Network simulation reveals significant contribution of network motifs to the age-dependency of yeast protein–protein interaction networks†
Cheng
Liang
,
Jiawei
Luo
* and
Dan
Song
College of Information Science and Engineering, Hunan University, Changsha, Hunan, China. E-mail: alcs417@hnu.edu.cn; luojiawei@hnu.edu.cn; dansongph@hnu.edu.cn
First published on 25th June 2014
Abstract
Advances in proteomic technologies combined with sophisticated computing and modeling methods have generated an unprecedented amount of high-throughput data for system-scale analysis. As a result, the study of protein–protein interaction (PPI) networks has garnered much attention in recent years. One of the most fundamental problems in studying PPI networks is to understand how their architecture originated and evolved to their current state. By investigating how proteins of different ages are connected in the yeast PPI networks, one can deduce their expansion procedure in evolution and how the ancient primitive network expanded and evolved. Studies have shown that proteins are often connected to other proteins of a similar age, suggesting a high degree of age preference between interacting proteins. Though several theories have been proposed to explain this phenomenon, none of them considered protein-clusters as a contributing factor. Here we first investigate the age-dependency of the proteins from the perspective of network motifs. Our analysis confirms that proteins of the same age groups tend to form interacting network motifs; furthermore, those proteins within motifs tend to be within protein complexes and the interactions among them largely contribute to the observed age preference in the yeast PPI networks. In light of these results, we describe a new modeling approach, based on “network motifs”, whereby topologically connected protein clusters in the network are treated as single evolutionary units. Instead of modeling single proteins, our approach models the connections and evolutionary relationships of multiple related protein clusters or “network motifs” that are collectively integrated into an existing PPI network. Through simulation studies, we found that the “network motif” modeling approach can capture yeast PPI network properties better than if individual proteins were considered to be the simplest evolutionary units. Our approach provides a fresh perspective on modeling the evolution of yeast PPI networks, specifically that PPI networks may have a much higher age-dependency of interaction density than had been previously envisioned.
Introduction
With the advent of modern proteomic and computational technologies, it is now feasible to construct and study a complete or near-complete protein–protein interaction (PPI) network of a eukaryotic organism such as budding yeast.1–3 Analysis of yeast PPI has revealed several broadly applicable network properties such as scale-free topology, hierarchical modularity, and small world. These properties form the basis for the observed PPI network architecture and have motivated intense theoretical efforts in modeling its origin and evolution.Initially, Barabasi and Albert proposed the preferential attachment (PA) model, in which a new node will gain connections with probability proportional to the degree of pre-existing nodes.4 While the PA model can generate networks with scale-free topology, it lacks the ability to simultaneously recapitulate network modularity, i.e. the high clustering coefficients that are usually observed in real biological networks (i.e. protein complexes or network motifs). Shortly after, the gene duplication–divergence model (DD) emerged as an alternative and more realistic model for network evolution.5–7 This model was inspired by the frequently occurring gene and genome duplication events: (1) randomly duplicating a node and retaining all its connections; (2) rewiring the connections of the duplicated node or the original node according to a certain probability distribution.8,9 Finally, the crystal growth (CG) model was proposed by Kim and Marcotte, which takes into account interaction preferences between proteins with similar age and phylogenetic lineage in the yeast PPI networks; as a result, module structures were introduced as a constraint for the newly added nodes.10 In essence, new nodes are added into pre-divided communities with the possibility of having a fixed number of new connections, analogous to the natural growth of crystal lattice. Interestingly, Kim and Marcotte found that proteins belonging to the same age groups are more likely to interact with each other than those from different age groups. To calibrate the interaction preferences between nodes of different age groups, they introduced a new metric called “interaction density”, where a positive (negative) value indicates whether nodes make connections more frequently with nodes from the same (or different) age groups.
Generally, there are two criteria for evaluating the validity of these theoretical models: (1) whether the model can recapitulate the global or local properties observed in real networks, and (2) whether the underlying principles of these models are biologically plausible.11 The aforementioned models have been either investigated on purely theoretical grounds, or applied and tested on real interaction data sets, which resulted in varying degrees of success in recapitulating the observed network topological properties. One important issue with these previous modeling approaches is that they consider individual proteins as elementary evolutionary units, i.e. the network can only grow or contract by one protein at a time. However, it is often observed that biological networks can grow with the addition of several proteins simultaneously. These associated proteins are called “network motifs”, which are recurrent and statistically significant sub-graphs or patterns in complex networks.12 These motifs are often considered as recurring building blocks that make up bigger and more complex networks; these motifs often carry out specific biological functions as a coherent unit. For example, transcriptional regulatory networks can often be decomposed into motifs that have unique topology and carry out specific cellular functions: e.g. single input motif (SIM), multiple-input motif (MIM), feed-forward loop (FFL).13 Many earlier studies have shown that gene and genome duplication events have contributed to the emergence of network motifs in transcriptional regulatory networks.14 In the context of protein–protein interaction (PPI) networks, due to the high frequency of these motifs, their composition and evolution can often be studied to offer clues on the evolution of the entire network.15,16 In addition, it has been suggested that the abundance and distribution of network motifs also contributed to the robustness of the network to small external perturbations.17 These observations suggested that network motifs can potentially function as an independent evolutionary unit and may be important in the growth of biological networks. Recently, Liu et al. reported that the clustered interacting proteins along with the interactions among them were incorporated into the existing PPI networks during a relatively short period of time.18 They also concluded that proteins of the same age group tend to form network motifs while those of different age groups tend to avoid such motif structures. Such correlative observations are intriguing and have significant evolutionary implications, although they have not been rigorously modeled and simulated.
Considering the advantages of network motifs, here we attempt to model network evolution based on the notion that network motifs can be considered as cohesive and distinct evolutionary units. In our model the concept of “network motif” is similar to but distinct from “protein complexes” or “functional modules”. The main distinction between these two concepts is that network motifs are strictly bound by conserved topological structures. In essence, network motifs often reflect functionality of protein complexes or functional modules without preconceived functional limitations. Compared with protein complexes and functional modules, network motifs have explicit topological definition that ignores functionality so that they can be clearly identified and enumerated in biological networks. We show that this new modeling approach can not only capture the topological properties of the real yeast PPI network, but also generate comparative insight between the different modeling approaches that attempts to understand the underlying evolutionary mechanisms. We believe that such a new approach can provide a different perspective on the evolution of the yeast PPI network.
Results and discussion
Network topology and age-dependency analysis of yeast PPI networks
We collected four independent sets of yeast PPIs generated from three different methods, literature curated (LC-Kim), IDBOS-Gavin, IDBOS-Krogan and Y2H-Union PPIs (Table S1, ESI†), and inspected both the network topology and the age-dependency of these networks. All four datasets are able to recapitulate known topological features such as power-law degree distribution, high clustering coefficients as well as short characteristic path length, as shown in various network topological indices (Fig. S1, ESI† and Table 1). Among the four datasets, the IDBOS-Gavin generated the densest yeast PPI network as demonstrated by its high clustering coefficient and average degree. LC and IDBOS-Krogan datasets are relatively sparse but they still maintain a high clustering coefficient (Table 1). Only the network derived from Y2H-Union is remarkably sparse probably due to the incomplete genome-wide Y2H analysis. Overall, the high clustering coefficient is indicative of high modular structure of the yeast PPI network, which is consistent with previous studies.19 Different assortativity–dissortativity trends are also observed in these datasets, which is likely caused by the false positives in the datasets.| Dataset | Average degree | Characteristic path | Clustering coefficient | Delta D |
|---|---|---|---|---|
| LC-Kim | 7.3 | 4.0 | 0.50 | 0.54 |
| IDBOS-Gavin | 12.3 | 5.8 | 0.63 | 0.47 |
| IDBOS-Krogan | 4.2 | 4.9 | 0.63 | 0.88 |
| Y2H-Union | 2.7 | 5.6 | 0.046 | 0.70 |
In order to gain a deeper insight into the age-dependency of interaction density, we used a heat map to show the interaction preference between the four age groups classified by Kim and Marcotte, i.e. “ABE”, “AE/BE”, “E” or “Fu”, according to the taxonomic distribution of constituent domains among archaea (A), bacteria (B), eukaryotes (E) and fungi (Fu). Specifically, all four datasets not only show an overall high positive delta D value, but also exhibit dense interaction density tendencies within the same age group (diagonal) and sparse tendencies between different age groups (off-diagonal) in each column. This clearly demonstrates the existence of strong interaction preference between proteins (Fig. 1). The robustness of the positive delta D pattern regardless of the source of the data also suggests that the strong age preference is a genuine feature of yeast PPI networks, which is not caused by any artifacts in the data.
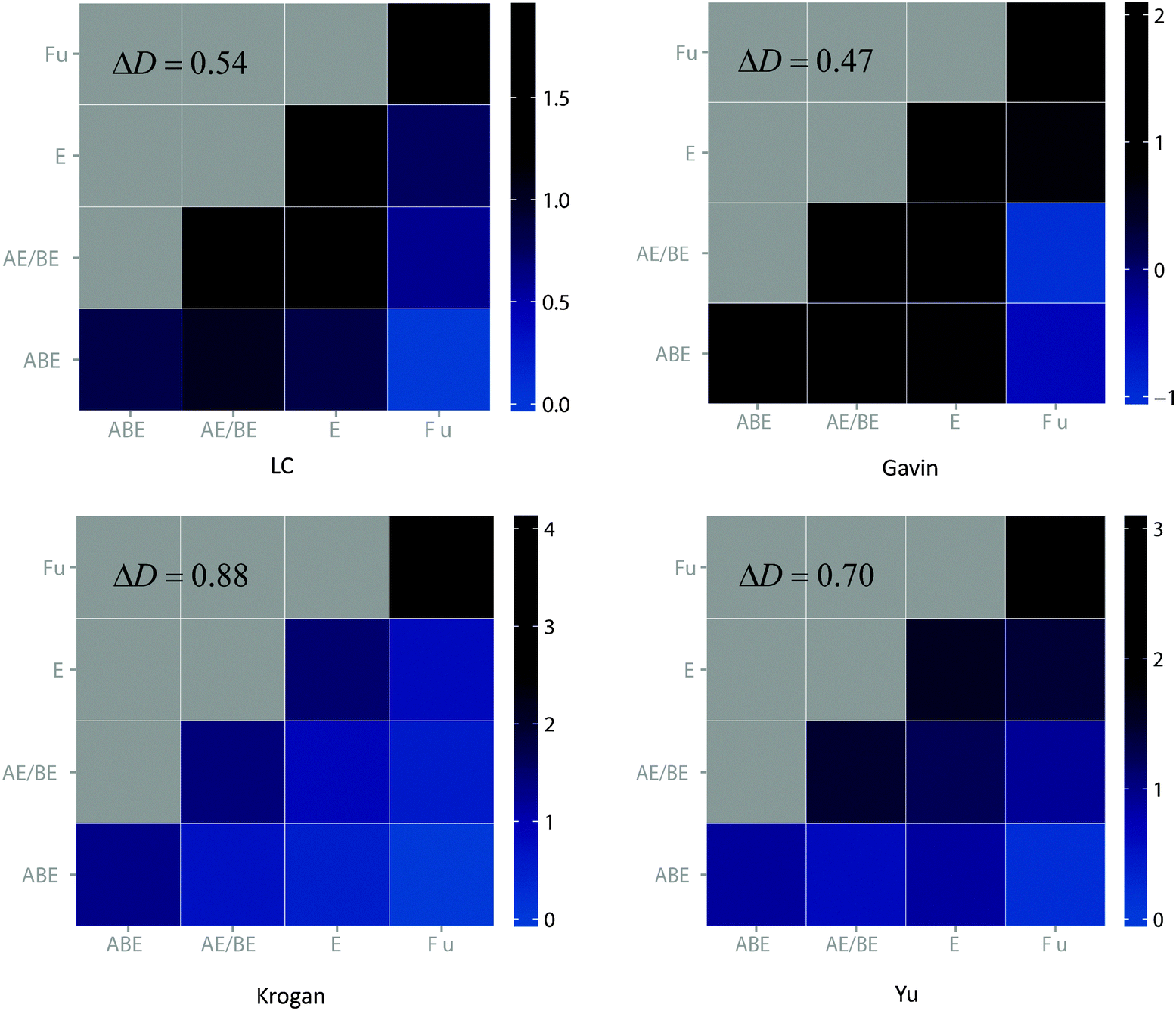 | ||
| Fig. 1 The age-dependency of interaction density of the four yeast PPI networks. | ||
Since the networks constructed from all four PPI datasets share very similar topological properties and interaction preference between proteins, we considered these properties as standard features of yeast PPI networks, and used them as benchmark features in evaluating the prediction performance of various models. The definition of these parameters can be found in Materials and methods. Particularly, the age-dependency of the interaction density pattern (delta D) will be discussed in detail and considered as the main criterion for our evaluation.
