Capture, release and culture of circulating tumor cells from pancreatic cancer patients using an enhanced mixing chip†
Weian
Sheng
a,
Olorunseun O.
Ogunwobi
b,
Tao
Chen
c,
Jinling
Zhang
a,
Thomas J.
George
*d,
Chen
Liu
*b and
Z. Hugh
Fan
*ace
aInterdisciplinary Microsystems Group, Department of Mechanical and Aerospace Engineering, University of Florida, P.O. Box 116250, Gainesville, FL 32611, USA. E-mail: hfan@ufl.edu; Fax: +1 352 392 7303; Tel: +1 352 846 3021
bDepartment of Pathology, Immunology and Laboratory Medicine, University of Florida, P.O. Box 100275, Gainesville, FL 32610, USA. E-mail: liu@pathology.ufl.edu
cDepartment of Chemistry, University of Florida, P.O. Box 117200, Gainesville, FL 32611, USA
dDepartment of Medicine, University of Florida, P.O. Box 100278, Gainesville, FL 32610, USA. E-mail: thom.george@medicine.ufl.edu
eJ. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, P.O. Box 116131, Gainesville, FL 32611, USA
First published on 30th October 2013
Abstract
Circulating tumor cells (CTCs) from peripheral blood hold important information for cancer diagnosis and disease monitoring. Analysis of this “liquid biopsy” holds the promise to usher in a new era of personalized therapeutic treatments and real-time monitoring for cancer patients. But the extreme rarity of CTCs in blood makes their isolation and characterization technologically challenging. This paper reports the development of a geometrically enhanced mixing (GEM) chip for high-efficiency and high-purity tumor cell capture. We also successfully demonstrated the release and culture of the captured tumor cells, as well as the isolation of CTCs from cancer patients. The high-performance microchip is based on geometrically optimized micromixer structures, which enhance the transverse flow and flow folding, maximizing the interaction between CTCs and antibody-coated surfaces. With the optimized channel geometry and flow rate, the capture efficiency reached >90% with a purity of >84% when capturing spiked tumor cells in buffer. The system was further validated by isolating a wide range of spiked tumor cells (50–50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in 1 mL of lysed blood and whole blood. With the combination of trypsinization and high flow rate washing, captured tumor cells were efficiently released. The released cells were viable and able to proliferate, and showed no difference compared with intact cells that were not subjected to the capture and release process. Furthermore, we applied the device for detecting CTCs from metastatic pancreatic cancer patients' blood; and CTCs were found from 17 out of 18 samples (>94%). We also tested the potential utility of the device in monitoring the response to anti-cancer drug treatment in pancreatic cancer patients, and the CTC numbers correlated with the clinical computed tomograms (CT scans) of tumors. The presented technology shows great promise for accurate CTC enumeration, biological studies of CTCs and cancer metastasis, as well as for cancer diagnosis and treatment monitoring.
000) in 1 mL of lysed blood and whole blood. With the combination of trypsinization and high flow rate washing, captured tumor cells were efficiently released. The released cells were viable and able to proliferate, and showed no difference compared with intact cells that were not subjected to the capture and release process. Furthermore, we applied the device for detecting CTCs from metastatic pancreatic cancer patients' blood; and CTCs were found from 17 out of 18 samples (>94%). We also tested the potential utility of the device in monitoring the response to anti-cancer drug treatment in pancreatic cancer patients, and the CTC numbers correlated with the clinical computed tomograms (CT scans) of tumors. The presented technology shows great promise for accurate CTC enumeration, biological studies of CTCs and cancer metastasis, as well as for cancer diagnosis and treatment monitoring.
Introduction
Pancreatic cancer is the fourth leading cause of cancer deaths in the United States, with the poorest 5-year survival rate (6%) for all cancer stages. Over 90% of pancreatic cancers progress to become metastatic.1,2 The poor prognosis of pancreatic cancer patients is related to the early dissemination of the disease and the lack of early detection.3 Circulating tumor cells (CTCs) are tumor cells disseminated from primary tumors which subsequently travel through the blood circulation to distant organs. CTCs are thus responsible for the initiation of metastasis and the in transit spread of cancer to distant sites.4,5 Therefore, CTCs hold the key to track metastasis, and they can be used for cancer diagnosis and monitoring of cancer status. Clinical studies have demonstrated that CTCs are correlated with disease progression for a wide range of cancers, such as breast, colorectal and prostate cancer.6,7 While biopsy is the current gold standard of cancer diagnosis, it involves removal of tissues or cells from the body and examination by experienced surgeons and pathologists.8 The invasive nature of biopsy prevents patients from being tested in an ongoing or repetitive basis. CTC examination, on the other hand, is much less invasive, with only 5–10 mL of patient blood needed; it is like a blood test for cancer. CTC monitoring is regarded as “liquid biopsy” or “live biopsy” of a tumor,9 which enables noninvasive cancer diagnosis and real-time monitoring of therapeutic response.However, CTCs are extraordinarily rare, with only a few CTCs circulating amidst billions of blood cells, making their isolation and characterization a tremendous technical challenge.10 Thus, high-efficiency and high-purity isolation of CTCs from patient blood is urgently needed to obtain accurate information of CTCs. Currently, only FDA-approved technology is the CellSearch system (Janssen Diagnostics, LLC, Raritan, NJ). Unfortunately, this system is limited by low efficiency, low purity and high cost, and does not fully address the issue of isolating the extremely low-abundance of CTCs.11,12 Recently, microfluidic devices with high affinity ligands, including antibodies13–18 and aptamers,19–21 have provided distinctive opportunities for efficient and specific isolation of CTCs from patient blood. Microfluidic devices, due to their large surface area-to-volume ratio and short diffusion distance, can substantially increase the interaction between cells and the ligand-coated surface.22–24
Staggered herringbone micromixers have been developed for fluid mixing in microchannels25 and have been exploited for enhancing the cell capture.17,26 Yet, limited research has been reported on the optimization of herringbone mixers for high-performance cell capture. Different from mixing solutions through transverse flow, inducing cell-surface interactions requires cells with nearly zero diffusivity for advection to microchannel surface.27 Herein, we have developed a geometrically enhanced mixing (GEM) chip for high-performance CTC capture (high efficiency, purity, throughput and cell viability). With experimental optimization of the herringbone micromixers, we achieved capture of spiked tumor cells with >90% capture efficiency and >84% purity. In addition, the time required to process 1 mL blood sample is <17 min, much faster than those reported in literature.13,17,26 Since very limited work has been done on cellular studies after CTC capture,28 we have investigated the release, the viability and the culture of the captured cells. Captured cells can be efficiently released with the combined methods of trypsinization and high flow rate washing. Experiments also showed that released cells grew as well as intact cells that had not been subjected to the capture and release process. Further, we applied the device for isolation of CTCs from pancreatic cancer patients, with CTCs observed in 17 of 18 patient samples. We also demonstrated the potential of using CTC enumeration as a surrogate for radiographic monitoring of chemotherapy response in pancreatic cancer patients. Our device sensitivity enables isolation and enumeration of CTCs from pancreatic cancer patients, a disease where invasive biopsies are difficult and the commercial CellSearch system has proven to be inefficient.29 Compared with reported efforts, this work demonstrated a systematic study of the following aspects: geometric optimization of a micromixer for enhanced target CTC capture, release and re-culture of captured tumor cells, cell viability before and after release, cell binding behaviors after release and re-culture, isolation and counting of understudied pancreatic CTCs, comparison of CTC enumeration with CT scans for monitoring chemotherapy response in pancreatic cancer patients. A comprehensive study of these aspects would further improve CTC isolation performance, help understand post-capture processing of CTCs and push forward CTC isolation for cancer diagnosis. A comparison of this work with published studies is detailed in Table S1.†
In this study, we first developed a geometrically enhanced mixing chip (GEM chip) based on patterned herringbone or chevron structures. The mixer design was inspired by several groups,25–27 and the dimensions were optimized for high-efficiency and high-purity cell capture. As shown in Fig. 1, the GEM chip is the same size as a microscope slide (3 in × 1 in), having 8 parallel channels with uniform flow to form a high throughput device. Each channel is 2.1 mm wide, 50 μm deep, with 50 μm deep herringbone grooves repeating over a total length of 50 mm. The staggered herringbone grooves disrupt streamlines and induce chaotic mixing and microvortex, which maximize collisions and interactions between target cells and device surfaces, leading to increased cell capture efficiency. The groove width and the groove pitch were carefully selected for high-performance cell capture, as discussed in detail in Results and discussion.
 | ||
| Fig. 1 a) Picture of the 3 in × 1 in microfluidic GEM chip, consisting of eight parallel channels with single inlet and outlet. b) Micrograph (4× bright field) of the staggered herringbone grooves inside a channel, showing their asymmetry and periodicity, scale bar = 200 μm. c) A narrow groove design based on reported herringbone (HB) chip,26 with 50 μm groove width, purple dots show cells captured inside a channel. d) Cross-sectional view of the wide groove GEM chip, with channel depth of 50 μm and groove depth of 50 μm; the groove pitch is set to be 200 μm, and the groove width is chosen to be 120 μm. | ||
Experimental section
Microfluidic device fabrication
The microfluidic GEM chip consists of a polydimethylsiloxane (PDMS) structure bonded to a 3′′ × 1′′ glass microscope slide. The PDMS structure was fabricated using two-layer soft lithography, according to literature.30,31 The two-layer SU-8 structure (a main channel layer and a herringbone mixer layer) was fabricated via two spin-coating and exposure steps and a single developing step. The device layout was designed in AutoCAD and then sent to CAD/Art Services, Inc. (Brandon, OR) to produce a high resolution transparency photomask. Silicon wafers were first spin-coated with 50 μm thick SU-8 2035 photoresist (MicroChem, Newton, MA) as the main channel layer. After soft baking, UV light exposure, and post exposure baking, another layer of SU-8 was added to form the herringbone mixer layer. With precise alignment between the main channel and the mixer, a second exposure was performed to create the herringbone mixer pattern. After development, a silicon master patterned with the complementary structures was obtained. PDMS structures were fabricated by casting a liquid PDMS precursor against the master using Sylgard 184 reagents (Dow Corning, Midland, MI), according to the manufacturer's instructions. Inlet and outlet wells were created at the channel ends by punching holes in the PDMS sheet. The channel depth, which was controlled by the spin speed of the SU-8, was measured using a Dektak 150 profilometer.Cell culture
L3.6pl cells32 (human pancreatic cancer) were obtained from Dr. Jose Trevino's lab (Department of Surgery, University of Florida). BxPC-3 cells (CRL-1687, human pancreatic adenocarcinoma) and MIAPaCa-2 cells (CRL-1420, human pancreatic carcinoma) were purchased from American Type Culture Collection (ATCC). Cells were cultured in DMEM medium (ATCC) supplemented with 10% fetal bovine serum (FBS; heat-inactivated; GIBCO) and 100 units mL−1 penicillin–streptomycin (PS, Cellgro, Manassas, VA) and incubated at 37 °C under 5% CO2 atmosphere. Cells were grown as adherent monolayers in 60 mm × 15 mm culture dishes to 90% confluence, subsequently detached with 0.05% trypsin–0.53 mM EDTA (0.05%, Cellgro) and re-seeded at a lower concentration.Reagents and buffers
Biotinylated anti-EpCAM (Anti-Human CD326, eBioscience, San Diego, CA) immobilized on device surface was used as the CTC capture agent. Anti-cytokeratin FITC (CAM 5.2, conjugated with fluorescein isothiocyanate, BD Biosciences, San Jose, CA) and anti-CD45 PE (conjugated with phycoerythrin, BD Biosciences) were used to label CTCs and white blood cells, respectively. DAPI (4′,6-diamidino-2-phenylindole, Invitrogen, Carlsbad, CA), which stains DNA in cell nuclei, was used to label all nucleated cells bound to the device (i.e., white blood cells and CTCs). Dulbecco's phosphate buffered saline with calcium and magnesium (PBS, Fisher Scientific, Hampton, NH) was used to wash cells. A buffer containing 10 mg mL−1 (1%) bovine serum albumin (BSA, Fisher Scientific) and 0.05% Tween-20 (Fisher Scientific) in PBS was used for rinsing the unbound molecules from the channel surface, and resuspending cells for cell capture. BSA and Tween-20 in PBS was used to fully passivate the surfaces to reduce nonspecific adsorption of cells in the channels.Flow cytometry analysis was used to test the binding capabilities of anti-EpCAM to pancreatic cancer cell lines. Fluorescence measurements were performed with a FACScan cytometer (BD Immunocytometry Systems, San Jose, CA). Briefly, 200000 cells were incubated with 10 μg mL−1 biotinylated anti-EpCAM in 200 μL of PBS (containing 0.1% BSA) for 20 min on ice. After incubation, the cells were washed three times with PBS. Then streptavidin phycoerythrin (SA PE)–Cy5 (Invitrogen) was added and incubated for another 20 min. After washing, 10000 counts were measured in the flow cytometer to determine the fluorescence. The cells incubated with SA PE–Cy5 alone were used as a negative control to determine nonspecific binding. Fig. S1 and S2† show the strong binding of the anti-EpCAM antibody with L3.6pl cells and BxPC-3 cells, respectively. Fig. S3† shows no binding between anti-EpCAM and MIAPaCa-2 cells, indicating that MIAPaCa-2 cells can be used as a negative control.
Capture of spiked tumor cells in microfluidic devices
Immediately before experiments, cells were detached from the culture dish and then rinsed with PBS and resuspended at 106 cells mL−1. By following the manufacturer's instructions, the target cells and control cells were stained with Vybrant DiI (red) and Vybrant DiD (blue) cell-labeling solutions (Invitrogen), then rinsed with PBS, and resuspended at 106 cells mL−1 in the PBS containing BSA and Tween-20. Labeled cells were stored on ice and further diluted or spiked into blood to the desired concentrations before experiments.Anti-coagulant-containing human whole blood from healthy participants was purchased from Innovative Research (Novi, MI), and used for all “spike-in” experiments. For some experiments, CTC capture from whole blood samples was preceded by red blood cell lysis performed as previously described.33 Briefly, lysed blood was obtained by treating whole blood with red blood cell (RBC) lysing buffer, prepared by adding 155 mM (8.3 g L−1) ammonium chloride in 0.01 M Tris–HCl buffer, with pH = 7.5. Different concentrations of cancer cell lines were then spiked in whole blood or lysed blood.
To initiate cell capture experiments, one channel volume (~100 μL) of 1 mg mL−1 avidin (Invitrogen) in PBS was first introduced into the device, followed by incubation for 15 min and then three rinses with PBS. Then, one channel volume of biotinylated anti-EpCAM (20 μg mL−1) was introduced into the device and incubated for 15 min, followed by three rinses with the PBS containing BSA and Tween-20. Finally, 1 mL of cell mixture or blood sample was pumped into the device at a flow rate of 1 μL s−1 (or other flow rates specified in the text). At the end of the experiment, the microchannel was washed three times with PBS, followed by acquiring fluorescent images for the determination of the number of cells captured.
Instrument setup
The cell suspension or blood sample was introduced into the device by pumping using a syringe pump (KD Legato 111, KD Scientific, Holliston, MA) with a BD syringe connected to the inlet of the device via polymer tubing and a female luer-to-barb adapter (IDEX Health & Science, Oak Harbor, WA). To avoid cell settling, a tiny magnetic stirring bar was placed inside the 1 mL syringe, with a stir plate beneath the syringe. The magnetic stirring bar kept cells in suspension while the cell mixture or blood was being pumped through the device. An Olympus IX71 fluorescence microscope (Olympus America, Melville, NY) with an automated ProScan stage (Prior Scientific, Rockland, MA) was used to image and count the captured cells on the device.Cell release and re-culture
Cell release was achieved by trypsin and high flow rate washing. After cell capture inside the channel, proteolytic enzyme trypsin (0.25%) was introduced into the device and incubated for 5 min at 37 °C. Then, cell culture medium was pumped into the device at a flow rate of 5 μL s−1 to dislodge the bound cells. The release flow rate was much higher than the cell capturing flow rate of 1 μL s−1. Released cells were collected in a new cell culture dish (60 mm × 15 mm size), with a total volume of 4 mL culture medium. Then the cells were put into the incubator for propagation in culture.To test the viability of cells captured by the device, propidium iodide (PI) and acridine orange (AO) staining (Invitrogen) assays were performed. PI is a membrane-impermeant stain that labels only dead cells with red fluorescence. AO is a membrane-permeable dye that binds to nucleic acids of all cells and induces green fluorescence. By following the manufacturer's instructions, PI–AO working solution was prepared to contain 2 μM PI and 2 μM AO in PBS. After incubating the working solution with cells for 10 min, fluorescent images were taken to evaluate the viability of the captured cells (Fig. S4a†).
Patient blood specimen collection and processing
Blood samples of patients with metastatic pancreatic cancer were obtained from the University of Florida Health Cancer Center after informed consent through a University of Florida Institutional Review Board (IRB)-approved protocol. Blood samples from normal healthy participants were obtained through the Gainesville LifeSouth Community Blood Center following a University of Florida IRB-approved protocol. Specimens were collected into BD Vacutainer tubes containing anti-coagulant sodium heparin and were processed within 6 hours after being drawn. CTC capture was performed by the same protocols as described above. Unlike the pre-stained tumor cells spiked in blood, CTCs from patients' blood were not labeled. Three-color immunocytochemistry (DAPI, FITC anti-cytokeratin, PE anti-CD45) was conducted to identify CTCs from nonspecifically captured blood cells. Cell staining began with cell fixation and permeabilization by incubation for 20 min with 4% paraformaldehyde and 0.2% Triton X-100, respectively. Then, a mixture of 10 μg mL−1 PE anti-CD45, 10 μg mL−1 FITC anti-cytokeratin and 500 nM DAPI were introduced into the device and incubated for 20 min. After washing, the microfluidic device was examined under the fluoresce microscope. Only cells that were DAPI positive, CD45 negative, cytokeratin positive, with the appropriate size and morphology were counted as CTCs (DAPI+, CD45−, cytokeratin+). Cell debris, red blood cells (DAPI−) and white blood cells (DAPI+, CD45+, cytokeratin−) and “double positive” cells (both CD45+ and cytokeratin+, with DAPI+) were excluded from counting. CTC capture purity was defined as the ratio of the number of CTCs captured to the total number of nucleated cells (DAPI+) bound to the device. For another sets of experiments, we released the specifically captured CTCs along with nonspecifically captured leukocytes into culture dish (instead of staining and counting). And fresh medium was added once a week (with leukocytes washed away). We observed a few cells (probably CTCs) adhered to the culture dish after 1 week of culture. However, these adhered cells did not proliferate, even after 4 months of culturing (unlike the spiked tumor cells which grew into clusters within 2 weeks).Results and discussion
Target cell capture from a homogenous cell mixture
The performance of the device was first evaluated by sorting a mixture of pancreatic cancer cell lines: target L3.6pl cells (EpCAM+) and control MIAPaCa-2 cells (EpCAM−). Flow cytometry results show that L3.6pl cells bind strongly with anti-EpCAM, while MIAPaCa-2 cells do not bind with anti-EpCAM (Fig. S1 & S3†). This means that L3.6pl cells express a significant number of EpCAM receptors, while MIAPaCa-2 cells express negligible surface EpCAM, which is consistent with data already reported in literature.34 To start the cell capture, biotinylated anti-EpCAM was first immobilized on the surface of microchannel. Then a cell mixture containing 106 L3.6pl cells (stained with Vybrant DiI, red) and 106 MIAPaCa-2 cells (stained with Vybrant DiD, blue) per mL sample was introduced into the microchannel. Fig. 2a shows a representative image of the cell mixture prior to sorting, with same number of target cells and control cells. Fig. 2b shows a typical image after the cell mixture was processed through the device, with L3.6pl cells in the majority, while most control MIAPaCa-2 cells were removed by washing. Fig. 2a & b clearly indicate that significant enrichment of target cells can be achieved using the antibody-coated microfluidic device. | ||
| Fig. 2 Representative image of a) 1:1 mixture of target L3.6pl cells (red) and control MIAPaCa-2 cells (blue) before sorting; b) L3.6pl cells (red) and MIAPaCa-2 cells (blue) after sorting. Target cells were efficiently captured while most control cells were removed. | ||
After the initial experiments, different flow rates were used to study the effects of flow rate on cell capture efficiency, defined as the ratio of the number of target cells captured to the number of target cells initially introduced. As shown in Fig. 3a, the capture efficiency of L3.6pl cells was >90% at low flow rates, but decreased dramatically at flow rates above 1 μL s−1, primarily due to the reduced interaction time between the cells and antibody-coated surfaces as well as the increased shear stress at higher flow rates. To obtain both efficient capture and sufficient throughput, an optimal flow rate of 1 μL s−1 was chosen, with a flow velocity of 0.75 mm s−1 and maximum shear stress of 0.38 dyn cm−2 at the wall. As shown in Fig. 3b, the capture efficiency was (90 ± 2)% for L3.6pl cells and (92 ± 4)% for BxPC-3 cells at 1 μL s−1.
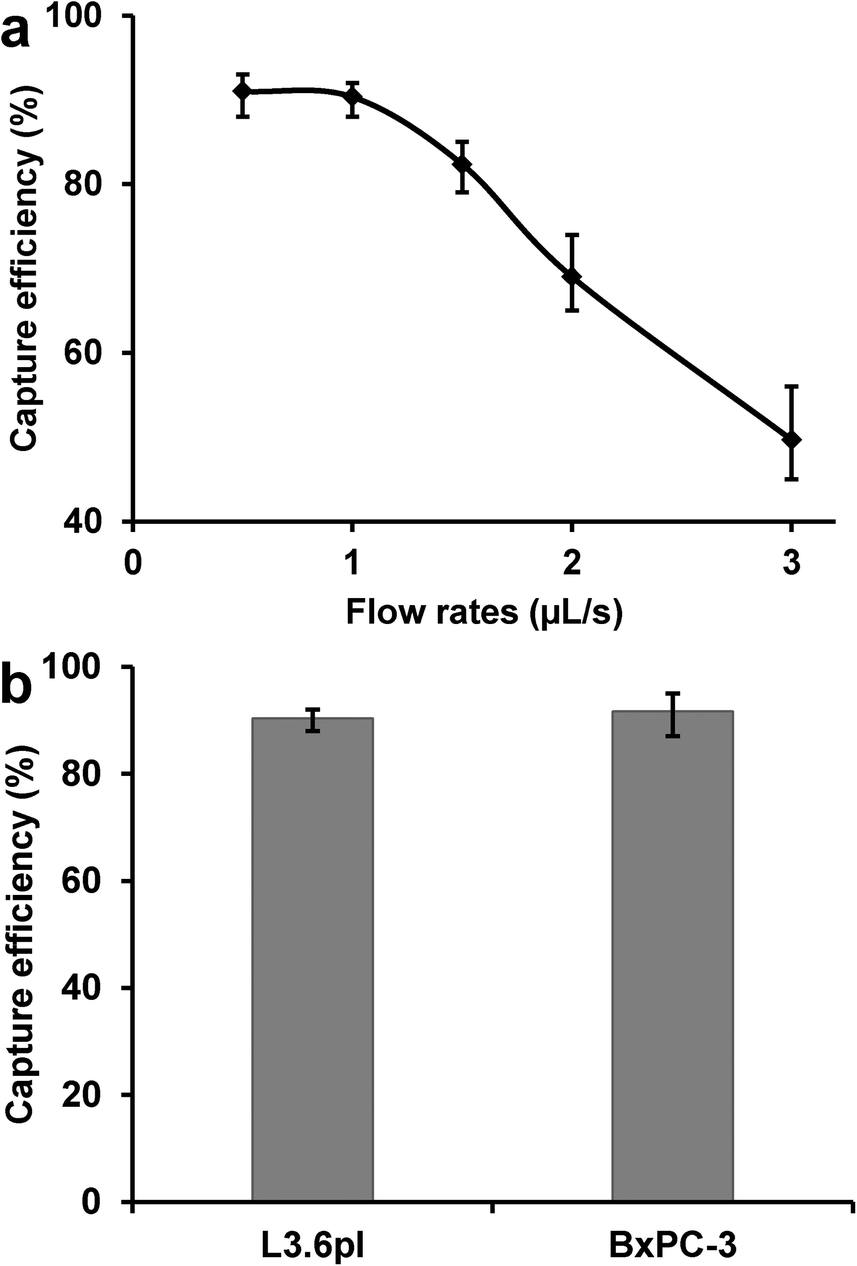 | ||
| Fig. 3 a) L3.6pl cell capture efficiency as a function of flow rate; reduced capture occurred at a high flow rate because of a larger shear force and the reduced interaction time between cells and antibody-coated surfaces. b) Capture efficiency of L3.6pl cells and BxPC-3 cells at the optimal flow rate of 1 μL s−1, with >90% capture efficiency for both types of cells. Error bars show range (n = 3). | ||
Micromixer device optimization for high-performance cell capture
When we used the traditional micromixer design dimensions (HB chip, Fig. 1c)26 for pancreatic tumor cell capture, we found that non-target cells were easily trapped in the device (causing low CTC capture purity) and cells were not captured on the same focus plane (making imaging and counting difficult). We suspected that cell trapping took place in narrow grooves (with high aspect ratio) as illustrated in Fig. 1c, and hypothesized that an increased groove width would give better purity. Thus we made two new designs by increasing the groove width from 50 μm (narrow groove, Fig. 1c) to 80 μm and 120 μm (wide groove, Fig. 1d). Experimental results proved that a wider groove with increased groove pitch achieved high purity cell capture, while maintaining cell capture efficiency. As shown in Fig. 4, with a groove width of 120 μm, we obtained a capture purity of 84%, while the traditional 50 μm groove width yield only 61% purity. In addition, the capture efficiency for the wide groove design was not reduced, even slightly higher, which agrees with simulation study by Forbes et al.27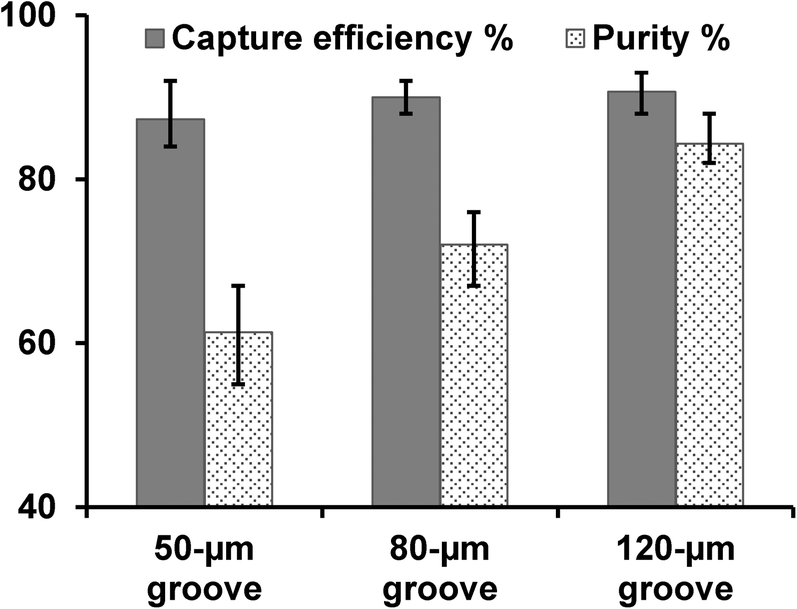 | ||
| Fig. 4 Comparisons of capture efficiency and purity of L3.6pl cells with different groove width: 50 μm (conventional narrow groove HB chip), 80 μm, and 120 μm (wide groove GEM chip). Capture purity is defined as the ratio of the number of target cells captured to the number of total cells captured. Error bars represent range (n = 3). | ||
Tumor cell capture from lysed blood and whole blood
To test cell capture under more physiological conditions and to mimic CTC capture from patient blood, we conducted a series of experiments in which labeled L3.6pl cells were spiked in lysed or whole blood. Samples were prepared by spiking 50–50000 L3.6pl cells in 1 mL lysed blood or whole blood. After being pumped through the micromixer device, as many as ~92% of L3.6pl cells were captured from lysed blood (Fig. 5a), and ~89% of L3.6pl cells were captured from whole blood (Fig. 5b), proving that the device and the conditions are suitable for capturing CTCs from patient blood specimens with or without prior red blood cell lysis.
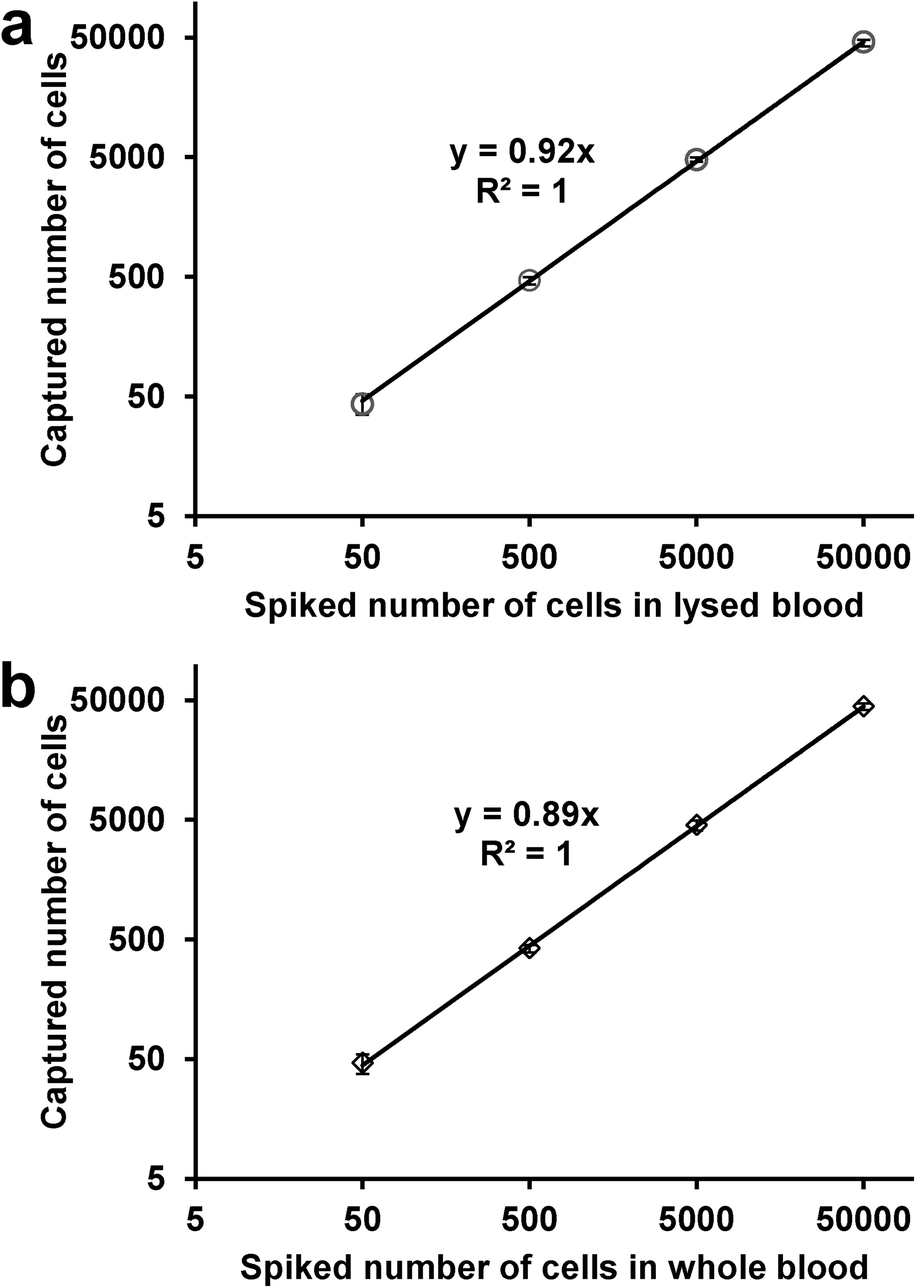 | ||
| Fig. 5 Regression analysis of the number of the L3.6pl cells captured by the microfluidic device versus the number of the cells spiked in 1 mL of a) lysed blood, b) whole blood. The x-axis indicates the number of spiked cells, y is the number of captured cells. Error bars show range (n = 3). | ||
Cell release and cell viability
The detachment and release of captured cells in antibody-coated microchannels were achieved by using a combination of trypsinization (enzymatic release)14 and high flow rate washing (high shear stress).16 Detached cells were collected in a cell culture dish with fresh medium for propagation in cell culture. As shown in Fig. 6a, the release efficiency of L3.6pl cells increased to >60% by using the combined releasing method, while high flow washing alone gave only ~30% release. The release efficiency is defined as the ratio of the number of cells released to the number of cells captured. The trypsin release and shear stress-based release procedures cause minimum cell damage as proved by cell viability assay and flow cytometry. PI–AO assay was used to test the viability of released cells, with >85% cells remaining viable after the capturing and release process (Fig. 6b), making the isolated tumor cells suitable for subsequent cellular analysis. Flow cytometry tests also showed that released L3.6pl cells retain their binding with anti-EpCAM, as shown in Fig. S4b.†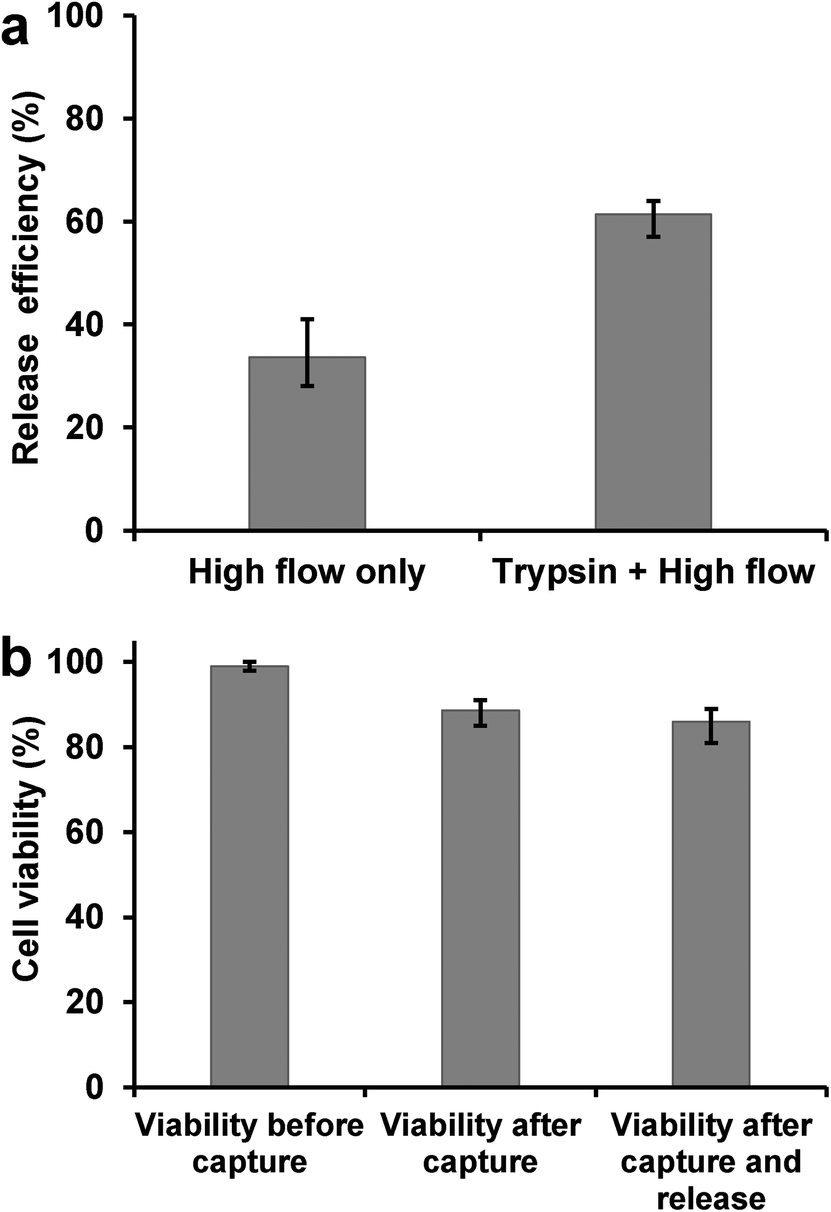 | ||
| Fig. 6 a) By high flow rate washing alone, a release efficiency of 34% was obtained; with a combination of trypsinization and high flow rate washing, the release efficiency reached 62% for L3.6pl cells. b) Cell viability before cell capture process (extracted directly from culture) is ~99%. Cell viability immediately after cell capture in device is ~89%; after release the viability is ~86%, without significant difference. The high viability indicates that released cells are suitable for subsequent cell culture. Error bars represent range (n = 3). | ||
Re-culture of captured cells
To determine whether isolated tumor cells can be cultured, 5000 L3.6pl cells were spiked into whole blood and subjected to the capture and release process as discussed above. The released cells were then seeded into cell culture dishes for propagation in culture. As comparison, 5000 intact L3.6pl cells (not subjected to the culture and release process) were directly seeded for culture with the same conditions. Results showed that both adhered well and proliferated on the culture dishes, forming large clusters and colonies by day 9 (shown in Fig. 7) and growth to confluence with longer time (14 days), although the captured cells took a little longer to reach confluence than intact cells. Then we were able to trypsinize these cells and seed them to other culture dishes, where they grew as adherent monolayers. The isolated cells have successfully undergone multiple (>8) passages without loss of viability or detectable changes in behavior. Flow cytometry tests indicated that the isolated cells maintain binding behavior with anti-EpCAM, as shown in Fig. 7c. These results clearly demonstrate that tumor cell lines isolated from whole blood retain both their viability and their proliferation ability, which are crucial for CTC cellular analysis.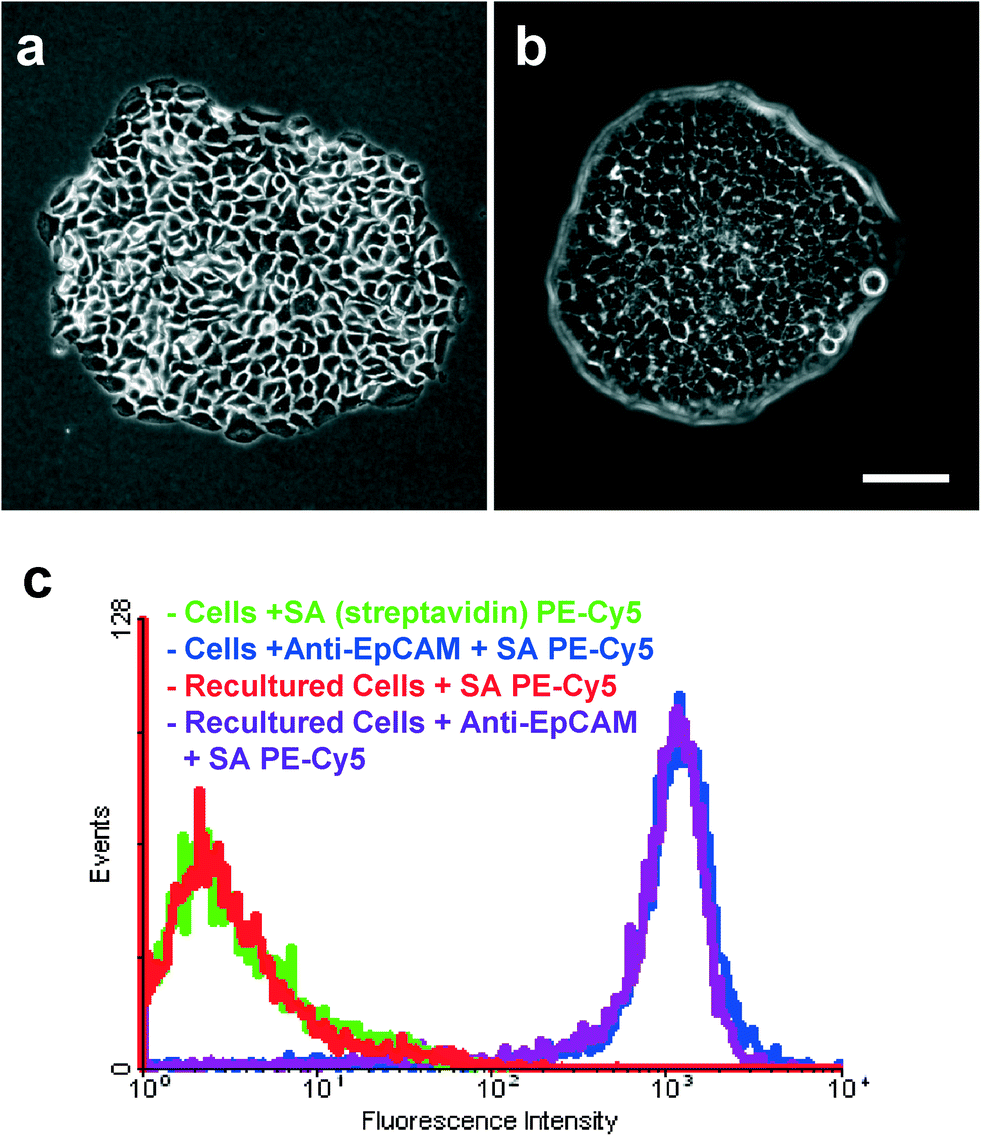 | ||
| Fig. 7 Phase contrast micrograph (10×) of a) re-cultured BxPC-3 cells; b) re-cultured L3.6pl cells after 9 days of growth. Scale bar = 100 μm. c) Flow cytometry test showing that the captured and then recultured cells maintained their binding capability with anti-EpCAM, without any differences compared to intact cells. | ||
Isolation of CTCs from patients with pancreatic cancer using the GEM chip
Blood samples from patients with metastatic pancreatic cancer (stage IV) were analyzed for CTC enumeration using the above-optimized device and conditions. Since EpCAM has been known to be overexpressed in pancreatic adenocarcinoma, anti-EpCAM was used as the capture agent.35 Milliliters of patient blood were pumped through the antibody-coated device. After fixation and permeabilization, three-color immunocytochemistry was utilized to identify and count CTCs from nonspecifically captured white blood cells, using FITC-labeled anti-Cytokeratin (CK, green), PE-labeled anti-CD45 (red) and DAPI (blue) for staining. As shown in Fig. 8, CTCs are DAPI+/CK+/CD45− cells, while WBCs are DAPI+/CK−/CD45+ cells. A significant population of “double positive” cells with both hematopoietic and epithelial markers (CK+/CD45+) were found in quite a few patient samples (average ~2 “double positive” cells in 1 mL patient blood). Since the origin and significance of these cells are under debate, we temporarily excluded them from CTC counting.26,36 However, detailed numbers of “double positive” cells were presented in Table S2 in ESI.† For the 18 pancreatic cancer patient samples processed, CTCs were found in 17 cases (>94%), with an average number of 3 CTCs per mL of blood, as shown in Table 1 (detailed information shown in Table S2†). To examine the possibility of false positives, we investigated capturing CTCs from whole blood of normal healthy individuals. Similar volumes of blood were run through our device using the same protocol. Table S3† shows the results from blood samples of nine healthy donors. Zero CTCs were detected from blood samples of all normal healthy individuals studied, thus showing a false positive rate of zero. Additionally, we found much fewer “double positive” cells in healthy donors' blood than in patient blood, indicating that most of the “double positive” cells could be the heterogeneous CTCs or the nonspecific binding of anti-CD45 to CTCs. Further studies with additional markers are required to understand and explain these “double positive” cells. | ||
| Fig. 8 Fluorescence microscope images (40×) of CTCs captured from patient blood: a) a representative image of CTCs, with DAPI+, cytokeratin+ and CD45−; b) typical image of white blood cells (WBCs), with DAPI+, CK−, and CD45+. Scale bar = 10 μm. | ||
| Patient sample | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 |
| CTCs per mL | 2 | 4 | 5 | 2 | 1 | 0 | 3 | 2 | 2 | 5 | 3 | 2 | 1 | 4 | 4 | 7 | 3 | 1 |
For capturing CTCs from patient blood, much more capture of leukocytes was observed than the spiking experiments using healthy donor's blood. For 1 mL of blood processed, ~3500 leukocytes were captured for spiking experiments using healthy samples, while >24000 of leukocytes were captured for patient samples. This could due to complexity of patient blood conditions. The high purity of the GEM chip shows more advantage over the traditional mixing chip when enumerating patient CTCs. The GEM chip would have been able to detect an average of ~23 CTCs from 7.5 mL blood, much higher than the cut-off number of CellSearch system. Considering that CellSearch is inefficient for pancreatic cancer, the GEM chip reported here could become a powerful tool for CTC enumeration in pancreatic cancer. In addition, with a flow rate of 1 μL s−1 (3.6 mL h−1), 1 mL blood sample can be processed within 17 min, which gives sufficient throughput for clinical applications.
We also released the captured patient CTCs and then attempted to culture them in vitro, using the above-mentioned protocol for re-culturing spiked tumor cells. Similar release efficiency was obtained for patient CTCs. However, CTCs were not able to proliferate or propagate in the culture dish, although they were found adhered to the culture dish. We processed 12 pancreatic patient blood samples (with 5–10 mL volume for each sample), but CTCs from them were not proliferating till now. We suspect that during the progression of metastasis, CTCs, shed from a primary tumor and entered into bloodstream, might lose their ability to proliferate.
Monitoring anti-cancer treatment response using CTCs
To demonstrate the unique clinical potential of our device and system, we evaluated the relation between the CTC number and tumor size in patients with pancreatic cancer undergoing chemotherapy. Three patients with stage IV metastatic pancreatic cancer (deemed unresectable) were included in the analysis. Each patient received identical standard treatments with the identical palliative chemotherapy and with X-ray computed tomography (CT) scans done at the same intervals. Blood samples were collected at baseline and at the first day of each subsequent treatment cycle. CTCs were captured and counted using the device and methods discussed above. Investigators were blinded to the demographic and clinicopathological characteristics of the patients. The number of CTCs captured at different treatment cycles is plotted in Fig. 9a–c. In general, the CTC number decreased with continuation of treatment and modeled the CT scan results (which represent standard clinical response measurements). The CTC number correlated proportionally with CT scan-measured tumor size in each of the three patients. Fig. 9d & e show that tumor size decreased as treatment progressed for patient #3, which was reflected by the trend of CTC number in Fig. 9c. CT scan data from patient #1 and patient #2 also indicated either reduced primary tumor size or reduced metastatic tumor burden (data not shown). Together, these results indicate that CTC quantification using our device correlates with clinical response and findings from CT imaging, but causes significantly less harms to patients than standard clinical radiographic measurements. With the noninvasive nature of our approach, it could provide a powerful tool for monitoring early response or failure to cancer treatment and potentially early cancer diagnosis and relapse prediction.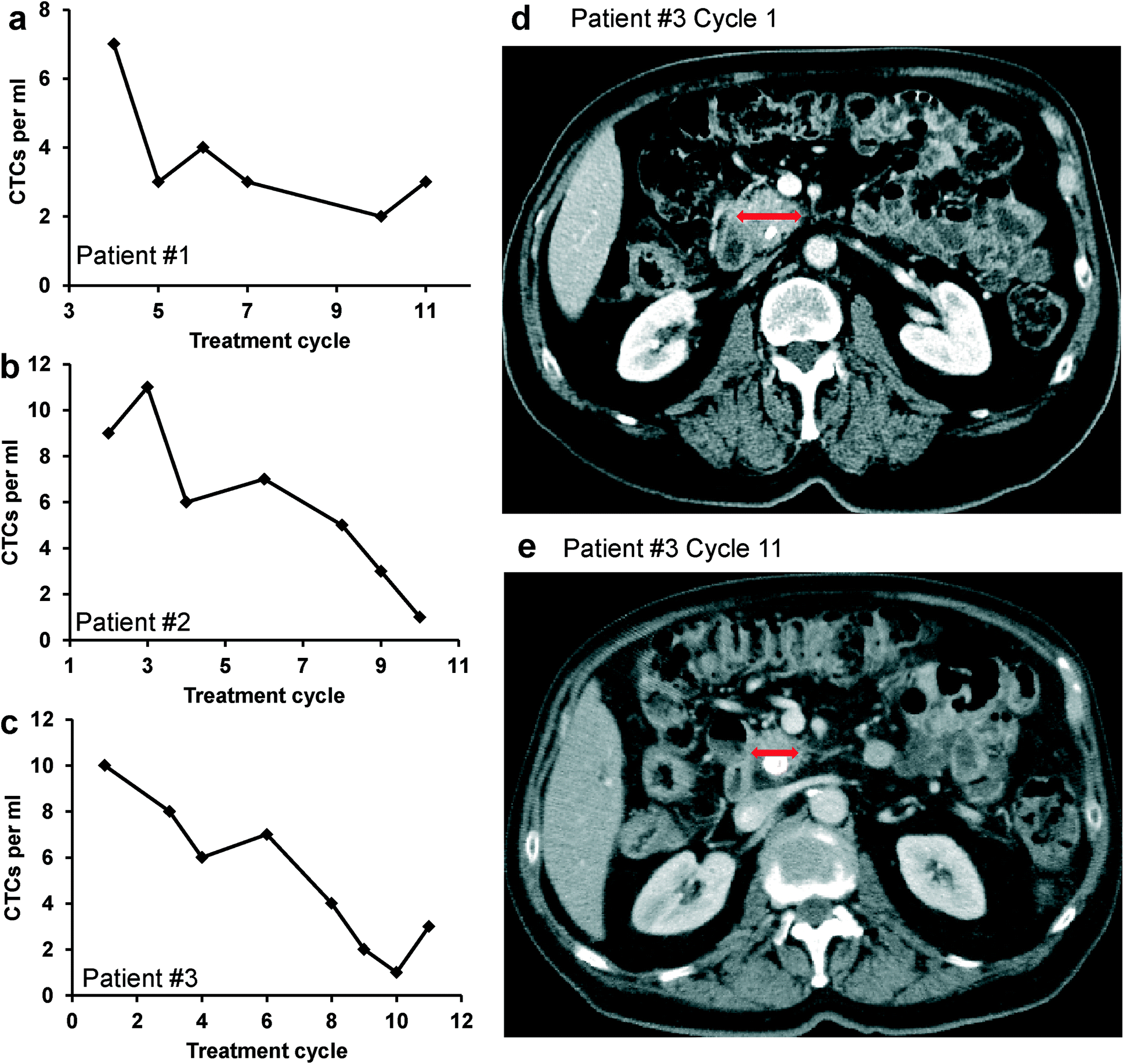 | ||
| Fig. 9 a–c) The number of CTCs per mL of blood from pancreatic cancer patients at different treatment cycles for three patients: a) patient #1; b) patient #2; c) patient #3. d–e) CT scan image of patient #3 at d) the beginning of the treatment (cycle 1); e) the latter stage of treatment (cycle 11); the red arrows indicate regression of the primary pancreatic cancer. Each treatment cycle is 14 days. | ||
Conclusion
In summary, we demonstrated an efficient CTC capture platform based on a geometrically enhanced mixing (GEM) chip. The device achieved >90% capture efficiency, >84% purity with a throughput of processing 3.6 mL blood in 1 hour. The system was then utilized to isolate CTCs from pancreatic cancer patient blood samples, with CTCs detected in 17 of 18 samples. We also successfully demonstrated positive correlation in monitoring anti-cancer treatment response using the CTC numbers obtained from our device. In addition, the captured cells were released from device with >61% release efficiency, and with >86% viability. Furthermore, we demonstrated the ability to culture the captured cells, a critical requirement for post-isolation cellular analysis. Although it is extremely challenging to culture the isolated CTCs from patient blood and to develop a new cell line, our system shows the possibility to culture spiked tumor cells, after the sophisticated capture and release process, while maintaining their viability and proliferation capability. Therefore, our CTC capture system shows great potential for efficient CTC enrichment, isolation, and cellular/genetic analysis, leading to now feasible “liquid biopsy” of pancreatic cancer. Our future efforts include further improving CTC capture purity, culturing the captured CTCs from patient, cellular and genetic study of isolated CTCs.Acknowledgements
This work was supported in part by National Cancer Institute of NIH (K25CA149080) and the University of Florida. Olorunseun O. Ogunwobi is supported by the NIH T32 grant (5T32 CA009126-31A1) and the American Association for Cancer Research AACR-FNAB Fellows grant in Translational Pancreatic Cancer Research (grant #12-30-14-OGUN). Chen Liu is also supported by NIH grants (R01CA133086, R01AA019976 and K26RR023976). We acknowledge Dr. Jose Trevino's lab in Department of Surgery for providing L3.6pl cells, Dr. Weihong Tan's lab in Department of Chemistry for accessing flow cytometers, Dr. Kathryn R. Williams for manuscript review, Dr. Xin Xu, Dr. Xiangjun Zheng and Mr. Chris Cassano for insightful discussions, Ms. Sydney Shaouy and Mr. Ram Mirchandani for assistance with PDMS device fabrication.References
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed
.
- K. Tjensvoll, O. Nordgård and R. Smaaland, Int. J. Cancer, 2014, 134, 1–8 CrossRef PubMed
- P. Cen, X. Ni, J. Yang, D. Y. Graham and M. Li, Biochim. Biophys. Acta, Rev. Cancer, 2012, 1826, 350–356 CrossRef CAS PubMed
- K. Pantel, R. H. Brakenhoff and B. Brandt, Nat. Rev. Cancer, 2008, 8, 329–340 CrossRef CAS PubMed
- K. Pantel, C. Alix-Panabieres and S. Riethdorf, Nat. Rev. Clin. Oncol., 2009, 6, 339–351 CrossRef CAS PubMed
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. M. M. Terstappen and D. F. Hayes, N. Engl. J. Med., 2004, 351, 781–791 CrossRef CAS PubMed
- M. C. Miller, G. V. Doyle and L. W. M. M. Terstappen, J. Oncol., 2010, 2010, 617421 CrossRef PubMed
- M. A. Eloubeidi, V. K. Chen, I. A. Eltoum, D. Jhala, D. C. Chhieng, N. Jhala, S. M. Vickers and C. M. Wilcox, Am. J. Gastroenterol., 2003, 98, 2663–2668 Search PubMed
- E. Crowley, F. Di Nicolantonio, F. Loupakis and A. Bardelli, Nat. Rev. Clin. Oncol., 2013, 10, 472–484 CrossRef CAS PubMed
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS PubMed
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. J. Tibbe, J. W. Uhr and L. W. M. M. Terstappen, Clin. Cancer Res., 2004, 10, 6897–6904 CrossRef PubMed
- S. Riethdorf, H. Fritsche, V. Müller, T. Rau, C. Schindlbeck, B. Rack, W. Janni, C. Coith, K. Beck, F. Jänicke, S. Jackson, T. Gornet, M. Cristofanilli and K. Pantel, Clin. Cancer Res., 2007, 13, 920–928 CrossRef CAS PubMed
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed
- A. A. Adams, P. I. Okagbare, J. Feng, M. L. Hupert, D. Patterson, J. Göttert, R. L. McCarley, D. Nikitopoulos, M. C. Murphy and S. A. Soper, J. Am. Chem. Soc., 2008, 130, 8633–8641 CrossRef CAS PubMed
- J. P. Gleghorn, E. D. Pratt, D. Denning, H. Liu, N. H. Bander, S. T. Tagawa, D. M. Nanus, P. A. Giannakakou and B. J. Kirby, Lab Chip, 2010, 10, 27–29 RSC
- X. Zheng, L. S.-L. Cheung, J. A. Schroeder, L. Jiang and Y. Zohar, Lab Chip, 2011, 11, 3269–3276 RSC
- S. Wang, K. Liu, J. Liu, Z. T. F. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss, Y.-K. Lee, L. W. K. Chung, J. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K. F. Shen and H.-R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef CAS PubMed
- P. G. Schiro, M. Zhao, J. S. Kuo, K. M. Koehler, D. E. Sabath and D. T. Chiu, Angew. Chem., Int. Ed., 2012, 51, 4618–4622 CrossRef CAS PubMed
- W. Sheng, T. Chen, R. Kamath, X. Xiong, W. Tan and Z. H. Fan, Anal. Chem., 2012, 84, 4199–4206 CrossRef CAS PubMed
- W. Sheng, T. Chen, W. Tan and Z. H. Fan, ACS Nano, 2013, 7, 7067–7076 CrossRef CAS PubMed
- J. A. Phillips, Y. Xu, Z. Xia, Z. H. Fan and W. Tan, Anal. Chem., 2009, 81, 1033–1039 CrossRef CAS PubMed
- J. Chen, J. Li and Y. Sun, Lab Chip, 2012, 12, 1753–1767 RSC
- S. K. Arya, B. Lim and A. R. A. Rahman, Lab Chip, 2013, 13, 1995–2027 RSC
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P.-I. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 5, 179ra147 CrossRef PubMed
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647–651 CrossRef CAS PubMed
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed
- T. P. Forbes and J. G. Kralj, Lab Chip, 2012, 12, 2634–2637 RSC
- J. H. Kang, S. Krause, H. Tobin, A. Mammoto, M. Kanapathipillai and D. E. Ingber, Lab Chip, 2012, 12, 2175–2181 RSC
- L. Khoja, A. Backen, R. Sloane, L. Menasce, D. Ryder, M. Krebs, R. Board, G. Clack, A. Hughes, F. Blackhall, J. W. Valle and C. Dive, Br. J. Cancer, 2012, 106, 508–516 CrossRef CAS PubMed
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491–502 CrossRef CAS PubMed
- A. Mata, A. J. Fleischman and S. Roy, J. Micromech. Microeng., 2006, 16, 276 CrossRef
- T. Nakamura, I. J. Fidler and K. R. Coombes, Cancer Res., 2007, 67, 139–148 CrossRef CAS PubMed
- O. O. Ogunwobi, W. Puszyk, H.-J. Dong and C. Liu, PLoS One, 2013, 8, e63765 CAS
- L. Ren-Heidenreich, P. A. Davol, N. M. Kouttab, G. J. Elfenbein and L. G. Lum, Cancer, 2004, 100, 1095–1103 CrossRef CAS PubMed
- G. Moldenhauer, A. V. Salnikov, S. Lüttgau, I. Herr, J. Anderl and H. Faulstich, J. Natl. Cancer Inst., 2012, 104, 622–634 CrossRef CAS PubMed
-
M. Lustberg, K. Jatana, M. Zborowski and J. Chalmers, in Minimal Residual Disease and Circulating Tumor Cells in Breast Cancer, ed. M. Ignatiadis, C. Sotiriou and K. Pantel, Springer Berlin Heidelberg, 2012, vol. 195, ch. 9, pp. 97–110 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc51017d |
| This journal is © The Royal Society of Chemistry 2014 |