The pan-cancer analysis of gene expression patterns in the context of inflammation†
Xuexin
Yu‡
a,
Baofeng
Lian‡
abc,
Lihong
Wang
d,
Yan
Zhang
a,
Enyu
Dai
a,
Fanlin
Meng
a,
Dianming
Liu
a,
Shuyuan
Wang
a,
Xinyi
Liu
a,
Jing
Wang
a,
Xia
Li
*a and
Wei
Jiang
*a
aCollege of Bioinformatics Science and Technology, Harbin Medical University, China. E-mail: jiangwei@hrbmu.edu.cn; lixia@hrbmu.edu.cn
bSchool of Life Science and Biotechnology, Shanghai Jiao Tong University, China
cShanghai Center for Bioinformation Technology (SCBIT), China
dInstitute of Cancer Prevention and Treatment, Harbin Medical University, China
First published on 24th June 2014
Abstract
Although several studies have investigated the essential roles of inflammation in tumor progression, not many have systematically analyzed gene expression patterns across diverse cancers in the context of inflammation. In this study, in order to better understand the inflammatory scenario, we initially constructed the inflammatory timeline (IT) based on two gene expression profiles during inflammatory progression (inflammatory bowel disease and Helicobacter pylori infection). Then, we separately identified the differentially expressed genes (DEGs) from 25 cancer-related microarray data. By comparing the distributions of DEGs in the IT, we identified three novel pan-cancer gene expression patterns. In the first pattern, the up-regulated genes in cancers were over-expressed in the early phase of inflammation, while the down-regulated genes were over-expressed in the late phase of inflammation. The second pattern was the opposite of the first one. The third pattern appeared to be transitional between the first and second patterns. We found that some cancers with different tissue origins have similar gene expression patterns. Finally, we identified two sets of tissue-independent inflammatory signatures that were over-expressed in early and late phases of inflammation, respectively. The dominant biological processes of early inflammatory signatures were cell proliferation, DNA replication, and DNA repair, whereas the late inflammatory signatures were reflective of innate immune response, neutrophil migration, and antigen processing. These inflammatory signatures may be useful to predict gene expression patterns in human cancers. Therefore, the pan-cancer analysis of gene expression patterns in the context of inflammation provides a novel insight into cancers and an unprecedented opportunity to develop new therapies.
Introduction
Although inflammation has been linked to cancers as early as 150 years ago, ever since Virchow suggested that cancer tended to originate from the sites of chronic inflammation,1 the progress of research on the relationship between inflammation and cancer has been rather gradual. In 2000, Hanahan and Weinberg proposed six hallmarks of cancer as a logical framework for explaining tumor complexity, and they also suggested that inflammatory responses enhanced tumorigenesis and progression by assisting the initial neoplasms to acquire hallmark capabilities.2 In 2009, Mantovani et al. surmised inflammation as the seventh hallmark of cancer, and they found that the microenvironment of most tumor tissues contained inflammatory components, even if the neoplastic tissues were not causally related to the inflammatory process.3Importantly, recent clinical studies have demonstrated the association between inflammation and cancer. For example, individuals with inflammatory bowel disease were found to have a 10-fold higher risk of developing colorectal cancer than those without. Through anti-inflammatory therapy,4,5 the incidence of colon cancer reduced greatly.6,7 Furthermore, inflammation caused by bacterial and viral infection also increases cancer risk. In the gastrointestinal tract, Helicobacter pylori infection is a leading cause of adenocarcinoma and mucosa-associated lymphoid tissue.8,9 In the hepatic system, carriers of hepatitis B and hepatitis C virus (HBV and HCV, respectively) were predisposed to hepatocellular carcinoma (HCC). Moreover, HCV-positive men have a 20-fold higher risk of developing HCC than HCV-negative subjects.10–12
Several transcription factors and inflammatory cytokines play important roles in cancer-related inflammation, such as nuclear factor-kappa B (NF-κB), tumor necrosis factor (TNF-α), and interleukin-6 (IL-6). However, current research has mainly focused on explaining the mechanism of only one of these types of inflammation-mediated carcinogenesis. Thus far, there has been no systematic analysis between various inflammations and cancers. In recent times, The Cancer Genome Atlas (TCGA) Pan-Cancer analysis project has analyzed the shared molecular features and relevant functional roles across cancers of disparate organs, which has helped clinicians to extrapolate therapy from one tumor type to others with a similar genomic profile.13,14 The Pan-Cancer project revealed that different tumors have several shared features such as somatic copy number alterations, mutations, and epigenomic alterations.14–16 Interestingly, Isaac et al. analyzed the links between ten distinct developmental processes and a series of human cancers. They classified all cancers de novo based on the gene expression signatures in the context of various developmental processes and found similar gene expression patterns in different tumor types, which depicted the tumor landscape in a very novel and comprehensive manner.17 With this background, we hope to describe the overall potential relationship between inflammations and tumors based on gene expression.
In the present study, in order to summarize the inflammatory landscape, we used principal component analysis (PCA) to construct inflammatory timelines (ITs) to evaluate the progression in inflammatory bowel disease (IBD) and H. pylori infection (Hp). As the species of IBD time course data was Mus musculus, we defined the human orthologs in the mouse through NCBI and got the equivalent gene symbol for each human/mouse ortholog pair. In addition, we identified three gene expression patterns with tissue-independent features and consequently obtained three corresponding cancer groups after generalizing the distribution of differentially expressed genes (DEGs) in the IT for all cancers. By comparing the functions of DEGs for each cancer group with that of two inflammatory signatures, we found that the functions of up-regulated DEGs for the first cancer group were similar to the function of the early-phase inflammatory signature, while the functions of up-regulated DEGs in the second group were similar to the function of the late-phase inflammatory signature. Thus, our study provides a novel insight into cancers in the context of inflammatory progression.
Results
Pan-cancer gene expression patterns
We firstly identified the DEGs of all cancer data sets using SAM. The number distribution of DEGs is shown in Fig. 1, and detailed information on DEGs is provided in Table S1 (ESI†). Next, in order to summarize the inflammatory landscape, we used PCA to construct ITs (refer Materials and methods). By projecting these DEGs on ITs, we drew frequency plots and calculated the probability distributions of 25 cancer data sets in two ITs (Fig. S1 and S2, ESI†). Finally, based on the probability distribution curves of up- and down-regulated genes, we used two regression lines (upE and upL) describing the former curve and the other two regression lines (downE and downL) for the latter curve (refer Materials and methods); we clustered all cancers based on the constructed 8-dimensional vectors (4 slopes of regression lines × 2ITs). Subsequently, three groups of cancer datasets emerged, which corresponded to the three major gene expression patterns. The cancers in the same group had similar gene expression patterns (Fig. 2A).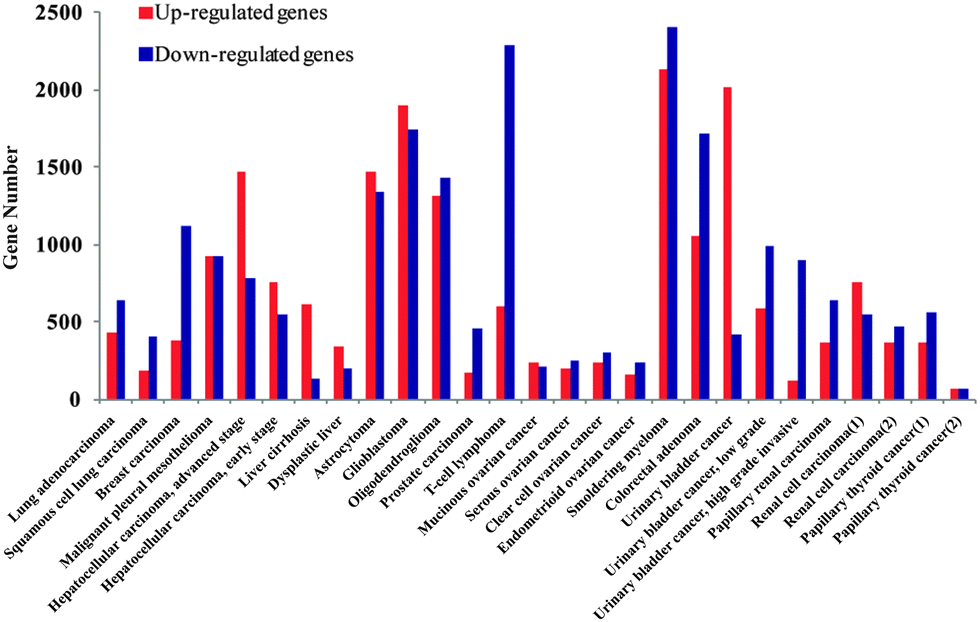 | ||
| Fig. 1 The number distribution of DEGs for all cancer data sets. The red bar shows the number of up-regulated genes and the blue bar describes the number of down-regulated genes. | ||
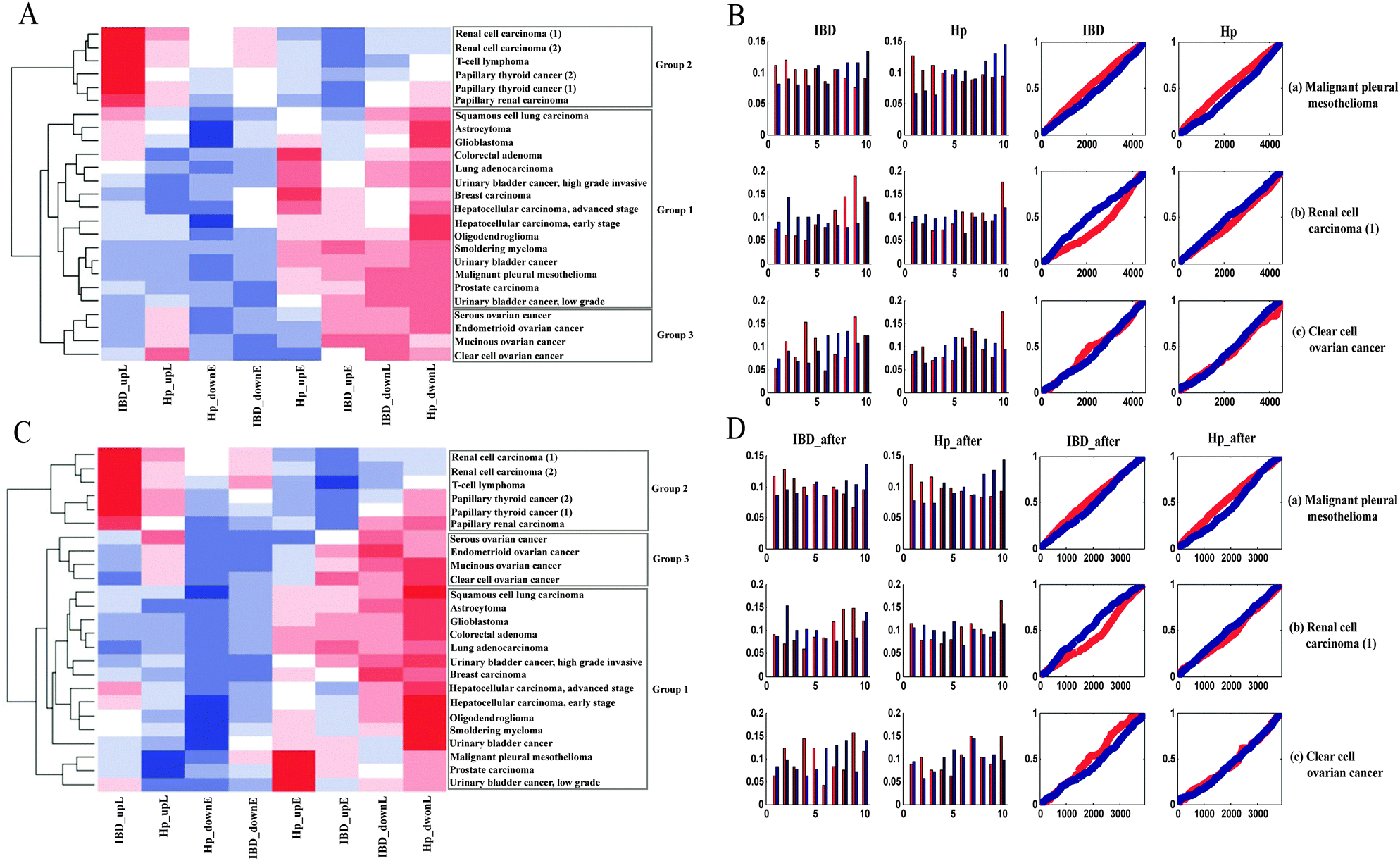 | ||
| Fig. 2 (A) Clustering result of probability distribution slope values. The heatmap represents the three cancer groups and the color of each cell describes the slope value of the regression line in a specific inflammatory context for each cancer. (B) Probability distributions and frequency plots for 3 representative cases of most tumors: (a) malignant pleural mesothelioma(b) renal cell carcinoma; (c) clear cell ovarian cancer. (C) Clustering result after inflammation-related gene subtraction. Abbreviations and colors are the same as in Fig. 2A. (D) Comparison of frequency plots after inflammation-related gene subtraction: (a) malignant pleural mesotheliomas(b) renal cell carcinoma; (c) clear cell ovarian cancer. | ||
Group 1 contained tumors with an “early” gene expression pattern, which meant up-regulated genes were preferentially expressed at the early IT, while down-regulated genes were activated at the late IT. For example, the frequency plot in Fig. 2B shows an early peak for up-regulated genes of malignant pleural mesothelioma, followed by a decline towards the late phase of the IT, which meant that these up-regulated genes were preferentially expressed in the early intestinal inflammation process. When compared to up-regulated genes, down-regulated genes presented an inverse pattern, which meant that down-regulated genes were preferentially activated in the late inflammatory stage. This observation was confirmed by the probability distribution. The slope of upE was larger than the slope of upL, while the slope of downL was larger than that of downE. A similar expression pattern was evident in Hp infection. This group encompassed 60% of all cancer datasets and contained tumors of varied tissues, including smoldering myeloma, oligodendroglioma, breast cancer, and hepatocellular carcinoma. Group 2 contained three independent data sets of renal carcinoma, two independent data sets of papillary thyroid and a T-cell lymphoma. The gene expression pattern of Group 2 was reverse of the Group 1 pattern, namely, “late” gene expression pattern. For instance, the up- and down-regulated genes of renal cell carcinoma were preferentially active in late and early ITs, respectively (Fig. 2B). In addition to the above two expression patterns, the tumors in Group 3, which included mucinous ovarian cancer, clear cell ovarian cancer, endometrioid ovarian cancer, and serous ovarian cancer, displayed an ambiguous relationship between cancer and inflammation. In the case of clear cell ovarian cancer (Fig. 2B), the ambiguity of the gene expression pattern was not satisfactorily explained by the inflammatory gradient, and hence, we surmised that it may be a transition pattern between Group 1 and 2 cancers.
Apparently, the identified gene expression patterns were tissue-independent, because each of them included many cancers with different tissue origin. The “early” gene expression pattern was a widespread feature across most of the cancers. In addition, by projecting the DEGs onto different ITs, we got similar frequency plots and probability distributions, which indicated that the pan-cancer gene expression patterns were stable in different inflammatory backgrounds.
Inflammation-related genes have no dominant influence on the gene expression patterns
