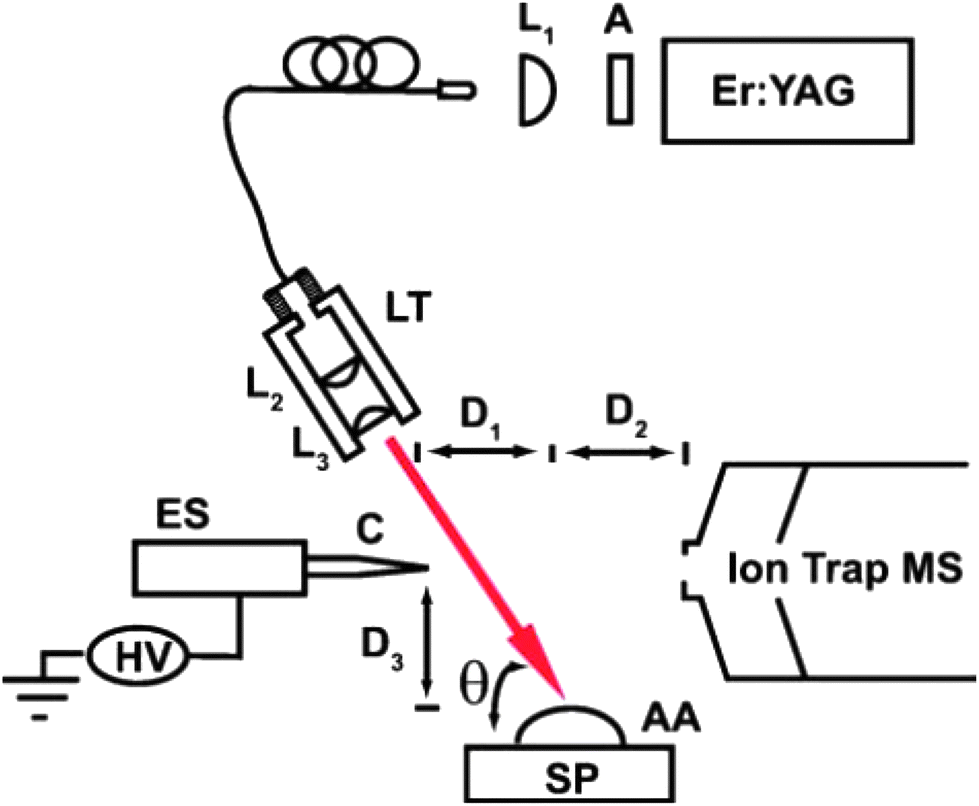 | ||
| Fig. 4 Schematic of the IR LADESI ion source: ES, nanoelectrospray; C, fused-silica capillary; HV, high voltage supply; L1, L2, and L3, planoconvex lenses; A, attenuator; LT, lens tube; SP, sample plate; AA, aqueous analyte; D1, distance between the capillary tip and ablation point; D2, distance between the ablation point and skimmer cone; D3, distance between the sample plate to capillary tip; h is the laser incidence angle. The drawing is not to scale. Reprinted with permission from ref. 84. Copyright © 2008, The Royal Society of Chemistry. | ||
Recently, IR-LADESI has been coupled to atmospheric pressure drift tube ion mobility spectrometry (DTIMS), providing ion mobility spectral fingerprints for medicine samples of dubious quality.87 The technique can be applied to high-throughput screening of pharmaceuticals, and has potential for distinguishing between falsified and genuine medicines rapidly. Proof of principle results were shown for the analysis of intact antimalarial tablets collected from certain regions in SE Asia, having piperaquine, chloroquine and artesunate as the stated APIs.
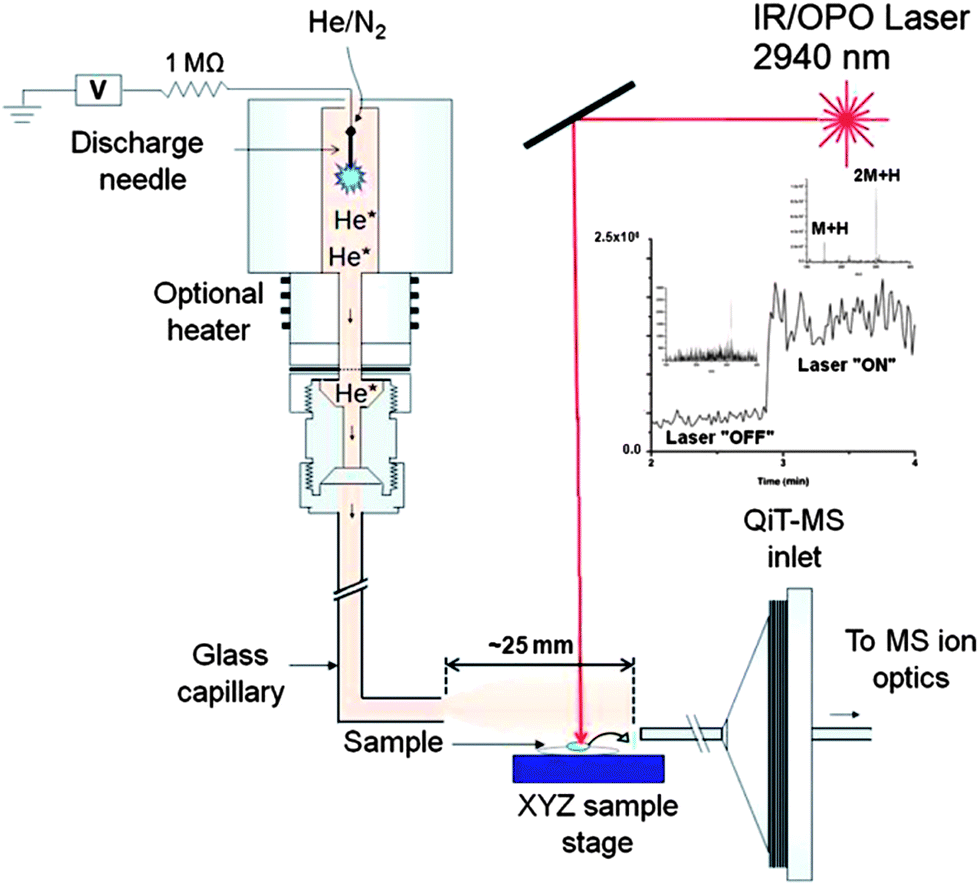 | ||
| Fig. 5 Schematic of the IR-LAMICI ion source coupled to a quadrupole ion trap mass spectrometer. The inset shows the total ion current trace observed in the analysis of a Tylenol tablet with the laser turned off and on. Reprinted with permission from ref. 93. Copyright © 2010, American Chemical Society. | ||
A counterfeit artesunate antimalarial tablet collected in the Republic of Cameroon was analyzed by IR-LAMICI in order to explore its capabilities compared to DART and DESI.93 The wrong active ingredient chloroquine was found in the sample as the protonated molecule, while the expected artesunate was not detected. This fact was confirmed by means of both DART and DESI MS analysis. The advantage of the IR-LAMICI over DART as an ion source lies in its ability to be used for imaging applications. Besides, there is no need for solvents as in DESI MSI.
2.3. Multimode techniques
DEMI is a multimode ambient source that combines the benefits of both DESI and DART-MICI.94 This ion source can be operated in three unique modes, i.e. plasma mode (MICI-only), spray mode (DESI-only), and dual mode (DEMI). While operation of the ion source in the MICI-only mode allows the analysis of low-polarity, low-molecular weight compounds, the DESI-only mode is well suited for the analysis of high-molecular weight, high-polarity compounds. Operation of the ion source in the DEMI mode expands the range of target analytes that can be simultaneously and directly detected in comparison to each of the individual ionization sources. This approach circumvents drawbacks associated with individual techniques such as limited polarity range and limited mass range, typical of DESI and DART. The DEMI set-up integrates a glow discharge source that generates He metastables95 with a DESI-type ion source having a resistive flow-through heater added to the nebulizer gas line67 (Fig. 6).94 The glass transport capillary receives the analytes that are being transported toward the mass spectrometer inlet after dissolution, thermal desorption, or aerosolization by the nebulizer gas. Before entering, the less polar molecules are chemically ionized by mechanisms involving He metastables, in contrast to the high polarity analytes that are ionized by DESI. The feasibility of the DEMI ionization mechanism to provide complementary ionic species, e.g. protonated and ammoniated species in the MICI-only mode, and sodiated species in the DESI-only mode, within the same spectrum increases the identification certainty in the screening of unknown samples. The latter fact was exemplified by analyzing falsified artesunate tablets collected on the China/Myanmar (Burma) border,20 which were found to contain the wrong API artemisinin.94
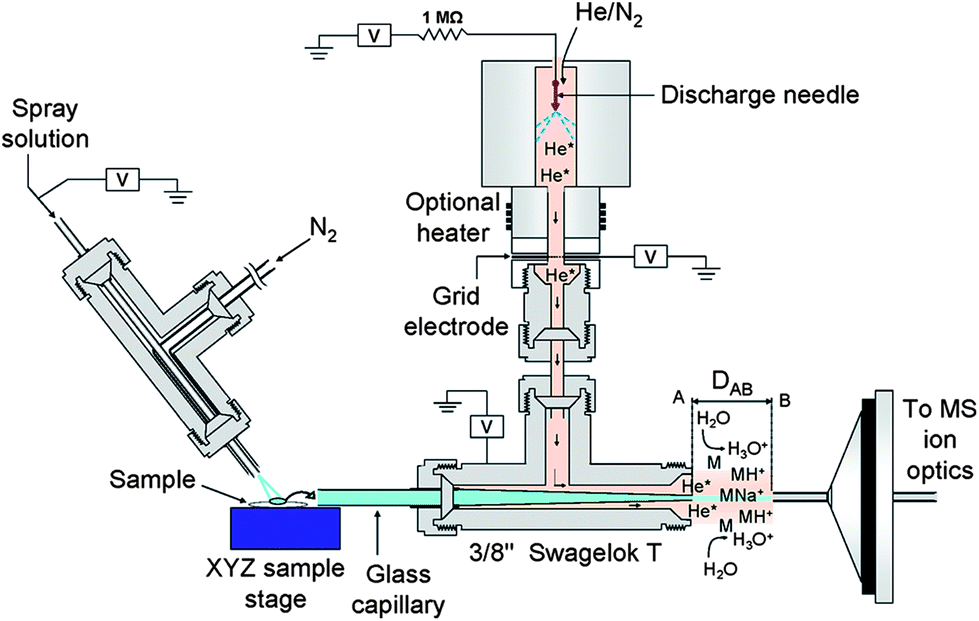 | ||
| Fig. 6 Schematic of the desorption electrospray/metastable induced ionization (DEMI) ion source coupled to a quadrupole ion trap mass spectrometer (only inlet shown). Reprinted with permission from ref. 94. Copyright © 2009, American Chemical Society. | ||
3. Concluding remarks
Classification of pharmaceutical samples as genuine, substandard, degraded or falsified is of vital importance to ensure public health. Many examples of pharmaceutical counterfeiting as well as efforts to detect them by ambient MS methodologies have been discussed in this review. Drug quality surveys and forensic efforts greatly benefit from the availability of high throughput ambient MS tools which are capable of reliably screening large numbers of samples without extensive sample preparation for the purposes of quantitation and identification of unknowns. Successfully decreasing the prevalence of poor quality drugs around the world will require implementation of these new types of technologies for rapid screening together with capacity building initiatives and development of robust and affordable field screening methods.Acknowledgements
The authors are grateful to the ACT consortium for funding their project to assess the quality of drugs in Africa via an award from the Bill and Melinda Gates Foundation to the London School of Hygiene and Tropical Medicine.References
- Countering the Problem of Falsified and Substandard Drugs, The National Academies Press, 2013 Search PubMed.
- WHO, http://whqlibdoc.who.int/hq/1999/WHO_EDM_QSM_99.1.pdf, 1999.
- J.-C. Wolff, L. A. Thomson and C. Eckers, Rapid Commun. Mass Spectrom., 2003, 17, 215–221 CrossRef CAS PubMed.
- P. N. Newton, A. A. Amin, C. Bird, P. Passmore, G. Dukes, G. Tomson, B. Simons, R. Bate, P. J. Guerin and N. J. White, PLoS Med., 2011, 8, e1001139 Search PubMed.
- A. Attaran, D. Barry, S. Basheer, R. Bate, D. Benton, J. Chauvin, L. Garrett, I. Kickbusch, J. C. Kohler, K. Midha, P. N. Newton, S. Nishtar, P. Orhii and M. McKee, BMJ [Br. Med. J.], 2012, 345, e7381 CrossRef PubMed.
- WHO, http://whqlibdoc.who.int/trs/WHO_TRS_961_eng.pdf, 2010.
- J. M. Caudron, N. Ford, M. Henkens, C. Mace, R. Kiddle-Monroe and J. Pinel, Trop. Med. Int. Health, 2008, 13, 1062–1072 CrossRef PubMed.
- P. N. Newton, M. D. Green, F. M. Fernández, N. P. J. Day and N. J. White, Lancet Infect. Dis., 2006, 6, 602–613 CrossRef CAS.
- F. M. Fernandez, D. Hostetler, K. Powell, H. Kaur, M. D. Green, D. C. Mildenhall and P. N. Newton, Analyst, 2011, 136, 3073–3082 RSC.
- WHO, http://www.who.int/medicines/services/expertcommittees/pharmprep/WHO-ACM-3IMPACTSurveyDataCollectionToolReport.pdf, 2010.
- W. C. Samms, Y. J. Jiang, M. D. Dixon, S. S. Houck and A. Mozayani, J. Forensic Sci., 2011, 56, 993–998 CrossRef CAS PubMed.
- WHO, http://www.who.int/mediacentre/factsheets/fs275/en/index.html, 2012.
- WHO, http://www.who.int/medicines/services/counterfeit/impact/TheNewEstimatesCounterfeit.pdf, 2006.
- A. K. Deisingh, Analyst, 2005, 130, 271–279 RSC.
- FDA, http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm216183.htm, 2010.
- P. N. Newton, R. McGready, F. Fernandez, M. D. Green, M. Sunjio, C. Bruneton, S. Phanouvong, P. Millet, C. J. M. Whitty, A. O. Talisuna, S. Proux, E. M. Christophel, G. Malenga, P. Singhasivanon, K. Bojang, H. Kaur, K. Palmer, N. P. J. Day, B. M. Greenwood, F. Nosten and N. J. White, PLoS Med., 2006, 3, e197 Search PubMed.
- M. de Veij, P. Vandenabeele, K. A. Hall, F. M. Fernandez, M. D. Green, N. J. White, A. M. Dondorp, P. N. Newton and L. Moens, J. Raman Spectrosc., 2007, 38, 181–187 CrossRef CAS.
- K. A. Hall, P. N. Newton, M. D. Green, M. De Veij, P. Vandenabeele, D. Pizzanelli, M. Mayxay, A. Dondorp and F. M. Fernandez, Am. J. Trop. Med. Hyg., 2006, 75, 804–811 CAS.
- P. Newton, Lancet, 2001, 358, 246 Search PubMed.
- P. N. Newton, F. M. Fernandez, A. Plancon, D. C. Mildenhall, M. D. Green, L. Ziyong, E. M. Christophel, S. Phanouvong, S. Howells, E. McIntosh, P. Laurin, N. Blum, C. Y. Hampton, K. Faure, L. Nyadong, C. W. Soong, B. Santoso, W. Zhiguang, J. Newton and K. Palmer, PLoS Med., 2008, 5, e32 Search PubMed.
- L. Nyadong, M. D. Green, V. R. De Jesus, P. N. Newton and F. M. Fernández, Anal. Chem., 2007, 79, 2150–2157 CrossRef CAS PubMed.
- F. M. Fernandez, R. B. Cody, M. D. Green, C. Y. Hampton, R. McGready, S. Sengaloundeth, N. J. White and P. N. Newton, ChemMedChem, 2006, 1, 702–705 CrossRef CAS PubMed.
- F. M. Fernandez, M. D. Green and P. N. Newton, Ind. Eng. Chem. Res., 2008, 47, 585–590 CrossRef CAS.
- P. N. Newton, M. D. Green and F. M. Fernández, Trends Pharmacol. Sci., 2010, 31, 99–101 CrossRef CAS PubMed.
- FDA, http://www.fda.gov/Drugs/DrugSafety/ucm291960.htm, 2012.
- MHRA, http://www.mhra.gov.uk/home/groups/ei/documents/websiteresources/con149816.pdf, 2010.
- WHO, http://apps.who.int/prequal/info_press/documents/Falsified_ZidolamN_23September2011.pdf, 2011.
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Science, 2006, 311, 1566–1570 CrossRef CAS PubMed.
- G. A. Harris, A. S. Galhena and F. M. Fernandez, Anal. Chem., 2011, 83, 4508–4538 CrossRef CAS PubMed.
- M. E. Monge, G. A. Harris, P. Dwivedi and F. M. Fernández, Chem. Rev., 2013, 113, 2269–2308 CrossRef CAS PubMed.
- H. Chen, G. Gamez and R. Zenobi, J. Am. Soc. Mass Spectrom., 2009, 20, 1947–1963 CrossRef CAS PubMed.
- G. J. Van Berkel, S. P. Pasilis and O. Ovchinnikova, J. Mass Spectrom., 2008, 43, 1161–1180 CrossRef CAS PubMed.
- A. Venter, M. Nefliu and R. G. Cooks, TrAC, Trends Anal. Chem., 2008, 27, 284–290 CrossRef CAS PubMed.
- A. S. Galhena, G. A. Harris, M. Kwasnik and F. M. Fernandez, Anal. Chem., 2010, 82, 9159–9163 CrossRef CAS PubMed.
- D. Suzzi, S. Radl and J. G. Khinast, Chem. Eng. Sci., 2010, 65, 5699–5715 CrossRef CAS PubMed.
- T. Cajka, K. Riddellova, P. Zomer, H. Mol and J. Hajslova, Food Addit. Contam., Part A, 2011, 28, 1372–1382 CrossRef CAS PubMed.
- S. E. Edison, L. A. Lin, B. M. Gamble, J. Wong and K. Zhang, Rapid Commun. Mass Spectrom., 2011, 25, 127–139 CrossRef CAS PubMed.
- E. Chernetsova, M. Bromirski, O. Scheibner and G. Morlock, Anal. Bioanal. Chem., 2012, 403, 2859–2867 CrossRef CAS PubMed.
- E. Crawford and B. Musselman, Anal. Bioanal. Chem., 2012, 403, 2807–2812 CrossRef CAS PubMed.
- R. L. Self and W. H. Wu, Food Control, 2012, 25, 13–16 CrossRef CAS PubMed.
- E. S. Chernetsova, E. A. Crawford, A. N. Shikov, O. N. Pozharitskaya, V. G. Makarov and G. E. Morlock, Rapid Commun. Mass Spectrom., 2012, 26, 1329–1337 CrossRef CAS PubMed.
- J. L. Rummel, A. M. McKenna, A. G. Marshall, J. R. Eyler and D. H. Powell, Rapid Commun. Mass Spectrom., 2010, 24, 784–790 CrossRef CAS PubMed.
- G. A. Harris, M. Kwasnik and F. M. Fernandez, Anal. Chem., 2011, 83, 1908–1915 CrossRef CAS PubMed.
- W. Eberherr, W. Buchberger, R. Hertsens and C. W. Klampfl, Anal. Chem., 2010, 82, 5792–5796 CrossRef CAS PubMed.
- S. Beissmann, W. Buchberger, R. Hertsens and C. W. Klampfl, J. Chromatogr., A, 2011, 1218, 5180–5186 CrossRef CAS PubMed.
- R. B. Cody, J. A. Laramee and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS PubMed.
- R. B. Cody, Anal. Chem., 2009, 81, 1101–1107 CrossRef CAS PubMed.
- G. A. Harris and F. M. Fernandez, Anal. Chem., 2009, 81, 322–329 CrossRef CAS PubMed.
- G. A. Harris, C. E. Falcone and F. M. Fernandez, J. Am. Soc. Mass Spectrom., 2012, 23, 153–161 CrossRef CAS PubMed.
- M. Vincenti and R. G. Cooks, Org. Mass Spectrom., 1988, 23, 317–326 CrossRef CAS.
- Z. Takats, I. Cotte-Rodriguez, N. Talaty, H. Chen and R. G. Cooks, Chem. Commun., 2005, 1950–1952 RSC.
- E. S. Chernetsova and G. E. Morlock, Mass Spectrom. Rev., 2011, 30, 875–883 CAS.
- G. A. Harris, D. M. Hostetler, C. Y. Hampton and F. M. Fernandez, J. Am. Soc. Mass Spectrom., 2010, 21, 855–863 CrossRef CAS PubMed.
- P. N. Newton, C. Y. Hampton, K. Alter-Hall, T. Teerwarakulpana, S. Prakongpan, R. Ruangveerayuth, N. J. White, N. P. J. Day, M. B. Tudino, N. Mancuso and F. M. Fernandez, Am. J. Trop. Med. Hyg., 2008, 79, 662–669 CAS.
- R. Helmy, W. Schafer, L. Buhler, S. Marcinko, B. Musselman, E. Guidry, H. Jenkins, F. Fleitz and C. J. Welch, Org. Process Res. Dev., 2010, 14, 386–392 CrossRef CAS.
- E. S. Chernetsova, P. O. Bochkov, G. V. Zatonskii and R. A. Abramovich, Pharm. Chem. J., 2011, 45, 306–308 CrossRef CAS.
- A. C. Moffat, R. B. Cody, R. D. Jee and A. J. O'Neil, J. Pharm. Pharmacol., 2007, 59, A26 CrossRef.
- L. Nyadong, G. A. Harris, S. Balayssac, A. S. Galhena, M. Malet-Martino, R. Martino, R. M. Parry, M. D. Wang, F. M. Fernandez and V. Gilard, Anal. Chem., 2009, 81, 4803–4812 CrossRef CAS PubMed.
- T. Ast, D. E. Riederer, S. A. Miller, M. Morris and R. G. Cooks, Org. Mass Spectrom., 1993, 28, 1021–1033 CrossRef CAS.
- H. Chen, J. Zheng, X. Zhang, M. Luo, Z. Wang and X. Qiao, J. Mass Spectrom., 2007, 42, 1045–1056 CrossRef CAS PubMed.
- J. P. Williams, V. J. Patel, R. Holland and J. H. Scrivens, Rapid Commun. Mass Spectrom., 2006, 20, 1447–1456 CrossRef CAS PubMed.
- R. Chen, S. Lilin, S. P. Yang, J. L. Shen-Tu, W. F. Liang, H. W. Chen, L. L. Zhang and Y. F. Huan, Res. J. Chem. Environ., 2010, 14, 11–18 CAS.
- X. Zhang, B. Jia, K. Huang, B. Hu, R. Chen and H. Chen, Anal. Chem., 2010, 82, 8060–8070 CrossRef CAS PubMed.
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef CAS PubMed.
- Z. Takats, J. M. Wiseman and R. G. Cooks, J. Mass Spectrom., 2005, 40, 1261–1275 CrossRef CAS PubMed.
- Y. S. Shin, B. Drolet, R. Mayer, K. Dolence and F. Basile, Anal. Chem., 2007, 79, 3514–3518 CrossRef CAS PubMed.
- L. Nyadong, E. G. Hohenstein, K. Johnson, C. D. Sherrill, M. D. Green and F. M. Fernandez, Analyst, 2008, 133, 1513–1522 RSC.
- A. P. Bruins, T. R. Covey and J. D. Henion, Anal. Chem., 1987, 59, 2642–2646 CrossRef CAS.
- L. Nyadong, S. Late, M. D. Green, A. Banga and F. M. Fernandez, J. Am. Soc. Mass Spectrom., 2008, 19, 380–388 CrossRef CAS PubMed.
- I. Cotte-Rodriguez, H. Chen and R. G. Cooks, Chem. Commun., 2006, 953–955 RSC.
- I. Cotte-Rodriguez, Z. Takats, N. Talaty, H. W. Chen and R. G. Cooks, Anal. Chem., 2005, 77, 6755–6764 CrossRef CAS PubMed.
- H. Chen, I. Cotte-Rodriguez and R. G. Cooks, Chem. Commun., 2006, 597–599 RSC.
- C. Ricci, L. Nyadong, F. Fernandez, P. Newton and S. Kazarian, Anal. Bioanal. Chem., 2007, 387, 551–559 CrossRef CAS PubMed.
- J. M. Wiseman, D. R. Ifa, A. Venter and R. G. Cooks, Nat. Protoc., 2008, 3, 517–524 CrossRef CAS PubMed.
- P. Nemes, A. A. Barton and A. Vertes, Anal. Chem., 2009, 81, 6668–6675 CrossRef CAS PubMed.
- V. Kertesz and G. J. Van Berkel, Rapid Commun. Mass Spectrom., 2008, 22, 2639–2644 CrossRef CAS PubMed.
- V. Kertesz, G. J. Van Berkel, M. Vavrek, K. A. Koeplinger, B. B. Schneider and T. R. Covey, Anal. Chem., 2008, 80, 5168–5177 CrossRef CAS PubMed.
- P. Ostman, S. J. Marttila, T. Kotiaho, S. Franssila and R. Kostiainen, Anal. Chem., 2004, 76, 6659–6664 CrossRef PubMed.
- V. Saarela, M. Haapala, R. Kostiainen, T. Kotiaho and S. Franssila, Lab Chip, 2007, 7, 644–646 RSC.
- M. Haapala, J. Pól, V. Saarela, V. Arvola, T. Kotiaho, R. A. Ketola, S. Franssila, T. J. Kauppila and R. Kostiainen, Anal. Chem., 2007, 79, 7867–7872 CrossRef CAS PubMed.
- D. B. Robb, T. R. Covey and A. P. Bruins, Anal. Chem., 2000, 72, 3653–3659 CrossRef CAS.
- T. J. Kauppila, A. Flink, M. Haapala, U.-M. Laakkonen, L. Aalberg, R. A. Ketola and R. Kostiainen, Forensic Sci. Int., 2011, 210, 206–212 CrossRef CAS PubMed.
- M. Thevis, Y. Schrader, A. Thomas, G. Sigmund, H. Geyer and W. Schaenzer, J. Anal. Toxicol., 2008, 32, 232–240 CrossRef CAS PubMed.
- Y. H. Rezenom, J. Dong and K. K. Murray, Analyst, 2008, 133, 226–232 RSC.
- M. Z. Huang, S. S. Jhang, C. N. Cheng, S. C. Cheng and J. Shiea, Analyst, 2010, 135, 759–766 RSC.
- S.-C. Cheng, Y.-S. Lin, M.-Z. Huang and J. Shiea, Rapid Commun. Mass Spectrom., 2010, 24, 203–208 CrossRef CAS PubMed.
- G. A. Harris, S. Graf, R. Knochenmuss and F. M. Fernandez, Analyst, 2012, 137, 3039–3044 RSC.
- S. Y. Lin, M. Z. Huang, H. C. Chang and J. Shiea, Anal. Chem., 2007, 79, 8789–8795 CrossRef CAS PubMed.
- C.-Y. Cheng, C.-H. Yuan, S.-C. Cheng, M.-Z. Huang, H.-C. Chang, T.-L. Cheng, C.-S. Yeh and J. Shiea, Anal. Chem., 2008, 80, 7699–7705 CrossRef CAS PubMed.
- M.-Z. Huang, H.-J. Hsu, C.-I. Wu, S.-Y. Lin, Y.-L. Ma, T.-L. Cheng and J. Shiea, Rapid Commun. Mass Spectrom., 2007, 21, 1767–1775 CrossRef CAS PubMed.
- J. Shiea, M.-Z. Huang, H.-J. Hsu, C.-Y. Lee, C.-H. Yuan, I. Beech and J. Sunner, Rapid Commun. Mass Spectrom., 2005, 19, 3701–3704 CrossRef CAS PubMed.
- P. Nemes and A. Vertes, Anal. Chem., 2007, 79, 8098–8106 CrossRef CAS PubMed.
- A. S. Galhena, G. A. Harris, L. Nyadong, K. K. Murray and F. M. Fernandez, Anal. Chem., 2010, 82, 2178–2181 CrossRef CAS PubMed.
- L. Nyadong, A. S. Galhena and F. M. Fernandez, Anal. Chem., 2009, 81, 7788–7794 CrossRef CAS PubMed.
- G. A. Harris, L. Nyadong and F. M. Fernandez, Analyst, 2008, 133, 1297–1301 RSC.
- Y. Song and R. G. Cooks, Rapid Commun. Mass Spectrom., 2006, 20, 3130–3138 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |