 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental design, validation and computational modeling uncover DNA damage sensing by DNA-PK and ATM†
R. J.
Flassig‡
*a,
G.
Maubach‡
b,
C.
Täger
b,
K.
Sundmacher
ac and
M.
Naumann
b
aProcess Systems Engineering Group, Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg, Germany. E-mail: flassig@mpi-magdeburg.mpg.de
bInstitute of Experimental Internal Medicine, Otto von Guericke University, Magdeburg, Germany
cProcess Systems Engineering Group, Otto von Guericke University, Magdeburg, Germany
First published on 9th May 2014
Abstract
Reliable and efficient detection of DNA damage constitutes a vital capability of human cells to maintain genome stability. Following DNA damage, the histone variant H2AX becomes rapidly phosphorylated by the DNA damage response kinases DNA-PKcs and ATM. H2AX phosphorylation plays a central role in signal amplification leading to chromatin remodeling and DNA repair initiation. The contribution of DNA-PKcs and ATM to H2AX phosphorylation is however puzzling. Although ATM is required, DNA-PKcs can substitute for it. Here we analyze the interplay between DNA-PKcs and ATM with a computational model derived by an iterative workflow: switching between experimental design, experiment and model analysis, we generated an extensive set of time-resolved data and identified a conclusive dynamic signaling model out of several alternatives. Our work shows that DNA-PKcs and ATM enforce a biphasic H2AX phosphorylation. DNA-PKcs can be associated to the initial, and ATM to the succeeding phosphorylation phase of H2AX resulting into a signal persistence detection function for reliable damage sensing. Further, our model predictions emphasize that DNA-PKcs inhibition significantly delays H2AX phosphorylation and associated DNA repair initiation.
1. Introduction
Cells are constantly affected by DNA damage, resulting from ionizing γ-irradiation (IR), genotoxic or replication stress and reactive oxygen species. DNA damage, including single and double strand breaks (DSB), base modification, deletions or point mutations, seriously affects genome stability and cell integrity if not properly detected and repaired by the DNA damage response (DDR).1Upon DNA damage detection, higher order chromatin has to be made accessible by various modifications before DSB can be repaired.2 Among several DNA-damage associated histone modifications, phosphorylation of H2AX is widely accepted as an indicator of DSB. H2AX becomes rapidly phosphorylated at serine 139 (γH2AX) to generate foci at the DSB site.3 The assembly of chromatin remodeling complexes at the DSB site greatly depends on γH2AX and enables the accessibility of the damaged DNA to repair proteins.4
Depending on the stimulus, γH2AX is induced by different members of the phosphoinositide 3-kinase like kinase (PIKK) family; ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs). ATR phosphorylates H2AX upon replicative stress,5 whereas ATM and DNA-PKcs are responsible for this phosphorylation upon DNA DSB, which are induced by IR.6 ATM and DNA-PKcs have been studied on a qualitative basis focusing on their impact of repair pathway choice for rebuilding damaged DNA either via rapid (classical) non-homologous end joining cNHEJ and/or slow homologous recombination repair (HR) pathway.7,8 As for the pathway choice, the interplay between ATM and DNA-PKcs regarding IR-induced H2AX phosphorylation remains puzzling. Because although ATM is required,9 DNA-PKcs can substitute for it.10
In this work we follow a model-based approach to analyze the contribution of DNA-PKcs and ATM to H2AX phosphorylation during the initial DNA damage sensing stage. Cucinotta et al.11 have created a dynamic model solely focused on DNA-PKcs to predict dose and dose-rate effects on γH2AX dynamics. Very recently, a mechanistic model describing DNA damage complexity dependent sub-pathway choice in cNHEJ repair has been presented.12 Although several other mechanistic models of DNA-PKcs and cNHEJ repair exist,13–16 mechanistic modeling of ATM dynamics in the context of DNA damage is rare.17
A computational model for ATM and DNA-PKcs interactions with regard to γH2AX activation integrating biochemical time course data is missing so far. We describe an iterative workflow to identify a predictive dynamic model involving ATM/DNA-PKcs mediated H2AX phosphorylation. Starting from several models, optimal experimental design (OED) was applied to optimize experiments for model identification. The identified model was used to analyze the dynamic contribution of ATM and DNA-PKcs to H2AX phosphorylation.
2. Results
2.1 Model identification
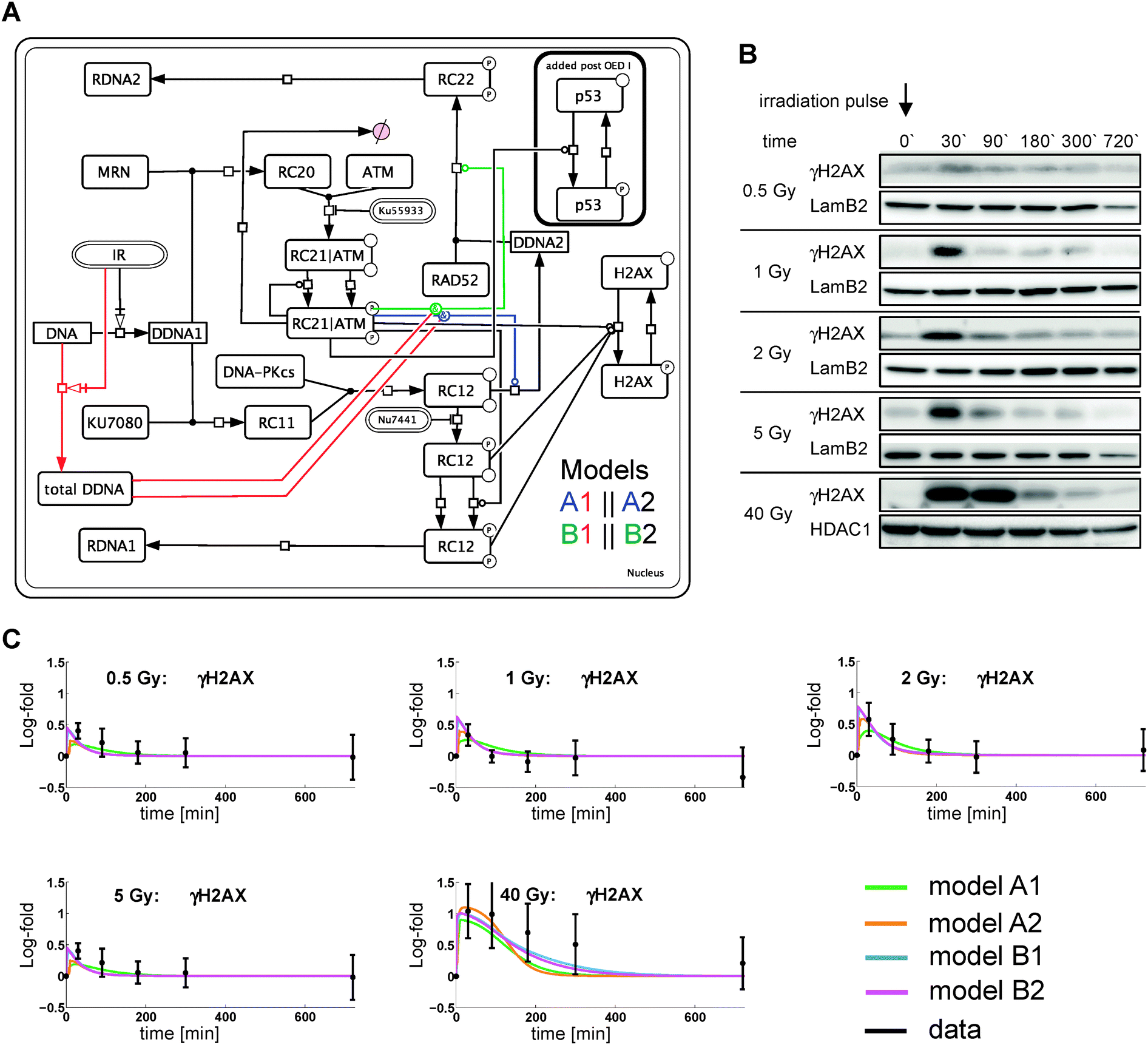 | ||
| Fig. 1 Network structure and initial data (OED 0). (A) The network structures of four different models based on meta-analysis is shown as an interaction graph. Interactions are modeled via state transitions (arrows with squares), enzyme catalysis (lines with circles) and complex formation (joined lines). Stimulus and inhibitors have round-edge boxes. Abbreviations: IR ionizing irradiation; DDNA1 initial, damaged DNA; RC11 Ku70/80 to DDNA1 association; RC12 Ku70/80-DNA-PKcs complex; RDNA1/2 repaired DNA (cNHEJ/aNHEJ or HR); RC20 MRN complex to DDNA1 association; RC21|ATM MRN-ATM complex at damage site; RC22 RAD52 mediated repair complex; DDNA2, unsuccessful cNHEJ repair moved to aNHEJ/HR. Four mechanisms have been considered for branching (A1, A2, B1, B2). A and B refer to the location of the catalytic activity of ATM and indices 1 and 2 refer to the kinetic law used. For model index 1 branching to DDNA2 is catalyzed by the total amount of damaged DNA. Index 2 does not use the total amount of damaged DNA. (B) MDCK cells were irradiated with different doses and the insoluble nuclear extracts were analyzed by immunoblot. Lamin B2 or HDAC1 served as loading control. (C) Model simulation and quantified experimental data for OED 0 using the estimated band intensities of γH2AX. Data represent mean ± 2SD of 3–5 independent experiments. | ||
Failure of DNA repair via cNHEJ potentially allows HR proteins to access the damage site. This is modeled by splitting the initial DSB pool (DDNA) into DDNA1 and DDNA2, whereby DDNA2 is associated to HR and/or alternative non-homologous end joining (aNHEJ).22 Phosphorylation of H2AX can be achieved by active DNA-PKcs or active ATM. We generated four alternative models describing various interplays between ATM, DNA-PKcs and γH2AX (Fig. 1A).
| OED | N data | N θ | N S | Fit statistics | Model A1 | Model A2 | Model B1 | Model B2 |
|---|---|---|---|---|---|---|---|---|
| 0 | 114 | 19 | 2 | χ 2 | 93.45 | 91.74 | 92.79 | 91.69 |
| p-Value χ2 | 4.09 × 10−01 | 4.59 × 10−01 | 4.28 × 10−01 | 4.60 × 10−01 | ||||
| p-Value AD3σ | 3.44 × 10−02 | 1.21 × 10−02 | 3.04 × 10−02 | 2.32 × 10−02 | ||||
| p-Value AD | 3.44 × 10−02 | 1.21 × 10−02 | 3.04 × 10−02 | 2.32 × 10−02 | ||||
| I | 147 | 19 | 7 | χ 2 | 135.98 | 131.53 | 125.84 | 125.64 |
| p-Value χ2 | 1.37 × 10−01 | 2.04 × 10−01 | 3.16 × 10−01 | 3.21 × 10−01 | ||||
| p-Value AD3σ | 1.38 × 10−01 | 1.84 × 10−01 | 9.22 × 10−02 | 5.64 × 10−02 | ||||
| p-Value AD | 2.12 × 10−01 | 1.84 × 10−01 | 9.22 × 10−02 | 5.64 × 10−02 | ||||
| II | 237 | 19 | 8 | χ 2 | 290.60 | 208.2 | 286.22 | 479.10 |
| p-Value χ2 | 1.35 × 10−04 | 4.83 × 10−01 | 2.60 × 10−04 | 0.00 | ||||
| p-Value AD3σ | 1.97 × 10−05 | 6.52 × 10−02 | 3.11 × 10−02 | 1.12 × 10−01 | ||||
| p-Value AD | 3.86 × 10−08 | 5.22 × 10−29 | 3.21 × 10−32 | 5.46 × 10−14 | ||||
To discriminate between models, we subsequently designed (i) an IR double-pulse (Fig. 2A–D) and (ii) an IR double-pulse in combination with kinase inhibitors (Fig. 3). The IR double-pulse was parameterized with 2 design variables, namely inter-pulse time D1 and second pulse dose D2, whereas the first pulse was fixed at 1 Gy. The objective was to maximize O = [Tred〈V〉〈S〉]T. Herein Tred is the reduced, modified T criterion to measure discriminative power,23 whereas 〈V〉, 〈S〉 represent mean model prediction variance and variance-entropy. The latter two criteria measure parameter information and distribution within the γH2AX signal (see Materials and methods).
 | ||
| Fig. 2 Parameterization of the stimulus design, design criteria and respective immunoblots. (A) Parameterization of the stimulus design for OED I/II. (B) Design criteria predicted from the model simulations are plotted over the feasible design space. The optimal design point for OED I DI* and corresponding criteria OI* = [Tred〈V〉〈S〉]T are indicated. (C) A representative immunoblot from an experiment based on DI* is shown. MDCK cells were irradiated as indicated and the insoluble nuclear extracts were analyzed by immunoblot. Lamin B2 served as loading control. (D) Corresponding model simulation describe the acquired data for γH2AX (model colors as in Fig. 1). Data represent mean ± 2SD of 3 independent experiments. (E) MDCK cells were irradiated as indicated. Inhibitors Ku55933 and Nu7441 were used at different concentrations and whole cell lysates were analyzed for p53-P and γH2AX. GAPDH served as loading control. Model simulation and quantified experimental data for p53-P are shown. Data of a single experiment. | ||
 | ||
| Fig. 3 (A) DII* is obtained as in Fig. 2(B). (B) MDCK were incubated with 1 μM of the indicated inhibitor and irradiated as indicated. The insoluble nuclear extracts were analyzed by immunoblot. HDAC1 served as loading control. (C) The corresponding model simulations compare the acquired data for γH2AX before and after OED II (mean ± 2SD of 2–4 independent experiments, model colors as in Fig. 1). | ||
For OED I, the optimal design DI* was chosen by trading off Tred, 〈V〉 and 〈S〉 (Fig. 2B). Recalibration of all models to data from OED 0 and I, and additional inclusion of p53-P data (Fig. 2E) from titration experiments did not allow for model discrimination (all p-values > α0.05 for both fit statistics; Table 1), but reduced prediction variances (Table 2).
| Criterion | OED I | OED II | ||
|---|---|---|---|---|
| Prediction | Final | Prediction | Final | |
| T*|T0* | 107.13|6.5 | 45.1|0.3 | 4.6 × 10−03|44.7 | 1.5 × 10−03|51.5 |
| T red*|Tred,0* | 0.05|3 × 10−3 | 0.02|1 × 10−04 | 28.2|0.3 | 9.3|0.3 |
| 〈V〉|〈V〉0 | 1.53|4 × 10−08 | 0.52|2 × 10−07 | 2.2|6 × 10−08 | 0.6|1 × 10−05 |
| 〈S〉|〈S〉0 | 7.05|2.26 | 7|2.29 | 20.1|7.5 | 5.1|3.1 |
Kinase inhibitors were employed for OED II to better dissect DNA-PKcs and ATM contributions. Titration of two highly specific inhibitors, namely Nu7441 and Ku55933 for DNA-PKcs and ATM, respectively, identified the optimal concentration for each. Further, we used the phosphorylation of p53 at S15 as a read-out to show the specificity of the inhibitors. Two successive pulses with different intensities (1 and 20 Gy) show in the immunoblot that the contribution of DNA-PKcs to this particular phosphorylation of p53 is marginal (Fig. 2E). This confirms earlier data.24,25
OED II was designed for three different inhibitor settings, namely Nu7441 and/or Ku55933. The estimated optimal design DII* potentially allowed for discrimination (Table 2, Tred* ≫ 1, Fig. 3A). The initial γH2AX peak showed a comparable reduction for both inhibitors. Phosphorylation of H2AX after the second pulse seemed to decay more rapidly for inhibited ATM compared to inhibited DNA-PKcs. Both inhibitors together showed synergistic effects on γH2AX (Fig. 3B).
According to the fit statistics of OED II (Table 1) only model A2 cannot be rejected in terms of χ2. However, we find significant AD p-values for all four models, whereas models A2 and B2 have non-significant AD3σp-values, which account only for residuals smaller than 3σ. This behavior may be attributed to outliers in one of the experimental conditions (Fig. 1C and 3C) owing to experimental variations or deficits of the models in describing experimental conditions of OED 0, I, II. We selected model A2 as the final model for further analysis, since it was the only model with p-values of χ2 and AD3σ statistics exceeding α0.05 for all 3 experimental runs.
2.2 Model predictions
 | ||
| Fig. 4 Model predictions for the dynamic contribution of DNA-PKcs and ATM to γH2AX. (A) Simulated time courses of active DNA-PKcs and ATM and resulting biphasic γH2AX activity for IR pulses of different dose levels (1 mGy to 100 Gy). At larger dose, ATM shows a damped oscillation as a result of a positive feedback (autophosphorylation), which contributes to peak level of γH2AX at doses above 10 Gy. (B) Model prediction of the corresponding dose response in terms of time points at maximal activity of γH2AX, DNA-PKcs and ATM. Thin lines indicate 95% confidence regions of the model predictions, estimated from simulations along the profile likelihood. (C) Ratio of maximal DNA-PKcs-P to ATM-P. Thin lines indicate 95% confidence region of the model predictions, estimated as in (B). | ||
According to the model predictions, phosphorylation of H2AX is biphasic, following a dose-independent temporal activation order: the first activation phase of γH2AX right after stimulation is associated to DNA-PKcs, whereat the second phase is linked to ATM-P (Fig. 4A). The γH2AX signal decays on the scale of hours and correlates with ATM-P. This dynamics of fast initial and prolonged response is known from coherent feed forward loops, which serve as a signal persistence detector.27 At doses below 1 dGy peak level of γH2AX is dominated by DNA-PKcs, whereas ATM dominates above 1 dGy (Fig. 4B and C). For larger dose levels, ATM auto-phosphorylation results into a prolonged activation phase, with γH2AX peak activity shifted from 10 minutes at 10 Gy to 40 minutes at 100 Gy.
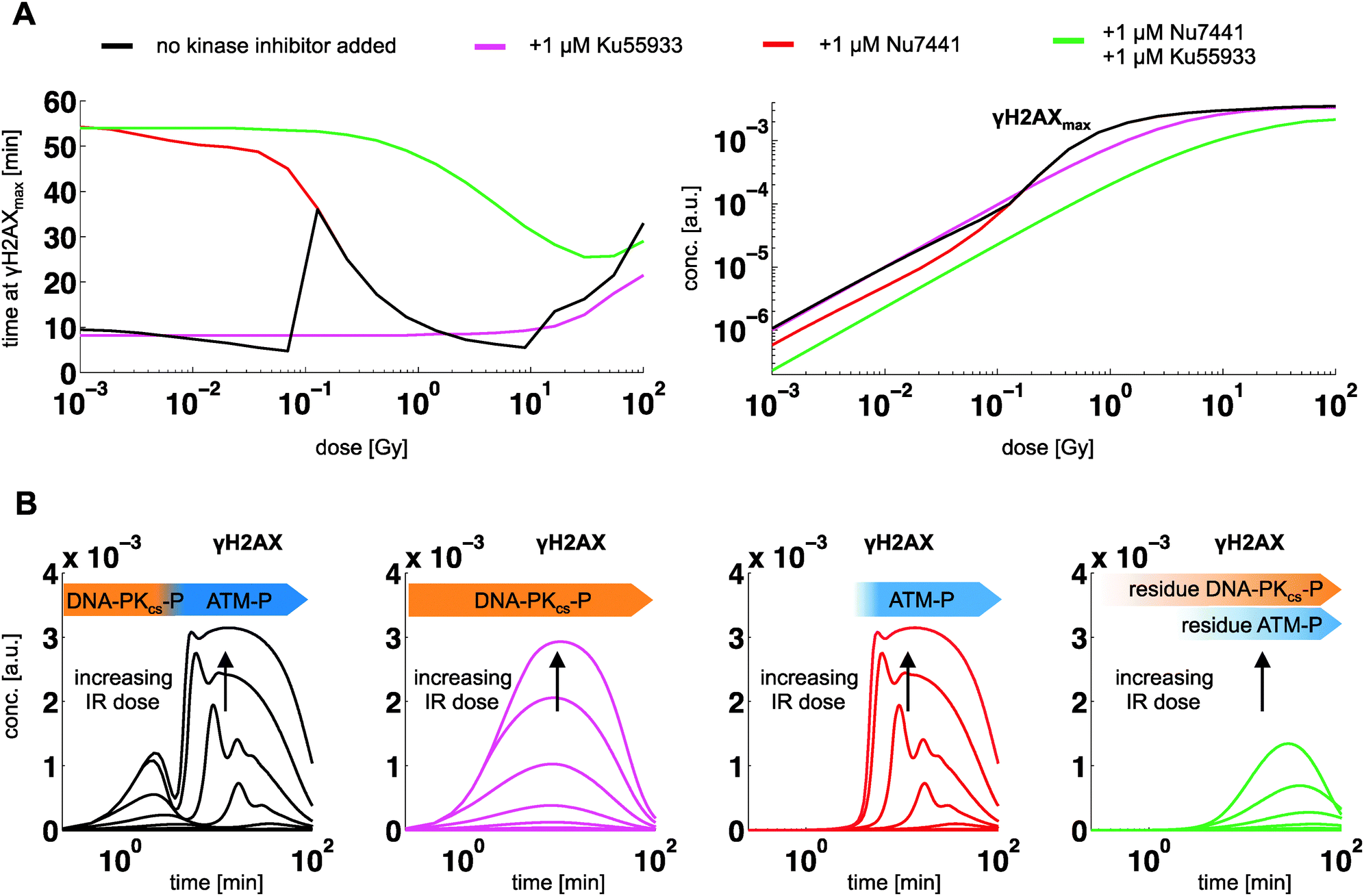 | ||
| Fig. 5 Model predictions for the dynamic contribution of DNA-PKcs and ATM to γH2AX at inhibition for different dose levels (1 mGy to 100 Gy). (A) Peak time and peak level of γH2AX for indicated inhibitors (color code), (B) corresponding time courses. | ||
3. Materials and methods
3.1 Cell culture and treatment with γ-irradiation
MDCK cells (ATCC CCL-34) were routinely cultured in RPMI-1640 supplemented with 10% fetal calf serum, glutamine and 100 U mL−1 penicillin and 100 μg mL−1 streptomycin, and incubated at 37 °C in a 5% CO2 humidified incubator. The MDCK cells were seeded at a density of 2 × 106 per 10 cm culture dish and cultured for 24 hours. The cells were irradiated with the Biobeam GM 2000 (Gamma-Service Medical GmbH, Germany) at a dose rate of 3.332 Gy min−1 using either single or double-pulse conditions. After a single pulse of 40, 5, 2, 1 or 0.5 Gy, the cells were harvested at 30, 90, 180, 300 and 720 minutes. The double-pulse consists of a single pulse of 1 Gy, followed 6 hours later by a second pulse of 20 Gy. The cells were harvested at 15, 35, 60, 160, 240, 370, 420 and 450 minutes. The inhibitors, Ku55933 (ATM, Tocris Bioscience, Germany) and Nu7441 (DNA-PKcs, Tocris Bioscience, Germany), used in the double-pulse setting, were added 30 minutes before first irradiation at a final concentration of 1 μM, either alone or together. The titration of the inhibitors were performed at 0, 0.1, 1, 10 and 0, 0.01, 0.1, 1 μM for Ku55933 and Nu7441, respectively. Both inhibitors belong to the class of ATP competitive inhibitors.10,283.2 Nuclear extraction, SDS-PAGE and Immunoblot
Cells were lysed in hypotonic cell lysis buffer (20 mM Tris/HCl pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 10 mM K2HPO4, 10% glycerol, 0.5 mM DTT) supplemented with 0.5 mM AEBSF, 1 mM sodium vanadate, 1 mM sodium molybdate, 10 mM sodium fluoride, 20 mM 2-phosphoglycerate and protease inhibitor mix (complete, Roche Germany). After addition of 1.25% NP-40, the cytosolic fraction was obtained by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 minutes. The nuclear pellet was resolved in 20 mM Tris/HCl pH 7.9, 420 mM KCl, 1.5 mM MgCl2, 10 mM K2HPO4, 10% glycerol, 0.2 mM EDTA, 0.5 mM DTT supplemented with the same inhibitors as before. The sample was incubated for 40 minutes on ice and centrifuged for 10 minutes at 13000 × g. The insoluble nuclear fraction was achieved by digesting the resulting pellet with nuclease (Calbiochem, Germany) at 37 °C for 30 minutes. The protein concentration was estimated using the BCA protein assay kit (Perbio Science, Germany). The samples were separated in Tris-Glycine gels (15%), transferred onto PVDF membranes (Millipore, Germany) and blocked for 1 h at room temperature with 5% skim milk in TBS-Tween (TBS-T). The primary antibodies were incubated overnight in 5% skim milk in TBS-T at 4 °C. The membranes were washed thrice in TBS-T. The appropriate HRP-conjugated secondary antibody was added at a dilution of 1:5000 in 5% skim milk in TBS-T for 1 hour at room temperature, followed by three washes in TBS-T. The membranes were developed using a chemiluminescence substrate (Millipore, Germany). The respective bands were visualized using the ChemoCam Imager (Intas, Germany), followed by the estimation of the band intensities using ImageJ.29
000 × g for 10 minutes. The nuclear pellet was resolved in 20 mM Tris/HCl pH 7.9, 420 mM KCl, 1.5 mM MgCl2, 10 mM K2HPO4, 10% glycerol, 0.2 mM EDTA, 0.5 mM DTT supplemented with the same inhibitors as before. The sample was incubated for 40 minutes on ice and centrifuged for 10 minutes at 13000 × g. The insoluble nuclear fraction was achieved by digesting the resulting pellet with nuclease (Calbiochem, Germany) at 37 °C for 30 minutes. The protein concentration was estimated using the BCA protein assay kit (Perbio Science, Germany). The samples were separated in Tris-Glycine gels (15%), transferred onto PVDF membranes (Millipore, Germany) and blocked for 1 h at room temperature with 5% skim milk in TBS-Tween (TBS-T). The primary antibodies were incubated overnight in 5% skim milk in TBS-T at 4 °C. The membranes were washed thrice in TBS-T. The appropriate HRP-conjugated secondary antibody was added at a dilution of 1:5000 in 5% skim milk in TBS-T for 1 hour at room temperature, followed by three washes in TBS-T. The membranes were developed using a chemiluminescence substrate (Millipore, Germany). The respective bands were visualized using the ChemoCam Imager (Intas, Germany), followed by the estimation of the band intensities using ImageJ.29
Antibodies used in this work were as follows: LaminB2 (sc-133722) and HDAC1 (sc-7872) were obtained from Santa Cruz (USA, CA). γH2AX (ab26350) was from Abcam (UK). The secondary anti-rabbit-HRP or anti-mouse-HRP antibodies were from Jackson ImmunoResearch Laboratories Inc. (USA, PA).
3.3 Building of a dynamic signaling model network of γH2AX activation
Initially, 4 dynamic models in the form of ordinary differential equation systems were derived from the network structures in Fig. 1A and implemented in MATLAB using the solver CVODES.30 Details on the choice of kinetic rate laws are given in the ESI† Section S2. After the poor discrimination performance of OED I, we extended the models to contain also p53. The tumor suppressor p53 is an important effector protein during DDR. Phosphorylation of p53 at Ser15 by ATM promotes its release from MDM2 and results in p53 activation.24,31 Activation of p53 by DNA-PKcs has also been described.32 However, DNA-PK−/− MEFs show normal p53 activation.25 We did not find evidence for a DNA-PKcs contribution to the p53 phosphorylation (Fig. 2E), which agrees with earlier data.33 Therefore, we implemented the p53 activation as an ATM-dependent process only. As described in detail in the ESI,† 19 kinetic and 8 scaling parameters were estimated by maximizing the likelihood function, whereas the variance has been estimated from data replicates. Parameter estimation was performed for each model in an iterative manner, according to the 3 datasets, OED 0/I/II. Optimization of the likelihood function was performed iteratively, using a hybrid strategy. We combined a genetic algorithm (‘ga’ function from the global optimization toolbox of MATLAB), which was used to obtain a population of suitable starting solutions for a local, gradient-based optimization. Here we used an interior-point algorithm (‘fmincon’ function from the optimization toolbox of MATLAB).Before analyzing DNA-PKcs, ATM and γH2AX dynamics with model A2, we performed an identifiability analysis based on the profile likelihood to assess the uniqueness of the model prediction and to also derive prediction uncertainty bands (see Fig. 4B and C). This analysis revealed that 8 kinetic parameters were not fully identifiable for the given optimization constraints, i.e. upper and lower bounds restricting the parameters to fall within 4 orders of magnitude. Six of these parameters were non-significant at the upper bound, whereas the other two were non-significant at the lower bound. One parameter was structurally non-identifiable. The non-identifiable parameters were not decisive for the question of kinase contribution to H2AX phosphorylation. More details on the identifiability analysis, parameter dependencies and impact on the prediction power are given in the ESI† in Section 2.
3.4 Experimental design criteria for model identification
Model identification is the process of comparing plausibility amongst models from a pool of competing computational models in the light of given experimental data. Plausibility is typically derived from some kind of lack-of-fit measure, for instance χ2 statistics. Experimental design for model identification aims at generating new experimental conditions and therefore data, to support this identification process in an optimal way using the models at hand. In the early phase of modeling a biochemical system with ODEs, parameters are typically very uncertain. Consequently, model predictions including design criteria are uncertain as well. Accounting for these uncertainties during design robustifies the optimal experiment against these uncertainties. In this work we use a multi criterion approach to identify optimal stimulus designs for model identification. We use three criteria that measure discriminative power, parameter information and its distribution along the time points of the model predictions for γH2AX. The discriminative power is measured with the reduced, modified T criterion,23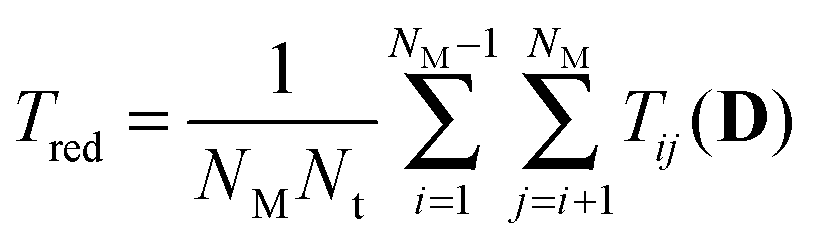 , with
, with 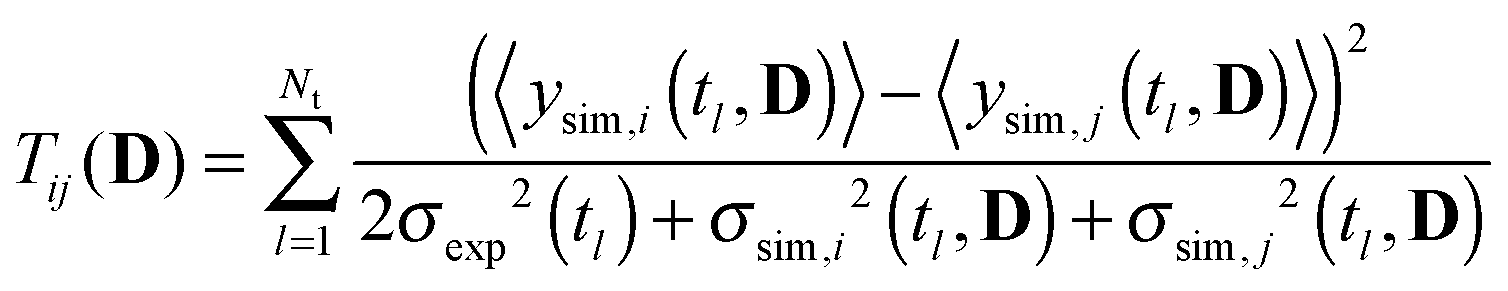 , where 〈ysim,i(tl, D)〉 represents the expected prediction of γH2AX of model i (total number NM) at time point tl (total number Nt). Measurement variances σexp2(tl) are interpolated sample variances averaged over all available experimental conditions. Expected model predictions and their variances σsim,i2(tl, D) have been derived with the sigma point method as shown in Flassig and Sundmacher.34 Expectation is taken with respect to the parameters, whereas parameter variance–covariances were derived from the χ2 Hessian. Parameter information was measured by the mean variance over time points of model predictions according to
, where 〈ysim,i(tl, D)〉 represents the expected prediction of γH2AX of model i (total number NM) at time point tl (total number Nt). Measurement variances σexp2(tl) are interpolated sample variances averaged over all available experimental conditions. Expected model predictions and their variances σsim,i2(tl, D) have been derived with the sigma point method as shown in Flassig and Sundmacher.34 Expectation is taken with respect to the parameters, whereas parameter variance–covariances were derived from the χ2 Hessian. Parameter information was measured by the mean variance over time points of model predictions according to  . Shannon's entropy is used to measure the variance distribution over time points and model predictions according to
. Shannon's entropy is used to measure the variance distribution over time points and model predictions according to  with normalized variances according to
with normalized variances according to 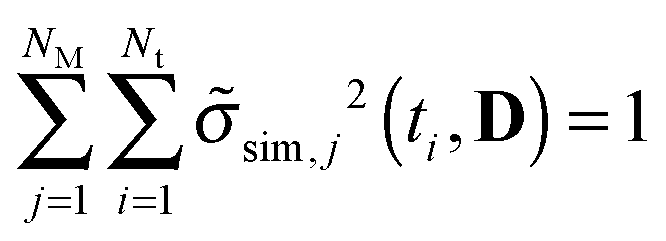 . In each experimental design, we chose the best design point as the trade-off between maximal Tred, 〈V〉 and 〈S〉. Maximal Tred yields best discrimination, maximal 〈V〉 ensures large sensitivity of the parameters and maximal 〈S〉 represents maximal homogenous variance distribution along time points and model predictions.
. In each experimental design, we chose the best design point as the trade-off between maximal Tred, 〈V〉 and 〈S〉. Maximal Tred yields best discrimination, maximal 〈V〉 ensures large sensitivity of the parameters and maximal 〈S〉 represents maximal homogenous variance distribution along time points and model predictions.
The evaluation of the objective in OED I was based on time points t = [0 15 35 60 160 240 370 420 450]T minutes. The first 6 time points were chosen from simulating OED 0 conditions to fully capture rising and falling flanks of the initial γH2AX peak, whereas the remaining time points were placed based on the estimated second signal peak. For OED II design criteria were evaluated at the time points used in OED I.
4. Conclusions
Here we report an iterative workflow combining experimental work, computational modeling and experimental design methodologies to shed light on the interplay of two PIKK family members (DNA-PKcs and ATM) to the rapid histone H2AX phosphorylation in the context of DNA damage sensing upon γ-irradiation. By performing optimized dynamic stimulation experiments, we generated an extensive set of time-resolved data to identify a computational model for analyzing DNA-PKcs-P, ATM-P and γH2AX dynamics. A parameter identifiability analysis revealed that the computational model can be used to predict internal state dynamics, e.g. phosphorylation of DNA-PKcs and ATM. With a predictive model at hand, we could then investigate the fast phosphorylation kinetics of DNA-PKcs, ATM and H2AX post irradiation without the need of direct kinase activity measurements, thus reducing confounding effects from experimental manipulations.Our model simulations show that H2AX phosphorylation is biphasic, with initial and succeeding phases associated to DNA-PKcs and ATM, respectively, in which the individual contributions to peak level of γH2AX are dose-dependent. It is tempting to link the dose-dependent biphasic response of γH2AX observed in silico to the known biphasic signaling responses of cNHEJ and HR, that is fast DNA-PKcs and slower ATM-related repair activity.22 In fact, following DNA-PKcs inhibition Davidson et al.35 have shown that HR activity is increased. Further, Neal et al.8 showed that DNA-PKcs enzymatic activity inhibits HR in a titratable fashion. From simulating DNA-PKcs inhibition we hypothesize that this is a consequence of delayed γH2AX activation, associated chromatin remodeling and DNA repair initiation of cNHEJ. We further conclude that DNA-PKcs and ATM have distinct roles in H2AX phosphorylation equipping cells with a signal persistence detection function, i.e. fast initial response (DNA-PKcs) and delayed signal attenuation (ATM). This ensures reliable damage detection and repair signaling.
Author contribution
RF performed data processing, statistical analysis and computational modeling. GM performed experiments and quantified data. CT performed the experiments. KS and MN designed the research concept. RF, GM, KS and MN wrote the manuscript.Acknowledgements
This work was supported by the German Federal Ministry of Education and Research (SysTec, 101-31P5900) and the Ministry of Education of Saxony-Anhalt (XD3639HP/0306).References
- A. Ciccia and S. J. Elledge, Mol. Cell, 2010, 40, 179–204 CrossRef CAS PubMed.
- M. J. Kruhlak, A. Celeste, G. Dellaire, O. Fernandez-Capetillo, W. G. Müller, J. G. McNally, D. P. Bazett-Jones and A. e. Nussenzweig, J. Cell Biol., 2006, 172, 823–834 CrossRef CAS PubMed.
- J. Yuan, R. Adamski and J. Chen, FEBS Lett., 2010, 584, 3717–3724 CrossRef CAS PubMed.
- M. Stucki and S. P. Jackson, DNA Repair, 2006, 5, 534–543 CrossRef CAS PubMed.
- I. M. Ward and J. Chen, J. Biol. Chem., 2001, 276, 47759–47762 CrossRef CAS PubMed.
- H. Wang, M. Wang, H. Wang, W. Bocker and G. Iliakis, J. Cell. Physiol., 2005, 202, 492–502 CrossRef CAS PubMed.
- M. Shrivastav, C. A. Miller, L. P. De Haro, S. T. Durant, B. P. C. Chen, D. J. Chen and J. A. Nickoloff, DNA Repair, 2009, 8, 920–929 CrossRef CAS PubMed.
- J. A. Neal, V. Dang, P. Douglas, M. S. Wold, S. P. Lees-Miller and K. Meek, Mol. Cell. Biol., 2011, 31, 1719–1733 CrossRef CAS PubMed.
- S. Burma, B. P. Chen, M. Murphy, A. Kurimasa and D. J. Chen, J. Biol. Chem., 2001, 276, 42462–42467 CrossRef CAS PubMed.
- I. Hickson, Y. Zhao, C. J. Richardson, S. J. Green, N. M. B. Martin, A. I. Orr, P. M. Reaper, S. P. Jackson, N. J. Curtin and G. C. M. Smith, Cancer Res., 2004, 64, 9152–9159 CrossRef CAS PubMed.
- F. A. Cucinotta, J. M. Pluth, J. A. Anderson, J. V. Harper and P. O'Neill, Radiat. Res., 2008, 169, 214–222 CrossRef CAS PubMed.
- Y. Li, P. Reynolds, P. O'Neill and F. A. Cucinotta, PLoS One, 2014, 9, e85816 Search PubMed.
- W. Friedland, P. Jacob and P. Kundrát, Radiat. Prot. Dosim., 2011, 143, 542–548 CrossRef CAS PubMed.
- R. Taleei and H. Nikjoo, Radiat. Res., 2013, 179, 530–539 CrossRef CAS PubMed.
- D. Dolan, G. Nelson, A. Zupanic, G. Smith and D. Shanley, PLoS One, 2013, 8, e55190 CAS.
- W. Friedland, P. Jacob and P. Kundrát, Radiat. Res., 2010, 173, 677–688 CrossRef CAS PubMed.
- K. Mouri, J. C. Nacher and T. Akutsu, PLoS One, 2009, 4, e5131 Search PubMed.
- E. Mladenov and G. Iliakis, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2011, 711, 61–72 CrossRef CAS PubMed.
- A. Kinner, W. Wu, C. Staudt and G. Iliakis, Nucleic Acids Res., 2008, 36, 5678–5694 CrossRef CAS PubMed.
- R. Poltz and M. Naumann, BMC Syst. Biol., 2012, 6, 125 CrossRef CAS PubMed.
- D. W. Chan, B. P.-C. Chen, S. Prithivirajsingh, A. Kurimasa, M. D. Story, J. Qin and D. J. Chen, Genes Dev., 2002, 16, 2333–2338 CrossRef CAS PubMed.
- J. A. Neal and K. Meek, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2011, 711, 73–86 CrossRef CAS PubMed.
- G. Buzzi-Ferraris and P. Forzatti, Chem. Eng. Sci., 1983, 38, 225–232 CrossRef CAS.
- C. E. Canman, D. S. Lim, K. A. Cimprich, Y. Taya, K. Tamai, K. Sakaguchi, E. Appella, M. B. Kastan and J. D. Siliciano, Science, 1998, 281, 1677–1679 CrossRef CAS.
- G. S. Jimenez, F. Bryntesson, M. I. Torres-Arzayus, A. Priestley, M. Beeche, S. Saito, K. Sakaguchi, E. Appella, P. A. Jeggo, G. E. Taccioli, G. M. Wahl and M. Hubank, Nature, 1999, 400, 81–83 CrossRef CAS PubMed.
- D. Davidson, L. Amrein, L. Panasci and R. Aloyz, Front. Pharmacol., 2013, 4 DOI:10.3389/fphar.2013.00005.
- S. Mangan and U. Alon, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11980–11985 CrossRef CAS PubMed.
- J. J. J. Leahy, B. T. Golding, R. J. Griffin, I. R. Hardcastle, C. Richardson, L. Rigoreau and G. C. M. Smith, Bioorg. Med. Chem. Lett., 2004, 14, 6083–6087 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- A. Hindmarsh, P. Brown, K. Grant, S. Lee, R. Serban, D. Shumaker and C. Woodward, ACM Trans. Math. Softw., 2005, 31, 363–396 CrossRef.
- S.-Y. Shieh, M. Ikeda, Y. Taya and C. Prives, Cell, 1997, 91, 325–334 CrossRef CAS.
- S. P. Lees-Miller, K. Sakaguchi, S. J. Ullrich, E. Appella and C. W. Anderson, Mol. Cell. Biol., 1992, 12, 5041–5049 CAS.
- F. S. Shaheen, P. Znojek, A. Fisher, M. Webster, R. Plummer, L. Gaughan, G. C. M. Smith, H. Y. Leung, N. J. Curtin and C. N. Robson, PLoS One, 2011, 6, e20311 CAS.
- R. J. Flassig and K. Sundmacher, Bioinformatics, 2012, 28, 3089–3096 CrossRef CAS PubMed.
- D. Davidson, Y. Coulombe, V. Martinez-Marignac, L. Amrein, J. Grenier, K. Hodkinson, J.-Y. Masson, R. Aloyz and L. Panasci, Invest. New Drugs, 2012, 30, 1248–1256 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data supporting the results of this article, a detailed description on data processing, model equations, parameter estimation, values and confidence intervals, identifiability and model analysis as well as the data are provided in the supplementary material. See DOI: 10.1039/c4mb00093e |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |