 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Yeast cells with impaired drug resistance accumulate glycerol and glucose†
Duygu
Dikicioglu
ab,
Sebnem
Oc
b,
Bharat. M.
Rash
c,
Warwick B.
Dunn
d,
Pınar
Pir
a,
Douglas B.
Kell
e,
Betul
Kirdar
b and
Stephen G.
Oliver
*ab
aCambridge Systems Biology Centre & Department of Biochemistry, University of Cambridge, CB2 1GA, Cambridge, UK. E-mail: sgo24@.cam.ac.uk; dd345@cam.ac.uk; pnarpir@gmail.com; Tel: +44 (0)1223 333 667
bDepartment of Chemical Engineering, Bogazici University, Bebek 34342, Istanbul, Turkey. E-mail: kirdar@boun.edu.tr; sebnem.oc@gmail.com; Tel: +90 212 359 7126
cFaculty of Life Sciences, University of Manchester, M13 9PT, Manchester, UK. E-mail: bharat.rash@manchester.ac.uk; Tel: +44 (0)161 275 5376
dManchester Centre for Integrative Systems Biology, University of Manchester, M1 7DN, Manchester, UK. E-mail: Warwick.Dunn@manchester.ac.uk; Tel: +44 (0)161 30 65146
eSchool of Chemistry and the Manchester Institute of Biotechnology, The University of Manchester, 131 Princess St, Manchester M1 7DN, UK. E-mail: dbk@manchester.ac.uk; Tel: +44 (0)161 306 4492
First published on 22nd October 2013
Abstract
Multiple drug resistance (MDR) in yeast is effected by two major superfamilies of membrane transporters: the major facilitator superfamily (MFS) and the ATP-binding cassette (ABC) superfamily. In the present work, we investigated the cellular responses to disruptions in both MFS (by deleting the transporter gene, QDR3) and ABC (by deleting the gene for the Pdr3 transcription factor) transporter systems by growing diploid homozygous deletion yeast strains in glucose- or ammonium-limited continuous cultures. The transcriptome and the metabolome profiles of these strains, as well as the flux distributions in the optimal solution space, reveal novel insights into the underlying mechanisms of action of QDR3 and PDR3. Our results show how cells rearrange their metabolism to cope with the problems that arise from the loss of these drug-resistance genes, which likely evolved to combat chemical attack from bacterial or fungal competitors. This is achieved through the accumulation of intracellular glucose, glycerol, and inorganic phosphate, as well as by repurposing genes that are known to function in other parts of metabolism in order to minimise the effects of toxic compounds.
Introduction
The major metabolic pathways, as well as the pathways for DNA repair and cell cycle control, are highly conserved throughout the eukaryotic kingdom.1 Hence, the functional and structural similarities between the cells of fungal pathogens and those of their human hosts make the treatment of fungal infections much harder, and the development of novel therapies much slower, than has been the case for bacterial infections through the use of antibiotics. The obstacles presented in treating a fungal infection are comparable to the specificity challenges that are faced in the treatment of cancers.2Populations of pathogenic fungi adapt to the presence of antifungal drugs by switching on general resistance mechanisms3 that are also used to deal with harmful chemicals in their environment. Interestingly, adaptations to chemical insults are similar between fungi and those observed in patients that develop resistance to chemotherapeutic agents, indicating that such mechanisms are highly conserved. In both fungi and humans, increases in the activity of cellular multidrug efflux pumps may be involved in resistance mechanisms.2,3 Such adaptations limit the therapeutic potential of both anti-fungal and anti-tumour drugs.4 Thus, preventing the development of drug resistance via efflux would result in more efficient uptake and utilization of drugs. The similarity of yeast's drug resistance mechanisms to those of humans makes it an ideal model with which to study these phenomena, and several studies have been conducted where S. cerevisiae is used as a model organism for the identification of novel mechanisms of resistance.4–7
Multidrug resistance (MDR) in S. cerevisiae is mediated by efflux pumps that belong to two major super-families of membrane transporters: the major facilitator super-family (MFS) and the ATP-binding cassette (ABC) super-family.8 In Saccharomyces cerevisiae, the MFS consists of the proton-coupled multidrug efflux system.9 Most of the 23 genes of this system are thought to be involved in multidrug resistance and their protein products contain either 12 or 14 predicted membrane-spanning segments and, on this basis, they are divided, respectively, into the DHA1 and DHA2 sub-families.10
The DHA1 sub-family includes the Qdr1, Qdr2 and Qdr3 transporters. Qdr1p confers resistance to the anti-fungal agents ketoconazole and fluconazole as well as to quinidine (an isomer of quinine) and to the carbamate herbicide, barban.9,11 Qdr2p also confers resistance to quinidine and barban, as well as to the anti-cancer agents, bleomycin and cisplatin.10,11 The range of compounds to which Qdr3p confers resistance is very similar to that of Qdr2p, the gene conferring resistance to a range of inhibitory compounds that are structurally and functionally unrelated, including the anti-malarial and anti-arrhythmic drug quinidine, barban, and the anti-cancer drugs cisplatin and bleomycin.11 Recent reports proposed a novel role for Qdr3p in polyamine homeostasis, specifically in that of spermine and spermidine but not of putrescine, through the maintenance of plasma membrane potentials. This contrasted with what was observed for Qdr2p, which conferred resistance to all three polyamines and this role was found to be associated with K+ homeostasis. The inability of QDR3 expression to rescue qdr2Δ strains from polyamine susceptibility was reported, suggesting different roles for the two transporters in conferring polyamine stress tolerance.12
The ABC super-family of MDR proteins includes transporters as well as the transcription factors that regulate them. ABC transporters utilize ATP hydrolysis to drive drug extrusion.9 Of the 30 genes encoding the ABC proteins in yeast, the ones involved in the facilitation of pleiotropic drug resistance (PDR) are very similar to the multidrug resistance routes that occur in mammalian cells, parasites, fungal pathogens, and bacteria. The PDR sub-family is the largest and best-characterized ABC sub-family in S. cerevisiae, consisting of transporter genes as well as transcription factors binding to the pleiotropic drug response elements (PDRE) on their target genes.13,14 Pdr1p and Pdr3p are the transcriptional regulators of the ABC transporter genes and neither is directly responsible for drug resistance.15 Pdr1p and Pdr3p take interchangeable roles in regulating the expression of genes with PDRE sites in their promoter regions; among these, it is PDR5 that encodes the transporter which is primarily responsible for the efflux of drugs from the cell. However, Pdr3p is also involved in the transport of retrograde signals from mitochondria to elevate the level of PDR5 transcription.16
Recent studies suggest a respiratory deficiency of qdr3Δ/qdr3Δ null mutants under glucose-depletion.17 Such a phenotype was not observed in either qdr1Δ or qdr2Δ mutants, indicating a wider role for Qdr3p, possibly involving mitochondrial functions. The present study aims to investigate how the respiratory involvement of a drug transporter gene, QDR3, might affect the cell's stress-dependent metabolic changes. To assess QDR3's wider roles or functions in metabolism, a homozygous diploid qdr3Δ/qdr3Δ deletion mutant was grown in glucose- or ammonium-limited continuous cultures since this mutant was previously reported to display conditional respiratory deficiency. The prospect of a respiratory involvement for QDR3 led the study further to include PDR3, another drug-resistance gene, in the analysis owing to the tight link between mitochondrial activity and PDR activity as previously reported.16 Transcriptome and metabolome data, as well as the flux distributions representing the optimum metabolic solution space yielding the presented phenotype, were therefore determined for qdr3Δ/qdr3Δ, and pdr3Δ/pdr3Δ, as well as for hoΔ/hoΔ, with the aim of understanding the involvement of these genes in mitochondrial and/or respiratory activity.
Drugs, and other xenobiotic compounds, have appeared during historical, rather than evolutionary, time. Therefore these export pumps have most likely evolved to allow yeast to cope with antibiotics or other toxic compounds produced by plants or competing microorganisms. Previous studies have focused on the drug and chemical resistance characteristics associated with proteins of the MDR family.9–12 Rather less attention has been paid to a system-based investigation of the roles of these proteins. We believe this to be the first integrative study to suggest additional and wider roles for the members of the MDR and PDR families.
Results and discussion
The responses to genetic perturbations resulting from the deletion of the QDR3 or PDR3 genes (which encode, respectively an MDR transporter and a transcription factor regulating the expression of genes encoding ABC transporters) were investigated using a systems-based integrative approach. Homozygous diploid deletion mutants of the QDR3, PDR3 and HO genes were grown at a constant growth rate of 0.1 h−1 in glucose- or ammonium-limited continuous cultures. Transcriptome, endo- and exo-metabolome profiles at steady state, as well as the distribution of the fluxes in the optimized solution space, were studied to elucidate the different metabolic routes that may relate to either the specific responsibilities of Qdr3p in multiple drug resistance or to a possible wider role in metabolic homeostasis as an extension of its nutrition-specific involvement in respiratory function. Therefore the response of these MDR gene deletants to different nutrient limitations was monitored in an attempt to clarify the involvement of these genes at the wider metabolic level, and to determine how the cell copes with the loss of these major detoxification effectors (ESI 1†).Intracellular metabolite accumulation to cope with the loss of drug resistance genes
In order to investigate how the cell's metabolism responds to cope with the loss of these drug resistance genes, which are involved in major mechanisms of cellular detoxification, the changes in the intracellular metabolite and transcriptome levels (ESI 2–4†) of the mutant cells were analysed and the data were integrated with the optimal flux distributions determined in silico (ESI 5†). The reaction fluxes in the metabolic pathways were best predicted in comparison to the experimentally determined transport fluxes through the maximization of the oxygen uptake in glucose-limited fermentations and of ethanol production in ammonium-limited fermentations (which had non-limiting concentrations of glucose in the extracellular environment).In contrast to our in silico predictions, we observed a higher intracellular glucose concentration in qdr3Δ/qdr3Δ null mutants when compared to wild-type levels, irrespective of the nutrient availability. This intracellular glucose accumulation was not accompanied by any significant changes in the expression levels of genes involved in the glucose-sensing, glucose-repression, glycolytic, or storage carbohydrate pathways, nor in the GPR/PKA cAMP pathways. However, HXT3, which encodes a low-affinity hexose transporter, was overexpressed in qdr3 mutants when glucose was abundant. Although this glucose transporter was reported to have induced expression levels at both low and high glucose concentrations, the qdr3 deletion reduced its expression under glucose limitation. Conversely, the expression levels of the high-affinity glucose transporter genes (HXT2, HXT4, and HXT7) were co-ordinately regulated and significantly higher under glucose limitation. In fact, it was previously reported that any one of HXT2, HXT6 or HXT7 would enable growth on 0.1% glucose.18HXT2 expression was observed to be high in qdr3Δ/qdr3Δ mutants growing on 0.2% glucose whereas HXT7 expression was high in pdr3Δ/pdr3Δ mutants under the same condition. Although HXT8 expression was reported to be induced at low levels of glucose and to be repressed to various degrees by glucose,18 we observed that its expression was repressed in the absence of QDR3 under glucose limitation and induced during glucose abundance for either genetic manipulation.
Previous reports indicated that the intracellular glucose concentration was very low when the extracellular glucose concentration was at or below the Km of the transport system.19 On the other hand, glucose was accumulated in the cell when the extracellular glucose concentration was higher than could be handled by the transport system.19 Based on this information, it could be suggested that the loss of drug transporter genes had changed the cell's ability to perceive the extracellular glucose concentration or regulate the expression of its repertoire of genes for hexose transporters. Previous reports indicated that HXT9 and HXT11 acted as MFS drug transporters in yeast18 and that these transporters were also acting in concert with the PDR system.20 Cells may increase the rate of facilitated glucose diffusion via the hexose transporters, by increasing their expression levels, in order to overcome the susceptibility of these drug-sensitive mutants in the case of any possible encounter with toxic compounds. Similar mechanisms were reported in mammalian cell lines, and possible roles proposed for facilitative hexose transporters in the development of drug resistance (Fig. 1).21
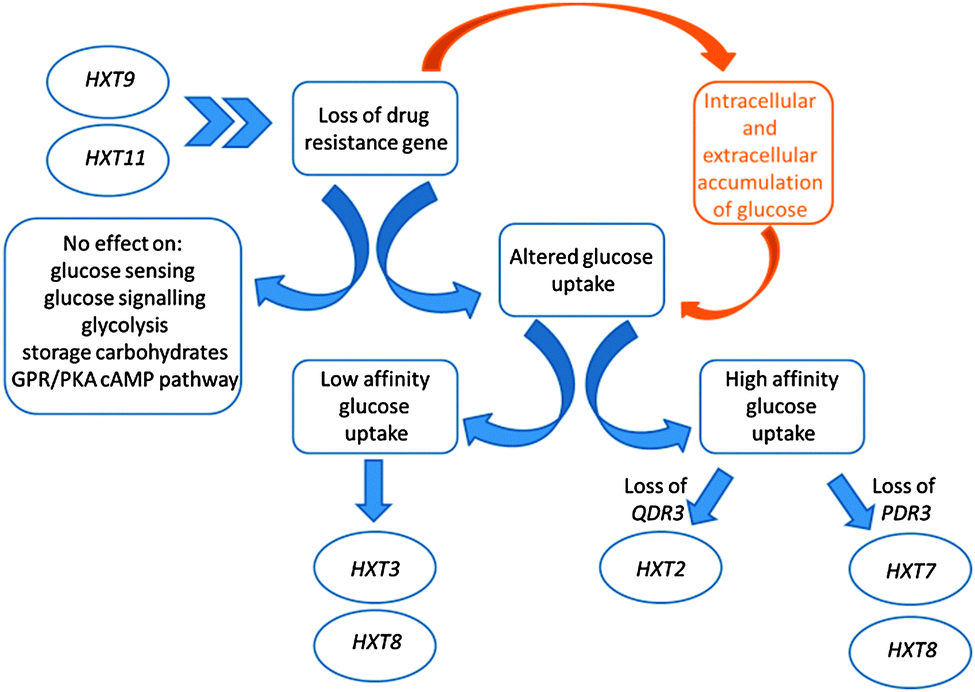 | ||
| Fig. 1 Alterations in the glucose metabolic processes in the absence of drug-resistance genes. The accumulation of glucose in response to the loss of resistance genes was shown to stem from the hexose transport system at the gene expression level rather than the glucose sensing, signalling, glycolysis, storage carbohydrates mechanisms, or the GPR/PKA cAMP pathway. A preferential selection of hexose transporters could be identified. The functioning of HXT8 was altered in response to loss of drug-resistance ability, whereas the gates encoded by HXT9 and HXT11 were opened to suppress the sensitivity to drugs induced by the loss of QDR3 and PDR3. Chevrons indicate the indirect effect that HXT9 and HXT11 have as MFS drug transporters. | ||
We observe that the transcription of genes encoding enzymes in the glycolytic pathway was slightly down-regulated in qdr3Δ/qdr3Δ mutants grown under glucose limitation and in pdr3Δ/pdr3Δ mutants grown under ammonium limitation. In both cases, this was accompanied by high levels of intracellular and extracellular inorganic phosphate. Previous reports indicated a rapid decrease in ATP and inorganic phosphate pools upon accumulation of phosphorylated glucose in the cell, consistent with the presence of a control mechanism over glycolysis by the intracellular, as well as extracellular, inorganic phosphate levels. However, the intracellular accumulation of phosphate and phosphorylation of glucose may be an indirect effect since a high concentration of phosphate does not, in itself, lead to phosphorylation of intracellular glucose.
The glycerol production and glycolytic pathways are linked to each other22 as glycerol synthesis liberates inorganic phosphate, thus rescuing a limitation on glycolysis.23 The intracellular glycerol accumulation and accompanying high levels of inorganic phosphate that we observed would indicate that the reduction in expression of the glycolytic pathway genes was caused by the cells' preference for accumulating unphosphorylated glucose in order to rescue the lost drug resistance function, rather than a requirement for keeping the glycolytic route accessible (Fig. 2).
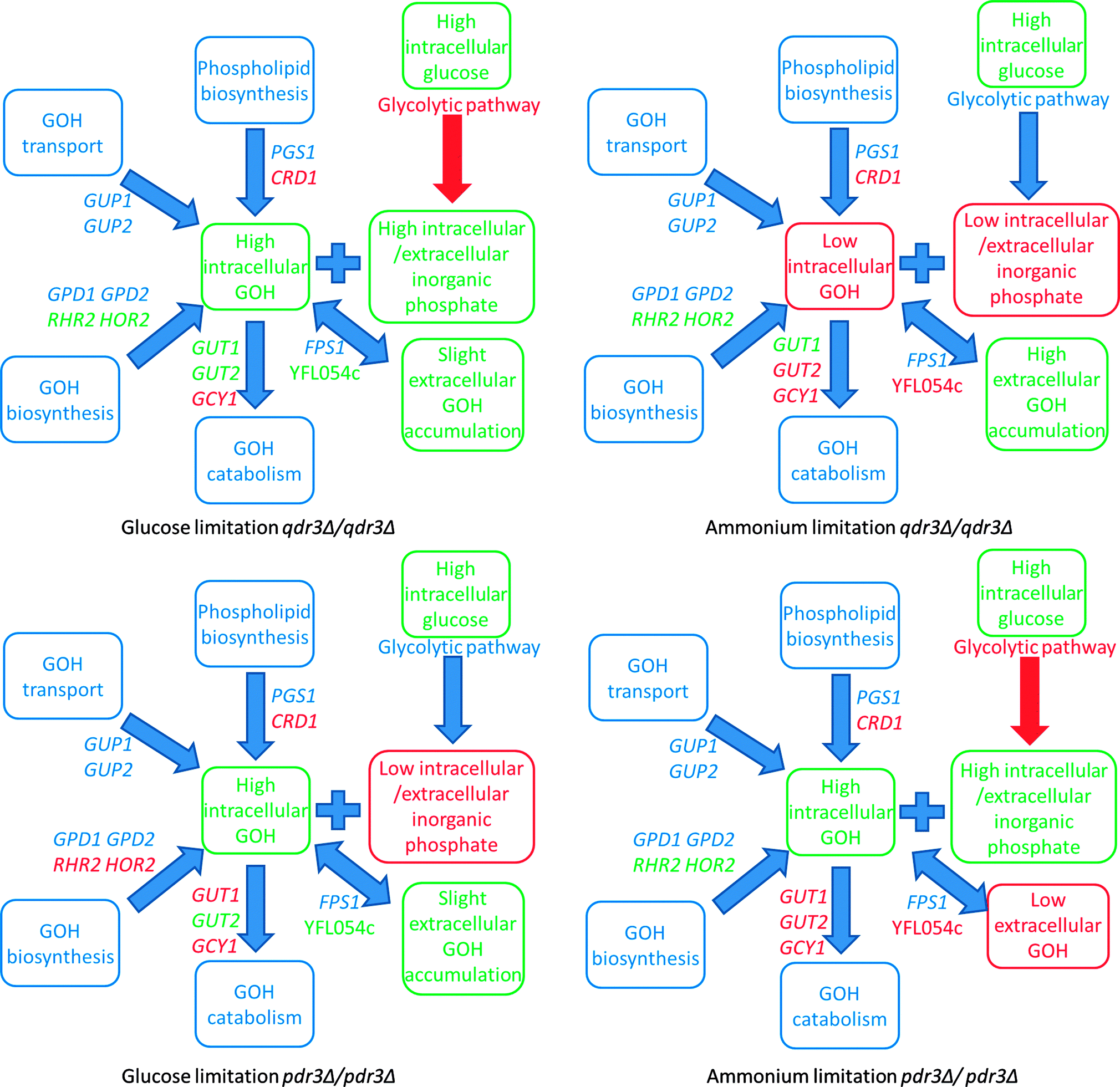 | ||
| Fig. 2 Different mechanisms involved in the production, degradation and transport of glycerol for qdr3Δ/qdrΔ and pdr3Δ/pdrΔ under glucose or ammonium limitation and the equilibrium between intracellular glucose, inorganic phosphate, and glycerol levels and transcriptional changes in the glycolytic pathway relating the balance between these metabolites. Green denotes a significant increase in the expression level of transcripts, metabolites or fluxes in the indicated pathways with respect to their corresponding wild-type values and red denotes a similar decrease. If no information was available, or the values remained constant, the relevant items are indicated in blue. GOH = glycerol. | ||
The proposed equilibrium between intracellular glucose, inorganic phosphate, and glycerol levels was further investigated taking into consideration other pathways and mechanisms leading to the production or degradation of glycerol as well as the transport routes.
Glycerol accumulates in yeast under stress conditions.24 The high osmolarity glycerol (HOG) pathway is one of the major effectors of osmoregulation and many genes regulated by this signalling cascade were reported to control the carbon flux in the glycolytic pathway for glycerol synthesis and reduced growth.25,26 However, in the present study, the expression level of the genes in the HOG signalling cascade was unaffected by either qdr3 or pdr3 deletions. Previous reports indicated that, although the intracellular accumulation of glycerol is essential for survival under hyperosmotic stress, a response at the gene expression level is not essential since adaptation that occurs solely at the level of the metabolic network can rescue yeast from osmotic stress.27
The changes in the expression levels of the genes in the metabolic network associated with the production, degradation or transport of glycerol were further investigated. The expression levels of the genes specifying proteins that transport glycerol into the cell also remained unchanged, whereas an up-regulation of RHR2 and HOR2 in the downstream path of the main glycerol biosynthetic pathway was observed. Glycerol production, as a by-product of cardiolipin biosynthesis, was reduced via the down-regulation of CRD1 (the gene encoding cardiolipin synthase), in both cases. Although the expression levels of both GUT1 and GUT2 were increased in response to glucose limitation in qdr3Δ/qdr3Δ, the down-regulation of GCY1 in an alternative pathway for glycerol catabolism indicated that the glycerol degradation pathway was not completely active and high intracellular glycerol levels were thus maintained in the organism. In all other cases, the gene expression levels of the enzymes involved in glycerol catabolism were reduced. Although the expression level of FPS1, the glycerol uptake and efflux mediator gated by changes in osmolarity, remained unchanged in both cases. A close homolog, YFL054c, mediating glycerol entry, was up-regulated under glucose limitation when there was only an incremental accumulation of glycerol in the extracellular environment.28 The Fps1p channel was previously reported to mediate the uptake of arsenite and antimonite in yeast29 and the preferential up-regulation of YFL054c, rather than FPS1, for the uptake of glycerol might indicate that, under the stated conditions, the cells might thus be trying to protect themselves from the risk of importing toxic compounds in the absence of a specific drug efflux pump; Qdr3p or those controlled by the facilitated drug transport regulator, Pdr3p (Fig. 2).
The expression of genes for the glycolytic pathway enzymes remained unchanged under ammonium limitation for qdr3Δ/qdr3Δ and under glucose limitation for pdr3Δ/pdr3Δ. In both cases, this was accompanied by low levels of intracellular and extracellular inorganic phosphate.
For qdr3Δ/qdr3Δ, there was no intracellular glycerol accumulation and the extracellular glycerol concentration was considerably higher under ammonium limitation, in contrast to the case under glucose limitation. Consequently, YFL054c was expressed less under ammonium limitation since it encodes a low-affinity glycerol transporter.28 YFL054c expression was previously reported to be more prone to ethanol stimulation28 and the glucose abundance might have caused the qdr3/qdr3 deletants to switch their metabolism towards fermentation and the production of ethanol, as previously observed.17 The outcome may be that this condition is the only case in which a significant increase in the expression of YFL054c was not observed (Fig. 2).
The intracellular and extracellular glycerol levels in the cell (as well as the expression levels of the genes involved in their transport, degradation, or release as a by-product of cardiolipin in phospholipid biosynthesis) display similar characteristics for either qdr3Δ/qdr3Δ or pdr3Δ/pdr3Δ under glucose limitation. RHR2 and HOR2, the downstream genes in glycerol biosynthesis, were down-regulated under glucose limitation in pdr3Δ/pdr3Δ. The mutant lacking the PDR3 gene suffered a depletion of the intracellular phosphate pool as well as the extracellular inorganic phosphate available under glucose limitation, as was the case for qdr3Δ/qdr3Δ under ammonium limitation (Fig. 2).
Furthermore, the metabolomics data provided no indication of diminishing or increasing extracellular glucose or glycerol levels during the intracellular accumulation of glucose and glycerol in drug-resistance gene mutants. This rules out the possibility that the MFS-MDR transporter Qdr3p or the transcriptional activator Pdr3p mediate (either directly or indirectly) the excretion or influx of these metabolites (ESI 4†). A slight accumulation of long-chain fatty acids was observed in the pdr3Δ/pdr3Δ diploid under ammonium limitation, suggesting the transcriptional activator's regulatory role on PDR transporters that appear to control membrane composition (ESI 4†). These observations at the metabolic level suggest that yeast perceives the loss of the QDR3 or PDR3 genes and the consequent reduction in detoxification ability as a potential threat to survival and prepares its metabolism accordingly by the intracellular accumulation of glucose and glycerol as well as the maintenance of the pool of inorganic phosphate.
The substrate specificity of the Qdr3 transporter and Pdr3 activator in this cellular protection mechanism was further investigated by growing homozygous diploid deletion mutants of the QDR3, PDR3 and HO genes in the presence of the Group I, II, VI, and VII ions: Na+, K+, Mg2+, Se4+, Cl−; the ion of a weak acid, acetate; the transition metal ions; Cu2+, Fe3+, Co2+, Zn2+; and the osmolarity regulator, sorbitol. The growth of the mutants on glucose or glycerol as the carbon source was monitored in micro-aerated or aerated batch cultures. The growth of the drug-sensitive deletants was not statistically different from that of control at a significance level of p = 0.05. The growth rate of the mutant cells was not reduced, ruling out the possibility that the loss of these drug-resistance genes affected the transport of the tested substances as additional substrates. Although the loss of the QDR3 transporter gene did result in a copper-sensitive phenotype, a recent report proposed copper as the main physiological substrate of Qdr2p, a close homolog of Qdr3p. While the copper uptake mechanism was not affected in the absence of QDR2, its extrusion was impaired – rendering the cells sensitive in the absence of QDR2. The function of Qdr2p in copper homeostasis was used to explain its role in oxidative stress response.30
These results indicated that the drug-sensitive mutants were observed to be significantly more resistant to the presence of osmotic stress that was created specifically by high extracellular glycerol concentrations (p-value < 0.005) but not by other osmotic stressors such as sorbitol or high ion concentrations (ESI 6†).
Maintaining high intracellular osmotic pressure to sustain growth rates in the absence of drug-resistance genes
The capability of qdr3Δ/qdr3Δ, and pdr3Δ/pdr3Δ to accumulate intracellular glycerol and unphosphorylated glucose while maintaining an available inorganic phosphate ion pool raised the possibility that these cells could be using this metabolic rearrangement to prepare a metabolic defence mechanism in anticipation of a possible chemical attack. In order to test this hypothesis, the mutants were grown in a micro-aerated environment under ammonium or glucose limitation with 10% NaCl or 1 M sorbitol present as the ionic and anionic osmotic stress inducers. At the stationary phase of population growth, the cells were then transferred into fresh media with a cocktail of barban, bleomycin, cisplatin and quinidine, all of which were previously reported to be exported from the cell by Qdr3p. The osmotic stress was removed from one set of samples and was maintained in another set at the same time that the drugs were introduced. The cells were allowed to grow for at least 24 hours in each environment in order to achieve maximum growth yield. The stationary phase cell concentration (as OD600) was then measured for each culture.Growth (OD600) was impaired significantly with respect to control in the absence of QDR3 or PDR3 under either nutrient limitation (p-value < 0.05). The addition of 10% NaCl impaired growth in all cell types under either glucose or ammonium limitation compared to their control growth profiles. However, in the presence of NaCl, the growth of the deletants was similar to that of the control. The growth of hoΔ/hoΔ control strain was lower in presence of 1 M sorbitol in the medium, whereas the growth of the qdr3Δ/qdr3Δ or pdr3Δ/pdr3Δ deletants was not significantly different. Similar to the observations in the presence of NaCl, the growth of the deletants were not significantly different from that of the control strain in the presence of sorbitol (Fig. 3 and Table 1). These results indicated that both the ionic (NaCl) and the anionic (sorbitol) sources of osmotic stress impaired the growth of the control strain more severely than that of the deletants, confirming the notion that the rearrangement of metabolism in the deletants may have a protective effect.
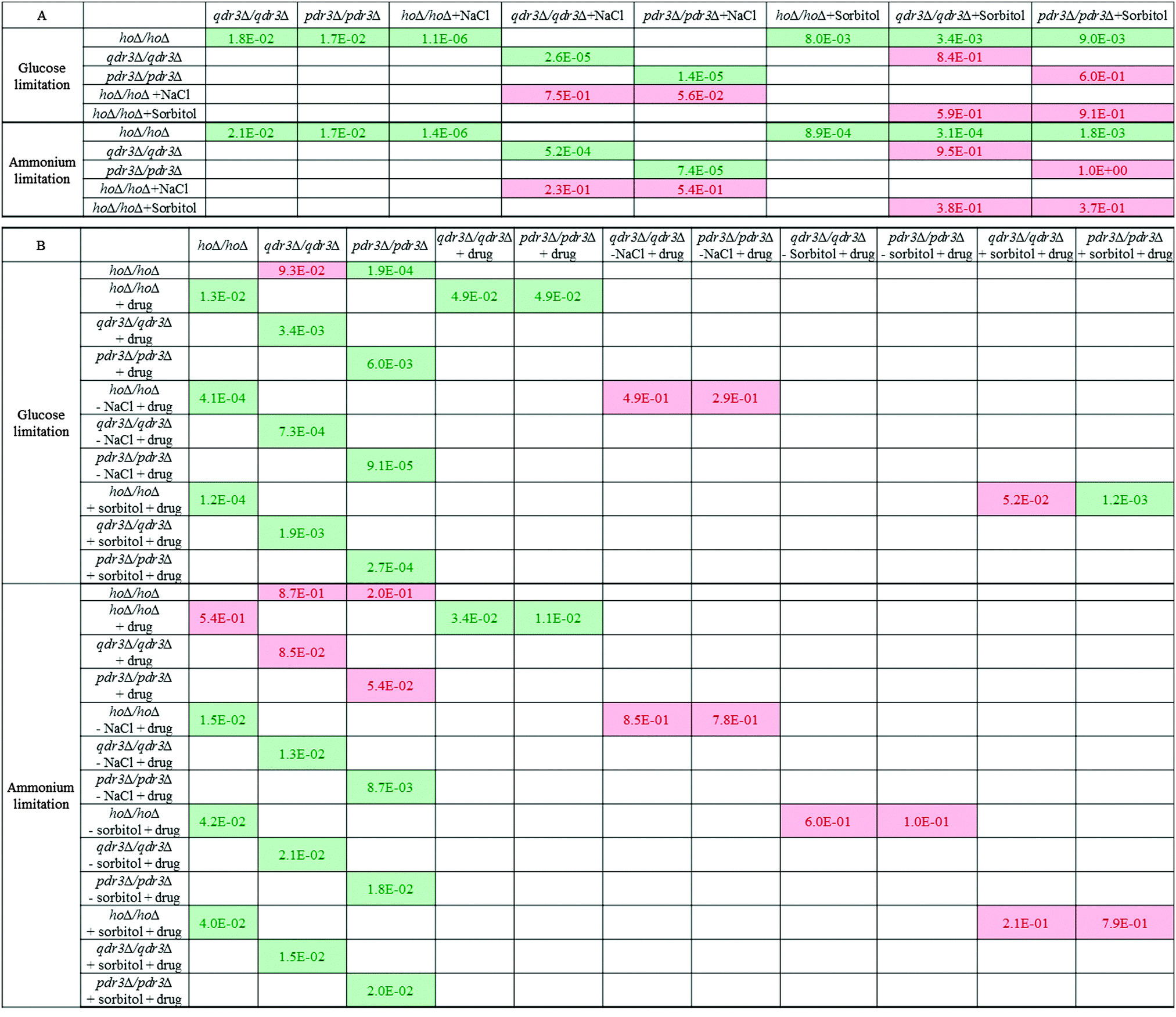 | ||
| Fig. 3 Population growth significance matrices pre- (A) and post-treatment (B) with drug cocktail. The significance of the differences in growth (measured as OD600) were evaluated based on replicate measurements under each specific condition using a significance threshold of α = 0.05. hoΔ/hoΔ, qdr3Δ/qdrΔ and pdr3Δ/pdrΔ were grown in microaerobic conditions under glucose or ammonium limitation. +NaCl and +sorbitol denote the presence of 10% NaCl or 1 M sorbitol in the medium, respectively, while –NaCl and −sorbitol denote that the fresh medium that the cells were transferred into was devoid of the osmotic stress constituent. The presence of drug treatment was denoted as +drug. The significant differences are highlighted in green and those identified to be insignificant were highlighted in red. | ||
| hoΔ/hoΔ | qdr3Δ/qdr3Δ | pdr3Δ/pdr3Δ | ||
|---|---|---|---|---|
| Synthetic medium under glucose limitation | ||||
| BT | Control | 2.60 | 2.03 | 2.16 |
| +NaCl | 0.49 | 0.47 | 0.37 | |
| +Sorbitol | 2.06 | 2.00 | 2.10 | |
| AT | Control | 6.05 | 5.64 | 5.60 |
| +Drug | 4.13 | 3.13 | 2.80 | |
| −NaCl + drug | 0.83 | 0.76 | 0.97 | |
| +NaCl + drug | 0.52 | 0.57 | 0.40 | |
| −Sorbitol + drug | 2.12 | 2.56 | 2.69 | |
| +Sorbitol + drug | 2.44 | 2.59 | 2.80 | |
| Synthetic medium under ammonium limitation | ||||
| BT | Control | 3.08 | 2.38 | 2.52 |
| +NaCl | 1.01 | 0.88 | 0.94 | |
| +Sorbitol | 2.43 | 2.36 | 2.52 | |
| AT | Control | 6.05 | 5.95 | 7.38 |
| +Drug | 5.72 | 4.93 | 5.14 | |
| −NaCl + drug | 1.68 | 1.79 | 1.59 | |
| +NaCl + drug | 0.72 | 0.92 | 0.97 | |
| −Sorbitol + drug | 3.81 | 3.68 | 3.24 | |
| +Sorbitol + drug | 3.68 | 3.18 | 3.62 | |
Placing the cells grown in the control culture in fresh control medium without introducing the drug cocktail or any osmotic stress caused the drug-sensitive deletants to recover from their growth deficiency under ammonium limitation whereas these mutants still displayed impaired growth in comparison to the wild type under glucose limitation. The presence or the absence of the drug cocktail, NaCl, sorbitol or combinations of these chemicals altered the growth significantly in comparison to the growth achieved in the control environment (p-value < 0.05) regardless of the strain type or nutrient limitation. Although the presence of drugs impaired growth in all strains, the drug-resistance gene mutants; qdr3Δ/qdr3Δ and pdr3Δ/pdr3Δ were affected significantly more severely.
Transferring the cells grown in 10% NaCl into fresh medium containing 10% NaCl followed by the introduction of the drug cocktail was a severe challenge to the cells and no additional growth was observed regardless of the type of nutrient limitation imposed and regardless of the strain (p-value > 0.05). Culture growth recovered significantly if the NaCl challenge was removed prior to the addition of the drug cocktail (p-value < 0.05) under either nutrient limitation. Similarly, growth was maintained in the cultures in the presence of sorbitol after the injection of the drug cocktail regardless of whether the osmotic stress induced by sorbitol was continued in the fresh culture or not (Table 1 and Fig. 3).
An interesting observation was that the final biomass concentration of the drug-sensitive deletants in the untreated control cultures as well as those cultures treated only with the drug cocktail was lower than that of control under glucose limitation. Conditioning the cells by inducing osmotic stress using 10% NaCl or sorbitol followed by the removal of this stress prior to the injection of the drug cocktail recovered the growth rates of the drug-sensitive deletants to that of the control strain. A similar situation was observed under ammonium limitation regarding the osmotic stress induced by NaCl. In the case of sorbitol, if this sugar alcohol was still present during drug treatment, the pdr3Δ/pdr3Δ cells showed a degree of drug tolerance at least equivalent to that of the control cells. In contrast, when sorbitol was removed prior to the addition of the drug cocktail, then both the qdr3Δ/qdr3Δ and pdr3Δ/pdr3Δ cultures showed a higher tolerance to the drug culture than did the control cells subjected to the same regime. The findings indicated a more severe response to drug treatment in the absence of QDR3 under all investigated conditions. These results imply that, in glucose-limited conditions, the high internal glycerol concentrations found in the drug-sensitive mutants can replace, to a marked extent, the protective effect against toxic chemicals of an externally added osmoticum.
Materials and methods
Strains, growth conditions and sampling
Three homozygous single-gene deletion mutants:| – hoΔ::kanMX4/hoΔ::kanMX4, |
| – qdr3Δ::kanMX4/qdr3Δ::kanMX4, and |
| – pdr3Δ::kanMX4/pdr3Δ::kanMX4 |
Four homozygous single deletion mutants:
| – hoΔ::kanMX4/hoΔ::kanMX4, |
| – qdr3Δ::kanMX4/qdr3Δ::kanMX4, |
| – pdr3Δ::kanMX4/pdr3Δ::kanMX4, |
Growth profiling under different osmotic pressure and drug treatment regimes
Single colonies from YPD-agar plates were inoculated into 5 ml pre-cultures grown in YPD. Glucose- and ammonium-limited cultures were inoculated from overnight grown pre-cultures normalizing the growth of different cell types (50 μl hoΔ/hoΔ equivalent into 5 ml culture volume in 50 ml conical tubes). 10% NaCl or 1 M sorbitol was used to induce osmotic stress. The cultures were allowed to grow until they reached the stationary phase and the optical density (OD600) values were recorded. The medium was discarded following centrifugation at 3000 rcf for 10 minutes. The cells were re-suspended in fresh medium and a drug cocktail of barban (0.01 mM), bleomycin (25 μg ml−1), cisplatin (20 μg ml−1) and quinidine (3 mM) was introduced at their indicated working concentrations. The optical density (OD600) values were recorded at the new stationary phase. The chemicals in the drug cocktail were purchased from Sigma with the following catalogue numbers: barban analytical standard (Supelco) (PS540), bleomycin sulphate from Streptomyces verticillus (B5507), crystalline cis-diammineplatinum(II) chloride (P4394), and quinidine anhydrous (Q3625).Transcriptome sampling RNA isolation and transcriptome analysis
Culture sampling and RNA extraction were performed as described previously.33 Total RNA was qualitatively assessed on an Agilent 2100 Bioanalyser (Agilent Technologies) and quantified using Nanodrop ultra-low-volume spectrophotometer (Nanodrop Technologies). cDNA was synthesized, and double-stranded cDNA was retrieved from ca. 15 μg of total RNA as described in the Affymetrix GeneChip® Expression Analysis Technical Manual, using appropriate kits. cDNA was checked for quality using the Agilent 2100 Bioanalyser and was quantified using Nanodrop. Biotin-labelled cRNA was synthesized and purified using clean-up kits and then quantified, using the Nanodrop spectrophotometer, before hybridization. Hybridization and loading onto Affymetrix Yeast2 arrays were carried out as described in the GeneChip® Expression Analysis Technical Manual. The chips were then loaded into a Fluidics station for washing and staining using Microarray Suite 5 with EukGe W S2v4 programme. Lastly, the chips were loaded onto the Agilent GeneArray scanner 2500 and another quality check was performed using Microarray Suite 5.34Data acquisition and analysis
The raw microarray data files were assessed, using dChip software, for outliers at the array level as well as at the probe-set level.35 RMA Express software was then used to normalize the data.36 The data were log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 transformed prior to analysis. In compliance with MIAME guidelines,37 the microarray data from this study has been submitted to ArrayExpress at the European Bioinformatics Institute under accession number [E-MTAB-707].
2 transformed prior to analysis. In compliance with MIAME guidelines,37 the microarray data from this study has been submitted to ArrayExpress at the European Bioinformatics Institute under accession number [E-MTAB-707].
The significance of the differences in expression levels under various conditions were evaluated using the paired two tail Student's t-test using a threshold of p < 0.05. A minimum of 1.5-fold change difference was ensured between different conditions in order to reduce the chance of false discoveries. Hierarchical Clustering Explorer (HCE) 3.038 was used to cluster genes with similar expression patterns. The significantly enriched functional categories and the process ontology terms of the genes falling into the same cluster were determined with AmiGO Term Finder tool39 using background correction based on hypergeometric distribution. The threshold p-value was selected as 0.05. A transcription factor (TF) list was compiled and the transcriptional regulatory network (TRN) was reconstructed using three sources; YEASTRACT,40,41 Lee et al. 2002,42 and Luscombe et al., 2004.43 The list of differentially expressed transcripts and key transcription factors are provided in ESI 2.†
The p-value for each transcript was obtained by a paired t-test between the drug-resistance gene deletants and the wild type. The size of the TRN was reduced by excluding the genes for which no expression data were available. Key transcription factors were determined for each deletion case using the reporter features algorithm.44 Transcription factors whose changes were significant at a p-value less than 0.05 were defined as “key”. Perturbation-responsive sub-networks (PRSs) were reconstructed and visualized via Cytoscape 2.8.2.45,46 The significant enrichment (p-value < 0.05) of process gene ontology (GO) terms in the perturbation-responsive sub-networks was determined using AmiGO (ESI 3†).39
Metabolomic procedures
For metabolic footprinting or exo-metabolomics, 1 ml samples were withdrawn from the fermentation broth onto ice and centrifuged at 4 °C at 14000 rpm for 4 minutes.47 The supernatant was stored at −80 °C until analysis. For metabolic fingerprinting, 5 ml of sample was rapidly quenched in 60% (v/v) methanol buffered with tricine at −50 °C and the endo-metabolites were extracted in 75% (v/v) boiling ethanol buffered with tricine at 80 °C as described.48 The vacuum-dried samples were stored at −80 °C until analysis. For both metabolic footprinting and fingerprinting, derivatization and identification of peaks via GC–ToF–MS were performed as described.49 A total of 54 unique metabolites were semi-quantitatively identified in the analysis among the 118 peaks that were detected. The samples for GC–MS analysis were spiked with 100 μl 0.18 mg ml–1 succinic d4 acid as the internal standard and the peak areas were normalized against that standard. Steady-state exometabolic concentrations of glucose, ethanol, ammonium, acetate, acetaldehyde and succinate were determined enzymatically using Boehringer-Mannheim kits and used as extracellular metabolic constraints in flux balance analysis. The metabolome data are provided in ESI 4.†
The optimum distributions of the fluxes under different genetic and environmental conditions were determined by linear optimization with the suitable biological objective functions using the metabolic model iFF708 and Yeast 5.50,51 The optimized fluxes were determined through the maximization of the oxygen uptake or ethanol production and the predicted transport and biomass fluxes were compared to those of experimentally determined values in selecting which objective function to use under the stated conditions. The solution space was constrained by the exometabolite concentrations for each deletant under each nutrient limitation at a fixed dilution rate of 0.1 h−1 at steady state in chemostat cultivations. In instances where many alternative optimal flux distributions were determined, the flux distribution minimizing overall intracellular flux distribution in order to maximize enzymatic efficiency was selected.52 The simulations were carried out in the MATLAB 6.0 or 7.0 (Mathworks, USA) environment. The results from steady-state simulations of flux distributions are provided in ESI 5.†
Conclusions
A systems-based investigation of the roles of QDR3 and PDR3; drug-resistance genes from two different families – QDR3 from the multidrug resistance family and PDR3, which regulates the expression of genes encoding members of the pleiotropic drug resistance family – was carried out through the integration of genome-wide transcriptional and metabolomic changes and the predicted metabolic fluxes for the homozygous deletion mutants grown in aerated continuous fermentations under glucose or ammonium limitation. The nutrient limitation in the environment was the dominant parameter in determining the hierarchical organization of the transcriptome and metabolome in response to the genetic perturbations. The cellular response at both the transcriptomic and endo-metabolomic levels was similar, whereas a slight difference was observed in the hierarchical organization of the exo-metabolome under glucose limitation.Although both a genetic perturbation and a nutrient limitation were imposed simultaneously, the gene expression levels of some stress-related transcription factors indicated that the cells did not perceive any stress. The present findings indicated that these drug-sensitive mutants re-organised their metabolism under glucose or ammonium limitation and the intracellular accumulation of metabolites was used as an alternative defence mechanism in the possible event of being exposed to a toxic compound in their environment. In this particular case, the yeast cells used the accumulation of intracellular glucose and glycerol as a pre-adaptive and defensive response to the loss of the drug resistance genes PDR3 or QDR3, when grown under glucose or ammonium limitation and of inorganic phosphate as an ion pool to assist the balance between the two accumulated metabolites.
The ability of yeast cells to anticipate the changes that might take place in their environment was previously studied in a model of the wine fermentation process and it was suggested that the yeast cells were able to cope with the stresses to which they were exposed in their natural order better than the artificial case in which the order was reversed.53 The results of the present study may suggest a similar mechanism. The yeast cells lacking functionality in their drug/toxin resistance metabolism were observed to accumulate glucose and glycerol intracellularly and use inorganic phosphate ion pools to equilibrate the charge potential between the two metabolites. This would then be considered as a priming period for an anticipated subsequent stress of being exposed to toxic compounds. Thus, this may be considered a defence mechanism that yeast has developed to cope with a reduced ability to detoxify its cytoplasm.
Acknowledgements
The authors gratefully acknowledge the financial support from BBSRC (Grant BB/C505140/1 to SGO), and the travel grants for DD kindly provided by the Research Council of Turkey (TUBITAK) through the BDP programme and the Turkish State Planning Organization DPT09K120520. The research was also financially supported by Bogazici University Research Fund through Project No. 10A05D4; 08A506 and TUBITAK through Project No. 106M444. Further support came from European Commission though the Coordination Action Project YSBN (Contract No. 018942 to both BK and SGO), UNICELLSYS Collaborative Project (Contract No. 201142 to SGO) and UK-Turkey HE& Industry Partnership Programme (Contract No. TR/012012/KP17 to both SGO and BK).Notes and references
- J. E. Kurtz, P. Dufour, J. P. Bergerat and F. Exinger, Curr. Pharmacogenomics, 2005, 3, 1 CrossRef CAS.
- M. E. Cardenas, M. C. Cruz, M. del Poeta, N. Chung, J. R. Perfect and J. Heitman, Clin. Microbiol. Rev., 1999, 12(4), 583 CAS.
- J. B. Anderson, Nat. Rev. Microbiol., 2005, 3, 547 CrossRef CAS PubMed.
- P. W. Schenk, M. Brok, A. W. M. Boersma, J. A. Brandsma, H. den Dulk, H. Burger, G. Stoter, J. Brouwer and K. Nooter, Mol. Pharmacol., 2003, 64(2), 259 CrossRef CAS PubMed.
- S. J. Brown, P. J. Kellett and S. J. Lippard, Science, 1993, 261, 603 CAS.
- P. W. Schenk, A. W. M. Boersma, J. A. Brandsma, H. den Dulk, H. Burger, G. Stoter, J. Brouwer and K. Nooter, Cancer Res., 2001, 6, 6982 Search PubMed.
- H. Burger, A. Capello, P. W. Schenk, G. Stoter, J. Brouwer and K. Nooter, Biochem. Biophys. Res. Commun., 2000, 269, 767 CrossRef CAS PubMed.
- B. Akache, S. MacPherson, M.-A. Sylvain and B. Turcotte, J. Biol. Chem., 2004, 279(27), 27855 CrossRef CAS PubMed.
- P. A. Nunes, S. Tenreiro and I. Sa-Correia, Antimicrob. Agents Chemother., 2001, 45(5), 1528 CrossRef CAS PubMed.
- R. C. Vargas, S. Tenreiro, M. C. Teixeira, A. R. Fernandes and I. Sa-Correia, Antimicrob. Agents Chemother., 2004, 48(7), 2531 CrossRef CAS PubMed.
- S. Tenreiro, R. C. Vargas, M. C. Teixeira, C. Magnani and I. Sá-Correia, Biochem. Biophys. Res. Commun., 2005, 327, 952 CrossRef CAS PubMed.
- M. C. Teixeira, T. R. Cabrito, Z. M. Hanif, R. C. Vargas, S. Tenreiro and I. Sá-Correia, Microbiology, 2011, 157, 945 CrossRef CAS PubMed.
- H. Wolfger, Y. M. Mamnun and K. Kuchler, Res. Microbiol., 2001, 152, 375 CrossRef CAS.
- R. Ernst, R. Klemm, L. Schmitt and K. Kuchler, Methods Enzymol., 2005, 400, 460 CAS.
- W. S. Moye Rowley, Gene, 2005, 354, 15 CrossRef CAS PubMed.
- F. Devaux, E. Carvajal, S. Moye-Rowley and C. Jacq, FEBS Lett., 2001, 515, 25 CrossRef.
- D. Dikicioglu, P. Pir, Z. I. Onsan, K. O. Ulgen, B. Kirdar and S. G. Oliver, Appl. Environ. Microbiol., 2008, 74(18), 5809 CrossRef CAS PubMed.
- S. Ozcan and M. Johnston, Microbiol. Mol. Biol. Rev., 1999, 63(3), 3554 Search PubMed.
- B. Teusnik, J. A. Diderich, H. V. Westerhoff, K. van Dam and M. Walsh, J. Bacteriol., 1998, 180, 556 Search PubMed.
- A. Nourani, M. Wesolwski-Louvel, T. Delaveau, C. Jacq and A. Delahodde, Mol. Cell. Biol., 1997, 17, 5453 CAS.
- J. C. Vera, G. R. Castillo and O. M. Rosen, Mol. Cell. Biol., 1991, 11, 3407 CAS.
- L. Neves, R. Oliveira and C. Lucas, FEMS Yeast Res., 2004, 5(1), 51 CrossRef CAS PubMed.
- K. Luyten, J. Albertyn, W. F. Skibbe, B. A. Prior, J. Ramos, J. M. Thevelein and S. Hohmann, EMBO J., 1995, 14(7), 1360 CAS.
- L. Li, Y. Ye, L. Pan, Y. Zhu, S. Zheng and Y. Lin, Biochem. Biophys. Res. Commun., 2009, 387, 778 CrossRef CAS PubMed.
- A. Blomberg, FEMS Microbiol. Lett., 2000, 182, 1 CrossRef CAS.
- J. H. Parmar, S. Bhartiya and K. V. Venkatesh, Mol. Biosyst., 2011, 7, 1138 RSC.
- J. Bouwman, J. Kiewiet, A. Lindenbergh, K. van Eunen, M. Siderius and B. M. Bakker, Yeast, 2011, 28(1), 43–53 CrossRef CAS PubMed.
- R. Oliveira, F. Lages, M. Silva-Graca and C. Lucas, Biochim. Biophys. Acta, 2003, 1613, 57 CrossRef CAS.
- R. Wysocki, C. C. Chery, D. Wawrzycka, M. van Hulle, R. Cornelis, J. M. Thevelein and M. J. Tamas, Mol. Microbiol., 2001, 40(6), 1391 CrossRef CAS.
- G. Rios, M. Cabedo, B. Rull, L. Yenush, R. Serrano and J. M. Mulet, FEMS Yeast Res., 2013, 13, 97 CrossRef CAS PubMed.
- C. B. Brachmann, A. Davies, G. J. Cost, E. Caputo, J. Li, P. Hieter and J. D. Boeke, Yeast, 1998, 14, 115 CrossRef CAS.
- F. Baganz, A. Hayes, D. Marren, D. C. J. Gardner and S. G. Oliver, Yeast, 1997, 13, 1563 CrossRef CAS.
- A. Hayes, N. Zhang, J. Wu, P. R. Butler, N. C. Hauser, J. D. Hoheisel, F. L. Lim, A. D. Sharrocks and S. G. Oliver, Methods, 2002, 26, 281 CrossRef CAS.
- J. A. Wishart, A. Hayes, L. Wardleworth, N. Zhang and S. G. Oliver, Yeast, 2005, 22, 565 CrossRef CAS PubMed.
- C. Li and W. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 31 CrossRef CAS.
- B. M. Bolstad, R. A. Irizarry, M. Astrand and T. P. Speed, Bioinformatics, 2003, 19(2), 185 CrossRef CAS PubMed.
- A. Brazma, P. Hingamp, J. Quackenbush, G. Sherlock, P. Spellman, C. Stoeckert, J. Aach, W. Ansorge, C. A. Ball, H. C. Causton, T. Gaasterland, P. Glenisson, F. C. P. Holstege, I. F. Kim, V. Markowitz, J. C. Matese, H. Parkinson, A. Robinson, U. Sarkans, S. Schulze-Kremer, J. Stewart, R. Taylor, J. Vilo and M. Vingron, Nat. Genet., 2001, 29, 365 CrossRef CAS PubMed.
- J. Seo, M. Bakay, Y. W. Chen, S. Hilmer, B. Shneiderman and E. P. Hoffman, Bioinformatics, 2004, 20, 2534 CrossRef CAS PubMed.
- S. Carbon, A. Ireland, C. J. Mungall, S. Shu, B. Marshall and S. Lewis, Bioinformatics, 2009, 25(2), 288 CrossRef CAS PubMed.
- P. T. Monteiro, N. Mendes, M. C. Teixeira, S. d'Orey, S. Tenreiro, N. Mira, H. Pais, A. P. Francisco, A. M. Carvalho, A. Lourenço, I. Sá-Correia, A. L. Oliveira and A. T. Freitas, Nucleic Acids Res., 2008, 36, D132 CrossRef CAS PubMed.
- M. C. Teixeira, P. Monteiro, P. Jain, S. Tenreiro, A. R. Fernandes, N. P. Mira, M. Alenquer, A. T. Freitas, A. L. Oliveira and I. Sá-Correia, Nucleic Acids Res., 2006, 34, 446 CrossRef PubMed.
- T. I. Lee, N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J. B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford and R. A. Young, Science, 2002, 298, 799 CrossRef CAS PubMed.
- N. M. Luscombe, M. M. Babu, H. Yu, M. Snyder, S. A. Teichman and M. Gerstein, Nature, 2004, 431, 308 CrossRef CAS PubMed.
- A. P. Oliveira, K. R. Patil and J. Nielsen, BMC Syst. Biol., 2008, 2, 17 CrossRef PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498 CrossRef CAS PubMed.
- D. I. Broadhurst and D. B. Kell, Metabolomics, 2006, 2(4), 171 CrossRef CAS.
- J. Allen, H. M. Davey, D. Broadhurst, J. K. Heald, J. J. Rowland, S. G. Oliver and D. B. Kell, Nat. Biotechnol., 2003, 21(6), 692 CrossRef CAS PubMed.
- J. I. Castrillo, A. Hayes, S. Mohammed, S. J. Gaskell and S. G. Oliver, Phytochemistry, 2003, 62, 929 CrossRef CAS.
- G. A. Pope, D. A. MacKenzie, M. Defemez, M. A. M. M. Aroso, L. J. Fuller, F. A. Mellon, W. B. Dunn, M. Brown, R. Goodacre, D. B. Kell, M. E. Marvin, E. J. Louis and I. N. Roberts, Yeast, 2007, 24, 667 CrossRef CAS PubMed.
- B. D. Heavner, K. Smallbone, B. Barker, P. Mendes and L. P. Walker, BMC Syst. Biol., 2012, 6, 55 CrossRef PubMed.
- J. Förster, I. Famili, P. Flu, B. O. Palsson and J. Nielsen, Genome Res., 2003, 13, 244 CrossRef PubMed.
- H. P. J. Bonarius, G. Schmid and J. Tramper, Trends Biotechnol., 1997, 15, 308–314 CrossRef CAS.
- A. Mithchell, G. H. Romano, B. Groisman, A. Yona, E. Dekel, M. Kupiec, O. Dahan and Y. Pilpel, Nature, 2009, 460, 220 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2mb25512j |
| This journal is © The Royal Society of Chemistry 2014 |