Complementarity between distance- and probability-based methods of gene neighbourhood identification for pathway reconstruction
Junha
Shin
,
Tak
Lee
,
Hanhae
Kim
and
Insuk
Lee
*
Department of Biotechnology, College of Life Science and Biotechnology, Yonsei University, Seoul, Korea. E-mail: insuklee@yonsei.ac.kr; Fax: +82 822 362 7265; Tel: +82 822 2123 5559
First published on 23rd October 2013
Abstract
Identifying gene neighbourhoods using either distance- or probability-based measures has proven effective in retrieving co-functional links. We report that these two approaches are highly complementary, with differential sensitivity for the core pathway links. We demonstrate that integrating these measures improves prediction of both pathways and phenotypes.
The layout of a genome represents an evolutionary footprint, reflecting functional and regulatory constraints upon genes that operate in the same pathways and functional modules. As a result, genomic context has proven useful in discovering the functional coupling of genes.1 The operon, in which genes belonging to the same biological pathway are encoded in a co-transcriptional unit, is one example of a genomic context that has been helpful for identifying functional links in prokaryotes. In the past, co-functional links were often inferred based on the conventional definition of the operon: co-transcriptional genes transcribed in the same direction, with intergenic regions between adjacent genes measuring a few hundred base pairs or less.2,3 More recently, it has been shown that neighbouring genes transcribed in opposite directions can be functionally associated as well,4 significantly increasing the number of co-functional links inferred based on gene clusters.5
The above criteria for identifying gene neighbourhoods are based on the chromosomal distance between orthologs of two query genes in a prokaryotic genome. An alternative approach is to calculate the probability of a given gene neighbourhood being conserved across prokaryotic genomes.6 Therefore, the identification of gene neighbourhoods is based on either distance-based measures or probability-based measures of evolutionary conservation between two genes. We will refer to these alternative approaches to gene neighbourhood identification as ‘distance-based gene neighbourhood’ (DGN) and ‘probability-based gene neighbourhood’ (PGN). Previous studies have employed only DGN or PGN measures to identify co-functional links. No study has yet compared these measures for the purpose of pathway reconstruction. Do they identify similar co-functional links? Are they equally good at predicting functional links, including in higher eukaryotes such as animals and plants? Would the integration of DGN and PGN measures improve prediction of pathways and phenotypes? To address these important questions, we have systematically compared these alternative methods of gene neighbourhood identification and assessed their predictive power, analyzing each alone and then integrating the two.
First, we used 1748 prokaryote genomes (122 archaea and 1626 bacteria) to search for conserved gene neighbourhoods between query gene pairs. For the DGN measure, we took the median value of chromosomal distance between orthologs of the two query genes with PBLAST E-values <1 across the 1748 reference prokaryote genomes. Each median distance value was normalized using the number of genomes in which orthologs of the two genes co-occurred, giving greater weight to gene pairs conserved in a larger number of prokaryote genomes. For the PGN measure, we used the probability score proposed by Bowers et al., as described below.6 The probability of two genes being separated by fewer than d genes in a genome containing N genes is:
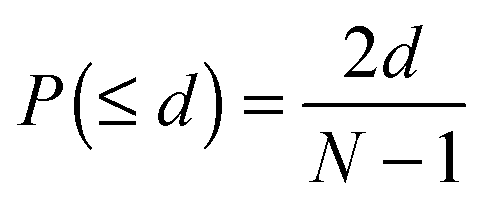
We calculate the product of the above probability across the m reference genomes containing orthologs of the two query genes:

To calculate the likelihood that two genes belong to the same conserved neighbourhood, we determine the probability of obtaining a value of X that is smaller than the observed value:

Using only a single representative genome from each genus in the reference genome set (66 archaea and 480 bacteria), we found that PGN performed better.
Subsequently, we determined whether both DGN and PGN could effectively infer co-functional links between human genes. The predictive power of gene neighbourhood scores for such links was determined using the percentage of annotated gene pairs that share any gene ontology biological process (GO-BP) term.7 Both DGN and PGN scores proved effective predictors of known functional links in humans (Fig. 1).
 | ||
| Fig. 1 The predictive power of distance- and probability-based measures of gene neighbourhood for co-functional links between human genes is demonstrated by the relationship between the percentage of linked gene pairs (y-axis) that share any Gene Ontology biological process term and the distance or probability scores (x-axis). Both DGN and PGN are effective predictors. | ||
Next, we constructed functional gene networks based on both DGN and PGN predictors for four widely divergent species: Escherichia coli, Saccharomyces cerevisiae, Arabidopsis thaliana, and Homo sapiens. We used the same benchmarking methods, based on log likelihood score schemes, as were employed in our previous work on network construction.8–10 We compared DGN and PGN networks in the four different species using precision–recall curves, which show the relationship between the likelihood of co-functional links, based on GO-BP annotation, and the coverage of coding genomes (Fig. 2A). In E. coli, S. cerevisiae, and A. thaliana, DGN performed slightly better than PGN. By contrast, PGN performed significantly better in humans. To determine whether integration of the two measures could improve prediction of co-functional links, we integrated the two networks using the weighted sum method employed in our previous network construction work.8–10 Naïve Bayes integration provided the optimal performance for A. thaliana, but for all other three species integration performed best with taking correlation between DGN and PGN into account. We found that integration of DGN and PGN improved pathway prediction in all four species tested.
 | ||
| Fig. 2 (A) Precision-recall curves for co-functional gene networks using DGN, PGN, and an integrated method (INT), depicted by the log likelihood score (LLS, y-axis) and the percentage coverage of coding genomes (x-axis). (B) Venn diagrams show that DGN and PGN networks exhibit high complementarity (i.e., a small intersection) for links (L), yet low complementarity (i.e., a large intersection) for genes (G) in all four species tested. | ||
The improvement obtained via integrating the DGN and PGN networks implies information complementarity between these two alternative measures of a gene neighbourhood. As expected, we found high complementarity for links, albeit not for genes, between DGN and PGN in all four species tested (Fig. 2B). This limited link complementarity was confirmed by analyzing only links validated by GO-BP (data not shown). The modest extension of genome coverage conferred by network integration is consistent with the low level of gene complementarity between the two measures. These results suggest that DGN and PGN identify largely non-overlapping functional links for similar pathways and that integration improves the completeness of networks for those pathways.
Genomic neighbourhoods were used to construct a co-functional network covering 80–90% of the coding genome of E. coli (Fig. 2A). Unfortunately, the impressive genome coverage of co-functional gene networks obtained using gene neighbourhoods did not extend to eukaryotes. Gene neighbourhood networks covered no more than 40%, 10%, and 15% of the coding genomes of S. cerevisiae, A. thaliana, and humans, respectively. Nevertheless, gene neighbourhood networks in all three of these eukaryotes retrieved 2000–3000 genes, suggesting that the total number of coding genes with co-functional links that can be identified based on genomic vicinity is similar from bacteria to higher eukaryotes, including humans.
To learn more about the origin of the complementarity between the two measures of gene neighbourhood, we investigated evolutionary and network properties of DGN and PGN-identified links. First, we found that the linked genes identified by PGN co-occurred in a relatively small number of reference genomes compared to those identified by DGN (Fig. 3A). Next, we observed contrasting modularity between the two network types: the network clustering co-efficient11 for PGN is high, while that for DGN is low (Fig. 3B). In the equation used to calculate the probability of a gene neighbourhood, even a few reference genomes with two orthologs interrupted by many genes (i.e., a large d value) can significantly increase the probability that the observed genomic vicinity has occurred by chance. Therefore, when using the PGN measure, only genomic vicinities that are highly conserved will be interpreted as co-functional links between genes. By contrast, DGN, which is based on the median distance scores across all reference genomes, proves robust in the presence of a modest number of orthologous gene pairs that have lost genomic vicinity. This loss can occur as a result of attenuated functional constraint under natural selection, due to processes such as functional repurposing.12 Therefore, we hypothesized that the PGN measure tends to capture the links between genes that belong to the highly conserved pathway cores, while the DGN measure does not. To test this hypothesis, we assessed enrichment of known protein complexes in DGN and PGN networks. Because protein complexes generally comprise the core of biological processes, if PGN primarily retrieves core pathway links, it will show greater enrichment for protein complexes than DGN. Using yeast protein complexes annotated by the Munich Information Center for Protein Sequences protein interaction resource on yeast (MPact)13 and human protein complexes annotated by the Comprehensive Resource of Mammalian protein complexes (CORUM),14 we found that the links identified by PGN were more enriched for protein complexes than those identified by DGN. In yeast, for example, protein complexes accounted for 14% and 4% of the top 3000 links identified by PGN and DGN measures, respectively (Fig. 4). This bias towards links comprising the core parts of pathways is also consistent with the high clustering coefficient (i.e., high modularity) observed in PGN networks (Fig. 3B).
 | ||
| Fig. 3 Comparison of DGN and PGN networks using (A) the distribution of the number of reference prokaryote genomes in which orthologs of two query genes co-occurred (i.e., were conserved together in the same prokaryote genome), and (B) the clustering coefficient (i.e., modularity) for a given network size. In the box-and-whisker plots, the boundaries of the box represent the first and third quartiles, the whiskers represent the 10th and 90th percentiles, and the black circles represent individual outliers. | ||
 | ||
| Fig. 4 Enrichment of DGN and PGN networks for yeast (S. cerevisiae) and human (H. sapiens) protein complexes, based on the Munich Information Center for Protein Sequences protein interaction resource on yeast (MPact) and the Comprehensive Resource of Mammalian protein complexes (CORUM), respectively. Enrichment was measured using the percentage of linked gene pairs that share any MIPS complex term (y-axis) for a given network size (x-axis). | ||
Co-functional gene networks have been used to predict phenotypes in various organisms, including humans.15 We investigated whether the high complementarity of the two measures of gene neighbourhood could improve phenotype prediction in higher eukaryotes, in particular with regard to human diseases. As for the pathway prediction test described above (Fig. 2B), only 10% of human coding genes could be functionally coupled using gene neighbourhood links. All linked genes were ancient, conserved across disparate phyla, from bacteria to human. We assessed whether these ancient co-functional links could predict human diseases using disease ontology (DO) annotation.16 Phenotype predictive power was measured by determining the area under the receiver operating characteristic curve (AUC) of network-based predictions, using the Gaussian field label propagation algorithm.17,18 Using an integrated DGN/PGN approach, we found 15 DO terms that were highly predictive (AUC ≥ 0.7) (Table 1). Using only DGN or PGN results in a loss of 6–7 of these disease terms. Our results suggest that the complementarity of these two gene neighbourhood measures improves phenotype predictions as well as pathway predictions, including in higher eukaryotes such as humans.
| a High prediction powers (AUC ≥ 0.7) are denoted by a gray background. |
|---|

|
In summary, we demonstrate information complementarity between two alternative measures of gene neighbourhood: DGN and PGN. This complementarity is rooted in the greater sensitivity of PGN to core pathway links. Linkage complementarity improves prediction of not only co-functional links but also phenotypes, including human diseases. Therefore, we propose integration of distance-based and probability-based measures as an optimal way to use gene neighbourhoods in pathway reconstruction.
Acknowledgements
This work was supported by the National Research Foundation of Korea (2010-0017649, 2012M3A9B4028641, 2012M3A9C7050151) and the Next-Generation BioGreen 21 Program (SSAC, PJ009029) to I.L.References
- M. Y. Galperin and E. V. Koonin, Nat. Biotechnol., 2000, 18, 609–613 CrossRef CAS PubMed.
- T. Dandekar, B. Snel, M. Huynen and P. Bork, Trends Biochem. Sci., 1998, 23, 324–328 CrossRef CAS.
- R. Overbeek, M. Fonstein, M. D'Souza, G. D. Pusch and N. Maltsev, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2896–2901 CrossRef CAS.
- J. O. Korbel, L. J. Jensen, C. von Mering and P. Bork, Nat. Biotechnol., 2004, 22, 911–917 CrossRef CAS PubMed.
- L. J. Jensen, M. Kuhn, M. Stark, S. Chaffron, C. Creevey, J. Muller, T. Doerks, P. Julien, A. Roth, M. Simonovic, P. Bork and C. von Mering, Nucleic Acids Res., 2009, 37, D412–D416 CrossRef CAS PubMed.
- P. M. Bowers, M. Pellegrini, M. J. Thompson, J. Fierro, T. O. Yeates and D. Eisenberg, Genome Biol., 2004, 5, R35 CrossRef PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 CrossRef CAS PubMed.
- I. Lee, B. Ambaru, P. Thakkar, E. M. Marcotte and S. Y. Rhee, Nat. Biotechnol., 2010, 28, 149–156 CrossRef CAS PubMed.
- I. Lee, U. M. Blom, P. I. Wang, J. E. Shim and E. M. Marcotte, Genome Res., 2011, 21, 1109–1121 CrossRef CAS PubMed.
- I. Lee, S. V. Date, A. T. Adai and E. M. Marcotte, Science, 2004, 306, 1555–1558 CrossRef CAS PubMed.
- D. J. Watts and S. H. Strogatz, Nature, 1998, 393, 440–442 CrossRef CAS PubMed.
- A. Frost, M. G. Elgort, O. Brandman, C. Ives, S. R. Collins, L. Miller-Vedam, J. Weibezahn, M. Y. Hein, I. Poser, M. Mann, A. A. Hyman and J. S. Weissman, Cell, 2012, 149, 1339–1352 CrossRef CAS PubMed.
- U. Guldener, M. Munsterkotter, M. Oesterheld, P. Pagel, A. Ruepp, H. W. Mewes and V. Stumpflen, Nucleic Acids Res., 2006, 34, D436–D441 CrossRef PubMed.
- A. Ruepp, B. Waegele, M. Lechner, B. Brauner, I. Dunger-Kaltenbach, G. Fobo, G. Frishman, C. Montrone and H. W. Mewes, Nucleic Acids Res., 2010, 38, D497–D501 CrossRef CAS PubMed.
- I. Lee, Anim. Cells Syst., 2013, 17, 75–79 CrossRef.
- L. M. Schriml, C. Arze, S. Nadendla, Y. W. Chang, M. Mazaitis, V. Felix, G. Feng and W. A. Kibbe, Nucleic Acids Res., 2012, 40, D940–D946 CrossRef CAS PubMed.
- S. Mostafavi, D. Ray, D. Warde-Farley, C. Grouios and Q. Morris, Genome Biol., 2008, 9(Suppl 1), S4 CrossRef PubMed.
- P. I. Wang and E. M. Marcotte, J. Proteomics, 2010, 73, 2277–2289 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |