Identification of pro- and anti-proliferative oligosaccharides of heparins
Qi Ying
Lean
a,
Rahul P.
Patel
a,
Niall
Stewart
a,
Sukhwinder Singh
Sohal
b and
Nuri
Gueven
*a
aSchool of Pharmacy, University of Tasmania, Hobart, TAS, Australia. E-mail: nguven@utas.edu.au; Fax: +61-3-6226-2870; Tel: +61-3-6226-1715
bSchool of Medicine, University of Tasmania, Hobart, TAS, Australia
First published on 5th December 2013
Abstract
Heparins, unfractionated heparin (UFH) and low molecular weight heparins (LMWHs), are heterogeneous mixtures of anticoagulant and non-anticoagulant oligosaccharides. In addition to their well-known anticoagulant effect, heparins have shown to mediate a wide range of non-anticoagulant effects, including the modulation of cellular growth. However, contradictory results have been reported with regard to their effects on cellular proliferation, with some studies suggesting anti-proliferative while others indicating pro-proliferative effects. This study investigated the proliferation of human colonic epithelial cancer cells in the presence of UFH and LMWHs (enoxaparin and dalteparin). In our experimental setting, all heparins caused a dose-dependent reduction in cellular growth, which correlated well with the induction of cell cycle arrest in the G1 phase and which was not associated with significant changes in cell viability. The effects on cellular proliferation of 14 different oligosaccharides of enoxaparin obtained through ion-exchange chromatography were also assessed. Surprisingly, only two oligosaccharides showed distinctive anti-proliferative effects while the majority of oligosaccharides actually stimulated proliferation. Interestingly, the smallest oligosaccharide devoid of any anticoagulant activity showed the strongest anti-proliferative effect. Notably, heparins are currently standardised only according to their anticoagulant activity but not based on other non-anticoagulant properties. Our results indicate that slight differences in the composition of heparins' non-anticoagulant oligosaccharides, due to different origins of material and preparation methods, have the potential to cause diverse effects and highlight the need for additional characterisation of non-anticoagulant activities.
Insight, innovation, integrationBy combining an ion exchange chromatography technique with cell culture assays, we describe for the first time that the oligosaccharides of a commonly used anti-coagulant agent, enoxaparin, have opposing activities that are distinct from enoxaparin's well-known anti-coagulant activity. We observed that some enoxaparin oligosaccharides strongly inhibited cellular proliferation, while others from the same mixture stimulated it. The results obtained from combining this separation technique with in vitro assays can now rationalize the contrasting results described in the literature. More importantly, this approach highlights the potential that even well-known drugs might only be partially characterized, with the possibility for variations between different production batches and manufacturers. |
Introduction
Unfractionated heparin (UFH) and its derivatives, low-molecular-weight heparins (LMWHs), have been the subject of considerable research interest with regard to their structural and biological properties.1,2 UFH has been clinically used as an anticoagulant for more than 65 years.3 It belongs to the family of glycosaminoglycans and it is a heterogeneous mixture of structurally unidentified anionic oligosaccharides.4 UFH is prepared by extraction from animal tissues including bovine lung and porcine intestinal mucosa5 with an average mass of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 20000 Dalton (Da).3,6 Despite the widespread use of UFH, it is associated with a number of problems including drug-induced adverse effects, low bioavailability and unpredictable dose response.6 These problems have led to the development of LMWHs, which are obtained by controlled enzymatic or chemical depolymerisation of UFH.5,6 LMWHs have an average molecular weight ranging from 4000 to 6000 Da.7 In the current clinical settings, LMWHs have replaced the UFH to a large extent because of their favourable pharmacokinetic properties and fewer adverse effects.6 All LMWHs are distinct from each other depending on the tissue source of the parent UFH, and the method and conditions employed for its preparation.5
000 to 20000 Dalton (Da).3,6 Despite the widespread use of UFH, it is associated with a number of problems including drug-induced adverse effects, low bioavailability and unpredictable dose response.6 These problems have led to the development of LMWHs, which are obtained by controlled enzymatic or chemical depolymerisation of UFH.5,6 LMWHs have an average molecular weight ranging from 4000 to 6000 Da.7 In the current clinical settings, LMWHs have replaced the UFH to a large extent because of their favourable pharmacokinetic properties and fewer adverse effects.6 All LMWHs are distinct from each other depending on the tissue source of the parent UFH, and the method and conditions employed for its preparation.5
It has been now well recognised that in addition to their well-known anticoagulant activity, heparins (UFH and LMWHs) possess a wide range of biological properties including their ability to modulate the cellular growth.2,8 Heparins have mostly shown inhibitory effects on the proliferative capacity of cells originating from different tissues and organisms.9,10 For example, multiple studies have demonstrated that heparins inhibited the proliferation of smooth muscle cells derived from bovine, porcine, rat and human tissues,9,11–14 human normal osteoblast cells,15,16 human bone marrow derived mesenchymal stromal cells17 and human mesenchymal stem cells derived from bone marrow.18 The mitogenic activity of rodent, bovine and human endothelial cells has been shown to be significantly inhibited in the presence of heparins.9,10,19 Although the dose response varied depending on the specific cell types used, most studies generally agreed that the inhibition of cell proliferation by heparins is concentration-dependent.9,13 Interestingly, this anti-proliferative property of heparins has also been observed in cancer cells, which has sparked interest in a potential therapeutic use of heparins for treating human malignancies.8 Multiple studies have reported that UFH and LMWHs, such as dalteparin, enoxaparin and nadroparin, can decrease the proliferation of human lung adenocarcinoma cells,20–22 murine mammary adenocarcinoma cells,23 human malignant mesothelioma cells,24 human osteosarcoma cells16 and human glioma-derived primary cells.25
Despite a number of studies reporting that heparins exert anti-proliferative effects, surprisingly, some studies have indicated that heparins also exert the opposite effect. For example, it has been reported that UFH can stimulate human intestinal cancer cell growth.26,27 Likewise, UFH has also been reported to increase the proliferation of primary rat intestinal cultures in a dose-dependent manner.28 In addition, some studies have reported no change in cell proliferation in the presence of heparins.29–31 Therefore, the current literature remains contradictory with regard to the cell growth regulation effects of heparins.
Heparins are heterogeneous mixtures of sulfated oligosaccharides and, along with the oligosaccharides which possess anticoagulant properties, they also have a large number of non-anticoagulant oligosaccharides.32 Anticoagulant oligosaccharides contain a unique pentasaccharide sequence responsible for the anticoagulant effects of heparins.7 In recent years, attention has shifted to the non-anticoagulant activities of heparins.2 Importantly, many studies have shown that the non-anticoagulant effects of heparins, such as anti-inflammatory activities, are independent of their anticoagulant activity and largely dependent on the presence of non-anticoagulant oligosaccharides.2,11 A number of studies have investigated the effects of heparins on angiogenesis.33 Interestingly, UFH has been shown to have a biphasic effect; some of the chemically synthesised oligosaccharides of heparins have an anti-angiogenic effect while some have a pro-angiogenic effect and importantly these effects are dependent on the size and sulfation patterns of oligosaccharides.33–35
However, the ability of heparins to modulate cellular growth has largely been studied with intact heparins rather than their individual unmodified oligosaccharides, with some studies indicating anti-proliferative effects and other claiming no or even pro-proliferative effects.22,26,30,31 Therefore, we hypothesised that intact heparins could have either an anti- or a pro-proliferative effect depending on the types of non-anticoagulant oligosaccharides present in the heterogeneous mixture of heparins. Hence, the present investigation examined and compared the cellular growth modulation effects of intact UFH, intact LMWHs (namely enoxaparin and dalteparin) and structurally unmodified oligosaccharides of enoxaparin (isolated using a novel ion-chromatography technique) on different human intestinal epithelial cell lines.
Materials and methods
Cell culture
Human colon carcinoma epithelial cell lines (HCT-116, HT-29 and DLD-1) were obtained from European Collection of Cell Cultures (ECACC) and cultured under standard conditions (37 °C, 5% CO2, 95% humidity) in modified McCoy's 5A medium or RPMI1640 medium. All culture media were supplemented with 2 mM L-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% heat inactivated fetal calf serum (FCS). When cells had reached confluence, they were harvested using trypsin (0.5 g L−1)/EDTA (0.2 g L−1). All chemicals and supplies were obtained from Sigma-Aldrich (Castle Hill, NSW, Australia) unless otherwise specified. All cell culture plastics were obtained from Corning (Tewksbury, MA, USA) unless otherwise specified.UFH and LMWHs
Heparin (DBL heparin sodium, Hospira, 5000 IU/0.2 ml, referred to unfractionated heparin (UFH) in the text), enoxaparin (Clexane, Sanofi Aventis, 20 mg/0.2 ml) and dalteparin (Fragmin, Pfizer, 2500 IU/0.2 ml) were diluted with phosphate buffered saline (PBS) when necessary to achieve final concentrations of 0 (for control), 0.01, 0.1, 1, 10, 100, 200, 400, 1000 μg ml−1 (with 160 IU = 1 mg heparin, 100 IU = 1 mg enoxaparin and 156.25 IU = 1 mg dalteparin).Isolation of enoxaparin oligosaccharides
Cell growth assay
A total of 2500 HCT-116 cells were seeded in 12 well-plates using culture media containing 0.5% and 10% FCS respectively. Final concentrations of UFH, enoxaparin, dalteparin (as indicated: 0.01–400 μg ml−1) were added into wells in triplicates. Cell growth was followed up to maximum two weeks with the treatment and media replacement biweekly before staining with trypan blue or protein quantification. For staining in 12 well-plates, cells were fixed with ice cold methanol at −20 °C for 10 minutes before being incubated with 0.4% w/v trypan blue for 5 minutes. Cells were washed twice with tap water and air-dried overnight.Protein quantification
Protein levels were measured using the Bio-Rad detergent-compatible protein assay (BioRad Laboratories, Gladesville, NSW, Australia). Bovine serum albumin (BSA) standards were prepared in sample buffer as serial dilutions between 0.08 and 5 mg ml−1. Cells were washed with PBS twice before being lysed in 100 μl per well of lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 0.5% Tween 20 and 0.2% Triton X-100) for 15 minutes at 4 °C. The content of each well was centrifuged in Eppendorf tubes (10 min, 14000 rpm, 4 °C, Eppendorf® Micro Centrifuge, 5417R, Eppendorf, NY, USA). For measuring protein content, 10 μl of supernatant and protein standards were pipetted in triplicate into a 96 well-plates before 25 μl of reagent AS (alkaline copper tartrate and surfactant) and 200 μl of reagent B (diluted folin) were added into each well. After 15 minutes, absorbance was read at 750 nm (Thermo Scientific Multiskan GO UV/Vis microplate spectrophotometer, SkanIt Software). Protein concentrations were calculated using the standard curve derived from the BSA standards.
Colony formation assay
Cells (HCT-116: 200, 500, 800, 2000 cells per dish; DLD-1: 800 cells per dish, HT-29: 800 cells per dish) were seeded into 10 cm tissue culture dishes (Nunc, Thermo Fisher Scientific, Victoria, Australia) to allow overnight adherence without drug interference. Cells were treated in four replicate dishes per condition with different concentrations (0.01–400 μg ml−1) of UFH, enoxaparin, dalteparin, or enoxaparin oligosaccharides in FCS containing media (0.5%, 2%, 5% or 10%). The cells were incubated for two weeks before being washed twice with PBS and fixed with 2% paraformaldehyde in PBS for 15 minutes. Subsequently, colonies were stained with 1 ml of 0.4% trypan blue or 0.1% w/v crystal violet (Oxoid, West Heidleberg, VIC, Australia) for 5 minutes before washed twice with tap water and air-dried. Colonies over 50 cells were manually scored using a microscope (Inverted Microsope, Model INV-100, Aktivlab, SA, Australia). Colony formation was expressed as a number of colonies or a percentage of control values.Cell viability assays
Cell cycle analysis
Cell cycle analysis was carried out using PI staining and flow cytometry. HCT-116 cells (4 × 105) were seeded in 6 well-plates and allowed to adhere for 7 hours followed by treatment with UFH, enoxaparin, dalteparin or enoxaparin oligosaccharides in triplicate per condition at concentration as indicated: 100 μg ml−1 or 400 μg ml−1. After 18 hours of drug exposure, cells were harvested by trypsinization and washed with PBS before being resuspended in 750 μl of PBS. Three millilitres of ice cold 95% ethanol were added steadily to the cells while vortexing and were further fixed overnight at 4 °C. Cells were washed with PBS twice before being resuspended in 500 μl of PBS and counted to adjust cell density to 1 × 106 cells ml−1. Cells were additionally treated with 5 μl of ribonuclease A (10 mg ml−1) before DNA was labelled with 50 μl of PI (1 mg ml−1) and incubated at 37 °C for 15 minutes. Fluorescence was measured using a flow cytometer (FACSCanto, Becton Dickinson, CA, USA). Ten thousand events per sample were collected and acquired in a list mode with the accompanying software (FACS Diva software, Becton Dickinson). Effects on the cell cycle were determined by changes in percentage of cell distribution at each phase of the cell cycle, and assessed by histograms (FlowJo software, version 10; Treestar, Ashland, USA). The proportion of cells in sub-G1, G1, S and G2 phases was quantified.Statistical analysis
All data are presented as mean ± standard deviation (SD) or as per cent change compared to control. By using GraphPad Prism (version 6, GraphPad Software Inc, CA, USA), statistical significance was evaluated using Student's t-test and one way analysis of variance (ANOVA), where appropriate, followed by Dunnett's multiple comparison tests to evaluate the difference between treatment groups and control. A p-value of <0.05 was considered statistically significant.Results
Serum dependency of the effect of heparins on cell growth
Since UFH was previously reported to inhibit cell proliferation,16 we attempted to compare its efficacy against enoxaparin under normal growth conditions (10% FCS). Surprisingly, no reduction of cell growth by UFH or enoxaparin in HCT-116 cells was detected using a colony formation assay. There was also no indication of a dose dependency under these conditions (Fig. 1A). To investigate if an abundance of growth factors in the serum could be masking any effects by UFH in our cell culture, the colony formation assay was performed in the absence or presence of UFH at different FCS concentrations (Fig. 1B). Here, UFH clearly showed a significant reduction (58.2% of control) of colony numbers at 5% FCS and a strong trend (74.2% of control) at 2% FCS. While at 10% FCS concentration no difference in colony numbers was detected, at 0.5% the lack of colonies prevented any analysis (Fig. 1B).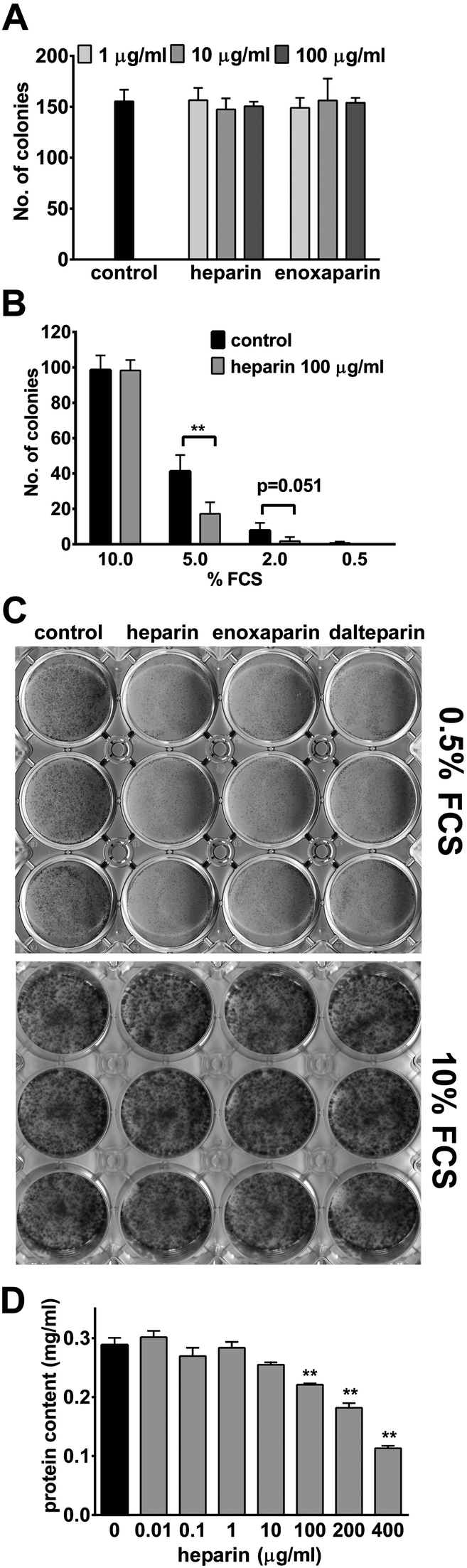 | ||
| Fig. 1 Effect of heparins on epithelial cell growth. (A) Colony formation of HCT-116 cells in the presence of 10% of FCS and heparin or enoxaparin. Data = mean of four individual samples per concentration of one typical experiment. Error bars = SD. (B) Colony formation of HCT-116 cells at 10%, 5%, 2% and 0.5% of FCS and 100 μg ml−1 of heparin. Data = mean of four individual samples per concentration of a typical experiment. Error bars = SD. **P < 0.01 versus control. (C) HCT-116 cells in the absence (control) or presence of 100 μg ml−1 of heparin, enoxaparin and dalteparin respectively, before stained with trypan blue. (D) HCT-116 cells were grown with heparin (0.01–400 μg ml−1) for 14 days before protein content per well was measured. Data represent the mean of triplicates from a representative experiment. Error bars = SD. **P < 0.01 versus control. | ||
This serum dependency was also observed when looking at cell growth in a mass culture at 0.5% FCS concentration (Fig. 1C) and 2% (data not shown). Under these conditions, HCT-116 cells were clearly able to proliferate in the absence of heparins. However, in the presence of all heparins, a significant reduction in cell numbers could be observed. Similar to the results above, at 10% FCS, no difference in cell growth was evident (Fig. 1C). Consequently, for all further experiments, FCS concentrations of 0.5% or 2% were used. The observed reduction in the cell number was also quantified by measuring protein content per well and again UFH dose-dependently reduced protein content per well up to 60.8% of control at the highest UFH concentration used (Fig. 1D).
Dose-dependent reduction of colony formation by heparins
To compare a possible dose-dependent reduction in colony formation by UFH against LMWHs, concentrations ranging from 0.01 to 400 μg ml−1 were employed (Fig. 2A–C). UFH and the two LMWHs, enoxaparin and dalteparin, dose-dependently reduced colony formation by up to 34.9%, 44.5% and 33.0%, respectively. Interestingly, significant reductions in colony numbers were detected at 10 μg ml−1 for enoxaparin (Fig. 2B), while significance was achieved for UFH (Fig. 2A) and dalteparin (Fig. 2C) only at concentrations at and above 100 μg ml−1. This effect was clearly not cell line specific since UFH and LMWHs induced comparable reductions in colony numbers in the 3 intestinal cancer cell lines used (Fig. 2D). We also observed the same response in the rodent retinal cell line RGC5 (data not shown). | ||
| Fig. 2 Inhibition of cellular colony formation by heparins. Colony formation of HCT-116 cells in the presence of heparin (A), enoxaparin (B), dalteparin (C) and 2% FCS. (D) Epithelial cells (HCT-116, DLD-1, HT-29) were exposed to 400 μg ml−1 in media (2% FCS). Data represent the average of four individual samples per concentration of a typical experiment. Error bars = SD. *P < 0.05, **P < 0.01 versus control. | ||
Inhibition of cell cycle progression by heparins
In order to explain the observed reduction of colony numbers by heparins, their effect on cell cycle progression was analysed (Fig. 3). Consistent with the results described above, after 18 h of treatment both UFH and LMWHs significantly increased the proportion of cells in the G1 phase and at the same time decreased cell numbers in S and G2 phases (Fig. 3A and B). | ||
| Fig. 3 Inhibition of cell cycle progression by heparins. Flow cytometric cell cycle analysis of HCT-116 cells after 18 hours exposure to 400 μg ml−1 heparin, enoxaparin, dalteparin in media (2% FCS). (A) Representative histograms of cell counts versus propidium iodide (PI) for control versus treatment groups. (B) Graphical representation of the percentage of cells in G1, S, G2 phases determined by gating. Data = mean of three individual samples per treatment of a typical experiment. Error bars = SD. *P < 0.05, **P < 0.01 versus negative control. | ||
Effect of heparins on cellular viability
Although the observed effects on cell cycle progression alone could explain the reduced colony numbers described above, heparins have also been reported to induce cell death.37 To investigate if cell death was also present in this experimental system, cellular viability was measured by the commonly used redox-dye WST-1 (Fig. 4A). In HCT-116 cells, after 24 h of treatment with UFH and LMWHs, no signs of cell death could be detected even at concentrations of up to 1 mg ml−1 (Fig. 4A). A second assay based on trypan blue exclusion was employed to detect possible delayed toxicities. Consistent with the prior results, this approach also failed to demonstrate any significant toxicity of heparins after 5 days of treatment in 3 different cell lines (Fig. 4B). Finally, Annexin V-FITC combined with propidium iodide (PI) staining was performed to detect the possible presence of apoptotic and/or necrotic cells (Fig. 4C). After treatment of HCT-116 cells with UFH or enoxaparin, no increased numbers of apoptotic (quadrants Q2 and Q3) or necrotic cells (quadrants Q1 and Q2) could be detected (Fig. 4C). | ||
| Fig. 4 Effect of heparins on cellular viability. (A) HCT-116 cells were treated with heparin, enoxaparin, dalteparin (0.01–1000 μg ml−1) for 24 hours before the cell viability was measured using WST-1. The fold change of the absorbance signal in negative control was designated as 1. Data represent the average of five replicates from four independent experiments. Error bars = SD. (B) Epithelial cells (HCT-116, DLD-1, HT-29) were incubated with heparin, enoxaparin, dalteparin at 400 μg ml−1 in media containing 2% of FCS for over 5 days. The cell viability was determined by trypan blue dye exclusion assay where viable cells without staining blue while non-viable cells appearing blue were counted. Error bars = SD. (C) HCT-116 cells were untreated or treated with heparin and enoxaparin at 400 μg ml−1 in media containing 2% of FCS. After 96 hours, Annexin V-FITC/PI staining was analysed by flow cytometry. Data represent samples of control, heparin and enoxaparin of a typical experiment. | ||
Differential effects of enoxaparin oligosaccharides on proliferation and the cell cycle
Heparins are a complex mixture of oligosaccharides with and without anticoagulant activity of different lengths or sizes, various degrees and distribution of sulfate groups and sulfation patterns.38 We therefore investigated if different oligosaccharides might harbour different activities with regard to the proliferative capacity of cells as observed in the previous results. For this purpose, enoxaparin was separated into 14 different oligosaccharides using ion-exchange chromatography based on size and charge (Fig. 5A). When these oligosaccharides were individually assessed for their effect on colony formation, distinct profiles were observed (Fig. 5B). While some oligosaccharides (2 and 11) potently and significantly reduced colony formation, others (6, 7, 8, 10 and 12) surprisingly increased colony numbers. It has to be noted that under these conditions, the intact enoxaparin, which contains all oligosaccharides, inhibited colony formation dose-dependently (grey bars) as described above (Fig. 5B). This differential effect of 2 selected oligosaccharides could also be demonstrated when looking at cell growth in a mass culture (Fig. 5C). While oligosaccharide 2 clearly reduced cell growth more potently compared to intact enoxaparin, oligosaccharide 7 resulted in a strong increase in the cell number. Finally, these two oligosaccharides were also tested for their effects on cell cycle progression (Fig. 5D). Consistent with the results from the colony formation assay, oligosaccharide 2 strongly increased the number of cells in the G1 phase while leading to a significant reduction in S and G2 phase cells. It is important to note that at the same concentration, this effect was more pronounced for oligosaccharide 2 compared to intact enoxaparin (Fig. 5D). In line with the previous data, oligosaccharide 7 significantly reduced the number of cells in G1, while effects on S and G2 phases did not reach statistical significance. There was no difference in sub-G1 peaks of treated cells compared to control (data not shown). | ||
| Fig. 5 Differential effects of enoxaparin oligosaccharides on proliferation and the cell cycle. (A) Ion-exchange chromatographic separation of enoxaparin oligosaccharides. Numbers and lines indicate enoxaparin oligosaccharides tested. (B) Effect of enoxaparin (E1: 10 μg ml−1; E2: 100 μg ml−1, E3: 200 μg ml−1) and its oligosaccharides (1–14: 100 μg ml−1) on HCT-116 epithelial cell colony forming capacity in media (2% FCS). The total numbers of colonies are presented as %-change versus negative control. Data represent the average of four individual samples per concentration of a typical experiment. Error bars = SD. *P < 0.05, **P < 0.01 versus negative control. (C) HCT-116 cells in the absence (control) or presence of 100 μg ml−1 of enoxaparin (enox.) or its oligosaccharides (2 and 7), respectively, before stained with trypan blue. (D) Flow cytometric cell cycle analysis of HCT-116 cells after 18 hours exposure to 100 μg ml−1 of enoxaparin and its oligosaccharides (2 and 7) in media (2% FCS). Graph depicts quantification of the percentage of cells in G1, S, and G2 phases. Data represent the average of three individual samples per treatment of a typical experiment. Error bars = SD. *P < 0.05, **P < 0.01 versus negative control. | ||
Discussion
Together with previous reports,1,2,8 the data presented here demonstrate that in addition to their anticoagulation activity, heparins can effectively modulate additional biological functions. We and others20,39 have demonstrated that under conditions of reduced serum concentration in the growth media, UFH and LMWHs can dose-dependently inhibit proliferation of different cell lines in vitro. In contrast to our experiments that are characterized by reduced serum levels, reduced cell proliferation by UFH in the presence of 10% or 20% FCS9,14,40 was reported. However, in these studies, to increase the sensitivity of cells to the anti-proliferative effect of UFH, cells were arrested by serum-starvation for up to 96 h before the exposure to heparins, which tests the effects of UFH on the re-entry of cells into the cell cycle as opposed to the inhibition of proliferating cells.Although the majority of studies described a significant inhibition of cellular proliferation by heparins, some reports also indicated that increased cell death might be associated with the observed effects in some cell lines.17,20,22 In our experimental setting, increased cell death could have accounted for the reduced colony formation but no significant levels of either necrosis or apoptosis could be detected in the cell lines tested with both UFH and LMWHs. At the same time, it cannot be completely excluded that some cells might respond to exposure to heparins with cell death.
The current literature suggests strongly that the observed cell cycle effects of heparins are not cell type specific, since inhibition of proliferation was demonstrated in cells from a multitude of tissues and organisms.14,16,22,41,42 In this context, it is important to note that different cell types might have different dose-effect-responses to UFH.9 Overall, a wide range of UFH and LMWHs concentrations have been investigated and, depending on the types of heparins and cell culture conditions used, effective concentrations varied only slightly. Most studies agreed that cellular proliferation was significantly inhibited by UFH at doses between 10 and 100 μg ml−1.9,10,13,14,39 For enoxaparin, 25–50 μg ml−1 were reported to significantly inhibit proliferation of epithelial cells,20,43 while the minimum inhibitory concentration of dalteparin was found at 10 IU ml−1 (approximately 64 μg ml−1).22 Our results describe reduced proliferation between 10 and 400 μg ml−1, with a significant reduction at 10 μg ml−1 for enoxaparin and 100 μg ml−1 for dalteparin or UFH, respectively, and largely mirror the effective dose ranges described in previous studies. Thus, the effectiveness of these compounds might be less dependent on the cell type but rather more dependent on the experimental setting and more importantly on the origin of the compound.
Despite the well-established, standardized and quality-controlled methods of heparin production, heparins (UFH and LMWHs) remain complex mixtures of heterogeneous oligosaccharides and we have shown here that these oligosaccharides contain different and interestingly even opposing biological activities. This observation for the first time rationalizes the variation in the responses reported by different laboratories when using heparins, which are available from different sources and manufacturers,3 and contain different anti-proliferative properties.44 Although all commercially available heparins are characterized by their specific anticoagulant activity, this does not guarantee a uniform composition of other activities, since they are currently not tested for. Thus, differences in composition other than the overall anticoagulant activity in different preparations are likely to contribute to the differences seen when testing the non-anticoagulant activities. In support of this hypothesis, it was suggested previously that the compositional differences in non-anticoagulant oligosaccharides between batches of enoxaparin could be responsible for the inconsistent patient response to treatment of an inflammatory skin condition.45,46 Furthermore our results can explain the conflicting literature with regard to the pro- and anti-proliferative effects that have been described for heparins.26,27,47 From the model, that enoxaparin consists of pro- and anti-proliferative oligosaccharides, it can be hypothesized that only small variations in the quantity and the presence of specific oligosaccharides can alter the overall proliferative response to enoxaparin and likely also other heparins.
Even though repeating disaccharides of alternating sequence of uronic acid and glucosamine serve as the building blocks of heparin chains, sulfation groups can be unevenly distributed along the chains.4 Depending on the sources of raw material and the conditions of the purification process, unique structural features of the final products can be generated.5,38 This may include the presence of more, less or inactive moieties, or uncharacterized heparin molecules from the natural occurrence of glycosaminoglycans in the extracted tissues.48 For example, in 2007 and 2008 hundreds of hypersensitive reactions were reported worldwide after administration of heparin preparations contaminated with an impurity known as oversulfated chondroitin sulfate.49 It is believed that this contaminant was introduced to certain batches of heparin during the extraction and manufacturing process.50 This highlights the compositional complexity and structural variability present in the commercially available heparin mixtures and thus potential for varying clinical responses.
In order to identify oligosaccharides that are responsible for an effect on cellular proliferation in this study, enoxaparin was fractionated using ion-exchange chromatography which generates molecules of varying size and sulfation status with and without anticoagulant activity.36 This study demonstrates that not all oligosaccharides result in significant changes to the mitogenic activity of cells. Oligosaccharide 2 of enoxaparin consists mainly of disaccharides, which are distinctively shorter than the required smallest sequence of pentasaccharides for antithrombin binding;7 oligosaccharide 7 represents hexasaccharides with only low anticoagulant activity36 that can significantly increase the mitogenic activity of cells. This suggests that specific structures of heparin oligosaccharides are responsible for their effects on proliferation.
In contrast to our data, dodecasaccharides or even larger fragments have been reported to possess anti-proliferative activity, while hexasaccharides were identified previously as the minimum chain length for any anti-proliferative activity,42,51 and disaccharides had shown only very low anti-proliferative effects.42 However, Garg and colleagues concluded that size per se does not determine the anti-proliferative activity of heparin oligosaccharides1 but rather a consequence of the different methods of oligosaccharide preparation from UFH. These methods are predominantly based on different chemical or enzymatic reactions, which significantly increases the structural heterogeneity of heparins, causing variations in chain length and the substitution of important chemical groups present on glucosamine and urinate residues.5,38 Therefore, even subtle changes in preparation could conceivably lead to different compositions and pharmacological profiles of heparins and thus different experimental results.
Even though a large body of evidence shows that heparins can influence cell growth, the details on how heparins regulate or modulate cell proliferation are still lacking. While further studies are needed to uncover the detailed mechanism by which heparins affect cellular proliferation, this study provides the framework for the standardized isolation of enoxaparin oligosaccharides with pro- and anti-proliferative activities. Furthermore, this standardized isolation of oligosaccharides is a crucial pre-requisite for exploring other potential therapeutic uses of heparin oligosaccharides.
Conclusion
Using a powerful chromatographic technique, this study has for very first time demonstrated that different oligosaccharides obtained from intact heparin have different and even opposing effects. For example, out of all oligosaccharides tested, oligosaccharide 2 (disaccharide) showed the largest anti-proliferative effect while oligosaccharide 7 (hexasaccharide) profoundly stimulated proliferation. Since oligosaccharide 2 is too small to have any anticoagulant activity and oligosaccharide 7 has only minimal anticoagulant activity, the effects of heparins on cellular proliferation appear to be largely independent of their anticoagulant activity. All commercially available heparins have comparable anticoagulant activities; however, their non-anticoagulant oligosaccharide content greatly differs among the different heparin preparations. The composition of non-anticoagulant oligosaccharides generally depends on the source of isolation and the type of preparation employed. Therefore, depending on the structural characteristics of non-anticoagulant oligosaccharides, heparins can have either pro- or anti-proliferative effects. This study provides the platform for future studies determining the structural characteristics of non-anticoagulant oligosaccharides responsible for the cellular proliferative effects of the parent heparin, as well as investigating their effects on cellular proliferation in vivo.Abbreviations
| Da | Dalton |
| FCS | Fetal calf serum |
| HPLC | High performance liquid chromatography |
| IC | Ion-exchange chromatography |
| LMWH | Low molecular weight heparin |
| PBS | Phosphate buffered saline |
| SD | Standard deviations |
| UFH | Unfractionated heparin |
Acknowledgements
The authors are grateful to Professor Gregory Peterson (School of Pharmacy, University of Tasmania, Australia) for helpful review of the manuscript; Terry Pinfold, Jessica Kling, Jocelyn Darby, Mark Cozens, Farzaneh Atashrazm and Greg Woods (Menzies Research Institute, Tasmania, Australia) for their technical flow cytometry support. This work was supported by the School of Pharmacy, University of Tasmania, Australia.References
- H. G. Garg, H. Mrabat, L. Yu, C. Freeman, B. Li, F. Zhang, R. J. Linhardt and C. A. Hales, Carbohydr. Res., 2008, 343, 2406–2410 CrossRef CAS PubMed.
- R. Lever and C. P. Page, in Heparin - A Century of Progress, ed. R. Lever, B. Mulloy and C. P. Page, Springer, Heidelberg, 2012, vol. 207, pp. 281–305 Search PubMed.
- B. Mulloy, E. Gray and T. W. Barrowcliffe, Thromb. Haemostasis, 2000, 84, 1052–1056 CAS.
- B. Casu, in Chemistry and Biology of Heparin and Heparan Sulfate, ed. H. G. Garg, R. J. Linhardt and C. A. Hales, Elsevier, Oxford, 1st edn, 2005, ch. 1, pp. 1–28 Search PubMed.
- R. J. Linhardt and N. S. Gunay, Semin. Thromb. Hemostasis, 1999, 25, 5–16 CrossRef CAS PubMed.
- J. Hirsh, T. E. Warkentin, S. G. Shaughnessy, S. S. Anand, J. L. Halperin, R. Raschke, C. Granger, E. M. Ohman and J. E. Dalen, Chest, 2001, 119, 64S–94S CrossRef CAS.
- E. Gray, B. Mulloy and T. W. Barrowcliffe, Thromb. Haemostasis, 2008, 99, 807–818 CAS.
- S. M. Smorenburg and C. J. F. Van Noorden, Pharmacol. Rev., 2001, 53, 93–106 CAS.
- J. J. Castellot, D. L. Cochran and M. J. Karnovsky, J. Cell Physiol., 1985, 124, 21–28 CrossRef CAS PubMed.
- R. Tiozzo, D. Reggiani, M. R. Cingi, P. Bianchini, B. Osima and S. Calandra, Thromb. Res., 1991, 62, 177–188 CrossRef CAS.
- H. G. Garg, N. Cindhuchao, D. A. Quinn, C. A. Hales, C. Thanawiroon, I. Capila and R. J. Linhardt, Carbohydr. Res., 2002, 337, 2359–2364 CrossRef CAS.
- M. Dufresne and R. Warocquier-Clérout, Cytotechnology, 2001, 37, 13–22 CrossRef CAS.
- V. Kanabar, S. J. Hirst, B. J. O'Connor and C. P. Page, Br. J. Pharmacol., 2005, 146, 370–377 CrossRef CAS PubMed.
- R. C. Patel, I. Handy and C. V. Patel, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 1439–1444 CrossRef CAS.
- H. J. Kock and A. E. Handschin, Clin. Appl. Thromb./Hemostasis, 2002, 8, 251–255 CrossRef CAS PubMed.
- D. Nikitovic, A. Zafiropoulos, G. N. Tzanakakis, N. K. Karamanos and A. M. Tsatsakis, Anticancer Res., 2005, 25, 2851–2856 CAS.
- H. Hemeda, J. Kalz, G. Walenda, M. Lohmann and W. Wagner, Cytotherapy, 2013, 15, 1174–1181 CrossRef CAS PubMed.
- A. Papathanasopoulos, D. Kouroupis, K. Henshaw, D. McGonagle, E. A. Jones and P. V. Giannoudis, J. Orthop. Res., 2011, 29, 1327–1335 CrossRef CAS PubMed.
- J. Sudhalter, J. Folkman, C. M. Svahn, K. Bergendal and P. A. D'amore, J. Biol. Chem., 1989, 264, 6892–6897 Search PubMed.
- W. Abu Arab, R. Kotb, M. Sirois and E. Rousseau, Can. J. Physiol. Pharmacol., 2011, 89, 705–711 CrossRef CAS PubMed.
- Y. Carmazzi, M. Iorio, C. Armani, S. Cianchetti, F. Raggi, T. Neri, C. Cordazzo, S. Petrini, R. Vanacore, F. Bogazzi, P. Paggiaro and A. Celi, Cell Proliferation, 2012, 45, 545–556 CrossRef CAS PubMed.
- X. Chen, W. Xiao, X. Qu and S. Zhou, Cancer Invest., 2008, 26, 718–724 CrossRef CAS PubMed.
- G. E. Bertolesi, L. Lauria de Cidre and A. M. Eijan, Tumor Biol., 1994, 15, 275–283 CrossRef CAS PubMed.
- A. Syrokou, G. Tzanakakis, T. Tsegenidis, A. Hjerpe and N. K. Karamanos, Cell Proliferation, 1990, 32, 85–99 Search PubMed.
- M. Balzarotti, F. Fontana, C. Marras, A. Boiardi, D. Croci, E. Ciusani and A. Salmaggi, Oncol. Res., 2006, 16, 245–250 CAS.
- G. Chatzinikolaou, D. Nikitovic, A. Asimakopoulou, A. Tsatsakis, N. K. Karamanos and G. N. Tzanakakis, IUBMB Life, 2008, 60, 333–340 CrossRef CAS PubMed.
- W. K. K. Wu, V. Y. Shin, Y. N. Ye, H. P. S. Wong, F. Y. Huang, M. K. C. Hui, E. K. Y. Lam and C. H. Cho, Anticancer Res., 2006, 26, 439–443 CAS.
- N. Flint, F. Cove and G. S. Evans, J. Cell Sci., 1994, 107, 401–411 CAS.
- J. M. Herbert and J. P. Maffrand, J. Cell. Physiol., 1989, 138, 424–432 CrossRef CAS PubMed.
- Y. Uzun, E. Akdogan, F. Ozdemir and E. Ovali, Bratisl. Lek. Listy, 2009, 110, 3–6 CAS.
- G. N. Solayar, P. M. Walsh and K. J. Mulhall, BMC Musculoskeletal Disord., 2011, 12, 247 CrossRef CAS PubMed.
- A. Pervin, C. Gallo, K. A. Jandik, X. Han and R. J. Linhardt, Glycobiology, 1995, 5, 83–95 CrossRef CAS.
- K. Norrby, APMIS, 2006, 114, 79–102 CrossRef CAS PubMed.
- S. A. Mousa and S. Mohamed, Oncol. Rep., 2004, 12, 683–688 CAS.
- S. A. Mousa, X. Feng, J. Xie, Y. Du, Y. Hua, H. He, L. O'Connor and R. J. Linhardt, J. Cardiovasc. Pharmacol., 2006, 48, 6–13 CrossRef CAS PubMed.
- M. D. Shastri, C. Johns, J. P. Hutchinson, M. Khandagale and R. P. Patel, Anal. Bioanal. Chem., 2013, 405, 6043–6052 CrossRef CAS PubMed.
- T. Guler, Z. A. Polat, E. Yayci, T. Atacag and A. Cetin, Arch. Gynecol. Obstet., 2013, 287, 217–222 CrossRef CAS PubMed.
- P. Bianchini, L. Liverani, F. Spelta, G. Mascellani and B. Parma, Semin. Thromb. Hemostasis, 2007, 33, 496–507 CrossRef CAS PubMed.
- S. Vannucchi, F. Pasquali, V. P. Chiarugi and M. Ruggiero, Biochem. Biophys. Res. Commun., 1990, 170, 89–95 CrossRef CAS.
- H. G. Garg, P. A. M. Joseph, B. T. Thompson, C. A. Hales, T. Toida, T. Imanari, I. Capila and R. J. Linhardt, Arch. Biochem. Biophys., 1999, 371, 228–233 CrossRef CAS PubMed.
- S. A. Kilfeather, S. Tagoe, A. C. Perez, K. Okona-Mensa, R. Matin and C. P. Page, Br. J. Pharmacol., 1995, 114, 1442–1446 CrossRef CAS.
- T. C. Wright, J. J. Castellot, M. Petitou, J. Lormeau, J.-C. Choay and M. J. Karnovsky, J. Biol. Chem., 1989, 264, 1534–1542 CAS.
- L. Ma, H. Qiao, C. He, Q. Yang, C. H. A. Cheung, J. R. Kanwar and X. Sun, Invest. New Drugs, 2012, 30, 508–517 CrossRef CAS PubMed.
- J. J. Castellot, J. Choay, J.-C. Lormeau, M. Petitou, E. Sache and M. J. Karnovsky, J. Cell Biol., 1986, 102, 1979–1984 CrossRef CAS.
- E. Hodak, G. Yosipovitch, M. David, A. Ingber, L. Chorev, O. Lider, L. Cahalon and I. R. Cohen, J. Am. Acad. Dermatol., 1998, 38, 564–568 CrossRef CAS.
- R. Rai, I. Kaur and B. Kumar, J. Am. Acad. Dermatol., 2002, 46, 141–143 CrossRef.
- E. Al-Ansari, H. Du, L. Yu, C. D. Ochoa, H. G. Garg, D. A. Quinn and C. A. Hales, Chest, 2007, 132, 1898–1905 CrossRef CAS PubMed.
- L. Liverani, G. Mascellani and F. Spelta, Thromb. Haemostasis, 2009, 102, 846–853 CAS.
- M. Guerrini, Z. Shriver, A. Bisio, A. Naggi, B. Casu, R. Sasisekharan and G. Torri, Thromb. Haemostasis, 2009, 102, 907–911 CAS.
- H. Liu, Z. Zhang and R. J. Linhardt, Nat. Prod. Rep., 2009, 26, 313–321 RSC.
- J. J. Castellot, D. L. Beeler, R. D. Rosenberg and M. J. Karnovsky, J. Cell. Physiol., 1984, 120, 315–320 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |