Isolation and mutational analysis of circulating tumor cells from lung cancer patients with magnetic sifters and biochips†
Christopher M.
Earhart
a,
Casey E.
Hughes
b,
Richard S.
Gaster
c,
Chin Chun
Ooi
d,
Robert J.
Wilson
a,
Lisa Y.
Zhou
e,
Eric W.
Humke
b,
Lingyun
Xu
f,
Dawson J.
Wong
g,
Stephen B.
Willingham
h,
Erich J.
Schwartz
i,
Irving L.
Weissman
hi,
Stefanie S.
Jeffrey
j,
Joel W.
Neal
be,
Rajat
Rohatgi
b,
Heather A.
Wakelee
be and
Shan X.
Wang
*agk
aDepartment of Materials Science and Engineering, Stanford University, Stanford, CA 94305, USA
bDivision of Oncology, Department of Medicine, Stanford University School of Medicine, Stanford, California 94305, USA
cDepartment of Bioengineering, Stanford University, Stanford, CA 94305, USA
dDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305, USA
eStanford Cancer Institute, Stanford, CA 94305, USA
fDepartment of Radiology, Stanford University, Stanford, CA 94305, USA
gDepartment of Electrical Engineering, Stanford University, California 94305, USA
hInstitute for Stem Cell Biology and Regenerative Medicine and the Ludwig Cancer Center, Stanford, CA 94305, USA
iDepartment of Pathology, Stanford University School of Medicine, Stanford, CA 94305, USA
jDepartment of Surgery, Stanford University, Stanford, CA 94305, USA
kGeballe Laboratory for Advanced Materials, Stanford University, McCullough Building, Room 351, 476 Lomita Mall, Stanford, CA 94305-4045, USA. E-mail: sxwang@stanford.edu; Tel: +1 650-723-8671
First published on 23rd July 2013
Abstract
Detection and characterization of circulating tumor cells (CTCs) may reveal insights into the diagnosis and treatment of malignant disease. Technologies for isolating CTCs developed thus far suffer from one or more limitations, such as low throughput, inability to release captured cells, and reliance on expensive instrumentation for enrichment or subsequent characterization. We report a continuing development of a magnetic separation device, the magnetic sifter, which is a miniature microfluidic chip with a dense array of magnetic pores. It offers high efficiency capture of tumor cells, labeled with magnetic nanoparticles, from whole blood with high throughput and efficient release of captured cells. For subsequent characterization of CTCs, an assay, using a protein chip with giant magnetoresistive nanosensors, has been implemented for mutational analysis of CTCs enriched with the magnetic sifter. The use of these magnetic technologies, which are separate devices, may lead the way to routine preparation and characterization of “liquid biopsies” from cancer patients.
Introduction
Routine capture and characterization of circulating tumor cells (CTCs) from peripheral blood of cancer patients has the potential to revolutionize solid tumor oncology, ushering in the era of noninvasive “liquid biopsies” (i.e. blood samples containing CTCs) as opposed to the invasive tissue biopsies for initial diagnosis and subsequent management of disease. CTC enrichment and characterization is especially challenging because these cells must be captured from blood at parts per billion levels.1–4In 2007, Nagrath et al. reported their groundbreaking development of the “CTC chip”, a microfluidic cell-capture platform with sensitivity superior to that of the FDA-approved Veridex “CellSearch” platform.5 Since then, a host of devices, many of which are microchip technologies, have been developed for CTC isolation and detection. These devices generally rely on differences in physical properties (e.g. size, rigidity) or expression of surface antigens (e.g. positive selection with the epithelial cell adhesion molecule (EpCAM)) between CTCs and background blood cells.4–16 Several devices, including the magnetic sifter, feature isolation from whole blood to simplify processing and reduce losses, a feature which is not currently available from Veridex.
Each microdevice platform possesses various advantages and limitations, and most need further development before widespread clinical adoption. Devices based on size selection rely on the ordinarily larger diameter and higher stiffness of CTCs as compared with peripheral blood cells.6–9 Size selection offers label-free and high-throughput capture, however, successful enrichment assumes that CTCs are predictable in size and stiffness, the latter of which has been hypothesized to be variable in epithelial to mesenchymal (EMT) transitions.17 Another class of microdevices involves flow through microchannels containing micropillars, nanowires, or patterned grooves, aimed at increasing the interaction between cells and antibody-functionalized surfaces.5,10–13 These devices have demonstrated sensitive detection of CTCs, but the planar nature of flow limits operating flow rates to approximately 1–2 ml hr−1 before capture efficiency suffers. Furthermore, harvesting of cells is challenging due to covalent immobilization of capture antibodies within the device. The device footprints are also in the order of ∼1000 mm2 and, while seemingly small, can require a large number of images to identify CTCs.5,11,12
Magnetic separation is an established method practised in both bulk16,18–21 and microchip platforms,15,22–24 and an FDA approved tool is available for enumeration of CTCs for prostate, breast and colorectal cancers.25,26 In magnetic separation, antibody-functionalized magnetic particles bind in suspension with target cells. Labeled cells are subjected to magnetic field gradients, introduced by permanent magnets or electromagnets, leading to capture. Magnetic approaches offer the same benefits of specificity as immobilized antibody-based approaches while allowing cell recovery by removal of the magnetic field. Bulk separators, however, often suffer from non-uniformities in capture and rinsing forces, as well as cell loss, due to non-uniform, dense capture matrices often incorporated to enhance field gradients. Magnetic microdevices can avoid these issues, but generally offer lower throughput due to the planar nature of flow.
In addition to enumeration, such devices also provide enriched CTCs for use in post-separation nucleic acid characterization of cancer mutations, typically using cells lysed on, or after elution from, various capture devices. Such detection of specific tumor mutations is quite important as it can inform proper selection of therapy. The identification of associated expressed mutant proteins can, in principle, provide more direct information regarding protein expression, which complements mRNA based methods. Recent progress in using giant magnetoresistive (GMR) sensors27–29 to quantitate cancer biomarker proteins with high-sensitivity makes this detection platform a suitable candidate for analysis of CTCs enriched by the magnetic sifter. We later show that the magnetic sifter's ability to release cells for downstream analysis can be exploited to detect the presence of an epidermal growth factor receptor (EGFR) mutation in a lung cancer patient’s CTCs by using EGFR mutation-specific antibodies in magnetically sensed antibody sandwich assays, enabling proteomic mutational analyses of tumor cells.30
In this context, we have adapted a magnetic sifter, a magnetic pore structure (Fig. 1) that uses a flow-through fluidic array configuration to yield large equivalent magnetic forces at each pore and uniform rinse flows, for cell separation. The separation principle of the magnetic sifter is shown in Fig. 1c. Target cells are labeled with magnetic nanoparticles via anti-EpCAM. The sample is then pumped through the magnetic sifter during application of an external magnetic field, whereupon labeled cells experience large magnetic capture forces directed towards the pore edges. Unlabeled cells pass through the chip, and captured cells can be imaged directly on the magnetic sifter array, and/or harvested by removing the field and rinsing. Previously, we reported a magnetic sifter device intended for individual magnetic nanoparticles and protein separation.31,32 In this work, the magnetic sifter has been re-engineered to enhance the purity, capture yield, and viability of CTCs retrieved from whole blood.
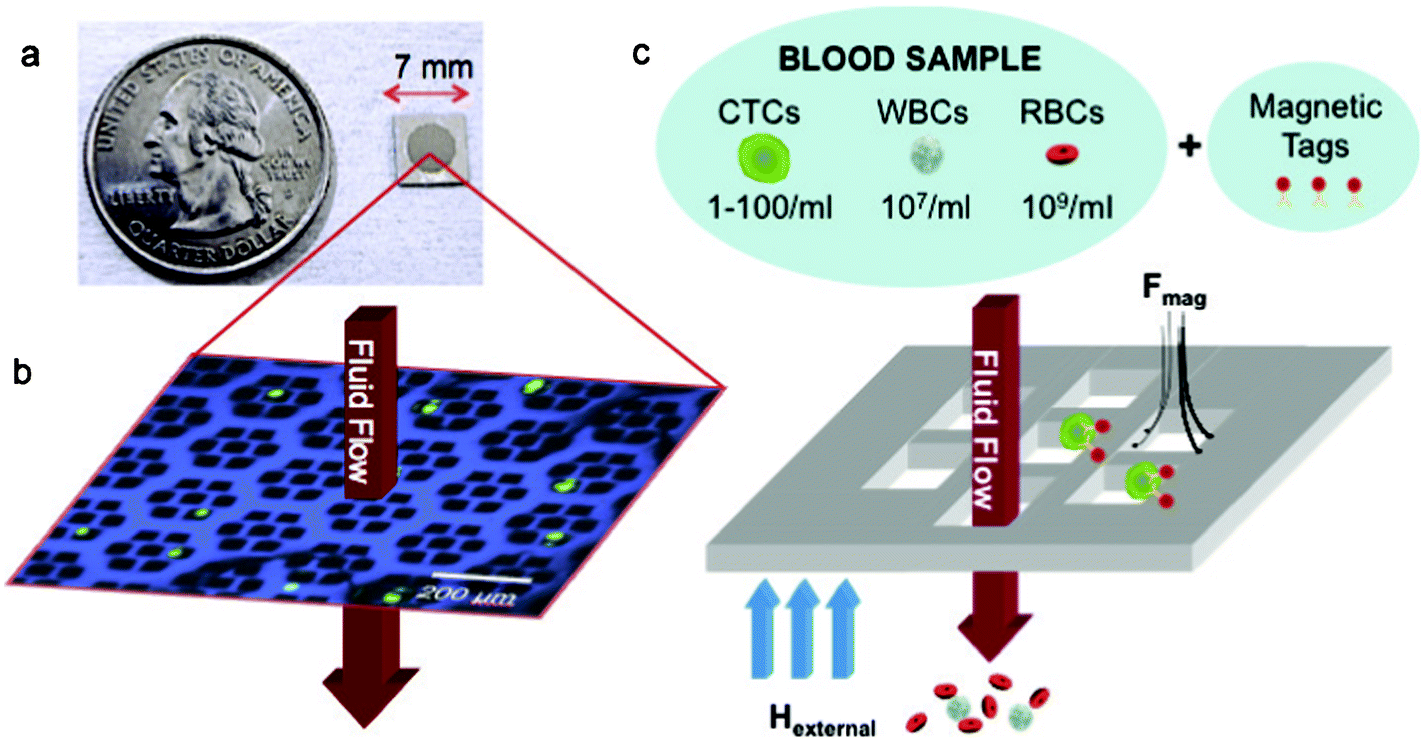 | ||
| Fig. 1 Overview of the magnetic sifter device. (a) Single magnetic sifter die and (b) optical micrograph showing a section of the patterned pore array (artificially colored blue) and cultured H-1650 lung tumor cells (green) captured by the magnetic sifter. Pores are 40 × 40 μm squares. (c) Capture principle. A whole blood sample is labeled with magnetic tags and pumped through the pores during the application of an external magnetic field. Magnetically labeled target cells are captured at the pore edges where high magnetic field gradients exist. Unlabeled cells pass through the pores. | ||
The magnetic sifter design offers attractive characteristics for CTC enrichment, including high capture efficiency at high flow rates due to extremely high field gradients at the pore edges; high throughput due to the high density of pores (∼200 pores/mm2); scalability via standard lithographic fabrication widely used in the semiconductor industry; a small capture area (19.6 mm2) for rapid imaging of captured cells; and lastly, harvesting of viable cells. Uniquely, our separation protocols eliminate cumulative losses from preparatory steps (lysis, centrifugation, washing, etc.) and utilize a vertical flow configuration, Supplementary Fig. 10, ESI,† that maintains macroscopic field and flow homogeneity while retaining sufficient shear flow to prevent blood cell flow problems (Supplementary Fig. 8 and Supplementary Fig. 9, ESI†).
Here we report the details of the fabrication and development of the magnetic sifter. In proof of concept experiments, we capture CTCs from lung cancer patients and subsequently characterize these with an EGFR-mutation-specific antibody and magneto-nanosensor.
Results
The magnetic sifter design and its cell capture behavior
The magnetic sifter is a magnetically active porous filter, composed of a silicon wafer with hexagonal arrays of 40 μm holes in a silicon nitride membrane and a 12 μm thick coating of a magnetically soft permalloy. The silicon wafer serves as the platform for photolithographic patterning of the membranes prior to being converted into a honeycomb structure to allow fluid flow while providing mechanical support. The magnetic layer is passivated with SiO2 to render the surface hydrophilic and reduce corrosion in an aqueous environment. Magnetic sifters retain initial magnetic properties after being stored 4 weeks in phosphate-buffered saline (PBS) and subjected to experimental use (Supplementary Fig. 1, ESI†). A standard 100 mm silicon wafer yields 120 magnetic sifters, each 7 mm × 7 mm in size.For quantitative separation experiments, the magnetic sifter die is loaded into a custom-made acrylic holder, which creates a watertight seal around the patterned area (Supplementary Fig. 2, ESI†). Blood samples are loaded into a well and pulled through the holder with a syringe pump. A small neodymium-iron-boron (NdFeB) permanent magnet magnetizes the magnetic sifter and is removed for elution of captured cells.
The capture behavior of the magnetic sifter has been studied with numerical simulations of magnetically labeled cell trajectories computed by a finite element-based simulation package (COMSOL Multiphysics™). In these combined electromagnetic and fluid dynamic simulations, magnetically labeled cells exhibit trajectories terminating at the edges and surfaces near the magnetic sifter pores, providing accessibility and positional consistency during imaging (Supplementary Fig. 3, ESI†).
To study flow and capture behavior, we developed a flow-cell for live observation of the separation process using a fluorescence microscope. In this configuration, small NdFeB magnets apply a field in the plane of the magnetic sifter surface, and the sample is delivered in a “lateral flow” configuration across the magnetic sifter surface. For magnetically-labeled lung tumor cells (H-1650) spiked in PBS, qualitative observations reveal uninhibited passage of magnetically-labeled non-small cell lung tumor cells (H-1650) through the pores at 5 ml hr−1 while the external magnetic field is absent. When the magnetic field is applied, tumor cells are captured near the pore edges. The cells can subsequently be eluted by removing the magnetic field and increasing the flow rate (Supplementary Movie 3, ESI†). When working with whole blood solutions in this lateral flow configuration, blood cell aggregation and clogging is observed at low flow rates (Supplementary Movies 1–2, ESI†) and described in detail in Supplementary Note 1, ESI†. Blood cell aggregation and non-uniformities in flow are avoided by using a “vertical flow” configuration (Supplementary Fig. 10, ESI†) in the quantitative separations described below.
Validation experiments using whole blood spiked with tumor cell lines
To evaluate the magnetic sifter for CTC separation, we applied the system to human whole blood spiked with tumor cell lines. The dependence of capture efficiency on sample flow rate was studied using H1650 cells (50–100 cells ml−1) that were pre-labeled with a fluorescent dye and spiked into whole blood obtained from healthy donors. The samples are processed with a wash-free magnetic labeling protocol using streptavidin-conjugated magnetic nanoparticles and biotinylated anti-EpCAM antibodies. The labeled sample is pumped through the magnetic sifter, followed by PBS to wash the magnetic sifter surface of residual blood cells. The magnetic sifter is then examined under a fluorescence microscope, and the number of tumor cells counted on the surface is compared with the number of cells spiked into the sample to determine capture efficiency (also referred to as capture yield). Flow rates ranging from 5–25 ml hr−1 were tested (Fig. 2b). The optimal flow rate was observed to be 10 ml hr−1, at which 95.7% of tumor cells are captured on the magnetic sifter surface. Beyond 10 ml hr−1, a linear decrease in capture efficiency with increasing flow rate is observed. Capture efficiency at the highest tested flow rate, 25 ml hr−1, is still appreciable at 74.3%.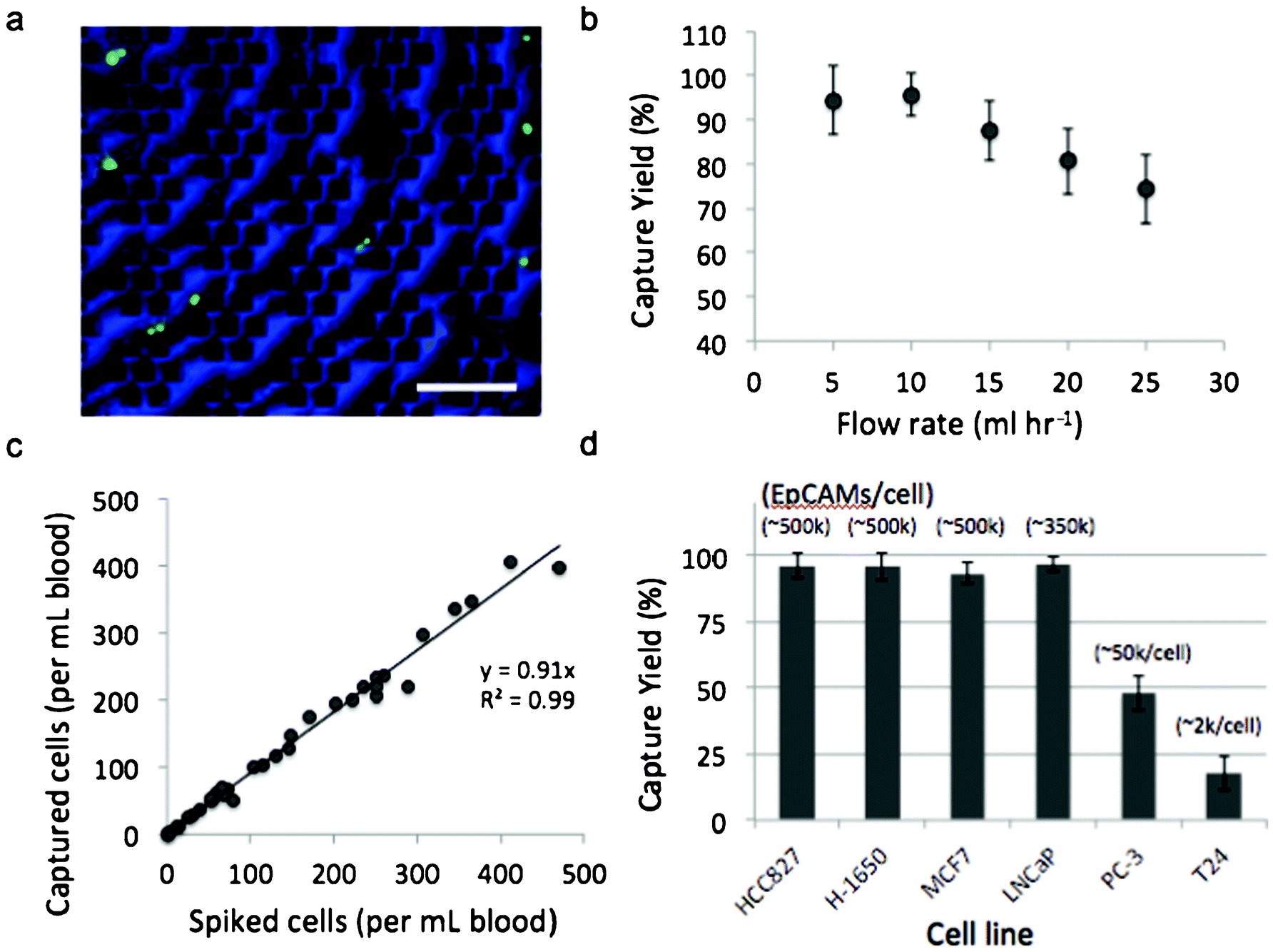 | ||
| Fig. 2 Capture efficiency results for 2 ml of spiked tumor cell samples. (a) Magnetically labeled H-1650 cells stained with Green CellTracker™ dye are captured and enumerated on the magnetic sifter surface (artificially colored blue). Scale bar is 200 μm. (b) Capture yield versus flow rate for low concentrations of H-1650 cells (50–100 H-1650 cells ml−1) captured from whole blood and enumerated on the magnetic sifter surface. (c) Regression analysis of capture yield for a range of H-1650 cell concentrations spiked into whole blood. (d) Capture yield results for six different cell lines with varying EpCAM expression. Separations in (c) and (d) were performed at the optimal flow rate of 10 ml hr−1. The error bars in (b) and (d) represent means ± s.d. | ||
To characterize the linearity of the capture efficiency with target cell concentration, whole blood samples were prepared with H-1650 concentrations ranging from 4–470 cells ml−1. Separations were performed at 10 ml hr−1, and magnetic labeling conditions were identical to those used above. Fig. 2c shows a regression analysis of capture efficiency for the range of concentrations. The average capture efficiency over the entire range is 91.4%, suggesting that the magnetic sifter capacity is not an issue for clinically relevant concentrations of CTCs.
To examine the impact of surface EpCAM expression on capture efficiency, we performed separations on six different cell lines with EpCAM expression levels ranging from ∼2000/cell to ∼500,000/cell (Fig. 2d). EpCAM expression levels were obtained from literature31 and examined by flow cytometry (Supplementary Fig. 4, ESI†). Capture efficiencies were evaluated by spiking 50–100 cells ml−1 into whole blood samples, followed by magnetic labeling and separation at 10 ml hr−1. The cellular level of surface EpCAM expression was found to have a significant impact on capture efficiency. For high EpCAM expressing cells (>100 k EpCAM/cell), capture efficiencies were measured to be greater than 90% for all cell types. For low expressing cells, including prostate cancer PC-3 cells (∼50,000 EpCAM/cell) and bladder cancer T24 cells (∼2,000 EpCAM/cell), capture efficiency was reduced to 48.0% and 17.7%, respectively.
Flow cytometry analysis revealed that EpCAM expression of cells was not uniform within the cultured cell lines (Supplementary Fig. 4, ESI†). Both H-1650 cells and MCF7 cells, for example, contain a high EpCAM expressing cell population (>100 k/cell) and a minority population (∼3–5%) of low EpCAM expressing cells (<20 k EpCAM/cell), suggesting that cells not captured may have low levels of magnetic labeling. Extension of these capture methods to even lower flow rates could enable the efficient capture of more weakly labeled cells, such as for intracellular cytokeratin labeling,33 a marker often used in CTC characterization with scanning cytometry, or for low EpCAM expression.
Release of captured cells is accomplished by removing the external magnet and flushing gently with buffer. The magnetic sifter was examined under a fluorescence microscope and on average 92.7 ± 6.1% of captured tumor cells were released. Cells are collected in a conical tube and centrifuged to remove the excess volume of the elution buffer. The cell pellet is resuspended and imaged in a hemocytometer for quantification of cell recovery. 89.6 ± 12.1% of tumor cells captured are collected in the eluted fraction, indicating high efficiency recovery. The viability of cells processed with the magnetic sifter was found to be unchanged as assessed by a LIVE/DEAD imaging kit and by re-culturing of eluted cells (Supplementary Fig. 5, ESI†).
Capturing and imaging of CTCs from patient samples
After validation with spiked samples, the magnetic sifter was tested on whole blood donated by patients with non-small cell lung cancer (NSCLC). All patients suffered from metastatic disease and had not yet received chemotherapy at the time of sample collection. Blood volumes ranging from 0.9–3.3 ml were analyzed. In addition, five samples (2 ml sample volume) from healthy donors were analyzed as controls.An immunofluorescence staining protocol was developed for identification and enumeration of CTCs captured on the magnetic sifter surface. A three-color scheme commonly employed for CTC identification was adopted, including anti-cytokeratin-fluorescein isothiocyanate (FITC) to label epithelial cells, anti-CD45-PE to identify white blood cells, and 4,6-diamidino-2-phenylindole (DAPI) to stain for nuclei.4 Staining was performed on chip while the captured cells were held by magnetic retention forces. Post-staining, the magnetic sifter was imaged with a fluorescence microscope equipped with an automated stage. Composite images of the three fluorescence channels were manually inspected. Cells that stained positive for cytokeratin, negative for CD-45, and positive for DAPI were scored as CTCs (Fig. 3). Our simple scoring criteria are significantly altered if pathologists impose stringent screening, rejecting arguable “false positives” from cells with somewhat weak DAPI signals (possibly apoptotic cells) or from cells with weak cytokeratin signals (possibly from non-specific staining). Digital methods are being developed to quantify such subjective enumeration criteria.34
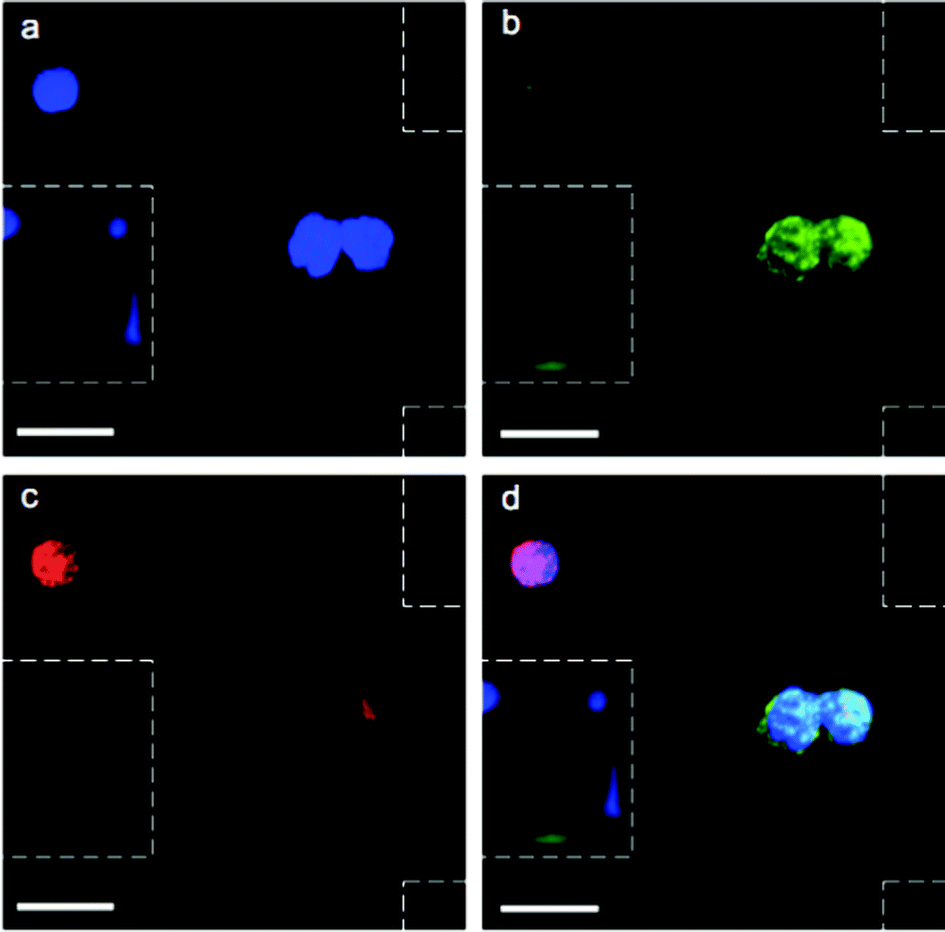 | ||
| Fig. 3 CTC staining and imaging. Images showing CTCs and a normal hematologic cell stained with (a) DAPI (blue), (b) CK-FITC (green), and (c) CD45-PE (red). A merged image of all three fluorescence channels is shown in (d), revealing a pair of CTCs and a single white blood cell in the vicinity of magnetic sifter pores (outlined by dashed boxes). Scale bars are 40 μm. | ||
CTC enumeration results were obtained for six patients with NSCLC (Fig. 4). Using scoring criteria consented by our pathologist, CTCs were detected for all six patients and ranged from 31–96 CTCs/ml. The five samples from healthy donors yielded only one cell scored as a CTC (Supplementary Fig. 6, ESI†), which may be a normal epithelial cell introduced during venipuncture, so a score of >1 CTC correctly classifies these few samples as being from cancer patients. No error bars were given for CTC counts because only the first author and one pathologist were involved in CTC scoring using one set of criteria. Additional galleries of CTC images and white blood cell counts can be found in the Supplementary Fig. 7, ESI† and Table 1 below, respectively. The ratio of the number of cells scored as CTCs, using non-stringent criteria, to the total number of imaged cells was 17.7 ± 9.3% for these cancer patient samples, indicating that CTCs were readily found on the sifter surface.
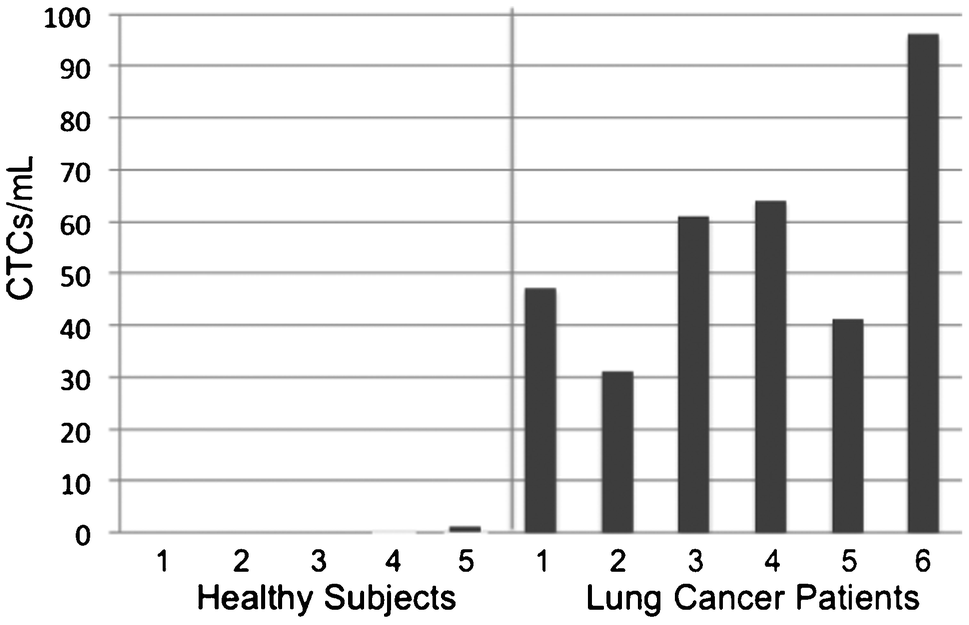 | ||
| Fig. 4 CTC Enumeration Results. Blood samples obtained from five healthy donors and six patients with metastatic non-small cell lung cancer were processed with the magnetic sifter, and captured CTCs were imaged on the magnetic sifter surface and enumerated. | ||
| Cohort | Total # of samples | Blood volume (ml) | CTCs/ml | WBCs captured per ml | CTCs/WBCs (%) | |||
|---|---|---|---|---|---|---|---|---|
| 5–25 | 25–50 | 50–100 | >100 | |||||
| Healthy subjects | 5 | 2.0 ±0.0 | 0 | 0 | 0 | 0 | 326 ± 165 | — |
| Lung cancer | 6 | 0.9–3.3 | 0 | 3 | 3 | 0 | 368 ± 299 | 17.7 ± 9.3 |
Mutant and wild type EGFR detection in CTC lysates with magneto-nanosensors
To demonstrate the magnetic sifter's utility in preparing samples for molecular analysis of CTCs, an assay was developed using a mutation-specific EGFR antibody and a magneto-nanosensor biochip to detect a mutated variant of EGFR (deletions in exon 19) in CTC lysates. The assay was first validated on tumor cell lines spiked into whole blood and processed with the magnetic sifter. Eluted cells underwent a membrane protein extraction protocol, and the lysate was applied to a magnetic biochip functionalized with antibodies against wild type (Wt EGFR) and mutated EGFR (Exon 19 Del), as shown in Fig. 5a. Lysates from a lung tumor cell line, HCC827, known to possess the Exon 19 Del mutation yielded a positive signal in the channel monitoring Exon 19 Del, while a lung tumor cell line (H1975) without the Exon 19 Del mutation yielded no significant signal (Fig. 5b) in the channel monitoring Exon 19 Del. Both cell types produced signals for wild-type EGFR, as the wild-type capture and detection antibodies bind to epitopes on EGFR that are not mutated.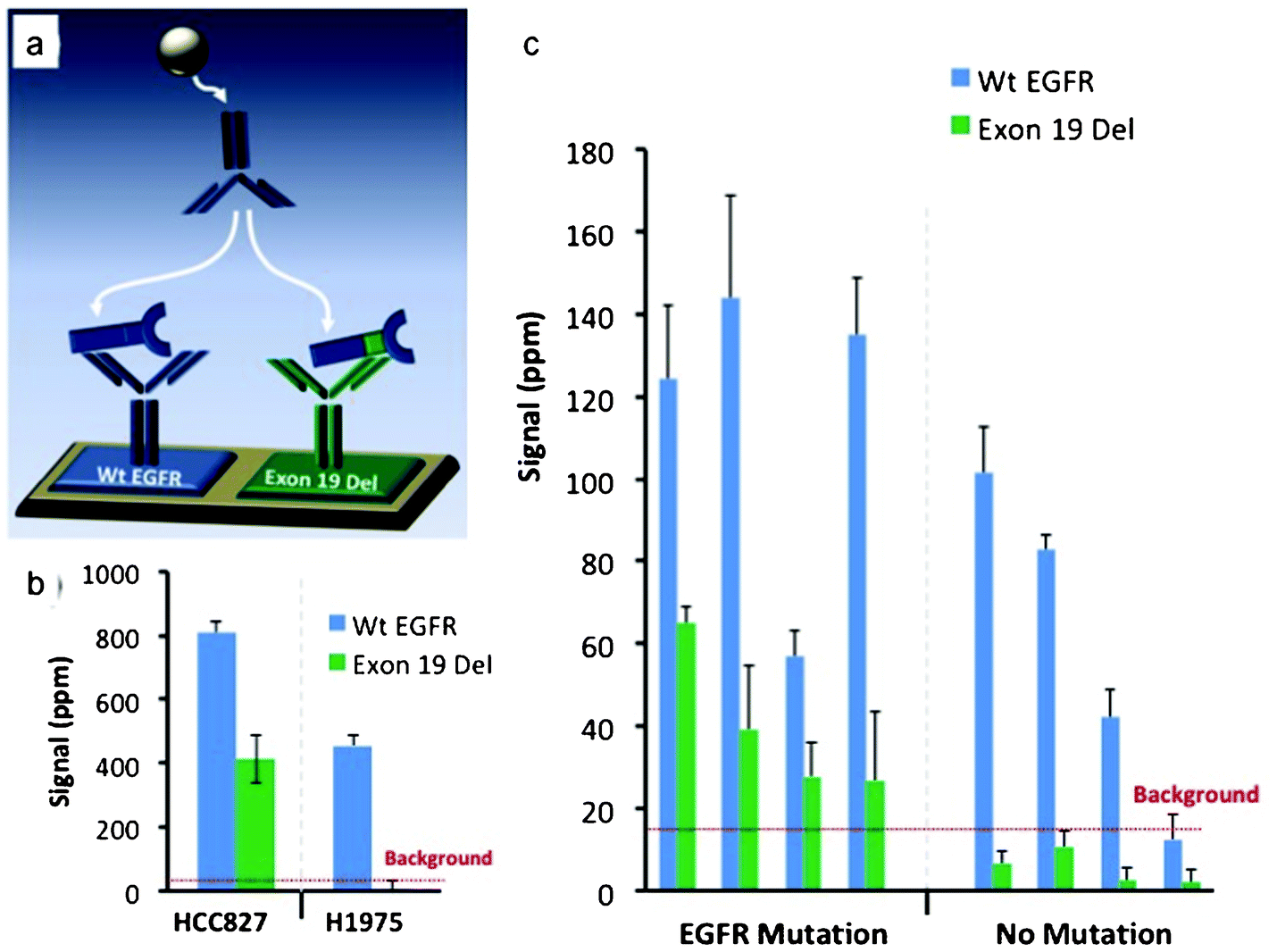 | ||
| Fig. 5 Detection of mutated and wild type EGFR with a magneto-nanosensor biochip. (a) Schematic showing capture antibody functionalization for two channels used in the assay. Channels are functionalized with anti-wild type-EGFR (Wt EGFR) or anti-Exon 19 Del-EGFR (Exon 19 Del). Following sample incubation, a biotinylated detection antibody (Wt EGFR) and streptavidin-coated magnetic nanoparticles are added, resulting in detectable signals in the underlying GMR biosensor. (b) Results from cell lines with (HCC827) and without (H1975) exon 19 deletions captured from whole blood samples with the magnetic sifter, subjected to cell lysis, and analyzed by the GMR biosensor chip. (c) Assay results from 8 patient samples, 4 with known exon 19 deletion mutations, and 4 without exon 19 deletion mutations. The red dotted line represents the cutoff for background signal. The error bars represent means ± s.d. | ||
Blood samples obtained from 8 patients with metastatic NSCLC and known mutational statuses were enriched for CTCs by the magnetic sifter, subjected to cell lysis, and tested on the magneto-nanosensor biochip (Fig. 5c). In each case, an average of 6.1 ± 2.1 ml of blood obtained from the patient was enriched for CTCs. In the 4 patients with known Exon 19 Del mutations, positive detection-where the signal was greater than twice the level of the background signal-of the mutant variant of EGFR was obtained. The 4 patients without the Exon 19 Del mutation did not yield a positive result in the Exon 19 Del channel. There is some overlap of error bars in this data, so more patient samples and statistical analysis would be necessary to establish confidence levels.
Discussion
The results from our experiments suggest the magnetic sifter is a strong candidate for routine preparation of CTC samples. The 3,808 pores per magnetic sifter each act as a microfluidic channel with high uniformity owing to the microfabrication techniques used to build the device. With a small active area (∼20 mm2), the magnetic sifter's capture efficiency compares well with data reported for existing microdevices, especially in the high flow rate (>10 ml hr−1) regime.5,8,11,12 In the case of 25 ml hr−1, a 1 ml sample containing ∼109 cells is passed through the magnetic sifter in under three minutes, translating to approximately 2000 cells passing through each magnetic sifter pore every second. Although the small size of the magnetic sifter allows for rapid imaging for enumeration of CTCs, the device area can be easily scaled up during fabrication to allow for even higher volumetric flow rates while maintaining the linear velocity of cells passing through the pores, and hence capture efficiency. Scaling the device to just 2.54 cm (1 inch) in diameter, for example, would correspond to an allowed increase in volumetric flow rate of about 25× while maintaining the capture efficiency reported in this work, but would also cause a corresponding increase in the