Since inflammation-related genes such as pro-inflammatory mediators and pro-inflammatory transcription factors may play an essential role in tumor cell proliferation, transformation, metastasis, and survival, we further determined whether the inflammation-related genes dominate the identified pan-cancer gene expression patterns. Firstly, we obtained the inflammation-related genes from a previous study18 and eliminated these genes from the DEGs. Next, by calculating the frequencies of the inflammation-related genes in the 10 IT segments, we found that these frequencies fluctuated slightly among these different sections (Fig. S3, ESI†). Fig. 2D illustrates the gene expression patterns after the subtraction of inflammation-related genes for the tumors shown in Fig. 2B. Finally, in order to demonstrate the stability of the gene expression patterns for these cancer datasets, all of the cancers were re-clustered using their new probability distribution slope values after subtraction of the inflammation-related genes (Fig. 2C). After comparing Fig. 2A and C, we found that the two clustering results were approximately equal. Thus, we concluded that inflammation-related genes are not causative in generating the pan-cancer gene expression patterns.Functional analysis of the gene expression patterns
We detected two inflammatory signatures and defined them as early (eIN450) and late inflammatory signatures (lIN450). The eIN450 and lIN450 represented the 450 genes (10% of all genes in the IT) that were consistently over-expressed in the early and late phase of inflammation across two inflammatory time courses (the first and last 450 genes of the integrated IT). Furthermore, using DAVID19 with significance level (p-value) <0.05, we compared the biological functions of three gene expression patterns with two inflammatory signatures. The up-regulated genes in Group 1 were enriched in similar biological processes such as cell cycle, cell division, and nuclear division in eIN450, while the functions enriched in the up-regulated genes in Group 2 were similar to those of lIN450, which included response to wounding, cell adhesion, inflammatory response, and defense response (Table 1). Although we treated Group 3 as a transition class, the main processes were similar to those of Group 2, including the response to endogenous stimulus and cell–cell adhesion.| BP | BP | ||
|---|---|---|---|
| Note: the significant BP terms for eIN450, lIN450 and the up- as well as down-regulated genes of group 1, 2 and 3 cancer data sets. For example, cell division is enriched in the up-regulated genes of 14 out of 15 data sets belonging to group 1. | |||
| eIN450 | Cell cycle | lIN450 | Response to wounding |
| Cell division | Inflammatory response | ||
| Cell proliferation | Wound healing | ||
| Nuclear division | Response to endogenous stimulus | ||
| DNA repair | Response to hormone stimulus | ||
| Chromatin modification | Cell adhesion | ||
| Cell migration | |||
| Group 1(15) | |||
| Up | Cell cycle (15) | Down | Cell migration (15) |
| Nuclear division (15) | Response to hormone stimulus (15) | ||
| Cell division (14) | Response to endogenous stimulus (15) | ||
| DNA replication (14) | Cell adhesion (14) | ||
| Group 2(6) | |||
| Up | Response to wounding (6) | Down | Regulation of cell cycle (2) |
| Cell adhesion (6) | Positive regulation of transcription, DNA-dependent (2) | ||
| Inflammatory response (5) | Cell cycle (1) | ||
| Defense response (5) | |||
| Group 3(4) | |||
| Up | Response to endogenous stimulus (4) | Down | Positive regulation of transcription, DNA-dependent (2) |
| Cell–cell adhesion (4) | Positive regulation of gene expression (2) | ||
| Response to wounding (3) | Positive regulation of transcription, DNA-dependent (2) | ||
| Positive regulation of cell communication (2) | |||
In order to explore the process of inflammation promoting cancer, we found some key genes from the inflammatory signatures, which should have both high frequency of occurrence in one cancer group and be located at the left end or the right end of the IT. Detailed information on frequency of occurrence and rank for inflammatory signature genes is provided in Table S2 (ESI†). For early inflammatory signatures, the key genes have high frequency of occurrence and low rank in ITs, and the functions of these key genes are correlated with DNA repair (MUDT1), DNA replication (TYMS, HMGB3), apoptotic death (BCL2), stem cell proliferation (H2AFX), and nuclear matrix gene (GENPF). Previous studies have shown that inflammation can enhance the tumor initiation and progression by producing growth factors and cytokines which could confer a stem cell-like ability upon tumor progenitors or stimulate stem cell expansion, thereby enlarging the cell pool.20,21 The up-regulated genes of Group 1 were preferentially expressed in the early phase of inflammation; most tumors within this group were much more aggressive than other group cases, such as glioblastoma, squamous cell lung carcinoma, and malignant pleural mesothelioma. As for the late inflammatory signatures, the key genes have high frequency of occurrence and high rank in Its; these genes were involved in neutrophil migration (HCK), secretory process (RAC2), TNF-receptor superfamily protein (FAS), T cell development (LCP2), and antigen processing (TAP1). Some researchers explained that the tumor microenvironment contains innate immune cells and adaptive immune cells after inflammatory responses, including neutrophils and T lymphocytes, respectively.22 These disparate cells could either communicate with each other directly or promote cytokine and chemokine production and act in an autocrine and/or paracrine manner to control and shape tumor growth.23 The up-regulated genes of Group 2 displayed a late gene expression pattern. The cancer forms in this group were much more indolent than those belonging to Group 1, e.g., renal cancer and thyroid cancer grow slowly. Group 3 contained four subtypes of ovarian cancer, and their gene expression pattern was a transition of the other two patterns. Tumors like renal cancer and ovarian cancer have poor outcome because they often metastasize, despite their slow growth rate.
Finally, these results suggested that the unique relationship between inflammation and tumors is a common feature across different cancer data sets.
Discussion
The inflammatory microenvironment is a critical component of cancers, despite some of the cancers not having a direct causal link with inflammation. Inflammatory responses can affect every aspect of cancer development and progression: they may contribute to DNA replication, tumor cell survival, angiogenesis, metastasis, and subversion of adaptive immunity.24,25 In this study, we performed a comparison of gene expression between inflammation and cancers. In addition, through the disparate gene expression patterns, we have identified three groups of cancers that possessed distinctly specific patterns. The first group depicts the early gene expression pattern; this group contains 60% cases of all cancer datasets. The second group was more similar to the late phase of inflammation, and the frequency plots for these two groups represented mirror images of each other, and the DEGs of the two groups also showed distinguishable biological processes. Of these, a small group of cancers may act as a transition phenotype between Group 1- and 2-type patterns. Clearly, the context of inflammation provided a meaningful background for identifying differences in gene expression across a large variety of cancers.Interestingly, we detected three pan-cancer gene expression patterns and two inflammatory signatures. As we selected two inflammatory time courses and 25 cancer cases, the results from these data reflected tissue-independence with respect to the above-mentioned patterns and signatures. Furthermore, from a functional perspective, the biological process enrichment analysis of inflammatory signatures and each cancer group genes explains the relationship of three distinct cancer groups and inflammation.
Our results suggest that there is potential for a deeper understanding of cancer through an inflammation-based perspective. In the present study, we solely analyzed gene expression data; we could integrate other types of data as well. Increasing evidence describes the link between inflammation and cancer from the standpoint of genomic instability;26 for example, the inflammatory cells and mediators can destabilize the cancer cell genome by a variety of mechanisms either directly inducing DNA damage or affecting DNA repair systems and altering cell cycle checkpoints, thereby resulting in acceleration of somatic evolution, promotion of cell proliferation, and invasion and evasion of host defenses. Owing to the limitation of inflammatory time course microarray data, we cannot further prove the robustness of the distribution patterns of cancer DEGs in different inflammations. Therefore, if we integrated other high-throughput genomic data and more inflammatory time course data in future analyses, such as exon-seq data, RNA-seq data, copy number variations data, and DNA methylation data, we could illustrate more clearly the molecular mechanism of cancer progression in the context of inflammation.
Materials and methods
Microarray data
From the Gene Expression Omnibus,27 we obtained 25 gene expression profiles for cancers such as malignant pleural mesothelioma and clear cell ovarian cancer and their corresponding normal controls. In addition, we also downloaded two time course microarray data during inflammatory progression. All cancer microarray data were detected in the same platform Affymetrix HG-U133A. Two inflammatory time series data—GSE22307 for IBD and GSE3556 for Hp infection—had 4 and 5 time points, respectively. Detailed information on these data can be found in Table S3 (ESI†).We used the median normalization method to normalize all data using the BRB-ArrayTools.28 As the species of GSE22307 was Mus musculus, we used the HomoloGene database of NCBI to define orthologs between human and mouse genome. Consequently, the intersection of genes across different platforms of inflammatory time series data and cancer data includes 4548 unique genes.
Identification of DEGs
The significance analysis of microarrays (SAM) is a very popular method for identifying differentially expressed genes.29 Furthermore, SAM uses permutations to estimate the percentage of genes identified by chance, the false discovery rate (FDR). In this study, we used SAM to detect DEGs. The genes with fold change > 1.5 and FDR < 0.05 were considered as DEGs.Construction of the ITs by PCA
In order to explore the relationship between tumor and inflammation from the gene expression aspect, we should extract the intersection of the genes between all DEGs and genes in two inflammatory time course expression profiles. Consequently, there were 4548 genes of inflammation data for constructing ITs. We used PCA to construct IT for each inflammatory time course. An IT is a single ordered gene axis according to the period of gene over-expression. Here, we selected the first principal component, which was most significantly associated with the IT, to rank the 4548 genes in a descending order, and placed these genes on the x-axis as per the order; this gene axis is called IT. Thus, over-expressed genes in the early phase of inflammation were placed on the left side of the IT, over-expressed genes in the late phase of inflammation occupied the right side, and all other genes with no obvious expression pattern occupied the central portion.Finally, we got two ITs for two inflammatory time course data; in order to summarize the characteristics of two ITs and select some important genes from IT for subsequent analysis, we integrated them into one IT as follows: calculating the mean rank for each gene of two ITs and ordering the 4548 genes as per the mean rank values. The last gene axis was the integrated IT.
Frequency plots and probability distributions of DEGs in IT
We divided the ITs into ten equally sized compartments (each segment approximately contained 450 genes) and computed the frequency of up-regulated and down-regulated genes mapping to these compartments for each cancer. For example, the red and blue bars in Fig. 3A represent up- and down-regulated genes, respectively.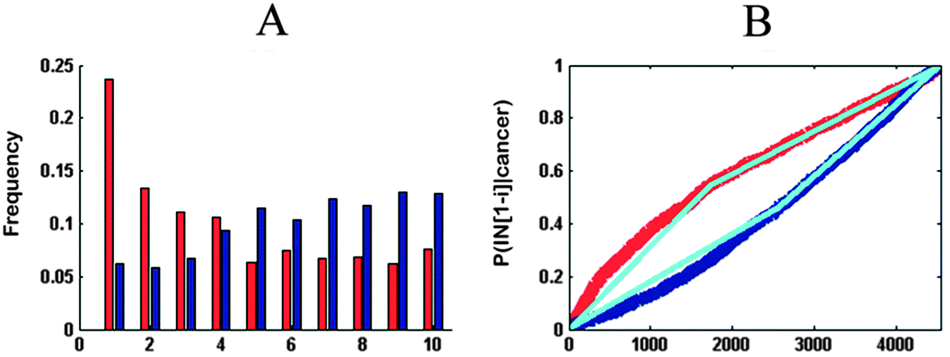 | ||
| Fig. 3 Frequency plot and probability distribution. The frequency plot shows a histogram-like representation of the frequency of up-regulated genes (red) and down-regulated genes (blue) in different segments of IT. The probability distribution curve illustrates the cumulative probability of up-regulated genes (red) or down-regulated genes (down) among the first i genes on the IT. The shape of each curve is summarized by two regression lines, and the slopes of these lines quantify the shape of each curve. | ||
For each of the cancer data sets, the probability distribution P(IN[1, 2, …, 4548]|cancer) described the cumulative probability of up-regulated genes or down-regulated genes among the first i genes on the IT, which was calculated as follows:
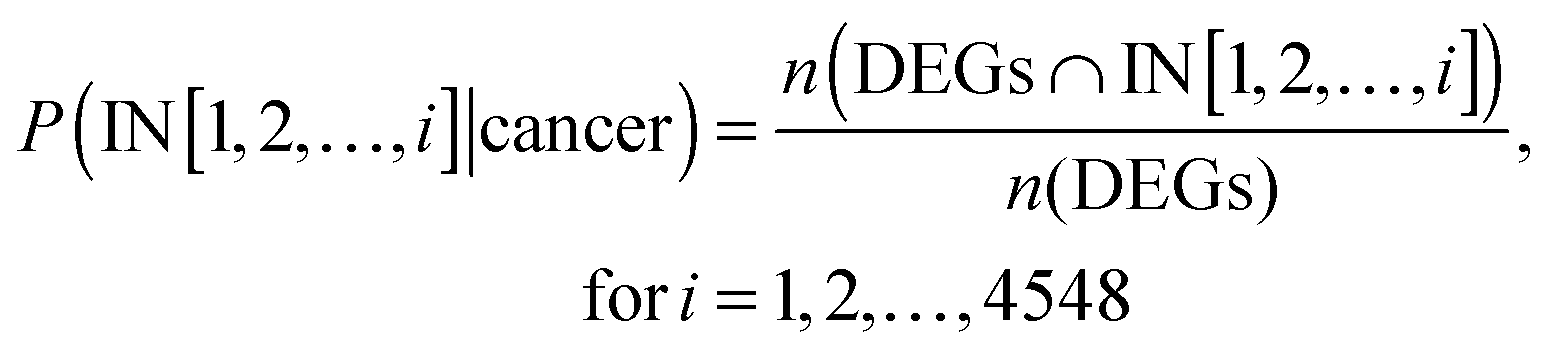
Finally, for each combination of cancer and inflammation, this method produced four regression lines: two lines describing the early and late probabilities for up-regulated genes (upE and upL, respectively; Fig. 3B) and the other two lines for the down-regulated genes (downE and downL, respectively; Fig. 3B). For each cancer, we can summarize its relationship to the two ITs using an 8-dimensional vector (4 regression line slopes × 2ITs). These vectors were applied for clustering cancers.
The flow chart of algorithm is presented in Fig. 4.
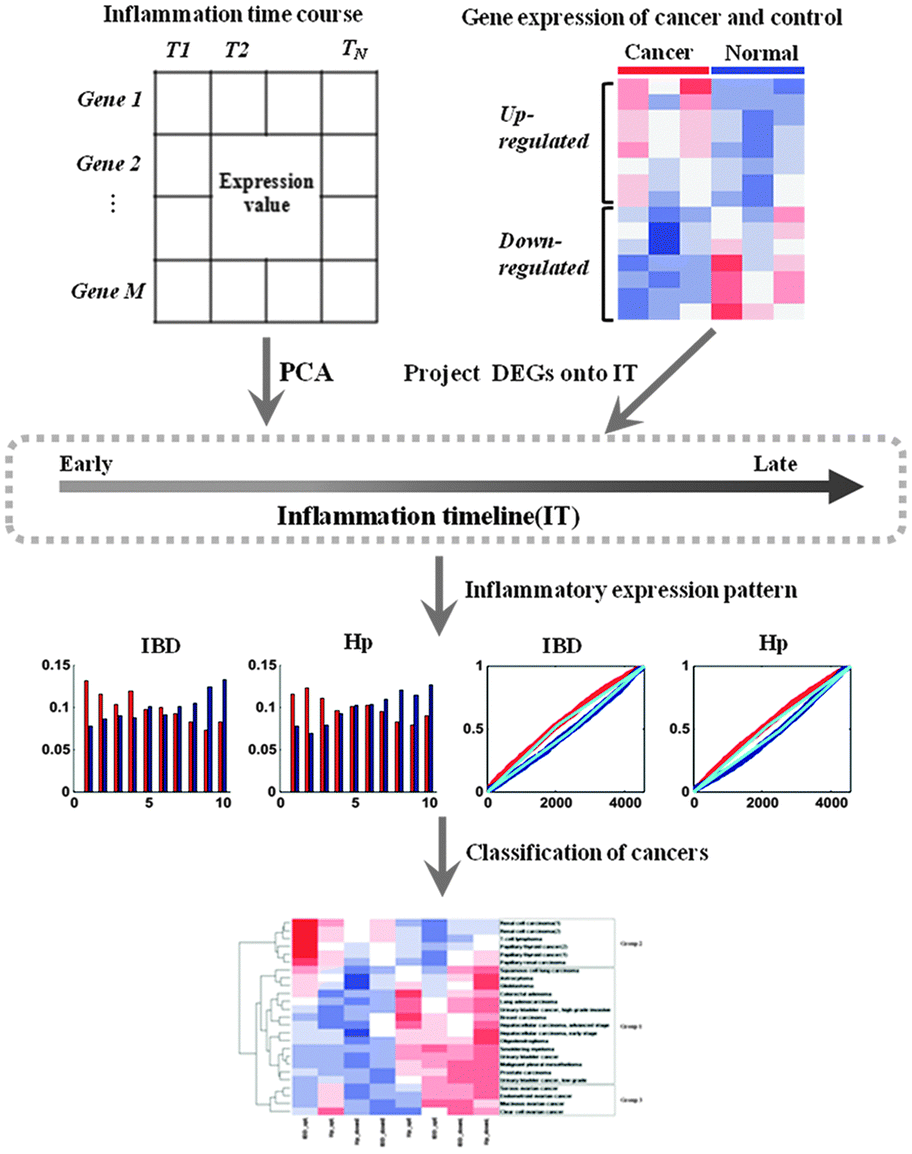 | ||
| Fig. 4 The flow chart of data analysis. | ||
Author contributions
Wei Jiang, Baofeng Lian and Xia Li participated in the design and coordination of the study. Xuexin Yu, Baofeng Lian and Wei Jiang carried out the algorithm construction. Lihong Wang, Yan Zhang, Enyu Dai, Fanlin Meng and Dianming Liu analyzed the results. Shuyuan Wang, Xinyi Liu, and Jing Wang participated in performance evaluation of the results. Wei Jiang, Xuexin Yu and Baofeng Lian wrote the manuscript. All authors have read and approved the manuscript and its contents, and are aware of responsibilities connected to authorship.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China [30900837 and 81202074] and the Foundation for University Key Teacher of the Education Department of Heilongjiang Province [1252G037].Notes and references
- F. Balkwill and A. Mantovani, Lancet, 2001, 357, 539–545 CrossRef CAS.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS.
- F. Colotta, P. Allavena, A. Sica, C. Garlanda and A. Mantovani, Carcinogenesis, 2009, 30, 1073–1081 CrossRef CAS PubMed.
- S. H. Itzkowitz and X. Yio, Am. J. Physiol.: Gastrointest. Liver Physiol., 2004, 287, G7–G17 CrossRef CAS PubMed.
- D. N. Seril, J. Liao, G. Y. Yang and C. S. Yang, Carcinogenesis, 2003, 24, 353–362 CrossRef CAS PubMed.
- G. A. Moody, V. Jayanthi, C. S. Probert, H. Mac Kay and J. F. Mayberry, Eur. J. Gastroenterol. Hepatol., 1996, 8, 1179–1183 CrossRef CAS PubMed.
- J. Eaden, K. Abrams, A. Ekbom, E. Jackson and J. Mayberry, Aliment. Pharmacol. Ther., 2000, 14, 145–153 CrossRef CAS.
- L. M. Coussens and Z. Werb, Nature, 2002, 420, 860–867 CrossRef CAS PubMed.
- M. Macarthur, G. L. Hold and E. M. El-Omar, Am. J. Physiol.: Gastrointest. Liver Physiol., 2004, 286, G515–G520 CrossRef CAS PubMed.
- A. Arzumanyan, H. M. Reis and M. A. Feitelson, Nat. Rev. Cancer, 2013, 13, 123–135 CrossRef CAS PubMed.
- D. Kremsdorf, P. Soussan, P. Paterlini-Brechot and C. Brechot, Oncogene, 2006, 25, 3823–3833 CrossRef CAS PubMed.
- H. Lu, W. Ouyang and C. Huang, Mol. Cancer Res., 2006, 4, 221–233 CrossRef CAS PubMed.
- K. Chang, C. J. Creighton, C. Davis, L. Donehower, J. Drummond, D. Wheeler, A. Ally, M. Balasundaram, I. Birol, Y. S. Butterfield, A. Chu, E. Chuah, H. J. Chun, N. Dhalla, R. Guin, M. Hirst, C. Hirst, R. A. Holt, S. J. Jones, D. Lee, H. I. Li, M. A. Marra, M. Mayo, R. A. Moore, A. J. Mungall, A. G. Robertson, J. E. Schein, P. Sipahimalani, A. Tam, N. Thiessen, R. J. Varhol, R. Beroukhim, A. S. Bhatt, A. N. Brooks, A. D. Cherniack, S. S. Freeman, S. B. Gabriel, E. Helman, J. Jung, M. Meyerson, A. I. Ojesina, C. S. Pedamallu, G. Saksena, S. E. Schumacher, B. Tabak, T. Zack, E. S. Lander, C. A. Bristow, A. Hadjipanayis, P. Haseley, R. Kucherlapati, S. Lee, E. Lee, L. J. Luquette, H. S. Mahadeshwar, A. Pantazi, M. Parfenov, P. J. Park, A. Protopopov, X. Ren, N. Santoso, J. Seidman, S. Seth, X. Song, J. Tang, R. Xi, A. W. Xu, L. Yang, D. Zeng, J. T. Auman, S. Balu, E. Buda, C. Fan, K. A. Hoadley, C. D. Jones, S. Meng, P. A. Mieczkowski, J. S. Parker, C. M. Perou, J. Roach, Y. Shi, G. O. Silva, D. Tan, U. Veluvolu, S. Waring, M. D. Wilkerson, J. Wu, W. Zhao, T. Bodenheimer, D. N. Hayes, A. P. Hoyle, S. R. Jeffreys, L. E. Mose, J. V. Simons, M. G. Soloway, S. B. Baylin, B. P. Berman, M. S. Bootwalla, L. Danilova, J. G. Herman, T. Hinoue, P. W. Laird, S. K. Rhie, H. Shen, T. Triche, D. J. Weisenberger, S. L. Carter, K. Cibulskis, L. Chin, J. Zhang, G. Getz, C. Sougnez, M. Wang, H. Dinh, H. V. Doddapaneni, R. Gibbs, P. Gunaratne, Y. Han, D. Kalra, C. Kovar, L. Lewis, M. Morgan, D. Morton, D. Muzny, J. Reid, L. Xi, J. Cho, D. Dicara, S. Frazer, N. Gehlenborg, D. I. Heiman, J. Kim, M. S. Lawrence, P. Lin, Y. Liu, M. S. Noble, P. Stojanov, D. Voet, H. Zhang, L. Zou, C. Stewart, B. Bernard, R. Bressler, A. Eakin, L. Iype, T. Knijnenburg, R. Kramer, R. Kreisberg, K. Leinonen, J. Lin, M. Miller, S. M. Reynolds, H. Rovira, I. Shmulevich, V. Thorsson, D. Yang, W. Zhang, S. Amin, C. J. Wu, C. C. Wu, R. Akbani, K. Aldape, K. A. Baggerly, B. Broom, T. D. Casasent, J. Cleland, C. Creighton, D. Dodda, M. Edgerton, L. Han, S. M. Herbrich, Z. Ju, H. Kim, S. Lerner, J. Li, H. Liang, W. Liu, P. L. Lorenzi, Y. Lu, J. Melott, G. B. Mills, L. Nguyen, X. Su, R. Verhaak, W. Wang, J. N. Weinstein, A. Wong, Y. Yang, J. Yao, R. Yao, K. Yoshihara, Y. Yuan, A. K. Yung, N. Zhang, S. Zheng, M. Ryan, D. W. Kane, B. A. Aksoy, G. Ciriello, G. Dresdner, J. Gao, B. Gross, A. Jacobsen, A. Kahles, M. Ladanyi, W. Lee, K. V. Lehmann, M. L. Miller, R. Ramirez, G. Ratsch, B. Reva, C. Sander, N. Schultz, Y. Senbabaoglu, R. Shen, R. Sinha, S. O. Sumer, Y. Sun, B. S. Taylor, N. Weinhold, S. Fei, P. Spellman, C. Benz, D. Carlin, M. Cline, B. Craft, K. Ellrott, M. Goldman, D. Haussler, S. Ma, S. Ng, E. Paull, A. Radenbaugh, S. Salama, A. Sokolov, J. M. Stuart, T. Swatloski, V. Uzunangelov, P. Waltman, C. Yau, J. Zhu, S. R. Hamilton, S. Abbott, R. Abbott, N. D. Dees, K. Delehaunty, L. Ding, D. J. Dooling, J. M. Eldred, C. C. Fronick, R. Fulton, L. L. Fulton, J. Kalicki-Veizer, K. L. Kanchi, C. Kandoth, D. C. Koboldt, D. E. Larson, T. J. Ley, L. Lin, C. Lu, V. J. Magrini, E. R. Mardis, M. D. McLellan, J. F. McMichael, C. A. Miller, M. O'Laughlin, C. Pohl, H. Schmidt, S. M. Smith, J. Walker, J. W. Wallis, M. C. Wendl, R. K. Wilson, T. Wylie, Q. Zhang, R. Burton, M. A. Jensen, A. Kahn, T. Pihl, D. Pot, Y. Wan, D. A. Levine, A. D. Black, J. Bowen, J. Frick, J. M. Gastier-Foster, H. A. Harper, C. Helsel, K. M. Leraas, T. M. Lichtenberg, C. McAllister, N. C. Ramirez, S. Sharpe, L. Wise, E. Zmuda, S. J. Chanock, T. Davidsen, J. A. Demchok, G. Eley, I. Felau, B. A. Ozenberger, M. Sheth, H. Sofia, L. Staudt, R. Tarnuzzer, Z. Wang, L. Omberg, A. Margolin, B. J. Raphael, F. Vandin, H. T. Wu, M. D. Leiserson, S. C. Benz, C. J. Vaske, H. Noushmehr, D. Wolf, L. V. Veer, E. A. Collisson, D. Anastassiou, T. H. Ou Yang, N. Lopez-Bigas, A. Gonzalez-Perez, D. Tamborero, Z. Xia, W. Li, D. Y. Cho, T. Przytycka, M. Hamilton, S. McGuire, S. Nelander, P. Johansson, R. Jornsten, T. Kling, J. Sanchez and K. R. Shaw, Nat. Genet., 2013, 45, 1113–1120 CrossRef CAS PubMed.
- G. Ciriello, M. L. Miller, B. A. Aksoy, Y. Senbabaoglu, N. Schultz and C. Sander, Nat. Genet., 2013, 45, 1127–1133 CrossRef CAS PubMed.
- T. I. Zack, S. E. Schumacher, S. L. Carter, A. D. Cherniack, G. Saksena, B. Tabak, M. S. Lawrence, C. Z. Zhang, J. Wala, C. H. Mermel, C. Sougnez, S. B. Gabriel, B. Hernandez, H. Shen, P. W. Laird, G. Getz, M. Meyerson and R. Beroukhim, Nat. Genet., 2013, 45, 1134–1140 CrossRef CAS PubMed.
- L. Omberg, K. Ellrott, Y. Yuan, C. Kandoth, C. Wong, M. R. Kellen, S. H. Friend, J. Stuart, H. Liang and A. A. Margolin, Nat. Genet., 2013, 45, 1121–1126 CrossRef CAS PubMed.
- K. Naxerova, C. J. Bult, A. Peaston, K. Fancher, B. B. Knowles, S. Kasif and I. S. Kohane, Genome Biol., 2008, 9, R108 CrossRef PubMed.
- W. Tan, M. A. Hildebrandt, X. Pu, M. Huang, J. Lin, S. F. Matin, P. Tamboli, C. G. Wood and X. Wu, J. Urol., 2011, 186, 2071–2077 CrossRef CAS PubMed.
- W. Huang da, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2009, 4, 44–57 CrossRef PubMed.
- K. Degenhardt, R. Mathew, B. Beaudoin, K. Bray, D. Anderson, G. Chen, C. Mukherjee, Y. Shi, C. Gelinas, Y. Fan, D. A. Nelson, S. Jin and E. White, Cancer Cell, 2006, 10, 51–64 CrossRef CAS PubMed.
- J. A. Lust, M. Q. Lacy, S. R. Zeldenrust, A. Dispenzieri, M. A. Gertz, T. E. Witzig, S. Kumar, S. R. Hayman, S. J. Russell, F. K. Buadi, S. M. Geyer, M. E. Campbell, R. A. Kyle, S. V. Rajkumar, P. R. Greipp, M. P. Kline, Y. Xiong, L. L. Moon-Tasson and K. A. Donovan, Mayo Clin. Proc., 2009, 84, 114–122 CrossRef CAS PubMed.
- K. E. de Visser, A. Eichten and L. M. Coussens, Nat. Rev. Cancer, 2006, 6, 24–37 CrossRef CAS PubMed.
- S. I. Grivennikov, F. R. Greten and M. Karin, Cell, 2010, 140, 883–899 CrossRef CAS PubMed.
- M. Philip, D. A. Rowley and H. Schreiber, Semin. Cancer Biol., 2004, 14, 433–439 CrossRef CAS PubMed.
- T. T. Tan and L. M. Coussens, Curr. Opin. Immunol., 2007, 19, 209–216 CrossRef CAS PubMed.
- S. Negrini, V. G. Gorgoulis and T. D. Halazonetis, Nat. Rev. Mol. Cell Biol., 2010, 11, 220–228 CrossRef CAS PubMed.
- R. Edgar, M. Domrachev and A. E. Lash, Nucleic Acids Res., 2002, 30, 207–210 CrossRef CAS PubMed.
- R. Simon, A. Lam, M. C. Li, M. Ngan, S. Menenzes and Y. Zhao, Cancer Inf., 2007, 3, 11–17 Search PubMed.
- V. G. Tusher, R. Tibshirani and G. Chu, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5116–5121 CrossRef CAS PubMed.
- strucchange [http://cran.r-project.org/web/packages/strucchange/].
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00258j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |