
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGene module based regulator inference identifying miR-139 as a tumor suppressor in colorectal cancer†
Jin
Gu‡
*a,
Yang
Chen‡
a,
Huiya
Huang
a,
Lingyun
Yin
a,
Zhen
Xie
a and
Michael Q.
Zhang
*ab
aMOE Key Laboratory of Bioinformatics, TNLIST Bioinformatics Division & Center for Synthetic and Systems Biology, Department of Automation, Tsinghua University, Beijing 100084, China. E-mail: jgu@tsinghua.edu.cn; michaelzhang@tsinghua.edu.cn
bDepartment of Molecular and Cell Biology, Center for Systems Biology, University of Texas at Dallas, Richardson, TX 75080, USA
First published on 30th September 2014
Abstract
Colorectal cancer is one of the most commonly diagnosed cancer types worldwide. Identification of the key regulators of the altered biological networks is crucial for understanding the complex molecular mechanisms of colorectal cancer. We proposed a gene module based approach to infer key miRNAs regulating the major gene network alterations in cancer tissues. By integrating gene differential expression and co-expression information with a protein–protein interaction network, the differential gene expression modules, which captured the major gene network changes, were identified for colorectal cancer. Then, several key miRNAs, which extensively regulate the gene modules, were inferred by analyzing their target gene enrichment in the modules. Among the inferred candidates, three miRNAs, miR-101, miR-124 and miR-139, are frequently down-regulated in colorectal cancers. The following computational and experimental analyses demonstrate that miR-139 can inhibit cell proliferation and cell cycle G1/S transition. A known oncogene ETS1, a key transcription factor in the gene module, was experimentally verified as a novel target of miR-139. miR-139 was found to be significantly down-regulated in early pathological cancer stages and its expression remained at very low levels in advanced stages. These results indicate that miR-139, inferred by the gene module based approach, should be a key tumor suppressor in early cancer development.
Introduction
Colorectal cancer is a kind of severe bowel disease and it is the third most commonly diagnosed cancer around the world. The molecular networks are usually significantly altered in cancer cells. To understand the complex regulatory mechanism of colorectal cancer initiation and progression, it is crucial to identify the key regulators of the altered molecular networks. MicroRNAs (miRNAs), a class of ∼22 nt endogenous small regulatory RNAs, are significantly differentially expressed between colorectal cancerous and adjacent normal tissues.1 But only a few of them are linked with the major alterations of the molecular networks in colorectal cancer. To infer the key miRNAs regulating the altered molecular networks in cancer, one simple way is to analyze the enrichment of their targets in the differentially expressed genes between cancerous and normal tissues.2 But due to the complexity of the cancer transcriptome, this kind of method frequently failed to find candidates due to low statistical significances. Multiple genes usually work cooperatively as functional gene modules.3 Differential network analysis can better identify the altered molecular networks by integrating gene expression data with network data (such as a protein–protein interaction network and a literature co-citation network).4,5The differential gene expression modules (DGEMs, or called active gene sub-networks), a class of “differential networks” consisting of a set of densely connected differentially expressed genes and their neighbors, can be treated as the major network alterations in cancer.4,6 Then, the key miRNAs can be inferred by analyzing their target enrichment in the DGEMs. Based on this hypothesis, we developed a gene module based master regulator inference (ModMRI) approach to identify the key miRNAs in colorectal cancer: firstly, the DGEMs were identified by the integrative analysis of multiple gene expression datasets and protein–protein interaction networks using ClustEx package.7 Then, a network-based permutation test was used to infer the key miRNAs whose target genes are significantly overlapped with the DGEMs. Among the inferred candidates, miR-101, miR-124 and miR-139 are frequently down-regulated in colorectal cancer.1,8 The following computational and experimental analyses suggest that miR-139 should act as an important tumor suppressor in early cancer pathologic stages.
Methods
Colorectal cancer gene expression data and gene network data
A combined gene expression dataset of colorectal cancer was constructed based on four microarray data series from the NCBI GEO database with at least 15 clinical cancer/normal samples and using Affymetrix Human Genome U133 Plus 2.0 Array (GSE20916, GSE21510, GSE22598, GSE23878; 90 normal and 107 cancer samples in total). The .CEL files of the four data series were processed and normalized using RMA package independently.9 Probe signals were mapped to gene expression values according to the latest probe annotations. Then, the raw gene expression values were transformed to ranks. To avoid the noises of the lowly expressed genes, only the genes ranked as top 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (∼50% of the genes on the microarray) in at least 30% samples were left for the following analysis.
000 (∼50% of the genes on the microarray) in at least 30% samples were left for the following analysis.
We extracted all binary interactions from HPRD (release 9)10 and BioGRID (version 3.1.86).11 To reduce the high false positives in PPI data, we only used the interactions annotated in both databases. By filtering the above “expressed” genes in microarray datasets, we got the background gene network with 4181 genes and 10261 edges. The edges were weighted by Spearman's co-expression levels according to the combined gene expression dataset.
A gene was regarded as a target of a miRNA family, if the gene has at least one conserved target site predicted by TargetScan (v6.0).12 The 3′-UTR lengths of genes were calculated from the 3′-UTR multiple alignment file from TargetScan resources. If a gene has multiple transcripts, the length of the longest 3′-UTR was used.
Gene module based key miRNA inference
The miRNAs, whose target genes were significantly enriched in the differential gene expression modules (DGEMs), were identified as key regulators. Firstly, differentially expressed genes were identified from the combined gene expression dataset. Secondly, the DGEMs were identified by finding the sub-networks consisting of a set of closely connected differentially expressed genes and their neighbors. Then, the key miRNAs were inferred by analyzing their target gene enrichments in the DGEMs. Finally, bootstrapping was implemented to get robust inferences. The flowchart of this gene module based master regulator inference (ModMRI) method can be found in Fig. 1.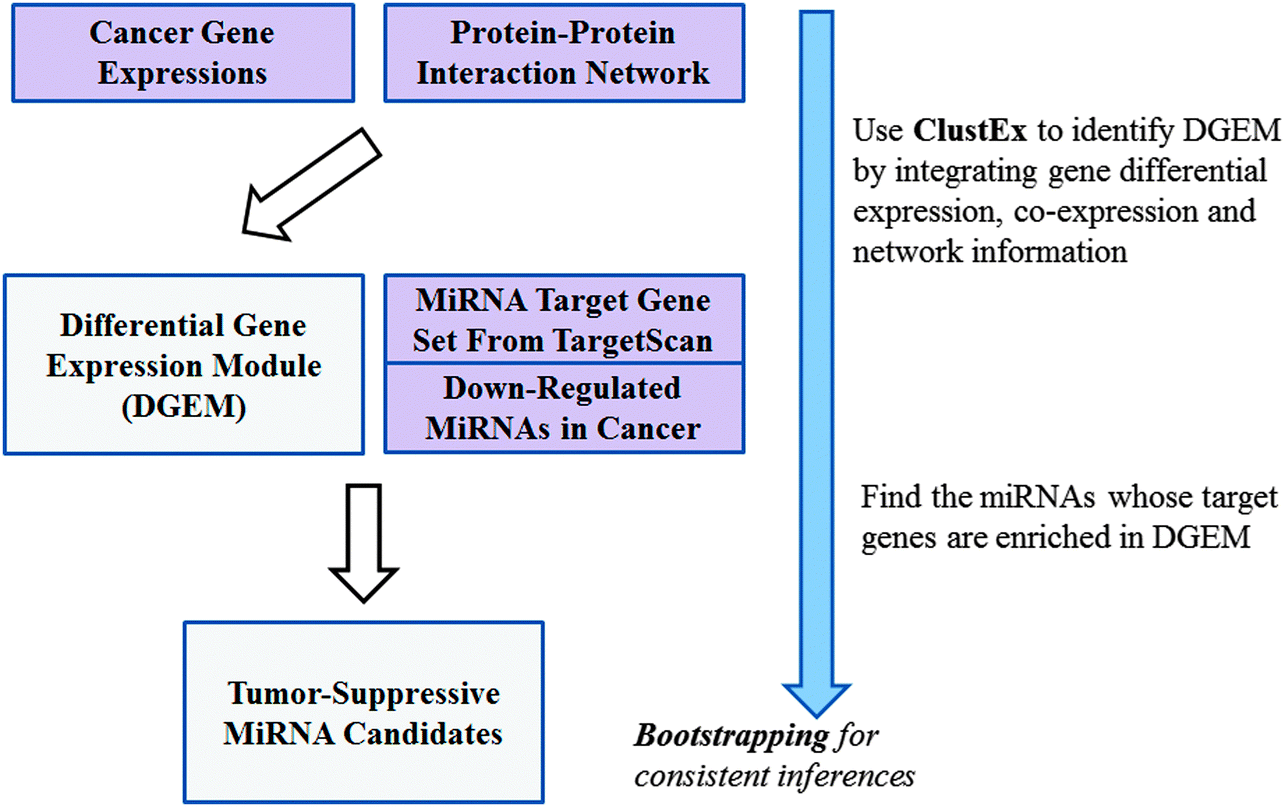 | ||
| Fig. 1 The flowchart of the module-based master regulator inference (ModMRI). | ||
000 randomized DGEMs. The numbers of the miRNA target genes in the randomized DGEMs were counted to estimate the background distribution of the number of the overlapped target genes. The p-value was calculated by comparing the original number with the corresponding background distribution. The p-values were multiple-test adjusted as q-values using fdrtool.13
Experimental validation of miR-139 cellular functions and target genes
A series of experiments were conducted to validate the cellular functions and target genes of miR-139 in colorectal cancer cells.:500, Abcam), and blotted with Goat anti Rabbit IgG (1:3000, Santa Cruz). Gene PCNA was used as loading control.
Results
The differential gene expression module in colorectal cancer
A large number of genes were significantly differentially expressed between colorectal cancer and noncancerous tissues. The gene module based master regulator inference (ModMRI) is an integrative gene network analysis for identifying the key regulators mediating these significant alterations of molecular networks. At first, ClustEx program was used to identify the differential gene expression modules (DGEMs) associated with colorectal cancer. The identified biggest or the principal DGEM has 822 genes including 305 differentially expressed seed genes. The module genes are significantly enriched in KEGG annotated “pathways in cancer” (q-value 1.4 × 10−17), “cell cycle” (1.4 × 10−14), “MAKP signaling pathway” (6.7 × 10−14) and “colorectal cancer” (2.8 × 10−10), which suggests that the identified DGEM is highly associated with colorectal cancer. The details of the module and the enriched cellular processes can be found in Supplementary File 1 (ESI†).The key miRNAs extensively regulating the differential gene expression modules
Then, several key miRNAs, including several known oncogenic and tumor-suppressive miRNA families in colorectal cancer, miR-17,14 miR-93,15 miR-10116 and miR-135,17 were identified by analyzing their target gene enrichments in the biggest DGEM (Table 1). Three of them, miR-101, miR-124 and miR-139, were found frequently down-regulated in colorectal cancer,1,8 which suggest that these miRNAs may act as tumor suppressors by extensively regulating the biggest DGEM. We also analyzed the miRNA target enrichments in the second biggest DGEM (609 genes), but no significant candidate was found.| miRFam | p-value | #Target | BS (%) | Expression |
|---|---|---|---|---|
| “p-value”: the p-value and the corresponding q-value. “#Target”: the number of targets overlapped with the DGEM and the total number of predicted targets; “BS”: the percentage of significant inferences in bootstrapping experiments; “Expression”: the miRNA expression patterns in ref. 1 and PhenomiR database. | ||||
| miR-135 | 0.0005 (0.049) | 57/198 | 50 | Up |
| miR-874 | 0.0021 (0.104) | 29/77 | 58 | — |
| miR-139 | 0.0042 (0.131) | 35/108 | 84 | Down |
| miR-17 | 0.0142 (0.200) | 91/358 | 50 | Up |
| miR-93 | 0.0160 (0.206) | 64/256 | 86 | Up |
| miR-124 | 0.0175 (0.211) | 116/500 | 52 | Down |
| miR-101 | 0.0365 (0.241) | 65/262 | 50 | Down |
miR-139 inhibited cancer cell proliferation and cell cycle progression
Among the three inferred tumor suppressors, miR-139 has the minimal p-value and highest bootstrapping re-sampling stability (Table 1). We chose to further study the cellular functions and target genes of miR-139 by a series of computational and experimental analyses. The 35 miR-139 target genes in the DGEM are significantly enriched in “regulation of cell proliferation” (11 out of the 35 target genes are annotated with the GO term, q-value < 0.05) (Supplementary File 2, ESI†). CCK-8 cell proliferation assays show that miR-139 significantly inhibited cancer cell proliferation (q-value < 0.01). The inhibition rates are comparable to the well-studied tumor suppressor miR-101 (Fig. 2A & B). FACS analysis indicates that miR-139 may inhibit cell proliferation by blocking cell cycle G1/S phase transition (Fig. 2C).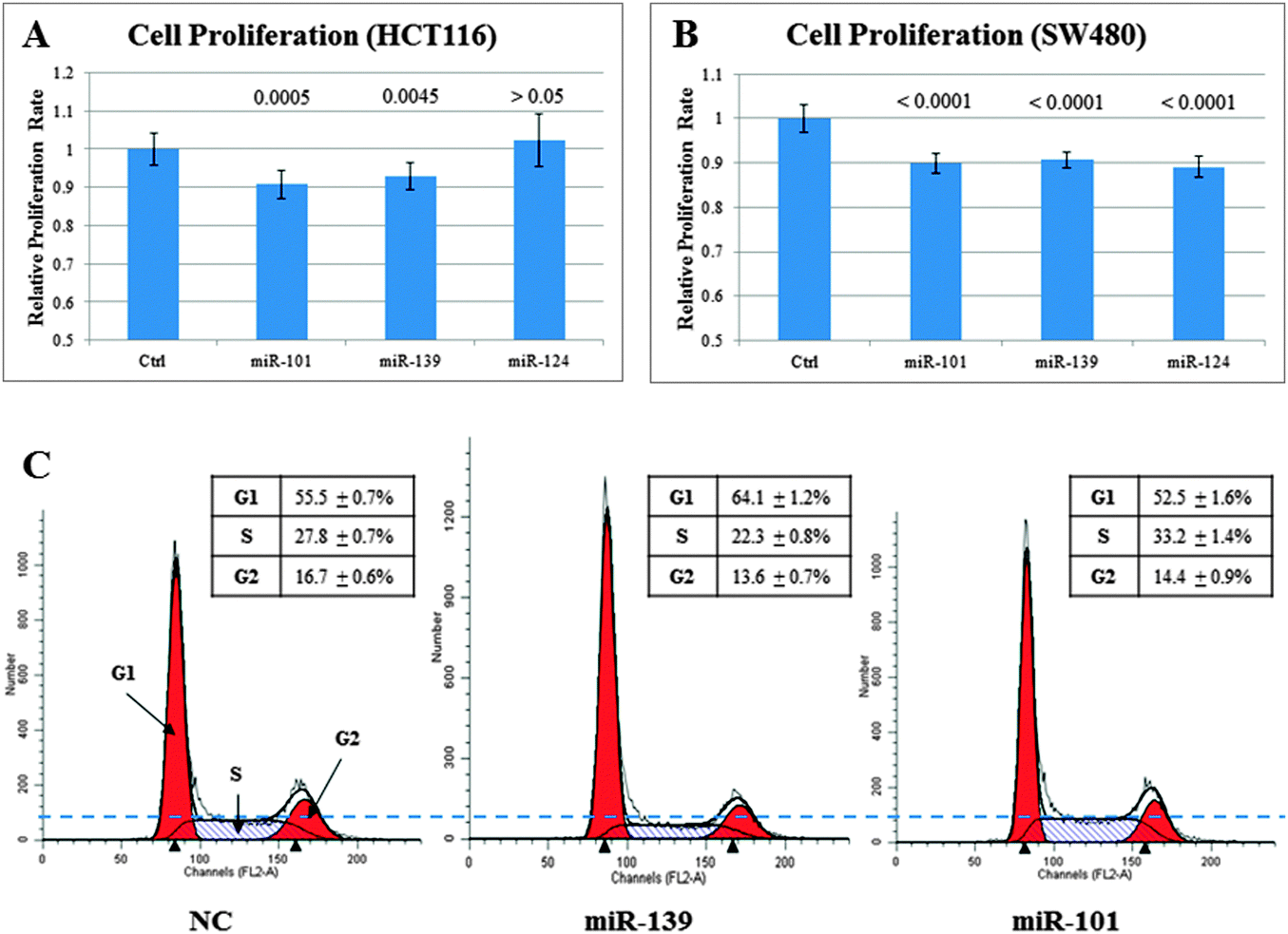 | ||
| Fig. 2 miR-139 can inhibit cancer cell proliferation and cell cycle G1/S phase transition. Colorectal cancer cell lines HCT116/SW480 were transfected with miRNA mimics. (A) The CCK-8 cell proliferation assays in HCT116. (B) The CCK-8 cell proliferation assays in SW480. The p-values of the t-test were adjusted by Bonferroni correction. (C) The FACS analysis of the cell cycle in HCT116. | ||
miR-139 directly targeted oncogene ETS1
miR-139 has 11 predicted target genes in the DGEM which are annotated as “regulation of cell proliferation”. Four of them, MNT, NOTCH1, ETS1 and JUN are with high TargetScan context+ scores and aggregate PCT. ETS1 and JUN are also involved in the KEGG “cancer signaling pathway”. The predicted conserved binding sites in the 3′-UTRs of the four genes were synthesized into luciferase reporters. It is observed that miR-139 mimics can significantly inhibit the luciferase activities of the reporters with the conserved ETS1 binding site, while the miR-139 cannot inhibit the reporters with a mutated ETS1 binding site (Fig. 3A & B, Fig. S1, ESI†). Western blot experiments show that miR-139 mimics can suppress ETS1 protein activity in a dose-dependent manner (Fig. 3C). These results indicate that miR-139 can directly suppress ETS1 activity via the conserved binding site in its 3′-UTR. ETS1 is known as an oncogenic transcription factor, which can promote cell cycle G1/S transition.18,19 It is suggested that miR-139 may inhibit cell cycle progression by subsequently suppressing the ETS1 mediated transcriptional program. | ||
| Fig. 3 ETS1 is a direct target of miR-139. (A) The conserved miR-139 binding site in ETS1 3′-UTR. (B) Dual luciferase assay experiments for the conserved miR-139 binding sites in target genes, 3′-UTRs. The p-values of the t-test were adjusted by Bonferroni correction. (C) Western blot for ETS1 protein activity with increasing dose of miR-139 mimics. PNAC is used as loading control. | ||
miR-139 expression was down-regulated in cancer tissues
We examined miR-139 expressions in two clinical studies (TCGA COAD dataset with 222 cancer and 8 non-cancerous samples;20 GSE28364 with 40 cancer and 40 non-cancerous samples21). The data show that miR-139 is significantly down-regulated in early cancer pathologic stages compared with adjacent non-cancerous tissues, and it remains at a very low expression level in advanced pathologic stages (Fig. 4). Reid et al. also reported that the miR-139 region has genomic loss in cancer samples.21 These results suggest that the loss of miR-139 is an important indicator of colorectal cancer. Similar expression patterns can be observed in several other solid tumors (Fig. S2, ESI†). | ||
| Fig. 4 miR-139 expressions in colorectal cancer tissues in different pathologic stages and adjacent normal tissues. (A) miR-139 expressions in the TCGA COAD dataset (HiSeq sequencing data). (B) miR-139 expressions in GSE28364 (qPCR data). | ||
Discussion
miR-139 is a broadly conserved miRNA in vertebrates. The module-based master regulator inference (ModMRI) predicted that miR-139 is a key regulator by targeting tens of genes in the DGEM of colorectal cancer. The following expression and functional analyses further suggested that miR-139 may act as a tumor suppressor by regulating cancer cell proliferation. Cellular functional assays validated that miR-139 can inhibit cancer cell proliferation and cell cycle G1/S transition. And ETS1, an oncogenic transcription factor promoting cell cycle progression, was verified as a direct target of miR-139. ETS1 is not differentially expressed in any of the four colorectal gene expression datasets, but it can be found by module-based analysis after incorporating the gene network information. Two large-scale expression datasets from colorectal cancer patients show that miR-139 was significantly down-regulated in early pathologic stages of colorectal cancer and remained at a very low expression level in advanced stages. A few other studies also reported that miR-139 can inhibit colorectal cancer cell proliferation.22,23 These results indicate that miR-139 should be a key tumor suppressor in early cancer development.Modularity is an important property of biological networks.4,24 Tens of genes work cooperatively as a functional module for different cellular processes. Inference of any single gene frequently fails to disturb the module activity due to complex network compensations and feedbacks. This study indicates that the module-based inference using miRNAs may be an alternative approach to disturb cancer cellular states.
Acknowledgements
Funding: this work is supported by National Basic Research Program of China [2012CB316503], National Natural Science Foundation of China [61005040, 61370035, 61105003 and 31301044] and Tsinghua National Laboratory for Information Science and Technology Cross-discipline Foundation.References
- Y. Ma, P. Zhang and J. Yang, et al. , Int. J. Cancer, 2012, 130, 2077–2087 CrossRef CAS PubMed.
- J. H. Hung, T. H. Yang and Z. Hu, et al. , Briefings Bioinf., 2012, 13, 281–291 CrossRef PubMed.
- L. H. Hartwell, J. J. Hopfield and S. Leibler, et al. , Nature, 1999, 402, C47–C52 CrossRef CAS PubMed.
- K. Mitra, A. R. Carvunis and S. K. Ramesh, et al. , Nat. Rev. Genet., 2013, 14, 719–732 CrossRef CAS PubMed.
- D. Pe'er and N. Hacohen, Cell, 2011, 144, 864–873 CrossRef PubMed.
- Z. Wu, X. Zhao and L. Chen, Mol. Cells, 2009, 27, 271–277 CrossRef CAS PubMed.
- J. Gu, Y. Chen and S. Li, et al. , BMC Syst. Biol., 2010, 4, 47 CrossRef PubMed.
- A. Ruepp, A. Kowarsch and D. Schmidl, et al. , Genome Biol., 2010, 11, R6 CrossRef PubMed.
- R. A. Irizarry, B. Hobbs and F. Collin, et al. , Biostatistics, 2003, 4, 249–264 CrossRef PubMed.
- T. S. Keshava Prasad, R. Goel and K. Kandasamy, et al. , Nucleic Acids Res., 2009, 37, D767–D772 CrossRef CAS PubMed.
- A. Chatr-Aryamontri, B. J. Breitkreutz and S. Heinicke, et al. , Nucleic Acids Res., 2013, 41, D816–D823 CrossRef CAS PubMed.
- A. Grimson, K. K. Farh and W. K. Johnston, et al. , Mol. Cell, 2007, 27, 91–105 CrossRef CAS PubMed.
- K. Strimmer, Bioinformatics, 2008, 24, 1461–1462 CrossRef CAS PubMed.
- Y. Ma, P. Zhang and F. Wang, et al. , Nat. Commun., 2012, 3, 1291 CrossRef PubMed.
- I. P. Yang, H. L. Tsai and M. F. Hou, et al. , Carcinogenesis, 2012, 33, 1522–1530 CrossRef CAS PubMed.
- A. Strillacci, M. C. Valerii and P. Sansone, et al. , J. Pathol., 2013, 229, 379–389 CrossRef CAS PubMed.
- R. Nagel, C. le Sage and B. Diosdado, et al. , Cancer Res., 2008, 68, 5795–5802 CrossRef CAS PubMed.
- A. K. Singh, M. Swarnalatha and V. Kumar, J. Biol. Chem., 2011, 286, 21961–21970 CrossRef CAS PubMed.
- Y. Zhang, L. X. Yan and Q. N. Wu, et al. , Cancer Res., 2011, 71, 3552–3562 CrossRef CAS PubMed.
- N. Cancer Genome Atlas, Nature, 2012, 487, 330–337 CrossRef PubMed.
- J. F. Reid, V. Sokolova and E. Zoni, et al. , Mol. Cancer Res., 2012, 10, 504–515 CrossRef CAS PubMed.
- H. Guo, X. Hu and S. Ge, et al. , Int. J. Biochem. Cell Biol., 2012, 44, 1465–1472 CrossRef CAS PubMed.
- T. Schepeler, A. Holm and P. Halvey, et al. , Oncogene, 2012, 31, 2750–2760 CrossRef CAS PubMed.
- A. L. Barabasi and Z. N. Oltvai, Nat. Rev. Genet., 2004, 5, 101–113 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00329b |
| ‡ The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint first authors. |
| This journal is © The Royal Society of Chemistry 2014 |