The age-dependency of network motifs and their contribution to the interaction preference observed in yeast PPI networks
Previous studies have investigated the age distribution of proteins that interact with each other or are members of the same subgraphs.10,18 To elucidate the interconnection tendency between proteins of the same/different age classes from the perspective of network motifs, here we took the same approach as in ref. 18 to analyze the four yeast PPI networks catalogued in this study. Briefly, we first enumerated all the subgraphs of size 3 and size 4 in each dataset and assigned an age to every protein according to ref. 10. In total we derived two distinct types of three-node subgraphs with three associated age patterns and six types of four-node subgraphs with five age patterns (Fig. 2). To calculate the statistical significance of the observed age patterns in these subgraphs, we shuffled the ages associated with each protein within each subgraph 1000 times, and recalculated the age patterns to yield an empirical p-value. The age distribution of a subgraph is considered as significantly enriched or depleted if the upper-tailed/lower-tailed p-value is less than 0.05. We then adopted a commonly used network motif discovery approach to find network motifs among these different types of subgraphs. In order to obtain reliable network motifs and eliminate the bias of the background null models as far as possible, we implemented two different randomization procedures to achieve our purpose (details can be found in Materials and methods). Eventually, we obtained four types of network motifs among all the subgraphs of size 3 and 4 across the four yeast PPI networks (Fig. 2 and Table 2). Similar to the observation reported in a previous study,18 we found that the majority of the enriched network motifs that are shared by all four datasets tend to consist of proteins of the same age classes while proteins of different age classes tend to avoid forming motifs; moreover, we observed that such age preference was stronger in more densely connected motifs (Table 2). Notably, subgraphs that are not enriched in the yeast PPI networks do not have obvious age preferences; this suggests that these enriched network motifs have a clear age-preference between proteins and the similar results across different datasets indicate that the conclusion above is robust to different data quality.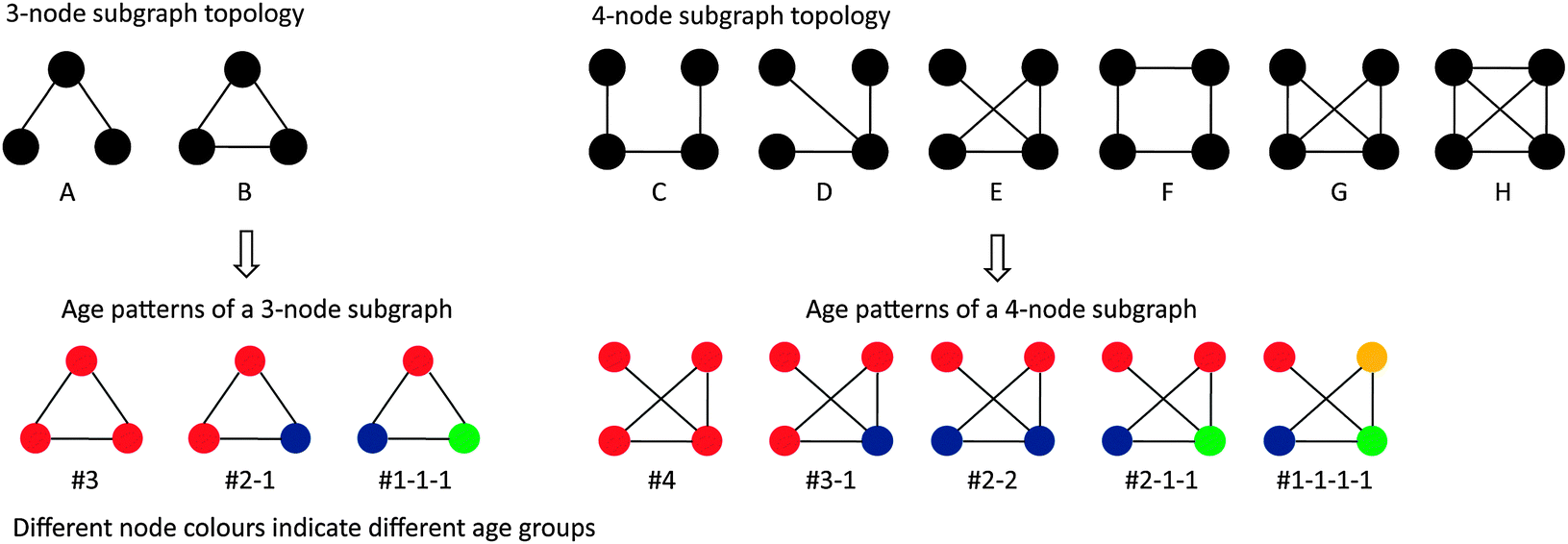 | ||
| Fig. 2 The age patterns for the 3-node and 4-node subgraphs. This figure is modified after Figure 2. Network motifs and evolutionary motif modes in Liu, et al., BMC Evol. Biol., 2011, 11, 133. | ||
| Subgraph typea | Motif or not | Empirical p-value | Dataset | ||
|---|---|---|---|---|---|
| #3 | #2–1 | #1–1–1 | |||
| a The subgraph types correspond to the labels in Fig. 2. | |||||
| A | No | 0.149 | 0.647 | 0.605 | LC-Kim |
| B | Yes | <10−3 | (<10−3) | (<10−3) | LC-Kim |
| A | No | 0.006 | 0.67 | 0.938 | IDBOS-Gavin |
| B | Yes | <10−3 | 0.929 | 0.97 | IDBOS-Gavin |
| A | No | 0.002 | 0.892 | 0.957 | IDBOS-Krogan |
| B | Yes | <10−3 | (<10−3) | (<10−3) | IDBOS-Krogan |
| A | No | 0.226 | 0.384 | 0.739 | Y2H-Union |
| B | Yes | <10−3 | 0.956 | 0.994 | Y2H-Union |
| Subgraph typea | Motif or not | Empirical p-value | Dataset | ||||
|---|---|---|---|---|---|---|---|
| #4 | #3–1 | #2–2 | #2–1–1 | #1–1–1–1 | |||
| C | No | 0.028 | 0.396 | 0.649 | 0.542 | 0.986 | LC-Kim |
| D | No | 0.913 | 0.773 | 0.745 | 0.165 | 0.568 | LC-Kim |
| E | Yes | <10−3 | <10−3 | 0.39 | (<10−3) | (<10−3) | LC-Kim |
| F | No | 0.091 | 0.324 | 0.378 | 0.67 | 0.996 | LC-Kim |
| G | Yes | 0.005 | 0.309 | 0.259 | 0.962 | (<10−3) | LC-Kim |
| H | Yes | <10−3 | <10−3 | (<10−3) | (<10−3) | (<10−3) | LC-Kim |
| C | No | 0.082 | 0.284 | 0.58 | 0.647 | 0.984 | IDBOS-Gavin |
| D | No | 0.131 | 0.209 | 0.265 | 0.787 | 0.943 | IDBOS-Gavin |
| E | Yes | 0.042 | 0.149 | 0.338 | 0.835 | 0.992 | IDBOS-Gavin |
| F | No | 0.488 | 0.447 | 0.249 | 0.594 | 0.948 | IDBOS-Gavin |
| G | Yes | 0.078 | 0.131 | 0.304 | 0.838 | 0.952 | IDBOS-Gavin |
| H | Yes | 0.046 | 0.261 | 0.59 | 0.713 | (<10−3) | IDBOS-Gavin |
| C | No | 0.028 | 0.59 | 0.867 | 0.473 | 0.761 | IDBOS-Krogan |
| D | No | 0.16 | 0.541 | 0.783 | 0.391 | 0.724 | IDBOS-Krogan |
| E | Yes | 0.004 | 0.381 | 0.719 | 0.765 | 0.88 | IDBOS-Krogan |
| F | No | 0.233 | 0.238 | 0.076 | 0.842 | 0.956 | IDBOS-Krogan |
| G | Yes | 0.037 | 0.674 | 0.631 | 0.506 | 0.913 | IDBOS-Krogan |
| H | Yes | <10−3 | 0.83 | 0.613 | 0.834 | 0.989 | IDBOS-Krogan |
| C | No | 0.317 | 0.274 | 0.174 | 0.751 | 0.735 | Y2H-Union |
| D | No | 0.017 | 0.004 | 0.614 | 0.982 | 0.935 | Y2H-Union |
| E | Yes | 0.366 | 0.162 | 0.216 | 0.748 | 0.981 | Y2H-Union |
| F | No | 0.976 | 0.55 | 0.054 | 0.608 | 0.478 | Y2H-Union |
| G | Yes | 0.022 | 0.173 | 0.534 | 0.86 | 0.899 | Y2H-Union |
| H | Yes | <10−3 | 0.698 | 0.788 | 0.985 | 0.355 | Y2H-Union |
After confirming the age-dependency of network motifs, we next investigated the degree to which these network motifs with proteins of similar age contribute to the observed interaction preference in the network. We removed the interactions within the age-homogeneous network motifs (i.e. age pattern 3# and 4#) and recalculated the delta D value. We then compared the significance of these interactions by randomly removing the same amount of interactions in the network. This process was repeated 1000 times to generate the empirical p-value. Clearly, after the removal of these interactions, the delta D drops much more significantly than the random deletion of interactions (Table S2, ESI†), which demonstrates that motifs with proteins of the same age largely contribute to the observed interaction preference in the network.
Proteins within network motifs are also enriched in protein complexes
Protein complexes are a group of proteins that physically associate with each other and carry out specific cellular functions. Studies have shown that fully connected motifs or motifs with proteins of the same age tend to exist in protein complexes.18 Here we analyzed the relationships between network motifs and protein complexes from a more general perspective. We investigated whether the nodes involved in network motifs are more likely to participate in protein complexes. In order to gain reliable insights, we tested two different datasets of protein complexes: SGD_complexes (http://www.yeastgenome.org/) and cyc_2008 (http://wodaklab.org/cyc2008/). There are 404 and 408 complexes in each dataset respectively. Both of them are manually curated and highly reliable, though SGD mainly includes complexes from high-throughput methods while cyc_2008 contains complexes from small-scale experiments. We first derived two lists of proteins which participate in each size of network motifs; then we calculated whether proteins involved in network motifs are also enriched in protein complexes by comparing their percentage in the protein complexes with randomly chosen proteins. We ran the process 1000 times to get an empirical p-value. As expected, proteins within network motifs are highly enriched while those outside of network motifs are actually depleted in both datasets of protein complexes. This observation holds for all the yeast PPI datasets except Y2H-Union (Fig. S2, ESI†). It is widely discussed in the literature that the PPI data derived from Yeast Two-Hybrid often have statistical properties that are different from those of PPI data derived from affinity purification or literature curation.20 This is mostly due to the distinct experimental technique employed in Y2H studies, i.e. it is more suitable for detecting pair-wise interactions than detecting protein complexes. This result strongly suggests that there is a tight relationship between network motifs and protein complexes as most of the proteins participating in network motifs also tend to take part in the protein complexes.Interactions between paralogs can only partially account for the age-dependency in network motifs
Yeast genome is known to have undergone a whole-genome duplication and many rounds of small scale duplications,8 which no doubt had shaped its PPI network; however a recent study showed that interactions between paralogs only partially contribute to the interaction preference among proteins of a similar age whereas the major contribution is from the interaction partners shared between paralogs.21 Here we also investigate whether the age preference of network motifs is the result of the interactions between paralogs. The DD model dictates that paralog pairs can usually be traced to the same origin and thus be assigned the same original age;18 therefore we can estimate their contribution to the interaction preference of network motifs by calculating the fraction of motifs that consist of paralogous pairs. We first derived a list of yeast gene paralogs from a previously published study,22 which contains 450 paralog pairs. We then further selected motifs that are covered by the paralog list, i.e. subgraph type B, E, G, H in Table 2. With these motifs, we further restricted inclusion based on age pattern 3# and 4# (Fig. 2), i.e. proteins within each motif are all from the same age groups. We found that, for each type of motifs, only a limited fraction of them constituted by paralog pairs contribute to the age pattern 3# and 4#, and this observation is almost consistent among the three yeast PPI networks (Table S3, ESI†). The results for the Y2H-Union dataset are not shown here since there are no motifs found to be covered by the paralog list and the protein age list simultaneously. However, this conclusion is restricted by the paralog list used here, which only covers a small portion of the proteins in these four yeast PPI datasets. Even so, these results imply that there might be other evolutionary constraints imposed on the age-dependency of network motifs apart from the duplication–divergence mechanism in yeast PPI networks.Simulation of canonical network growth models
We next simulated PPI network evolution using each of the four canonical models – PA (preferential attachment model), both symmetric and asymmetric DD (duplication divergence model) and CG (crystal growth model). The parameters of these models were calibrated as recommended by the authors in their respective publications (Table S4, ESI†). For each of these models, we ran the simulation pipeline 50 times and took the average value of the generated topological network parameters.As we can see (Table 3), networks generated by the PA model have the lowest clustering coefficient, indicating that the PA model is not able to explain the modularity that is frequently observed in the real yeast PPI networks. The symmetric DD model has relatively larger characteristic path length than the real yeast PPI network, while the asymmetric DD model has a smaller clustering coefficient compared to the real yeast PPI network. Only the CG model appears to have the network properties most resembling those of the real yeast PPI network. For node degree distribution P(k), all the models can recapitulate the power-law distribution; for clustering coefficient C(k), the PA model, the symmetric DD model and the CG model have distributions similar to those of the real PPI network. For the average degree of nearest neighbours k〈nn〉, all the models show a decreasing trend (Fig. 3). Similar to the observation in ref. 10, all the models except the CG model have a negative delta D value, which suggests that none of these models except the CG model is sufficient to recapitulate the interaction preference between proteins of real yeast PPI networks.
| Model | Characteristic path length | Clustering coefficient | Average degree | Delta D |
|---|---|---|---|---|
| PA model | 3.5 ± 0.02 | 0.02 ± 0.001 | 8.0 ± 1 × 10−14 | −0.8 ± 0.03 |
| Symmetric DD model | 7.6 ± 1.11 | 0.33 ± 0.02 | 4.1 ± 0.27 | −0.26 ± 0.04 |
| Asymmetric DD model | 4.5 ± 0.27 | 0.26 ± 0.02 | 6.6 ± 0.49 | −0.71 ± 0.04 |
| CG model | 4.3 ± 0.05 | 0.51 ± 0.01 | 8.0 ± 4 × 10−15 | 0.005 ± 0.09 |
| Network motif model, n = 3 | 5.6 ± 0.23 | 0.77 ± 0.002 | 10.0 ± 1 × 10−14 | 0.38 ± 0.03 |
| Network motif model, n = 3, 4 | 5.1 ± 0.22 | 0.77 ± 0.003 | 11.2 ± 0.04 | 0.29 ± 0.04 |
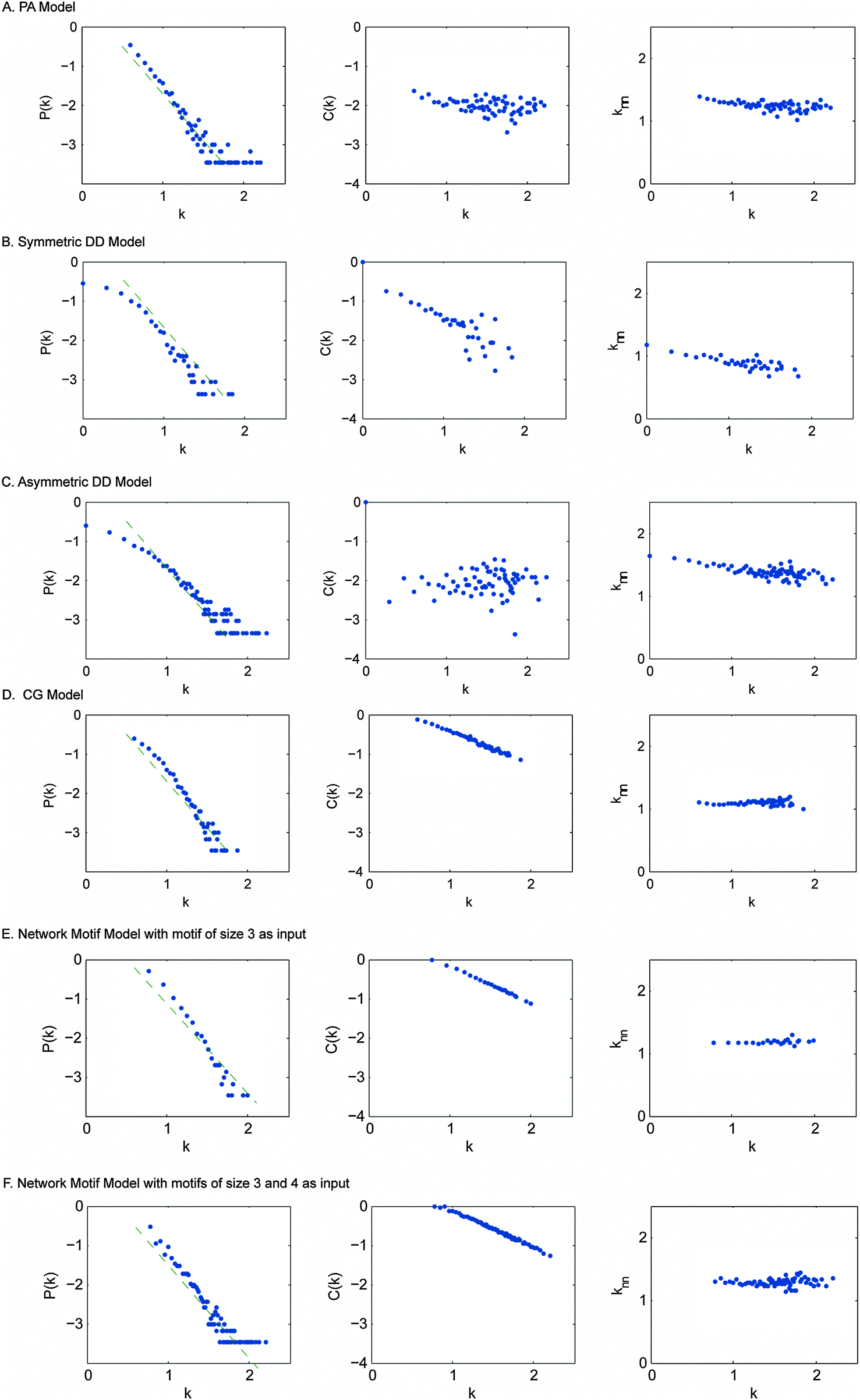 | ||
| Fig. 3 The P(k), C(k) and k〈nn〉 of all the models including our model. (A) PA model. (B) Symmetric DD model. (C) Asymmetric DD model. (D) CG model. (E) Network motif model with motif of size 3 as input. (F) Network motif model with motifs of size 3 and 4 as inputs. | ||
Network motif model can capture the primary characteristics of the yeast PPI network
To better address the age-dependency of real PPI networks, we developed an alternative model for PPI network evolution which takes network motifs as the basic evolutionary units. The key idea of our model is that during the network expanding process, network motifs or protein clusters instead of single proteins will be incorporated into the pre-existing network. A flow chart of our network motif model is illustrated in Fig. S3 (ESI†) and a detailed description of this model can be found in Materials and methods. Our model starts with a few seed nodes (N0 = 4) and at each time step a certain type of network motifs is attached to the existing nodes. As in the CG model, our model also takes two main steps in expanding the network: anchoring and extension. In the anchoring step, an anchor node is chosen according to the anti-preferential rule, which is based on the previously published observation that the number of interactions one protein can gain is restricted by its available interacting surfaces.10 Then, in the extension step, the anchor node as well as part of its neighbors further connects to the selected network motif. The number of connections made between members of network motifs and the existing nodes depends on the size of the selected network motif, and connections between neighbors of the anchor node and the members of the network motif are randomly created. Therefore, the network motif model is inherently cluster-oriented. Networks generated by the network motif model show a remarkable similarity to real PPI networks for all network properties (Table 3 and Fig. 3). The topology of our model shows scale-free, hierarchical modularity as well as short characteristic path length characteristics. These characteristics are robust regardless of the network sizes, e.g. N = 1000 and N = 3000 (data not shown).From our analysis, we found that only the CG model, the network motif model and the real yeast PPI networks have positive delta D values whereas the rest of the models have negative ones. Though the PA model and the DD model (including other DD-based models) are commonly used in simulating the evolution of yeast PPI networks, neither of them could generate the observed delta D value of the real networks. The positive delta D values obtained by the CG model and the network motif model proved that only these two models could generate networks with expected interaction density as the real PPI network. Notably, our model has the largest delta-D value which more closely mimics that of real yeast PPI networks. As a main criterion, we also investigated the interaction preference between different node groups for each model. Clearly, our model demonstrates a distinct tendency of interaction preference between nodes of the same age groups (diagonal) while other models except CG show an opposite trend (Fig. 4). Although CG has a similar tendency as that demonstrated by our model, it is less obvious in the younger age groups (like G3–G3 and G4–G4) compared to ours. Thus the CG model could only generate a moderate interaction preference between nodes while our model could potentially reflect the tendency as the real yeast PPI networks.
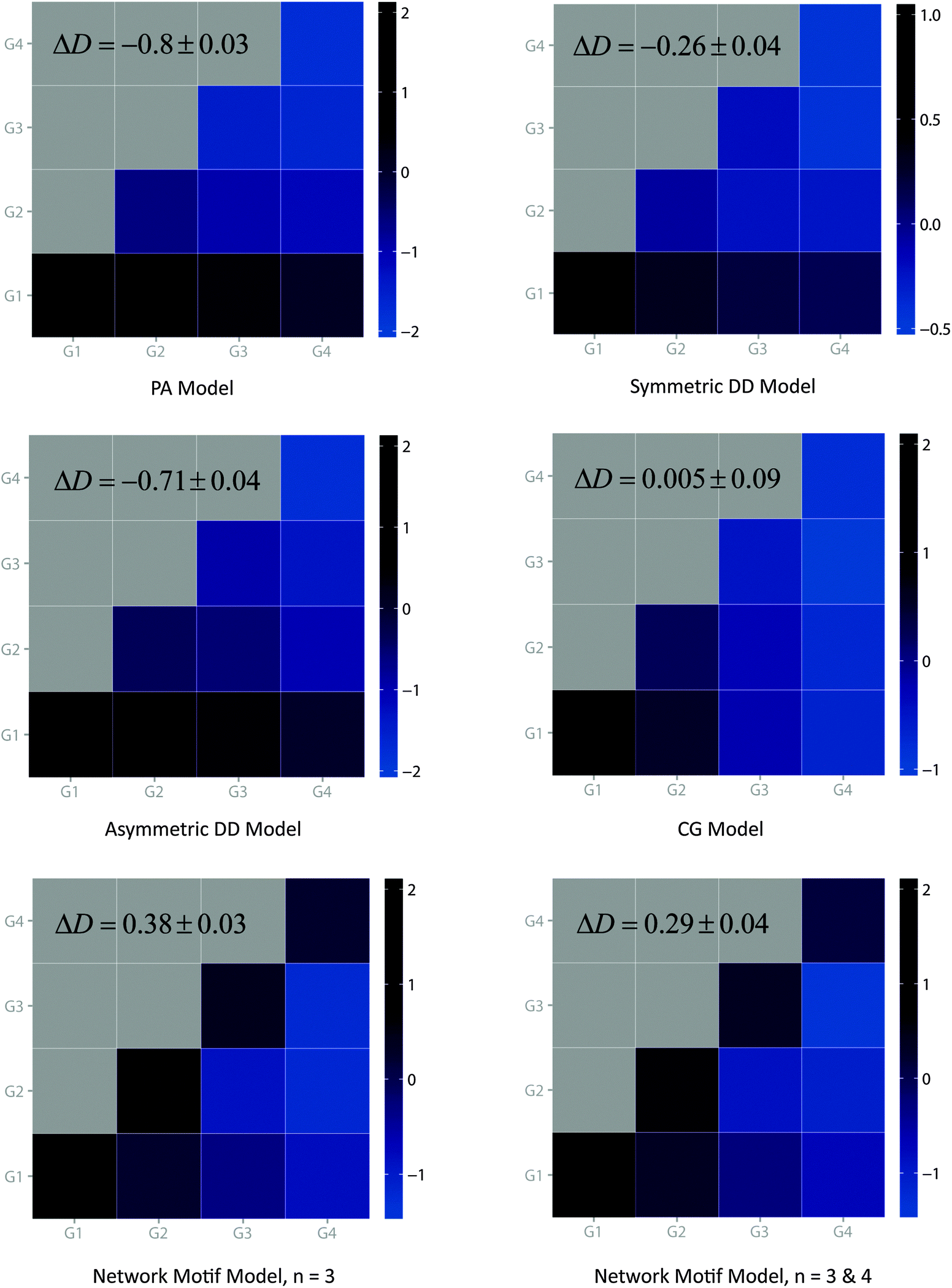 | ||
| Fig. 4 The age-dependency of interaction density of all the models. | ||
In summary, the network motif model can capture the primary topological properties of the yeast PPI networks (Table 3 and Fig. 3 and 4). The superiority of our model in capturing age preference of interaction density reflects the importance of the motif structures in the evolutionary process of the real yeast PPI networks.
Discussion
One of the hallmarks of the recent breakthroughs in systems biology is the (re)construction of protein–protein interaction (PPI) networks in living cells, which has transformed the way we think about how individual proteins function in the cell and how proteins cross-talk with each other. PPI networks have many unique topological properties, and how the networks have evolved into the current state has attracted many theoretical and experimental studies. Most of the earlier and contemporary modeling approaches only considered the evolutionary scenarios in which single proteins are added to the PPI network individually; however it is known that the expansion of genomes or proteomes is often accomplished by addition of multiple proteins simultaneously, e.g. by horizontal gene transfer of an entire operon or by segmental chromosomal duplications.As introduced by Kim et al., the interaction density is highly protein age-dependent in yeast PPI networks. By analyzing four yeast PPI networks from three different sources, we confirmed the strong age-dependency between PPIs. Then we further examined the age preference within network motifs. These network motifs are recognized as closely associated proteins that either make up a protein complex (or sub-complex) or constitute part of cellular pathways. Our analysis demonstrates that proteins within network motifs indeed tend to exist in protein complexes. Another property of these network motifs is that they are often over-represented in the PPI network; thus they can be considered as building blocks of such networks. However, network motifs are not always protein complexes as they do not carry specific functions other than conserved topological structures. Similarly, the common network motifs found throughout the four PPIs also illustrated a strong age preference and largely contributed to the observed age-dependency property in the yeast PPI networks. To verify whether this age preference is caused by the duplication divergence mechanism during the evolution of yeast PPI networks, we calculated the proportion of paralogs that are found in the common network motifs. Surprisingly, the paralog pairs have very limited contribution to the age preference, which indicates that the age preference within motifs might be formed by other evolutionary mechanisms.
Motivated by these biological observations, in this paper we proposed a new “network motif” based modeling approach, which simulates the evolution and growth of yeast PPI networks by addition of “network motifs”. An important underlying assumption of our model is that all the members within the same network motif have the same age. Building on this, our model adopted a two-step modeling strategy, anchoring and extension, iteratively attaching known network motifs to the anchor node and its neighbors. Unlike other existing models such as duplication–divergence models, our network motif model does not require special parameters to be tuned except the predefined network size. As a result, our model is fairly robust and insensitive to the initial starting states of the network and the parameters. In contrast, most of the current modeling approaches require custom fitting of a significant number of parameters (Table S4, ESI†); the topology of the resulting networks is often very sensitive to these parameters, e.g. the number of connections of a newly added node in the PA model23 and the divergence probability in the DD model. We showed that the new network-motif based modeling approach can successfully capture the principal topological properties of the yeast PPI network, such as power-law degree distribution, high clustering coefficients, short characteristic path length, and the age-dependency indicator – delta D value. The results demonstrated that our model can get much larger delta D values compared to the CG model, which means the networks generated by our model have stronger interaction preference between PPIs and most resemble the previously observed age-dependency of the interaction density pattern in real yeast PPI networks. In addition, our model is robust to different strategies of choosing the anchor node as well as the seed graph.
The motivation of this study is to show that it is a realistic and alternative approach to model the evolution of yeast PPI networks by addition of “network motifs” consisting of multiple proteins, which have occurred frequently in the evolutionary process. Such an integration process may take longer evolutionary time than the evolutionary step based on single genes as modeled in other methods. As demonstrated in this paper, it is clear that such a fresh modeling approach can indeed grab the main characteristics of the yeast PPI networks that we see today. However it is likely that the real yeast PPI network would have evolved through the addition of both single proteins and groups of protein (i.e. network motifs). From the analysis of all the existing models we can see that one single evolutionary mechanism is not sufficient to explain the diversities demonstrated by the real yeast PPI network. Even though the network motif model can better characterize the interaction preference in the yeast PPI networks, a major shortcoming of our model as well as the CG model is that low-degree nodes are not captured in the generated networks. So a mixed evolutionary mechanism that combines several of the existing plausible evolutionary scenarios would be a reasonable conjecture. Moreover, to obtain more accurate and complete results, we would recommend users to detect more reliable network motifs by removing the noise data in the datasets and use network motifs of different size to simulate their own networks. Our work presented here will open new avenues for research on network evolution modeling.
Materials and methods
Network motif discovery
Finding network motifs is a computationally intensive procedure that generally consists of two steps: finding topologically equivalent sub-graph classes in a given network, and determining which sub-graph classes occur statistically more frequently than what a null random graph model would predict. In particular, the randomization procedure has drawn significant criticism since the statistical outcomes largely depend on the background network distributions.24,25 In order to obtain reliable network motifs, we adopt a similar subgraph enumeration strategy as other motif discovery algorithms but implement two different randomization algorithms to check the consistency of the results. The first one is the commonly used Markov-chain algorithm,12 which starts with the real network and repeatedly swaps randomly chosen pairs of connections; the other is the sequential algorithm which only keeps the node degree sequence unchanged during the process regardless of any specific nodes' degree.26 We also adjust the results by multiple testing corrections and require the false discovery rate smaller than 0.05. Eventually we identified the same network motifs of size 3 and 4 using both strategies. These network motifs were then further analyzed and fed into our modeling algorithm for the further construction process.A network motif model for the yeast PPI network
Briefly our motif analysis pipeline consists of the following 4 steps (the corresponding flow chart is shown in Fig. S3, ESI†):Input: network motif file, specified network size that is reached at the end of simulation
Output: network of required size
Initialization: a seed network of four fully connected nodes
Step 1. Choose an anchor node in the network following the anti-preferential rule. This anchor node is to be connected to the network motifs selected later.
Step 2. Read the network motif storage file, choose a network motif and record its size n.
Step 3. Add the chosen motif into the network by connecting the network motif to the anchor node (selected in Step 1) and its neighbors. If the anchor node does not have n neighbors, then simply connect all its neighbors to the selected motif. Otherwise, randomly pick up n neighbors of its neighbors and connect them to the selected motif.
Step 4. Repeat this process till the network reaches the pre-defined size.
The above steps are repeated 50 times and all the results shown in the next section are averaged based on these 50 replications.
Having described the basic procedures in our simulation, next we explain the motivation and rationale behind each of these steps. In Step 1, the anti-preferential rule specifies that nodes are connected with a probability inversely proportional to the node's degree. We adopted this strategy based on the previously published observation that the number of interactions one protein can gain is restricted by its available surfaces.10 This is in contrast to the “preferential attachment” principle that had been used in some of the earlier network modeling studies, in which a new protein is preferentially added to a “hub” protein that already has many interacting partners, i.e. the probability of attachment is proportional to the node's degree.4 The “preferential attachment” model was successful in explaining the power-law distribution of network degrees, but it had difficulties in other aspects of the biological network modeling such as it failed to explain the modularity of the real network.11 In Step 2, we randomly choose a motif from the storage file and add it into the network. We only consider motifs of size 3 or 4 since it is more difficult for a large number of proteins to be incorporated into an existing network (e.g. horizontal gene transfer). We note that a smaller motif can be implicitly included as part of a larger motif in our process. Besides, all the network motifs have equal probabilities to be chosen on the basis of uniform distribution of the randomized process. Two reasons why we choose the motifs uniformly instead of taking their frequencies from the real yeast PPI data are because (1) motifs are only part of the entire set of subgraphs in the network and (2) uniform distribution does not introduce biased constraints on our model. In Step 3, we connect the external network motif with the anchor node following separate rules depending on whether the anchor node already has n connections (n is the size of the motif). If the anchor has fewer than n connecting neighbors, then we connect the anchor node and all of its neighboring nodes to all the nodes in the incoming motif; if the anchor node already has more than n neighboring nodes, then we randomly select n neighboring nodes from these nodes, and connect them and the anchor node to all the nodes of the imported network motif. The main idea of this strategy is that motifs are more likely to form or be integrated into bigger protein clusters, like protein complexes. By restricting the number of neighbors of the anchor nodes to be connected with the imported motifs, the current clusters will keep the probability to absorb other motifs due to the relatively low degree of other neighbors.
As mentioned above, we choose an anchor node in the existing network to attach the selected motif at each step. There are three ways to choose an anchor node: anti-preferential attachment, preferential attachment and random selection. The preferential attachment mechanism was based on the rationale that a new node tends to preferentially connect with the nodes with higher degrees.4 It has the advantage of power-law degree distribution but lacks modularity. Anti-preferential attachment was motivated by the rationale that a protein has only a limited amount of surface available to interact with other proteins; therefore when a new protein comes into the network it would have less chance to establish with a protein that already has many interacting partners.
We tested both models with all three different mechanisms for choosing the anchor node and found that our model can get a positive delta D value regardless of the strategies, whereas the CG model can only get a small positive delta D value with the anti-preferential mechanism (data not shown). In the meantime, we also found that our model yielded the largest delta D value with anti-preferential attachment, the second largest delta D value with random selection and the lowest delta D value with preferential attachment. Such observations suggest that our motif-based evolution model can capture the age preference of the yeast PPI network and is more robust compared to the CG model in choosing the anchor nodes.
where f(d) is how many times the shortest path length of d appears.
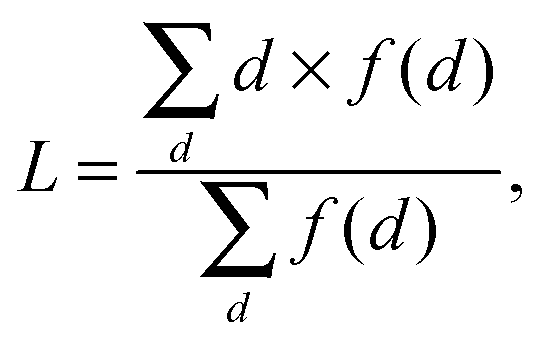
Clustering coefficient is the measurement of the cohesiveness between the neighbors of a node. Suppose the degree of node v is kv and the number of actual existing edges between its neighbors is Ev, then the clustering coefficient Cv is
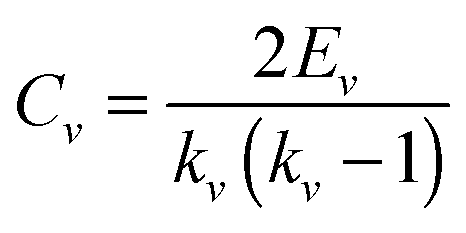
The clustering coefficient of a network is the average of the clustering coefficient of all nodes in this network, and C(k) is the average clustering coefficient of nodes with degree k.
Average degree is the average degree of all the nodes in the network.
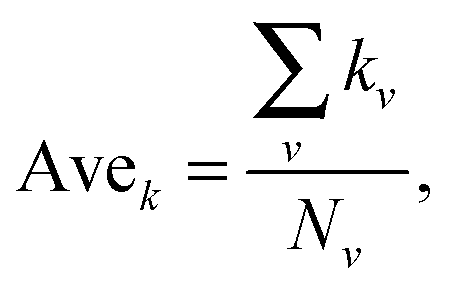
Degree distribution is the probability of nodes with degree k in the network.
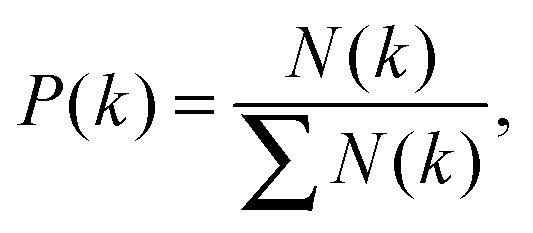
The age-dependency of interaction density Dm,n is defined as
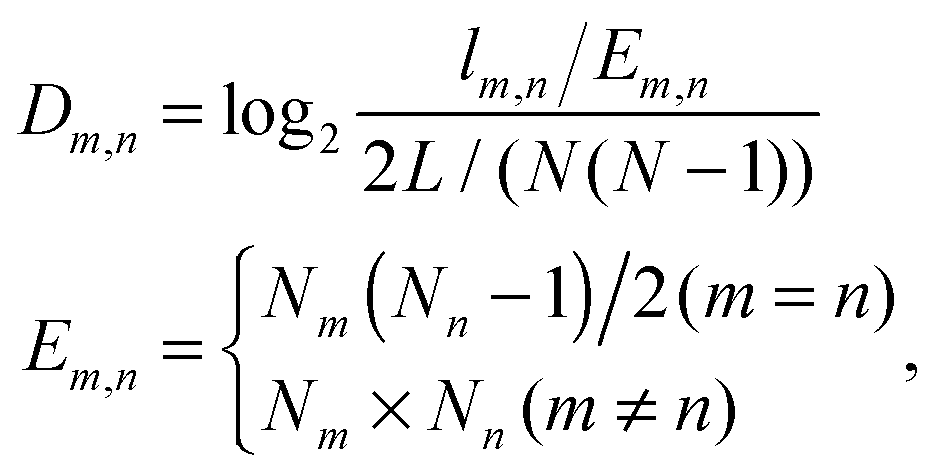
The average interaction density gradient ΔD is defined as
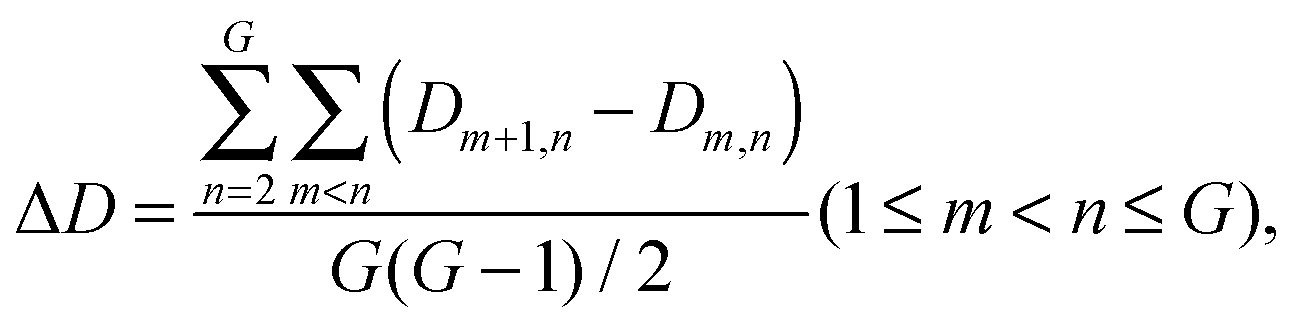
Competing interests
The authors declare that they have no competing interests.Authors' contributions
CL and JL initiated the study, CL and JL designed the project, CL and DS conducted the experiments, and CL and JL wrote the paper.Acknowledgements
We would like to thank John Li, Lee Zamparo and Prof. Qian Xiang for reviewing drafts of this manuscript. This work is supported by the National Natural Science Foundation of China (Grant No. 61240046) and the Hunan Provincial Natural Science Foundation of China (Grant No. 13JJ2017).References
- A. C. Gavin, P. Aloy, P. Grandi, R. Krause and M. Boesche, et al., Proteome survey reveals modularity of the yeast cell machinery, Nature, 2006, 440, 631–636 CrossRef CAS PubMed.
- N. J. Krogan, G. Cagney, H. Yu, G. Zhong and X. Guo, et al., Global landscape of protein complexes in the yeast Saccharomyces cerevisiae, Nature, 2006, 440, 637–643 CrossRef CAS PubMed.
- H. Yu, P. Braun, M. A. Yildirim, I. Lemmens and K. Venkatesan, et al., High-quality binary protein interaction map of the yeast interactome network, Science, 2008, 322, 104–110 CrossRef CAS PubMed.
- A. L. Barabasi and R. Albert, Emergence of scaling in random networks, Science, 1999, 286, 509–512 CrossRef PubMed.
- R. Pastor-Satorras, E. Smith and R. Sole, Evolving protein interaction networks through gene duplication, J. Theor. Biol., 2003, 222, 199–210 CrossRef CAS PubMed.
- A. Vazquez, M. Boguna, Y. Moreno, R. Pastor-Satorras and A. Vespignani, Topology and correlations in structured scale-free networks, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 046111 CrossRef.
- A. Vázquez, A. Flammini, A. Maritan and A. Vespignani, Modeling of Protein Interaction Networks, ComPlexUs, 2003, 1, 38–44 CrossRef.
- M. Kellis, B. W. Birren and E. Lander, Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae, Nature, 2004, 428, 617–624 CrossRef CAS PubMed.
- I. Ispolatov, P. L. Krapivsky and A. Yuryev, Duplication-divergence model of protein interaction network, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 061911 CrossRef CAS.
- W. K. Kim and E. Marcotte, Age-dependent evolution of the yeast protein interaction network suggests a limited role of gene duplication and divergence, PLoS Comput. Biol., 2008, 4, e1000232 Search PubMed.
- M. G. Sun and P. Kim, Evolution of biological interaction networks: from models to real data, Genome Biol., 2011, 12, 235 CrossRef PubMed.
- R. Milo, S. Shen-Orr, S. Itzkovitz, N. Kashtan and D. Chklovskii, et al., Network motifs: simple building blocks of complex networks, Science, 2002, 298, 824–827 CrossRef CAS PubMed.
- H. Yu, N. M. Luscombe, J. Qian and M. Gerstein, Genomic analysis of gene expression relationships in transcriptional regulatory networks, Trends Genet., 2003, 19, 422–427 CrossRef CAS PubMed.
- J. J. Ward and J. Thornton, Evolutionary models for formation of network motifs and modularity in the Saccharomyces transcription factor network, PLoS Comput. Biol., 2007, 3, 1993–2002 CAS.
- S. Wuchty, Z. N. Oltvai and A. Barabasi, Evolutionary conservation of motif constituents in the yeast protein interaction network, Nat. Genet., 2003, 35, 176–179 CrossRef CAS PubMed.
- R. J. Prill, P. A. Iglesias and A. Levchenko, Dynamic properties of network motifs contribute to biological network organization, PLoS Biol., 2005, 3, e343 Search PubMed.
- W. P. Lee, B. C. Jeng, T. W. Pai, C. P. Tsai and C. Y. Yu, et al., Differential evolutionary conservation of motif modes in the yeast protein interaction network, BMC Genomics, 2006, 7, 89 CrossRef PubMed.
- Z. Liu, Q. Liu, H. Sun, L. Hou and H. Guo, et al., Evidence for the additions of clustered interacting nodes during the evolution of protein interaction networks from network motifs, BMC Evol. Biol., 2011, 11, 133 CrossRef PubMed.
- A. L. Barabasi and Z. Oltvai, Network biology: understanding the cell's functional organization, Nat. Rev. Genet., 2004, 5, 101–113 CrossRef CAS PubMed.
- A. F. Altelaar, J. Munoz and A. Heck, Next-generation proteomics: towards an integrative view of proteome dynamics, Nat. Rev. Genet., 2013, 14, 35–48 CrossRef CAS PubMed.
- L. Fokkens, P. Hogeweg and B. Snel, Gene duplications contribute to the overrepresentation of interactions between proteins of a similar age, BMC Evol. Biol., 2012, 12, 99 CrossRef PubMed.
- G. Musso, M. Costanzo, M. Huangfu, A. M. Smith and J. Paw, et al., The extensive and condition-dependent nature of epistasis among whole-genome duplicates in yeast, Genome Res., 2008, 18, 1092–1099 CrossRef CAS PubMed.
- T. A. Gibson and D. Goldberg, Improving evolutionary models of protein interaction networks, Bioinformatics, 2011, 27, 376–382 CrossRef CAS PubMed.
- M. E. Beber, C. Fretter, S. Jain, N. Sonnenschein and M. Muller-Hannemann, et al., Artefacts in statistical analyses of network motifs: general framework and application to metabolic networks, J. R. Soc., Interface, 2012, 9, 3426–3435 CrossRef PubMed.
- M. Megraw, S. Mukherjee and U. Ohler, Sustained-input switches for transcription factors and microRNAs are central building blocks of eukaryotic gene circuits, Genome Biol., 2013, 14, R85 CrossRef PubMed.
- M. Bayati, J. H. Kim and A. Saberi, A Sequential Algorithm for Generating Random Graphs, Algorithmica, 2010, 58, 860–910 CrossRef.
- F. Hormozdiari, P. Berenbrink, N. Przulj and S. Sahinalp, Not all scale-free networks are born equal: the role of the seed graph in PPI network evolution, PLoS Comput. Biol., 2007, 3, e118 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00230j |
| This journal is © The Royal Society of Chemistry 2014 |