area imaged to identify CTCs.The ease with which CTCs are released from the magnetic sifter pores is promising for subsequent analytical methods. The elution step can be performed in less than one minute, and cells can be either viable or fixed, depending on whether imaging of the cells on chip is required and on the requirements of a subsequent analytical technique. The entire enrichment process, including magnetic labeling, separation, and elution, has been found to preserve cell viability. Results from preliminary attempts to culture spiked tumor cell lines enriched from healthy donor blood have suggested the magnetic sifter is a suitable platform for enabling the culturing of some patient CTCs which can have important implications in the field of personalized medicine (Fig. S4, ESI†).
One area of concern is the impact of cellular EpCAM expression on capture efficiency. Similar behavior has been reported previously.35,36 To account for lower and/or heterogeneous EpCAM expression, the use of additional targeting antibodies will likely need to be implemented in conjunction with anti-EpCAM. Improvements using this approach have been reported with an immunoaffinity-based device.36 The magnetic sifter platform is also expected to benefit from the development of additional candidate markers for CTC enrichment.
Our results suggest the magnetic sifter can serve as an enumeration device, although direct comparisons, using larger data sets, with existing technologies are needed and currently in progress. Sample processing with the magnetic sifter, including labeling, separation, and imaging of a 2 ml blood sample, requires ∼3 h to complete. Acquired images are then manually inspected for enumeration (∼1 h per sample). Automation of image processing and elimination of subjective scoring criteria via software will greatly increase the rate and consistency of sample analysis.
Increasing interest has been directed towards moving beyond enumeration to characterization of CTCs. Characterization of enriched CTCs has included genetic analysis by PCR, FISH, and DNA and RNA sequencing, and immunostaining of CTCs.4,5,9,12,13,16,37–39 The recent use of mutation-specific anti-EGFR antibodies to assess the mutational statuses of tumor cell samples has encouraged our use of magnetic biochips for mutational assessment. Further assay development with increased availability of mutation-specific antibodies, such as T790M mutation-specific antibodies, may enable practical monitoring of, and hence responding to, a patient's acquisition of a mutation and potential resistance to therapy following initial characterization of a tumor biopsy.40,41
Conclusion
It is widely believed that CTCs hold great promise as a cancer biomarker and as a means to study and understand complex metastatic processes. Although a host of rare cell separation technologies have been developed to address the technical challenge of obtaining a CTC sample, few devices have been able to efficiently transition from the laboratory setting to the clinic. While each method possesses distinct advantages over others, all methods face the unanswered question of which characterization or analytical method to apply to an enriched CTC sample in order to yield valuable clinical information. The work reported here has been aimed at developing a flexible platform capable of harvesting CTCs suitable for any subsequent analytical method that may lead to an improved clinical outcome. The high throughput capture and simple, efficient release of captured cells suggest the magnetic sifter is a strong candidate for use with a variety of post-capture CTC characterization approaches. By itself, the magnetic sifter shows significant promise as a rare cell enrichment tool, offering enumeration and imaging “on chip,” scalability, and high uniformity in capture and rinsing forces owing to the microfabrication techniques employed in building the device.The pairing of the magnetic sifter with a magnetic biochip for subsequent analysis of CTC lysates from patient samples demonstrates the utility of the magnetic sifter in the preparation of “liquid biopsies” from cancer patients. Furthermore, the detection of a mutated EGFR in a proteomic assay using a magnetic biochip represents a leap forward in the application of magnetic nanotechnologies to cancer research. By combining platforms into one integrated technology, CTCs can be captured in a longitudinal analysis for real-time monitoring of tumor marker expression at the initial time of diagnosis, monitoring response to therapy, and long-term follow up. With these techniques, mutational analyses of patients' CTCs via a “liquid biopsy” at the bedside may represent the future of personalized medicine for patients with detectable CTCs.
Experimental
Device fabrication
Fabrication of the magnetic sifter involves standard microfabrication techniques. First, a 2 μm thick low-stress silicon nitride (Si3N4) film is deposited by low-pressure chemical vapor deposition onto a standard double-polished (100) oriented single crystal silicon wafer. The Si3N4 membrane is then patterned via photolithography with a mask containing the arrays of 40 μm square pores. The pattern is etched through the front side of the membrane using NF3 plasma from an Applied Materials Technologies 8100 Hexode etcher. The backside of the wafer is then patterned via photolithography with a mask containing arrays of hexagonal holes (100 μm per side). The holes are etched completely through the silicon wafer to expose the Si3N4 membrane, by a deep reactive ion etching process in alternating SF6 and C4F8 plasmas (standard Bosch process) using an STS Deep RIE etcher. A 12 μm thick permalloy (Ni80Fe20, where the subscripts indicate atomic percent) film is then deposited onto the front side of the Si3N4 membrane using a Perkin-Elmer high vacuum RF-sputtering system. The magnetic layer is then passivated via RF-sputtering with a 30 nm thick layer of SiO2.Finite element simulations
Numerical simulations of magnetically-labeled cell trajectories were carried out with a finite element-based simulation package (COMSOL Multiphysics™ version 4.2a). The simulation geometry (Supplementary Fig. 3a, ESI†) consists of a single magnetic sifter pore array (seven 40μm × 40μm pores) with the magnetic properties of permalloy (Ni80Fe20). Periodic boundary conditions are applied through the entire simulation setup to reflect the fact that the magnetic sifter is a repeating tessellation of this array. On an 8-core Windows Server 2008 machine, the total required CPU hours was 175. The simulation proceeds by solving the fluidic flow profile and the magnetic field profile independently first, and then using the particle tracing module to follow the cell trajectories. The simulation also assumes that the cells do not affect the fluidic profile or magnetic field profile significantly regardless of its position or velocity.An inlet flow velocity corresponding to a volumetric flow rate across the magnetic sifter of 10 ml hr−1 was used, and the Navier–Stokes equation was solved to obtain the fluidic flow profile, assuming incompressible flow of water (density 1000 kg m−3 and viscosity 0.001 Pa s). Conservation of mass and momentum as expressed in the Navier–Stokes equations thus simplify to the following forms:
ρwater∇·![[u with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0075_20d1.gif) = 0 = 0 |
where ρ is density, u is fluid velocity, p is the fluid pressure, η is fluid viscosity, I is the identity matrix, S is the strain rate tensor, and F is the volume force tensor.
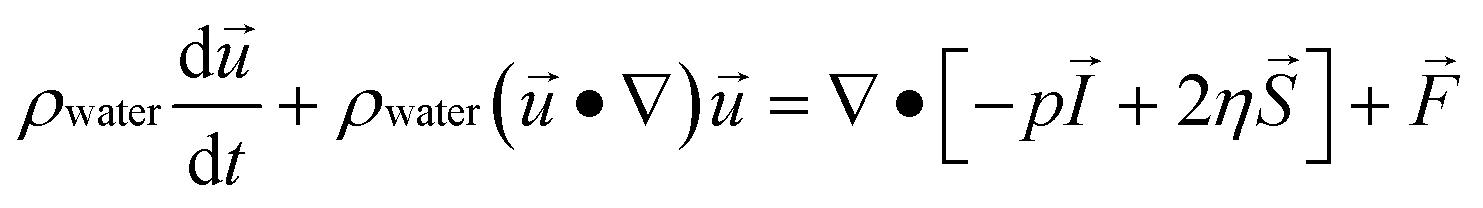
For the magnetic simulations, a uniform external field of 0.3 Tesla was applied in either the plane orthogonal (Supplementary Fig. 3b, ESI†) or parallel (Supplementary Fig. 3c, ESI†) to the magnetic sifter's patterned array. The external field is approximately three times greater than the field required to magnetically saturate the nanoparticles used in this work, which were experimentally measured by alternating gradient magnetometry to require ∼80,000 A m−1 (∼0.1 T) to reach saturation magnetization. Supplementary Fig. 12, ESI† shows a magnetic hysteresis loop obtained for counted, magnetically labeled cells that provides the nanoparticle saturation field and the average magnetic moment per cell, as well as demonstrating consistency with a Langevin function model. However, when the bias field is large enough to saturate the magnetization along the bias field direction, the calculations become independent of the bias field value. Since the simulation is magnetostatic, and no currents are present, COMSOL solved for the general field via the use of the magnetic scalar potential, and standard constitutive relations for water and permalloy were used as defined below:
![[H with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0048_20d1.gif) = −∇Vm = −∇Vm |
![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0042_20d1.gif) = μ0(+ = μ0(+ ![[M with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004d_20d1.gif) ) ) |
Magnetically labeled cells were treated as 20 μm diameter spheres of density 1080 kg m−3 with a saturation magnetic moment of 140 pico-emu. The individual cells were subject to drag from the fluid flow, gravitational forces in the positive z-direction, and magnetic forces, and their trajectories were solved (neglecting interparticle forces) via the following equation of motion in 10 ms time steps:
![[F with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0046_20d1.gif) mag = ( mag = (![[m with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006d_20d1.gif) ·∇) ·∇) |
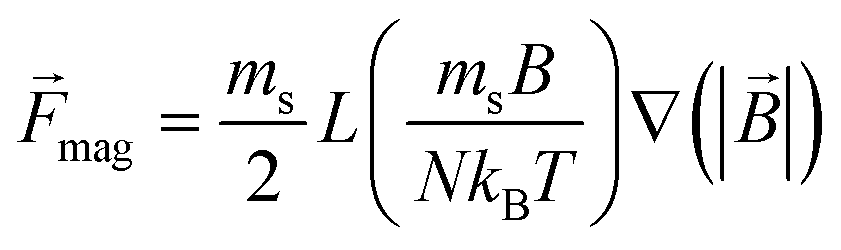
drag = 6πrcellη(− ![[small nu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0ea.gif) ) ) |
gravity = Vcell(ρcell − ρwater)![[g with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0067_20d1.gif) |
mcell![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) = mag + drag + gravity = mag + drag + gravity |
It is worth noting that while inertial and gravitational terms are included in this model, the viscous drag forces and the magnetic forces are the dominant terms. The gravitational forces are of the order 10−12 N for parameters listed here, while the magnetic forces are typically 2 to 3 orders of magnitude larger near the sifter at 10−9 to 10−10 N. Similarly, the Re number for this model is of the order 10−2, which puts it in the Stokes flow regime and suggests inertial forces should be negligible in effect. The inertial and gravitational terms have been retained however to facilitate expansion of the model to future instances where such terms might matter, such as when larger, denser magnetic microparticles such as Invitrogen's Dynal beads might be used.
Experimental apparatus
Magnetic sifters are loaded into custom-made laser-cut acrylic holders fitted with O-rings to create a fluid-tight seal around the patterned area of the magnetic sifter (Supplementary Fig. 2, ESI†). A polypropylene well is inserted into the holder above the magnetic sifter surface. The sample is manually fed into the well, 1.5 ml at a time, and pulled through the magnetic sifter by a syringe pump (New Era Pump Systems Inc.) with programmable pumping rates. A cylindrical neodymium-iron-boron magnet (19.1 mm diameter × 19.1 mm thickness, (BH)max = 320–340 kJ m−3) is positioned underneath the magnetic sifter holder to provide an external field of approximately 2.5 × 105 A m−1 to magnetize the magnetic sifter and magnetic nanoparticle labels during cell capture. The holder is removed from the magnetic field during the imaging and elution steps.Cell culture
All cell lines were obtained from ATCC (Manassas, VA, USA). H-1650, HCC827, LNCaP, PC-3, and T24 cell lines were maintained in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 0.05 mg ml−1 penicillin, 0.05 mg ml−1 streptomycin, 2 mM GlutaMAX, 1 mM sodium pyruvate, and 0.1 mM MEM non-essential amino acid supplement. The MCF7 cell line was maintained in high glucose DMEM media supplemented with 0.01 mg ml−1 bovine insulin, 10% FBS, 0.05 mg ml−1 penicillin, 0.05 mg ml−1 streptomycin, 2 mM GlutaMAX, 1 mM sodium pyruvate, and 0.1 mM MEM non-essential amino acid supplement. All cell lines were maintained in an incubator at 37 °C in 5% CO2.Spiked sample preparation and analysis
Tumor cell lines are prelabeled with CellTracker™ Green CMFDA dye (Invitrogen, Cat.# C2925), a probe which freely passes through the cell membrane and is converted to an extremely bright, cell-impermeant reaction product. The extreme brightness of this dye and its specificity towards cells allows for unequivocal determination of cell counts by visual inspection using a fluorescence microscope. To ensure accurate concentrations of spiked cells, a small droplet (∼2 μl) of tumor cell suspension is placed on the inside of a centrifuge tube cap, and left undisturbed for approximately one minute to allow cells to settle to the same focal plane. Settled cells are then counted by visual inspection. The cap is then sealed onto a centrifuge tube containing healthy donor blood, followed by a two-fold dilution in labeling buffer (PlusCellect Buffer, R&D Systems). Magnetic labeling of target cells is accomplished by the addition of biotinylated anti-EpCAM (Clone 9C4, Biolegend) to the spiked blood sample and incubation for 1 h with constant mixing at 4 °C. Without washing, 5 μl of magnetic nanoparticle stock (MagCellect Streptavidin Ferrofluid, R&D Systems, Cat. #MAG999) is added and the sample is incubated under constant mixing for an additional hour. The sample is then loaded into the well attached to the magnetic sifter holder and pumped through the magnetic sifter, followed by 1 ml of PBS to wash the magnetic sifter surface of peripheral blood cells. The magnetic sifter is then examined under a fluorescence microscope, and the number of tumor cells counted on the surface is compared with the number of cells spiked into the blood sample to determine capture efficiency. The patterned array of the sifter surface facilitates scanning and counting by visual inspection. All cell counts were confirmed independently by two investigators.For the handful of spiked samples with higher tumor cell concentrations (> 300 per ml, Fig. 2c), for which manual inspection becomes tedious, images were acquired of both the prepared sample droplet (containing settled tumor cells) as well as the sifter surface post-capture with a 4× magnification objective, such that the entire sample droplet or sifter surface is contained in a single field of view in a fluorescence image. Standard image thresholding and automated object/cell counting was applied to confirm cell counts obtained through manual inspection for these samples.
Quantification of surface EpCAM expression
Quantification of surface EpCAM expression was carried out by first generating a calibration curve by measuring fluorescence intensity of QuantiBRITE™ phycoerythrin (PE) Quantitation Kit beads (BD 340495) with known numbers of PE molecules per bead using a BD LSR Fortessa Analyzer (BD). Dissociated cells were then incubated with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PE conjugated monoclonal anti-EpCAM antibody (BD) and analyzed at the same settings and conditions as the calibration beads. Unlabeled cells were analyzed as negative controls. Median absolute EpCAM antibody binding for each cell line (Supplementary Fig. 4, ESI†) was determined from a calibration curve constructed from the QuantiBRITE bead data using FlowJo Data Analysis software calibration tool.
:1 PE conjugated monoclonal anti-EpCAM antibody (BD) and analyzed at the same settings and conditions as the calibration beads. Unlabeled cells were analyzed as negative controls. Median absolute EpCAM antibody binding for each cell line (Supplementary Fig. 4, ESI†) was determined from a calibration curve constructed from the QuantiBRITE bead data using FlowJo Data Analysis software calibration tool.
Patients and clinical samples
Patients with advanced lung cancer were recruited from the Stanford Cancer Center for participation under a protocol approved by the institutional review board (IRB), which includes informed consent. Healthy blood samples were obtained from the Stanford Blood Center under a separate IRB-approved protocol. Specimens from both patients with lung cancer and healthy donors were drawn into EDTA vaccutainer collection tubes (Becton-Dickinson) and processed within 12 h of the sample draw. Blood samples analyzed were drawn after discarding the first 10 ml to reduce the likelihood of epithelial cell contamination resulting from venipuncture.Fluorescence staining protocol and imaging of CTCs
Patient samples were magnetically labeled and separated at 10 ml hr−1 as described above. Following separation and washing with 2 ml of Superblock™ buffer (Thermo Scientific), fluorescence staining was achieved by first flowing through the magnetic sifter a 1 ml solution of 4% paraformaldehyde at 5 ml hr−1 to fix the captured cells, followed by 1 ml of 0.2% Triton X-100 surfactant (Sigma-Aldrich) in PBS at 5 ml hr−1 to permeabilize the cell membranes. 0.5 ml of SuperBlock™ T20 buffer containing 50 μl of anti-CD45-PE stock (Clone H130, BD Biosciences), 10 μl of anti-CK-FITC stock (CAM5.2 clone, BD Biosciences), and 5 μl of DAPI stock (Thermo Scientific) was then flowed through the magnetic sifter at 2 ml hr−1, followed by a wash with 3 ml Superblock™ T20 buffer at 10 ml hr−1.Following staining, magnetic sifters are removed from their holders and placed under a fluorescence microscope equipped with an automated stage (Leica DM5500B Upright Microcope and Digital Imaging System). A rectangular array of 414 locations containing the entire patterned area is imaged in three channels (FITC, PE, DAPI) with a 40× objective. Images from the three channels are merged to form 414 composite images for manual inspection and identification of CTCs.
EGFR assay with magneto-nanosensors
Blood samples were magnetically labeled and enriched for CTCs by the magnetic sifter. Following enrichment, cells were eluted into cold RIPA buffer (Cell Signaling Technologies) containing Halt protease and phosphatase inhibitors (Thermo Scientific) and subjected to ten cycles of 6 s (1 min total) of agitation in a chilled ultrasonic bath. Samples were then centrifuged at 14,000 g at 4 °C. The supernatant was collected and stored at −80 °C prior to detection with the magnetic biochip.The magneto-nanosensor biochip consists of an array of 64 magnetically responsive and individually addressable GMR sensors. Each sensor in the array covers a 100 × 100 μm2 area. Fabrication and surface functionalization of these biosensors are described previously.29 Capture antibodies to wild type EGFR (AF231, R&D Systems) and exon 19 deletion (E746-A750del) EGFR (mAb #2085, Cell Signaling Technology) were robotically spotted in 3 nL droplets at a concentration of 500 μg ml−1 over at least ten unique sensors in the array. In addition, 1% bovine serum albumin (BSA) was spotted on four unique sensors as a negative control, and an epoxy resin was deposited on four sensors in order to monitor systematic fluctuations in the electronics. After incubation with the sample of interest for 1 h, biotinylated detection antibody to EGFR (BAF 231, R&D Systems) was added at a concentration of 1 μg ml−1 for 30 min completing the sandwich assay. Finally, a 50 μl solution of streptavidin-coated magnetic nanoparticles (MACS 130-048-102, Miltenyi Biotec) was added. We monitored the real-time binding of the streptavidin-coated magnetic nanoparticles to the bound biotinylated detection antibody over the GMR sensors until the signal reached saturation. The GMR biosensor signals, represented as a change in magnetoresistance (MR) normalized to the initial MR and displayed in ppm, were recorded at the plateau of the binding curves. The background signal was defined as the average signal over the BSA control sensors plus 3 standard deviations.
Acknowledgements
Funding: This work was supported by Stanford Graduate Fellowship and Achievement Rewards for College Scientists fellowship (C.M.E.), Stanford Medical School Medical Scientist Training Program and National Science Foundation Graduate Fellowship (R.S.G.), Agency for Science Technology and Research fellowship (C.C.O.), Center for Cancer Nanotechnology Excellence (U54CA151459), Innovative Molecular Analysis Technologies (R33CA138330), and Physical Science Oncology Center (U54CA143907).Author contributions: C.M.E., R.S.G., R.R., H.A.W. and S.X.W. designed research. C.M.E., C.E.H., R.J.W., R.S.G., L.X., E.W.H., S.B.W., C.C.O., and D.J.W. performed research. C.M.E., R.S.G., R.J.W, S.B.W., and S.X.W. analyzed the results. C.M.E. fabricated the magnetic sifter devices. C.C.O. carried out finite element simulations. C.E.H. and E.W.H. carried out cell culture experiments. L.Y.Z, J.W.N., and H.A.W. obtained IRB approval and collected patient samples. C.M.E., S.X.W, and H.A.W. wrote the paper, and all the authors commented on the drafts.
Competing interests: C.M.E., R.S.G., R.J.W., and S.X.W. have related patent or patent applications assigned to Stanford University and out-licensed for potential commercialization.
References
- S. Mocellin, U. Keilholz, C. R. Rossi and D. Nitti, Trends Mol. Med., 2006, 12, 130–9 CrossRef CAS PubMed.
- K. Pantel, R. H. Brakenhoff and B. Brandt, Nat. Rev. Cancer, 2008, 8, 329–40 CrossRef CAS PubMed.
- P. Paterlini-Brechot and N. L. Benali, Cancer Lett., 2007, 253, 180–204 CrossRef CAS PubMed.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–82 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- V. Hofman, C. Bonnetaud, M. I. Ilie, P. Vielh, J. M. Vignaud, J. F. Fléjou, S. Lantuejoul, E. Piaton, N. Mourad, C. Butori, E. Selva, M. Poudenx, S. Sibon, S. Kelhef, N. Vénissac, J. P. Jais, J. Mouroux, T. J. Molina and P. Hofman, Clin. Cancer Res., 2011, 17, 827–35 CrossRef CAS PubMed.
- H. K. Lin, S. Zheng, A. J. Williams, M. Balic, S. Groshen, H. I. Scher, M. Fleisher, W. Stadler, R. H. Datar, Y.-C. Tai and R. J. Cote, Clin. Cancer Res., 2010, 16, 5011–8 CrossRef CAS PubMed.
- S. J. Tan, L. Yobas, G. Y. H. Lee, C. N. Ong and C. T. Lim, Biomed. Microdevices, 2009, 11, 883–92 CrossRef PubMed.
- S. Zheng, H. K. Lin, J. Q. Liu, M. Balic, R. H. Datar, R. J. Cote and Y. C. Tai, J. Chromatogr., A, 2007, 1162, 154–61 CrossRef CAS PubMed.
- A. A. Adams, P. I. Okagbare, J. Feng, M. L. Hupert, D. Patterson, J. Göttert, R. L. McCarley, D. Nikitopoulos, M. C. Murphy and S. A. Soper, J. Am. Chem. Soc., 2008, 130, 8633–41 CrossRef CAS PubMed.
- S. Wang, K. Liu, J. Liu, Z. T.-F. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss, Y. K. Lee, L. W. K. Chung, J. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K.-F. Shen and H. R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–8 CrossRef CAS PubMed.
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- S. L. Stott, R. J. Lee, S. Nagrath, M. Yu, D. T. Miyamoto, L. Ulkus, E. J. Inserra, M. Ulman, S. Springer, Z. Nakamura, A. L. Moore, D. I. Tsukrov, M. E. Kempner, D. M. Dahl, C.-L. Wu, A. J. Iafrate, M. R. Smith, R. G. Tompkins, L. V. Sequist, M. Toner, D. A. Haber and S. Maheswaran, Sci. Transl. Med., 2010, 2, 25ra23 CrossRef PubMed.
- D. Issadore, J. Chung, H. Shao, M. Liong, A. A. Ghazani, C. M. Castro, R. Weissleder and H. Lee, Sci. Transl. Med., 2012, 4, 141ra92 CrossRef PubMed.
- K. Hoshino, Y.-Y. Huang, N. Lane, M. Huebschman, J. W. Uhr, E. P. Frenkel and X. Zhanga, Lab Chip, 2011, 11, 3449–57 RSC.
- A. H. Talasaz, A. A. Powell, D. E. Huber, J. G. Berbee, K.-H. Roh, W. Yu, W. Xiao, M. M. Davis, R. F. Pease, M. N. Mindrinos, S. S. Jeffrey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3970–5 CrossRef CAS PubMed.
- D. Wirtz, K. Konstantopoulos and P. C. Searson, Nat. Rev. Cancer, 2011, 11, 512–22 CrossRef CAS PubMed.
- P. Balasubramanian, L. Yang, J. C. Lang, K. R. Jatana, D. Schuller, A. Agrawal, M. Zborowski and J. J. Chalmers, Mol. Pharmaceutics, 2009, 6, 1402–8 CrossRef CAS PubMed.
- L. Yang, J. C. Lang, P. Balasubramanian, K. R. Jatana, D. Schuller, A. Agrawal, M. Zborowski and J. J. Chalmers, Biotechnol. Bioeng., 2009, 102, 521–34 CrossRef CAS PubMed.
- M. Zborowski and J. J. Chalmers, Anal. Chem., 2011, 83, 8050–6 CrossRef CAS PubMed.
- S. Miltenyi, W. Müller, W. Weichel and A. Radbruch, Cytometry, 1990, 11, 231–8 CrossRef CAS PubMed.
- J. D. Adams, U. Kim and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18165–70 CrossRef CAS PubMed.
- J. H. Kang, S. Krause, H. Tobin, A. Mammoto, M. Kanapathipillaia and D. E. Ingber, Lab Chip, 2012, 12, 2175–2181 RSC.
- N. Xia, T. P. Hunt, B. T. Mayers, E. Alsberg, G. M. Whitesides, R. M. Westervelt and D. E. Ingber, Biomed. Microdevices, 2006, 8, 299–308 CrossRef CAS PubMed.
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. Terstappen and D. F. Hayes, N. Engl. J. Med., 2004, 351, 781–791 CrossRef CAS PubMed.
- M. Cristofanilli, D. F. Hayes, G. T. Budd, M. J. Ellis, A. Stopeck, J. M. Reuben, G. V. Doyle, J. Matera, W. J. Allard, M. C. Miller, H. A. Fritsche, G. N. Hortobagyi and L. W. M. M. Terstappen, J. Clin. Oncol., 2005, 23, 1420–30 CrossRef PubMed.
- R. S. Gaster, D. A. Hall, C. H. Nielsen, S. J. Osterfeld, H. Yu, K. E. Mach, R. J. Wilson, B. Murmann, J. C. Liao, S. S. Gambhir and S. X. Wang, Nat. Med., 2009, 15, 1327–1332 CrossRef CAS PubMed.
- R. S. Gaster, D. A. Hall and S. X. Wang, Lab Chip, 2011, 11, 950–6 RSC.
- S. J. Osterfeld, H. Yu, R. S. Gaster, S. Caramuta, L. Xu, S.-J. Han, D. A. Hall, R. J. Wilson, S. Sun, R. L. White, R. W. Davis, N. Pourmand and S. X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20637–40 CrossRef CAS PubMed.
- J. Yu, S. Kane, J. Wu, E. Benedettini, D. Li, C. Reeves, G. Innocenti, R. Wetzel, K. Crosby, A. Becker, M. Ferrante, W. C. Cheung, X. Hong, L. R. Chirieac, L. M. Sholl, H. Haack, B. L. Smith, R. D. Polakiewicz, Y. Tan, T.-L. Gu, M. Loda, X. Zhou and M. J. Comb, Clin. Cancer Res., 2009, 15, 3023–8 CrossRef CAS PubMed.
- C. M. Earhart, R. J. Wilson, R. L. White, N. Pourmand and S. X. Wang, J. Magn. Magn. Mater., 2009, 321, 1436–1439 CrossRef CAS PubMed.
- C. M. Earhart, E. M. Nguyen, R. J. Wilson, A. Y. Wang and S. X. Wang, IEEE Trans. Magn., 2009, 45, 4884–4887 CrossRef CAS.
- G. Deng, M. Herrler, D. Burgess, E. Manna, D. Krag and J. F. Burke, Breast Cancer Res., 2008, 10, R69 CrossRef PubMed.
- T. M. Scholtens, F. Schreuder, S. T. Ligthart, J. F. Swennenhuis, J. Greve and L. W. M. M. Terstappen, Cytometry, Part A, 2012, 81A, 138–148 CrossRef PubMed.
- C. G. Rao, D. Chianese, G. V. Doyle, M. C. Miller, T. Russell, R. A. Sanders and L. W. M. M. Terstappen, Int. J. Oncol., 2005, 27, 49–57 CAS.
- S. D. Mikolajczyk, L. S. Millar, P. Tsinberg, S. M. Coutts, M. Zomorrodi, T. Pham, F. Z. Bischoff and T. J. Pircher, J. Oncol., 2011, 2011, 252361 CrossRef PubMed.
- S. Maheswaran, L. V. Sequist, S. Nagrath, L. Ulkus, B. Brannigan, C. V. Collura, E. Inserra, S. Diederichs, A. J. Iafrate, D. W. Bell, S. Digumarthy, A. Muzikansky, D. Irimia, J. Settleman, R. G. Tompkins, T. J. Lynch, M. Toner and D. A. Haber, N. Engl. J. Med., 2008, 359, 366–377 CrossRef CAS PubMed.
- M. Yu, D. T. Ting, S. L. Stott, B. S. Wittner, F. Ozsolak, S. Paul, J. C. Ciciliano, M. E. Smas, D. Winokur, A. J. Gilman, M. J. Ulman, K. Xega, G. Contino, B. Alagesan, B. W. Brannigan, P. M. Milos, D. P. Ryan, L. V. Sequist, N. Bardeesy, S. Ramaswamy, M. Toner, S. Maheswaran and D. A. Haber, Nature, 2012, 487, 510–513 CrossRef CAS PubMed.
- M. Yu, A. Bardia, B. S. Wittner, S. L. Stott, M. E. Smas, D. T. Ting, S. J. Isakoff, J. C. Ciciliano, M. N. Wells, A. M. Shah, K. F. Concannon, M. C. Donaldson, L. V. Sequist, E. Brachtel, D. Sgroi, J. Baselga, S. Ramaswamy, M. Toner, D. A. Haber and S. Maheswaran, Science, 2013, 339, 580–584 CrossRef CAS PubMed.
- S. Kobayashi, T. J. Boggon, T. Dayaram, P. A. Janne, O. Kocher, M. Meyerson, B. E. Johnson, M. J. Eck, D. G. Tenen and B. Halmos, N. Engl. J. Med., 2005, 352, 786–92 CrossRef CAS PubMed.
- W. Pao, V. A. Miller, K. A. Politi, G. J. Riely, R. Somwar, M. F. Zakowski, M. G. Kris and H. Varmus, PLoS Med., 2005, 2, e73 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S12, Movies S1–S3. See DOI: 10.1039/c3lc50580d |
| This journal is © The Royal Society of Chemistry 2014 |