 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective synthesis of renewable bisphenols from 2,3-pentanedione and their application as plasticizers†
Wouter
Schutyser
a,
Steven-Friso
Koelewijn
a,
Michiel
Dusselier
a,
Stijn
Van de Vyver
ab,
Joice
Thomas
c,
Feng
Yu
c,
Maria Josefina
Carbone
d,
Mario
Smet
c,
Peter
Van Puyvelde
d,
Wim
Dehaen
c and
Bert F.
Sels
*a
aCentre for Surface Chemistry and Catalysis, KU Leuven, Kasteelpark Arenberg 23, 3001 Leuven, Belgium. E-mail: bert.sels@biw.kuleuven.be; Fax: +32 1632 1998; Tel: +32 1632 1593
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
cDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
dDepartment of Chemical Engineering, KU Leuven, Willem de Croylaan 46, 3001 Leuven, Belgium
First published on 24th February 2014
Abstract
2,3-Pentanedione (2,3-PD), a bio-based chemical derived from lactic acid, has the potential to serve as a precursor for the synthesis of novel bisphenols. We developed a solvent-free catalytic strategy for the condensation of phenol with 2,3-PD by using acid catalysts at temperatures ranging from 323 to 373 K. Various soluble and solid acids exhibit high activity, while a high chemoselectivity to bisphenol requires a high phenol to 2,3-PD molar ratio. Bisphenol yields as high as 84% are for instance reported in an excess of phenol in the presence of Nafion NR50. Recycling of the Nafion catalyst after washing with ethanol at room temperature is demonstrated. The regioselectivity in the bisphenol fraction is influenced by the acid strength. A clear trend is presented in which the regioselectivity towards the desired p,p′-isomers increases with increasing acid strength, showing p,p′/o,p′-isomer ratios as high as 100. A tentative mechanism is discussed based on the ionic versus non-ionic pathway. The purified 2,3-PD-derived p,p′-bisphenols are assessed as plasticizers for polyethylene terephthalate (PET), showing promising properties similar to that of the reference bisphenol A, but with a broader processing window due to the lower melting point and higher thermal stability under an inert atmosphere.
1. Introduction
The increasing demand for renewable materials has led to a faster search for biomass-derived substitutes for bisphenol A (BPA).1,2 BPA is an important monomer for the synthesis of polycarbonates and epoxy resins and it is a popular plasticizer for thermoplastic polymers.3,4 Despite its common use in food and drink packaging, controversy over its use arose since the discovery of its potential endocrine disruptor activity.5Two alternative bisphenols have recently received attention: (i) diphenolic acid (DPA) based on cellulose-derived levulinic acid6–11 and (ii) bisphenols based on lignin-derived creosol.2,12
This contribution explores the synthesis and potential applications of a BPA analogue based on biomass-derived 2,3-pentanedione (2,3-PD). 2,3-PD is produced on an industrial scale via a multi-step process combining hydroxyacetone and paraldehyde or through extraction out of dairy waste, but it can also be derived from lactic acid.13 Recent breakthroughs have allowed the direct catalytic conversion of trioses,14–19 sucrose14,19,20 and cellulose21,22 to lactic acid in water or lactates in aqueous/alcohol media. 2,3-PD is obtained through a Claisen-condensation of two such lactate molecules, followed by decarboxylation and dehydration.13,23–25 Condensation of 2,3-PD and phenol could thus ultimately open up opportunities to synthesize fully renewable bisphenols, since the phenolic substrates may be obtained from lignin (see Scheme 1).26–29
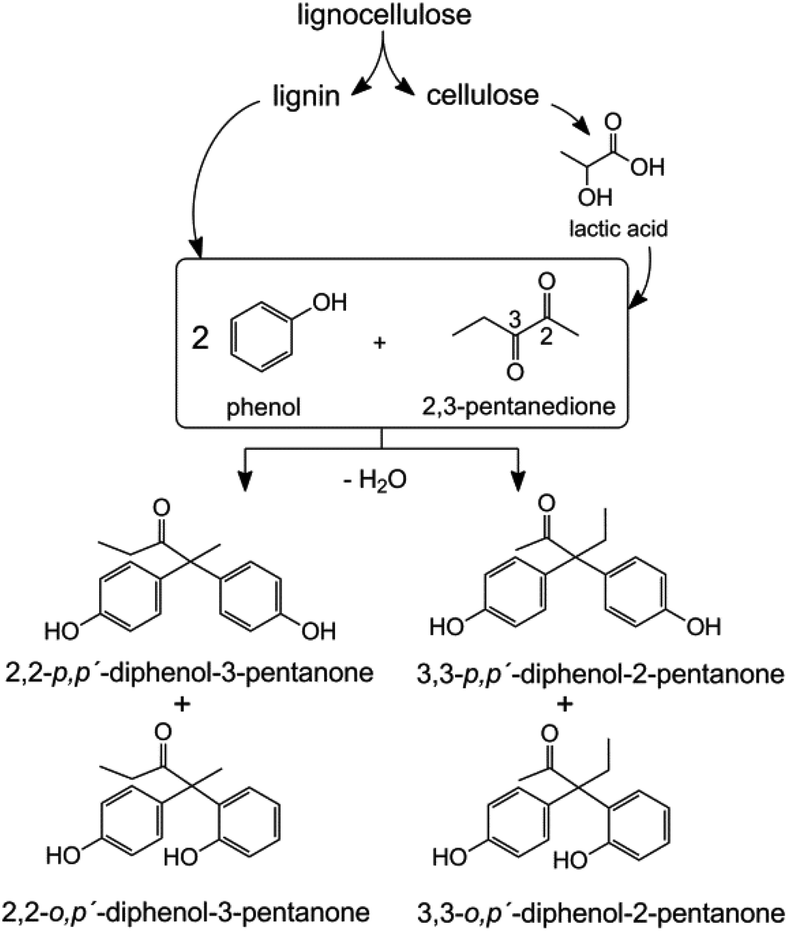 | ||
| Scheme 1 Synthesis of renewable bisphenols from phenol and 2,3-pentanedione (2,3-PD). | ||
The presence of the extra carbonyl functional group in the alternative bisphenols is expected to enhance their biodegradability30,31 and might offer additional functionality for the bio-based polymers. For the potential health effects, a reduced accumulation in body tissues is expected,32 and some studies indicate that more polar or electron-rich bisphenols—such as the ones studied here—are less able to penetrate the human skin33 and tend to bind less effectively to certain estrogen receptors.34
Bisphenols are generally produced by the acid-catalysed condensation of phenol with a ketone or aldehyde.3 The first step in the mechanism is the protonation of the ketone with formation of a carbocation intermediate. The reaction of this carbocation with phenol produces water and a monophenol intermediate, which condenses with a second phenol.
Acid-catalysed condensation of phenol with 2,3-PD has, to the best of our knowledge, not been reported. In this reaction, two pairs of bisphenol isomers can be produced by condensation of phenol on either the 2- or 3-position of 2,3-PD. The coordination of phenol groups determines the formation of p,p′- or o,p′-isomers, the former being the most useful precursor for the synthesis of polymers. A high p,p′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :o,p′ ratio of the isomers is of crucial importance in the synthesis of polycarbonates from BPA,4 as it influences physicochemical properties such as color stability35 and crystallinity.36 Separation of the p,p′- and o,p′-isomers requires energy-intensive procedures such as fractional distillation and crystallisation.3,4 Improving the isomer selectivity in the synthesis of bisphenols is therefore highly desirable.
:o,p′ ratio of the isomers is of crucial importance in the synthesis of polycarbonates from BPA,4 as it influences physicochemical properties such as color stability35 and crystallinity.36 Separation of the p,p′- and o,p′-isomers requires energy-intensive procedures such as fractional distillation and crystallisation.3,4 Improving the isomer selectivity in the synthesis of bisphenols is therefore highly desirable.
Thiols are typically added as a co-catalyst in the condensation reaction, as they increase the reaction rate and the p,p′-regioselectivity of the condensation step.3,37–42 Zeidan and Margelefsky et al. have suggested that the thiol additive activates the carbonyl of the ketone towards nucleophilic attack via a charged sulfur intermediate with an increased electrophilicity.38,40,42 It was proposed that the steric hindrance of the side chain of the thiol affected the approach of phenol, resulting in an increased regioselectivity. In the case of DPA synthesis, Van de Vyver et al. recently showed that the steric effects of various thiol co-catalysts play a predominant role in the condensation rate and para-selective introduction of phenol, regulating the regioselectivity.6,7 They also demonstrated an important contribution of an acid-catalysed isomerisation of p,p′-DPA to o,p′-DPA at high conversions of levulinic acid.
2. Experimental section
2.1. Characterization of the acid catalysts
Acid–base titrations of the acid catalysts were performed using a Metrohm 808 Titrando autotitrator and 801 stirrer at room temperature. In a typical experiment, 50 mg of the polymer was added to 10 mL of water containing 2 M NaCl. The resulting mixture was allowed to equilibrate overnight and titrated potentiometrically by dropwise addition of an aqueous solution containing 0.02 M NaOH.2.2. Condensation reactions and product analysis
Catalytic reactions were carried out in 10 mL glass vessels. Phenol and the acid catalyst were mixed with a magnetic stirrer. The vessel was flushed with N2 and heated to the desired temperature. Once the desired temperature was achieved, 2,3-PD was added with a syringe through the septum of the vessel. Samples of the reaction mixture were withdrawn periodically and analysed by GC. Prior to product analysis, approximately 0.05 g of the reaction mixture was weighed in a vial and mixed with 1 mL acetonitrile and 50 μL 2,4-dimethylphenol as external standards. Quantification of the products was performed using a GC equipped with a CP-SIL 5CB WCOT fused silica column. The formation of phenol oligomers from 2,3-PD condensation was studied by GC-MS with a HP5MS capillary column.2.3. Purification and characterization of the bisphenols
For the purification of the bisphenols, the unreacted phenol was first removed through multiple water extractions. The resulting sample was dissolved in dichloromethane in a sonication bath and purified by column chromatography (silica gel, 70–230 mesh) by eluting the column with dichloromethane and a solution of 10 vol% ethyl acetate in dichloromethane.1H, 13C NMR, COSY, DEPT and HSQC spectra were acquired on a Bruker Avance 400 MHz. The chemical shifts (δ) are reported in parts per million (ppm) referenced to tetramethylsilane (1H) or the internal NMR solvent signals (13C). For Fourier-transformed infrared (FT-IR) spectroscopy, KBr pellets of the purified bisphenols were prepared, and the FT-IR spectra were recorded on a Fourier Transform Infrared spectrometer. The elemental composition of the bisphenols was obtained through CHN analysis. Thermogravimetric analysis was performed with a TGA Q500 of TGA instruments. The samples were heated at 10 °C min−1 to 500 °C under a nitrogen atmosphere. Powder X-ray diffraction (XRD) patterns were recorded on a STOE STADI P Combi diffractometer with an image plate position sensitive detector (IP PSD) in the region 2θ = 5 to 60° (Δ2θ = 0.03°), and a scan of 1200 s. The measurements were performed in transmission mode at room temperature using CuKα1 radiation with λ = 1.54056 Å selected by means of a Ge(111) monochromator.
2.4. Plasticizer tests
For the plasticizer tests, PET pellets were grinded, physically mixed with a solution of bisphenol (BP(2,3-PD) or BPA) in dichloromethane, magnetically stirred and dried overnight at 35 °C. PET–bisphenol mixtures were prepared with different concentrations of bisphenol (2–5 wt%). The samples were analysed with Q2000 DSC equipment and subjected to the following non-isothermal protocol: heating at 10 °C min−1 from 20 °C to 250 °C, kept constant at this temperature for 5 min, and cooled down to 20 °C at 10 °C min−1. After 5 min the second heating started from 20 °C to 250 °C at 10 °C min−1. The calorimetric information showed in this work is obtained from the second heating section.3. Results and discussion
3.1. Catalyst screening and characterization of the bisphenols
Table 1 presents the catalytic results for the condensation of phenol and 2,3-PD, including the turnover frequency (TOF) of 2,3-PD and the rate of bisphenol formation (ROF) per acid site, the conversion of 2,3-PD, the bisphenol yield, the yield of monophenolic (MP) and diphenolic (DP) side-products, the carbon mass balance (CB) and the molar ratio of the p,p′- to the o,p′-bisphenol isomers. Note that all the reactions were performed with the same amount of Brønsted acid sites. Entries 1–9 show the results in the presence of sulfuric acid and various soluble and solid sulfonic acid-based catalysts after 72 h of reaction at 60 °C. Besides the four bisphenol isomers anticipated in Scheme 1, we observed an amount of side-products with one or two phenol groups.| Entry | Catalyst | Acid densityb (mmol H+ g−1) | Substrate | TOFc (h−1) | ROFd (h−1) | Ketone conv. (%) | Yield (%) | CBe (%) |
p,p′:o,p′f |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bisphenol | MP | DP | |||||||||
|
a Reaction conditions: 3.4 mmol ketone, 10.2 mmol phenol (3:1 ratio phenol–ketone), 0.12 mmol catalytic H+, inert N2 atmosphere, 60 °C, magnetic stirring for 72 h.
b Determined by acid–base titrations.
c Calculated as the molar amount of ketone converted per acid site per hour, at 1 h of reaction.
d Calculated as the molar amount of bisphenols formed per acid site per hour, at 1 h of reaction.
e See the ESI for the definition.
f The ratio of p,p′-bisphenol to o,p′-bisphenol.
g 3.4 mmol ketone, 23.8 mmol phenol (7:1 ratio phenol–ketone).
h 3.4 mmol ketone, 47.6 mmol phenol (14:1 ratio phenol–ketone).
i 0.24 mmol catalytic H+, 50 °C.
j 2nd run of the catalyst from entry 12.
k 100 °C, 24 h. Catalyst abbreviations: 1-propanesulfonic acid (PSA), p-toluenesulfonic acid (pTSA), trifluoromethanesulfonic acid (TFSA), nonafluorobutanesulfonic acid (NFBSA) and sulfonated hyperbranched poly(arylene oxindole)s (SHPAOs). Substrate abbreviations: 2,3-pentanedione (2,3-PD), acetone (Ac) and 2-pentanone (2-Pent).
|
|||||||||||
| 1 | Sulfuric acid | 20.4 | 2,3-PD | 11.1 | 5.1 | 100 | 57 | 3 | 11 | 71 | 17 |
| 2 | PSA | 7.7 | 2,3-PD | 13.5 | 6.6 | 100 | 58 | 3 | 14 | 75 | 16 |
| 3 | pTSA | 5.2 | 2,3-PD | 16.1 | 8.1 | 100 | 60 | 2 | 14 | 76 | 15 |
| 4 | TFSA | 6.6 | 2,3-PD | 16.3 | 9.1 | 100 | 67 | 1 | 8 | 76 | 55 |
| 5 | NFBSA | 3.2 | 2,3-PD | 17.7 | 8.0 | 100 | 66 | 1 | 9 | 76 | 61 |
| 6 | SHPAOs | 5.4 | 2,3-PD | 5.3 | 1.9 | 100 | 63 | 5 | 10 | 78 | 19 |
| 7 | Dowex 50WX2 | 4.1 | 2,3-PD | 10.1 | 5.5 | 100 | 68 | 8 | 14 | 90 | 12 |
| 8 | Amberlyst 15DRY | 4.3 | 2,3-PD | 2.8 | 0.9 | 72 | 33 | 10 | 8 | 71 | 11 |
| 9 | Nafion NR50 | 0.93 | 2,3-PD | 2.3 | 1.0 | 100 | 66 | 3 | 9 | 78 | 38 |
| 10g | Nafion NR50 | 0.93 | 2,3-PD | 2.4 | 1.0 | 100 | 76 | 3 | 10 | 89 | 52 |
| 11h | Nafion NR50 | 0.93 | 2,3-PD | 2.3 | 0.9 | 100 | 80 | 1 | 9 | 90 | 85 |
| 12h,i | Nafion NR50 | 0.93 | 2,3-PD | 1.6 | 0.5 | 100 | 84 | 1 | 7 | 92 | 104 |
| 13h,i,j | Nafion NR50 | 0.93 | 2,3-PD | / | / | 100 | 78 | 1 | 8 | 87 | 100 |
| 14k | Nafion NR50 | 0.93 | 2,3-PD | 12.2 | 4.6 | 100 | 56 | 3 | 14 | 73 | 11 |
| 15 | Nafion NR50 | 0.93 | Ac | 1.0 | 0.1 | 35 | 19 | 1 | 3 | 66 | 6 |
| 16 | Nafion NR50 | 0.93 | 2-Pent | 0.8 | <0.1 | 23 | 11 | 1 | 1 | 57 | 6 |
The as-obtained bisphenol product mixture was purified by water extraction and column chromatography. Gas chromatograms of the crude product mixture and the purified sample are shown in Fig. S1a and b of the ESI.† Fig. S2 of the ESI† shows the mass spectra of the obtained bisphenols. The structure of the 2,2- and 3,3-bisphenols was confirmed by NMR analyses of the purified compounds.
The 1H NMR spectrum in Fig. 1 shows the presence of two groups of singlet (1.71 and 1.93 ppm), triplet (0.57 and 0.85 ppm) and quartet (2.18 and 2.34 ppm) spin multiplicities in the aliphatic region. Integration of the 1H NMR signals revealed the molar ratio of 2,2- to 3,3-bisphenols to be approximately 3:1 for all catalysts. The preferential condensation of phenol on the carbon in the 2-position is suggested to be due to a combination of steric and electronic effects. The intensities of the two large doublet signals at 6.7 and 6.9 ppm in the aromatic region show the predominant formation of p,p′-isomers. More characterization data from 13C NMR, correlation spectroscopy (COSY), heteronuclear single-quantum correlation (HSQC) NMR, Fourier-transform infrared (FTIR) spectroscopy, elemental analysis and powder X-ray diffraction (of the purified crystalline p,p′-bisphenols) can be found in the ESI.†
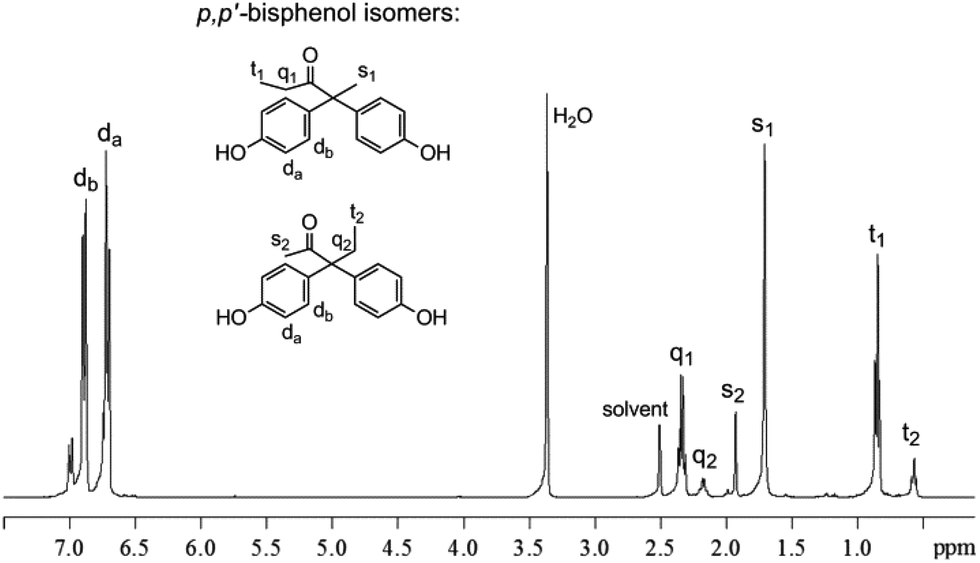 | ||
| Fig. 1 1H NMR spectrum of the renewable bisphenols obtained by TFSA-catalysed condensation of phenol with 2,3-PD in the 2- or 3-position. The following abbreviations are used for spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet. | ||
Fig. 2 illustrates the other identified products (4 compounds): two monophenol intermediates (a) and two diphenol side-products (b), next to two small unknowns. The two monophenol intermediates incorporate a phenol at the carbon in the 3-position of 2,3-PD (o-3-MP and p-3-MP). The monophenol products, in which phenol is attached to the 2-position of 2,3-PD (o-2-MP and p-2-MP), were only detected in very small amounts. Further identification data of the side-products can be found in the ESI† (1H, 13C, DEPT, HSQC and COSY NMR spectra).
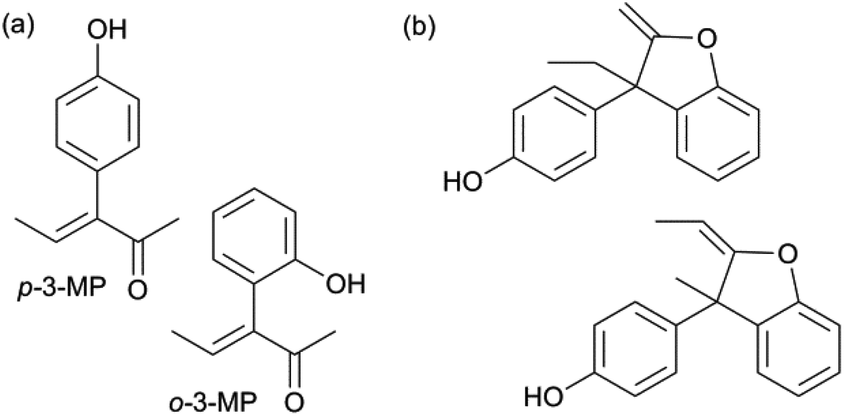 | ||
| Fig. 2 Identified monophenol intermediates (a) and diphenol side-products (b). | ||
The homogeneous catalysts p-toluenesulfonic acid (pTSA), trifluoromethanesulfonic acid (TFSA) and nonafluorobutanesulfonic acid (NFBSA) exhibit the highest activities, viz. the highest TOF and ROF (Table 1, entries 3–5), followed by 1-propanesulfonic acid (entry 2), sulfuric acid (entry 1) and Dowex 50WX2 (entry 7). The other polymer catalysts, Amberlyst 15 and Nafion NR50, show the lowest TOF and ROF (entries 8 and 9). Dowex 50WX2 and Amberlyst 15 are both polystyrene–divinylbenzene type polymer catalysts, while Nafion NR50 is a perfluorinated sulfonic acid resin.43 The different catalytic behaviour of Dowex 50WX2 and Amberlyst 15 is likely due to their different degrees of crosslinking (i.e., Amberlyst 15 is a highly cross-linked macroreticular resin, while Dowex 50WX2 is a gel-type resin with a low degree of crosslinking)44,45 since their acidity properties are very similar. The lower activity of Nafion NR50 might be explained by its relatively low surface area.43 Sulfonated hyperbranched poly(arylene oxindole)s (SHPAOs),46 a new class of acid catalysts with great potential in cellulose conversion47 and DPA synthesis,6,7 show an intermediate activity for this reaction (entry 6). Nearly all catalysts obtain complete conversion of the ketone substrate after 72 h of reaction.
Fig. 3 illustrates the time profiles of the conversion of 2,3-PD and the yield of the different bisphenol isomers and some side-products for the catalysts pTSA, Nafion NR50 and TFSA. The reactions catalysed by pTSA and TFSA are already near completion after 24 h (Fig. 3a and c), while Nafion NR50 requires 72 h to complete 2,3-PD conversion in the excess of phenol (Fig. 3b). The kinetic profiles of TFSA and pTSA also suggest that the monophenols are intermediates in the reaction network. The highest bisphenol yields (up to 68%) are obtained for TFSA, NFBSA, Dowex 50WX2 and Nafion NR50 (entries 4, 5, 7 and 9). The chemoselectivity to bisphenols was further increased by increasing the phenol to ketone ratio, and this increase is most pronounced for Nafion NR50. For instance, reactions in the presence of Nafion NR50 with a ratio of 7:1 or 14:1, instead of 3:1, yield respectively 76 and 80% of bisphenols at complete 2,3-PD conversion (compare entries 9, 10 and 11). Higher carbon balances, as obtained by summation of the products identified in Table 1, were noticed with increasing content of phenol, ranging from 78 to 90%. This is probably due to a limited condensation of 2,3-PD in an excess of phenol.
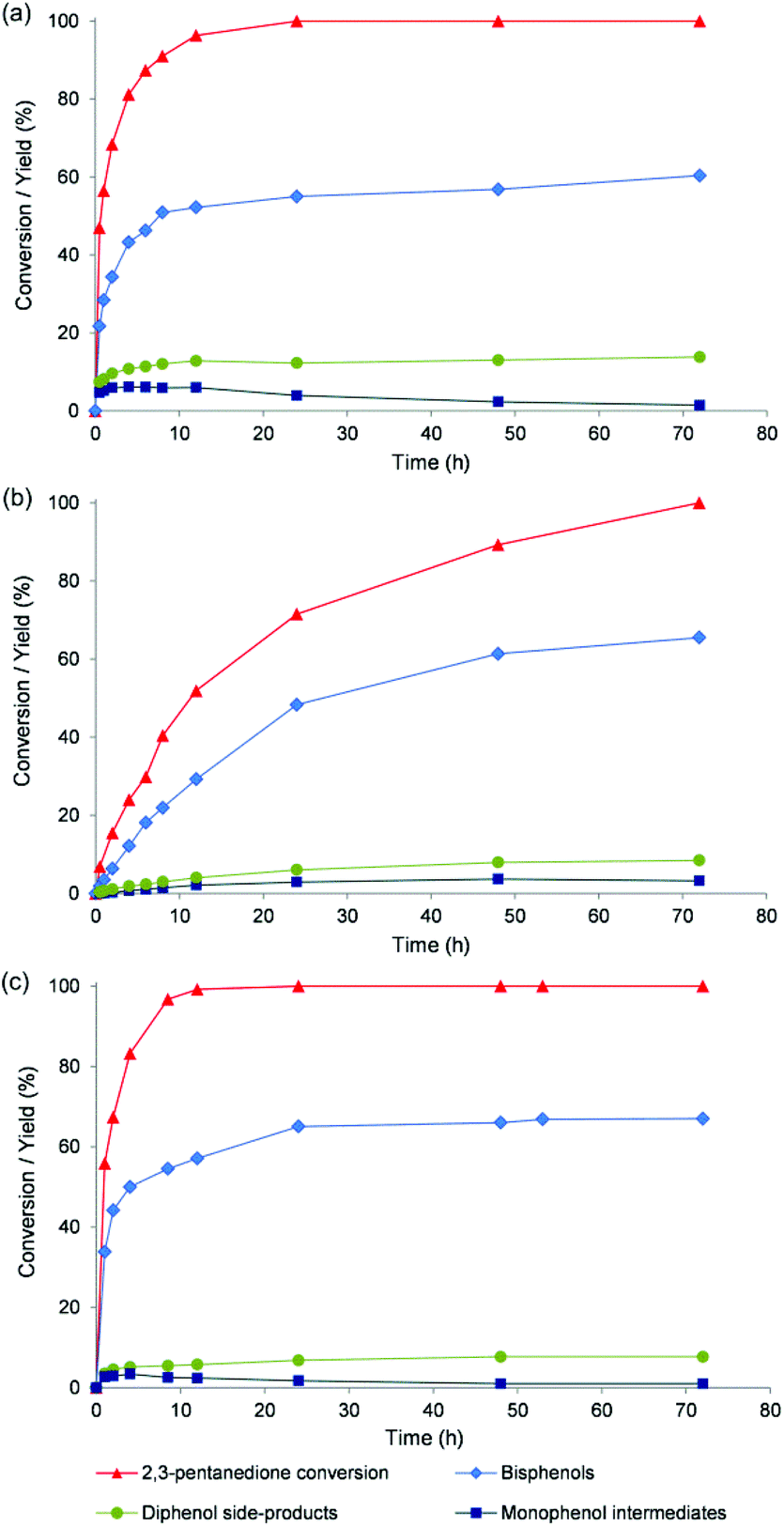 | ||
| Fig. 3 2,3-PD conversion, bisphenols and side-product (diphenolic and monophenolic) yield as a function of reaction time for pTSA (a), Nafion NR50 (b) and TFSA (c). Reaction conditions: see Table 1. | ||
Besides the reagent ratio, the temperature also has a significant effect on the final bisphenol yields. Increasing the reaction temperature from 60 to 100 °C (at a phenol:ketone ratio of 3) increases the TOF and ROF of Nafion NR50 in accordance with Arrhenius’ law, but the temperature increase causes a decrease in the maximum bisphenol yield (compare entries 9 and 14). Lowering the reaction temperature from 60 to 50 °C for instance shows an increase in bisphenol yield with Nafion NR50 up to 84% at a phenol:ketone ratio of 14 (compare entries 11 and 12). The total carbon balance of the latter product mixture reaches a high value of 92%. Nafion NR50 catalyst recycling was demonstrated by washing the spent catalyst of entry 12 at room temperature in ethanol before reuse. Only a slight decrease in bisphenol yield was noticed (from 84 to 78%, compare entries 12 and 13). A more detailed deactivation study is required to evaluate the stability of the catalyst under reaction conditions, for instance by monitoring the time-on-stream behaviour of the catalytic results in a continuous reactor set-up.
3.2. Influence of the catalyst and its acid strength on the regioselectivity
The p,p′:o,p′ ratios after 72 h of reaction at 60 °C range from 11 to 19 for all non-fluorinated catalysts (entries 1–3 and 6–8). Interestingly, the ratios are significantly higher for the fluorinated acid catalysts. For example, TFSA, NFBSA and Nafion NR50 show ratios of 55, 61 and 38, respectively (entries 4, 5 and 9). These p,p′:o,p′ isomer ratios from 2,3-PD are unexpectedly high, especially since the reactions were carried out in the absence of thiol promoters, which are typically used for boosting the regioselectivity in BPA synthesis to values above the thermodynamic equilibrium.7 Indeed, condensation reactions of phenol with monoketones like acetone or 2-pentanone in the presence of Nafion NR50 show ratios of about 6 (entries 15 and 16), in agreement with previous reports, showing the unique case of 2,3-PD.38,40 The presence of the adjacent ketone functions in 2,3-PD likely provides a suitable steric and electronic environment at the reactive center, which favors reaction with the p-position of phenol. Besides a significantly higher regioselectivity for the p,p′-isomers in reactions with 2,3-PD, the diketone also exhibits a much higher reactivity compared to acetone and 2-pentanone (compare entries 9, 15 and 16). The presence of the adjacent ketone functionality assists in a much faster reaction due to the higher electrophilicity of the protonated ketone, making the 1,2-diones interesting reactive substrates for condensation with phenols. Note the high TOFs for the reactions at 60 °C, even in the absence of thiols, which are usually required to speed up the reaction rate with monoketones.6 Classic ketone/phenol condensations such as BPA synthesis for instance are typically carried out at 70–90 °C with rate-accelerating agents like thiols.3
Fig. 4 shows the p,p′:o,p′ isomer ratio during bisphenol synthesis as a function of 2,3-PD conversion in the presence of various acid catalysts. All catalysts show a slight decrease of the isomer ratio with 2,3-PD conversion, likely due to acid-catalysed isomerization in agreement with previous observations.7 The isomer ratio is very important with regard to the use of these bisphenols as polymer building blocks, the p,p′-isomers being used preferably. Three different groups of catalysts may be roughly distinguished based on the isomer ratio: (i) low values around 20 for the common soluble and solid acids, (ii) intermediate values around 40 for Nafion NR50, and (iii) very high values around 80 and higher for the strongest (soluble) acids. These results indicate that the acid strength of the catalyst defines the isomer ratio.
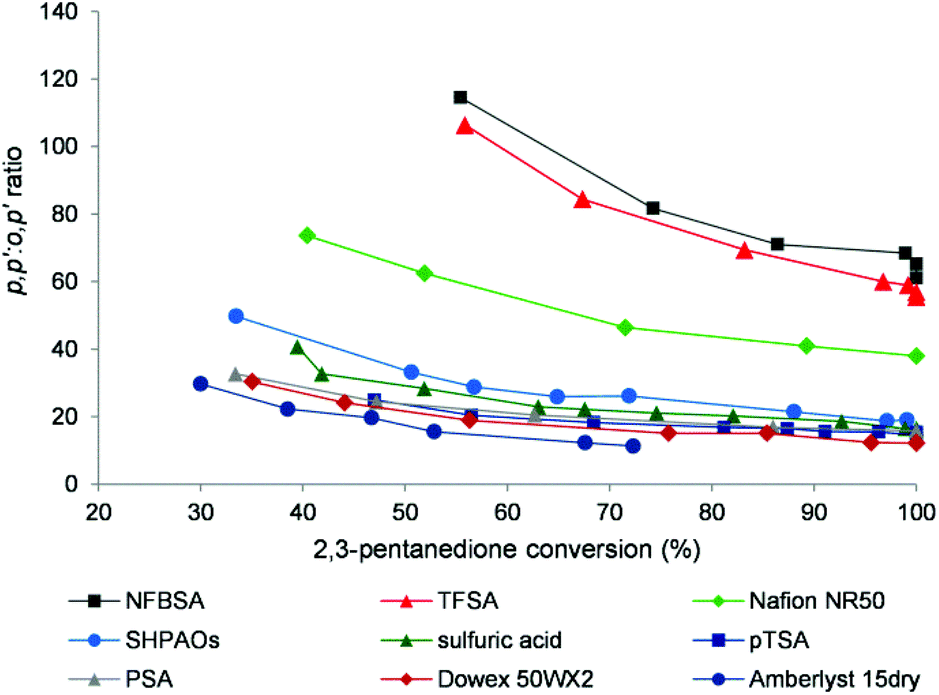 | ||
| Fig. 4
p,p′:o,p′ ratio of the bisphenols as a function of 2,3-PD conversion. Reaction conditions: 3.4 mmol 2,3-PD, 10.2 mmol phenol, 0.12 mmol catalytic H+, 60 °C. | ||
In an effort to evaluate the influence of the acid strength on the regioselectivity, a range of soluble acids with varying acidity were tested in the bisphenol synthesis. As a measure of the acid strength of the acids, their pKa values determined in acetonitrile (MeCN) were used, as reported in the literature.48–51 Although the reaction mixture and MeCN are very different in nature (phenol, the major component in the reaction media, is strongly protic, while MeCN is aprotic), and the media has a strong influence on the acidity,48 the pKa values in MeCN are used because MeCN is one of the only solvents in which pKa values of (very) strong acids are known with good accuracy, and because most of the acids used here are strongly acidic, reducing the solvent impact on the acid strength. Since no pKa (in MeCN) values of nonafluorobutanesulfonic acid (NFBSA), 2-naphthalenesulfonic acid (2-NSA) and 1-propanesulfonic acid (PSA) are available in the literature, it is suggested that these values are quite similar to TFSA, 1-naphthalenesulfonic acid and methanesulfonic acid (MSA), respectively.49,51
In Fig. 5a, an obvious correlation between the p,p′:o,p′ isomer ratio of the bisphenols at 55% 2,3-PD conversion and the pKa (MeCN) value of the acids is noticed. Increasing the catalyst acidity clearly results in an increased selectivity for the p,p′-bisphenol isomer during bisphenol synthesis. Sulfuric acid is not included in Fig. 5a since it possesses two protons with different acid strengths. The p,p′:o,p′ bisphenol ratio as a function of 2,3-PD conversion in the presence of different soluble acids is shown in Fig. S14 of the ESI.† The importance of the acid strength in the regioselectivity was also confirmed by condensing 2,3-PD with phenol in the presence of mixtures of TFSA and pTSA (Fig. 5b). Using defined mixtures of two acids with a great difference in acid strength offers a handy tool for varying the acid strength of a catalyst system.52 The total proton concentration was kept unchanged in the reaction, while their molar ratio was varied. Fig. 5b shows the p,p′:o,p′ ratio at 85% 2,3-PD conversion for TFSA/pTSA-catalysed bisphenol synthesis as a function of the molar fraction of TFSA in the catalyst mixture. The same trend is observed as in Fig. 5a; the p,p′-isomer preference increases with increasing acid strength of the catalytic system. This shift in regioselectivity requires some mechanistic consideration and this will be explained in the next section.
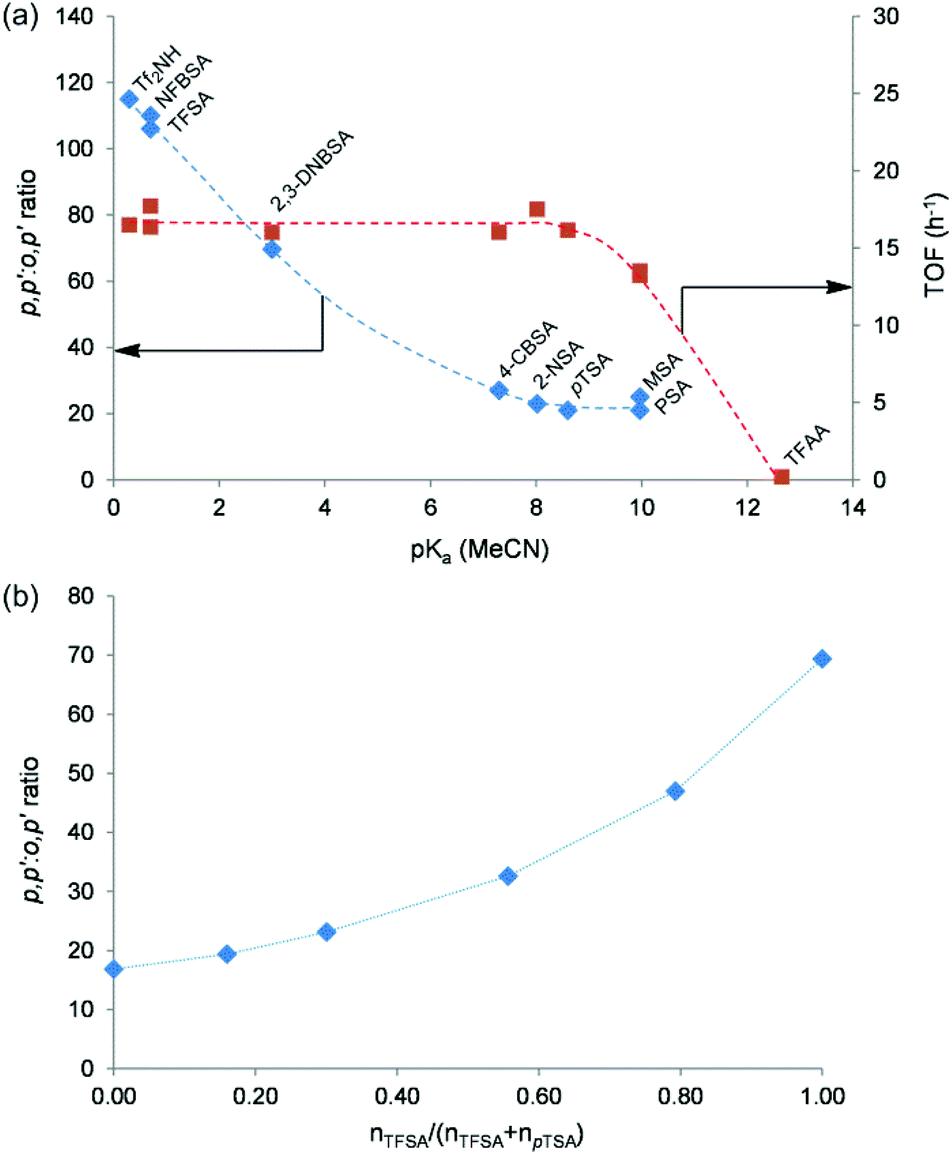 | ||
| Fig. 5 (a) p,p′:o,p′ ratio of the bisphenols at 55% 2,3-PD conversion and 2,3-PD TOF determined at 1 h of reaction in the phenol condensation of 2,3-PD catalysed by a range of soluble acids as a function of the pKa value of the acids in acetonitrile. Catalyst abbreviations: bis(trifluoromethanesulfonyl)imide (Tf2NH), nonafluorobutanesulfonic acid (NFBSA), trifluoromethanesulfonic acid (TFSA), 2,4-dinitrobenzenesulfonic acid (2,4-DNBSA), 4-chlorobenzenesulfonic acid (4-CBSA), 2-naphthalenesulfonic acid (2-NSA), p-toluenesulfonic acid (pTSA), methanesulfonic acid (MSA), 1-propanesulfonic acid (PSA) and trifluoroacetic acid (TFAA). (b) p,p′:o,p′ ratio of the bisphenols at 85% 2,3-PD conversion as a function of the molar fraction of TFSA in the TFSA/pTSA catalyst mixture. Reaction conditions: 3.4 mmol 2,3-PD, 10.2 mmol phenol, 0.12 mmol catalytic H+, 60 °C. | ||
As indicated in Fig. 5a, the acid strength has no influence on the reaction rate (expressed by the 2,3-PD TOF at 1 h of reaction) for acids with a pKa (MeCN) value smaller than 9. The reaction rate decreases for acids with a pKa (MeCN) value larger than 9 and even no reaction occurs when the acid is too weak, as is the case for trifluoroacetic acid (TFAA). Additional catalytic data for the reactions catalysed by the soluble acids indicated in Fig. 5a (bisphenol yield, the yield of monophenolic (MP) and diphenolic (DP) side-products and the carbon mass balance (CB) after 72 h of reaction) can be found in Table S2 of the ESI.†
3.3. Mechanistic proposal for the bisphenol synthesis
The preferential para-condensation of phenol with 2,3-PD in the presence of strong acids is suggested to be due to a more pronounced protonation of the carbonyl group, creating a distinct cationic (protonated ketone) and anionic species (acid anion). An equilibrium exists between the non-ionic 2,3-PD:HA and the ionic 2,3-PDH+:A− pair, with HA and A− respectively the acid and acid anion (see Scheme 2); a stronger acid will shift this equilibrium to the ionic pair. An analogue dependence of the acid strength on the protonation of a carbonyl function was recently reported for the acid-catalysed transesterification of ethyl acetate, where proton transfer from the acid to the carbonyl function occurred to a much lesser extent when changing from a strong acid (sulfuric acid) to a weak acid (trifluoroacetic acid (TFAA)). It was shown that the charge separation between the cation and acid anion was reduced when reducing the strength of the acid catalyst.53 An equilibrium reaction between a non-protonated and a protonated carbonyl group was also proposed by Sato et al. in the alkylation of phenol with propionaldehyde in supercritical water.54 The protonation of propionaldehyde, forming a 1-propanol cation, was enhanced by increasing the water density, i.e. by increasing the ionic character of the reaction medium. According to Sato et al., the 1-propanol cation reacts with both the ortho and para positions of phenol, while propionaldehyde reacts selectively with the ortho position due to an interaction of the propionaldehyde carbonyl group with the hydroxyl group of phenol. A shift of the equilibrium towards the cation therefore increases the para/ortho alkylation ratio.54 Sartori et al. noticed that in the AlCl3-catalysed alkylation of phenol with alkenes, reducing the protic acidity by adding potassium phenolate promoted the ortho-regioselective alkylation of phenol. They suggested that para-alkylation is characteristic for a charge-controlled process, in which a distinct cation and anion are formed, and that ortho-alkylation results from a H-bonding interaction between the phenol hydroxyl group and the alkene.55
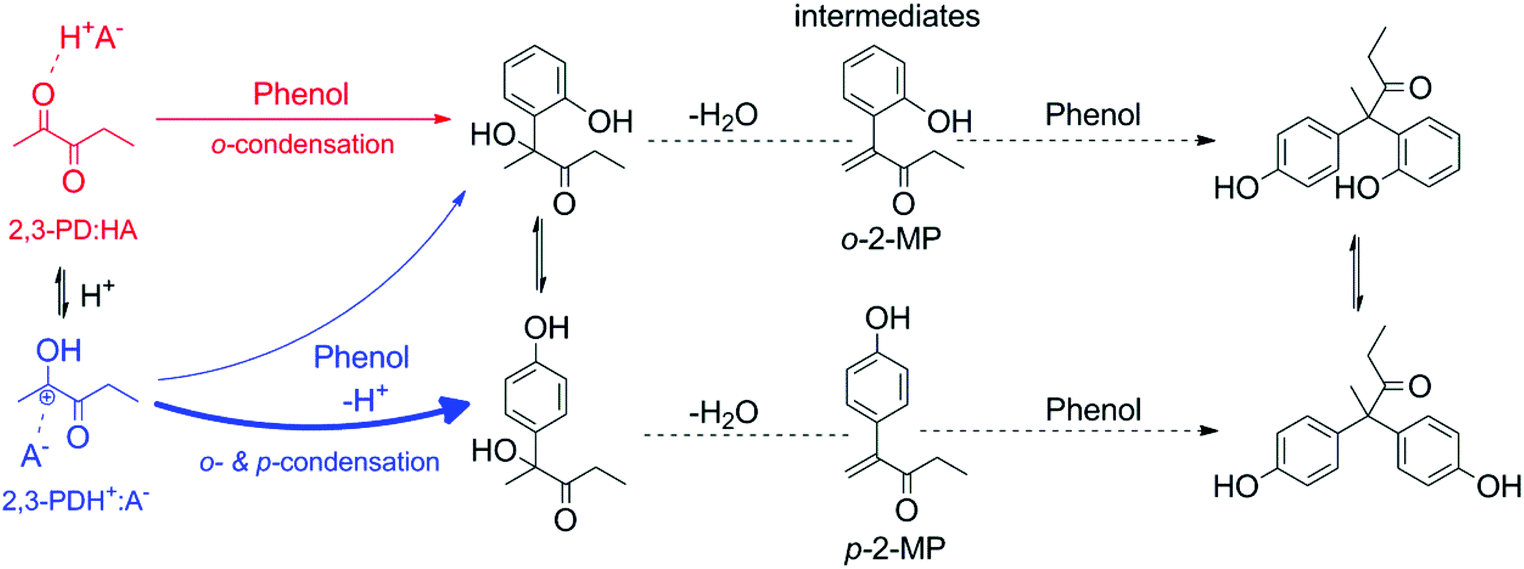 | ||
| Scheme 2 Proposed mechanism for the condensation of 2,3-PD and phenol to 2,2-p,p′-diphenol-3-pentanone and 2,2-o,p′-diphenol-3-pentanone. | ||
Based on these reports, it is now suggested that the ionic 2,3-PDH+:A− pair reacts preferentially with the para-position of phenol (indicated in blue in Scheme 2), while the nonionic 2,3-PD:HA pair selectively reacts with the ortho-position (indicated in red in Scheme 2) due to an interaction with the phenol hydroxyl group. Shifting the equilibrium towards the ionic pair by increasing the catalyst acid strength will thus increase the preference for para-condensation. This preference is only of importance in the first catalysed step since condensation with the second phenol addition is likely highly para-regioselective because of steric reasons.56
To verify the dependence of the reaction rate on the acid concentration for a strong and weaker acid, the influence of the amount of acid on the 2,3-PD conversion rate was determined for respectively TFSA and pTSA (Fig. 6). Both acid catalysts show a roughly linear relationship, indicating that the condensation reaction is first order in H+ concentration.
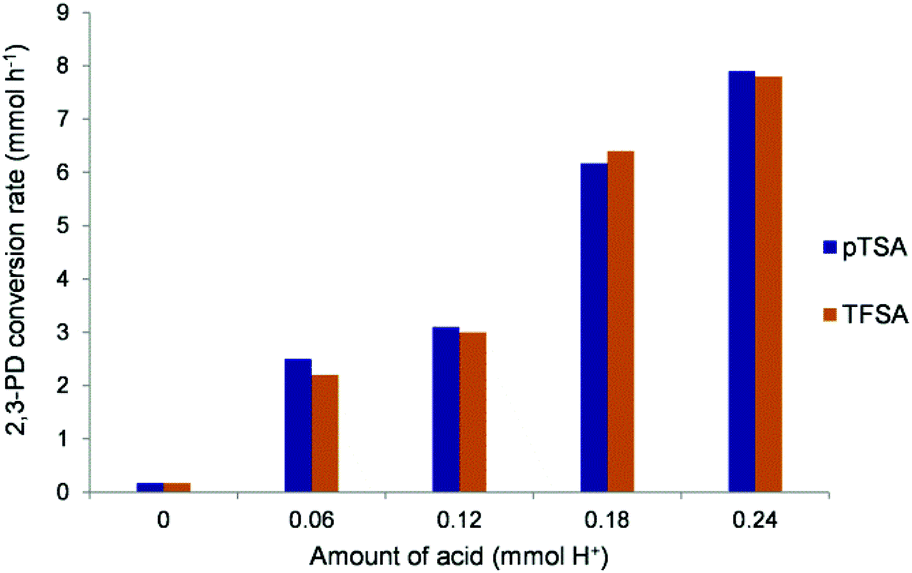 | ||
| Fig. 6 2.3-PD conversion rate determined at 30–40% 2.3-PD conversion for different amounts of catalytic H+ for pTSA and TFSA. Reaction conditions: 3.4 mmol 2,3-PD, 10.2 mmol phenol, 60 °C. | ||
Interestingly, the trend in Fig. 5a might give an indication of the acid strength of other acid catalysts under identical conditions, simply based on the experimental p,p′:o,p′ isomer ratio. For the acids in Table 1, sulfuric acid, SHPAOs, Dowex 50WX2 and Amberlyst 15 are expected to have a pKa (MeCN) value in the range of 7–10, while the value for Nafion NR50 is likely in the range of 3–4 (corresponding to a p,p′:o,p′ isomer ratio of 60 at 55% 2,3-PD conversion). Although Nafion NR50 features similar chemical properties to TFSA and NFBSA, its acid strength is considerably lower. Solid acids like Nafion NR50 are known to be weaker acids than their soluble analogues.57
3.4. Bisphenol properties and its use as a plasticizer
Finally, we have demonstrated the potential of these novel 2,3-PD based bisphenols as plasticizers for polyethylene terephthalate (PET). PET has a high glass transition temperature (Tg = 78 °C) and is therefore a good benchmark polymer for plasticizer screening. Catalytic reactions were performed on a larger scale with TFSA as a catalyst to produce several grams of the novel bisphenols. Highly pure 2,3-PD-based bisphenols (4 g, 99% purity, as ascertained by NMR spectroscopy) were obtained through purification of the reaction mixture by water extraction and column chromatography. First, the thermal stability of the bisphenols was determined by thermogravimetric analysis (TGA) (Fig. S8†). The TGA results indicate that the novel bisphenols possess a significantly higher thermal stability under N2 than BPA. For instance, the temperature for the maximum weight loss is 240 °C, while it is measured as 272 °C for the new bisphenol mixture. Additionally, the melting point of the bisphenols, with BPA as a reference, was determined via differential scanning calorimetry (DSC). The bisphenols have a significantly lower melting temperature than BPA (136 °C vs. 158 °C), allowing the processing of these compounds at a lower temperature. The combination of the lower melting temperature and the higher thermal stability under N2 creates a broader thermal processing window compared to BPA. For the plasticizer tests, PET pellets were ground into a powder and physically mixed with a solution of plasticizer in dichloromethane. This slurry was magnetically stirred and dried overnight at 35 °C. Different concentrations of the novel plasticizer were assessed (from 2 to 5 wt% in PET) and benchmarked against identically prepared BPA–PET mixtures and purely ground PET (Fig. 7). The Tg of the mixtures was determined by DSC.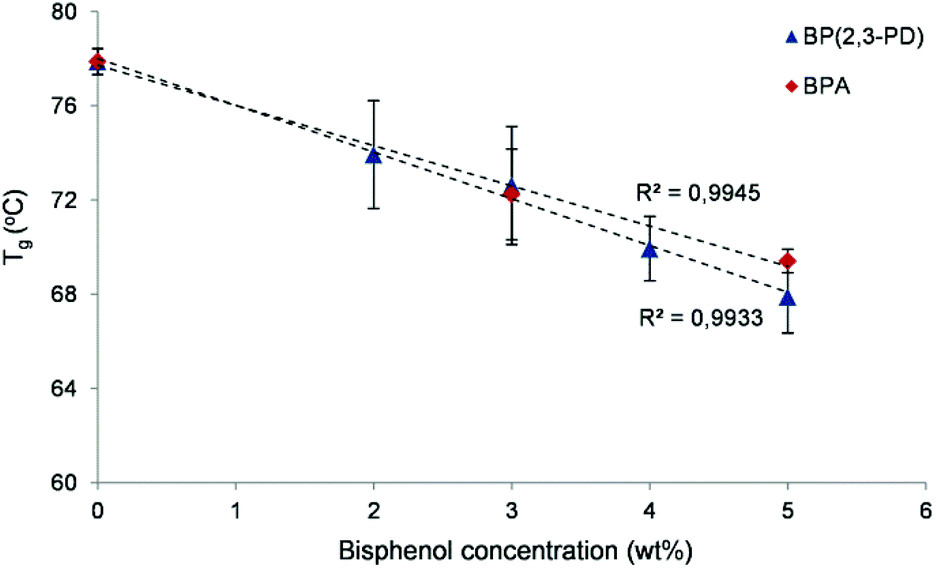 | ||
| Fig. 7 Glass transition temperature (Tg) of the bisphenol–PET mixtures as a function of the concentration of bisphenol (BP(2,3-PD) or BPA). Error bars indicate the standard deviation from four independent measurements. | ||
Fig. 7 illustrates a clear decrease of the Tg of PET for the 2,3-PD based bisphenolic plasticizer (BP(2,3-PD)), in the same order as that of the commonly used BPA reference. For instance, the Tg decreases by 10 °C by adding 5 wt% of the plasticizer. These preliminary experiments demonstrate the potential of these new bisphenols for plasticizing purposes. Further experiments are being carried out to examine the plasticizing effect on rheological and mechanical polymer properties. In addition to their use as polymer additives, the new bisphenols may have equally large potential as monomers for renewable polycarbonates, especially when one considers their low temperature synthesis, their high thermal stability and very high p,p:o,p ratios.
4. Conclusions
This contribution demonstrates the synthesis of renewable bisphenols from lignocellulosic feedstock, exemplified by the acid-catalysed condensation reaction of phenol with 2,3-pentanedione, obtained from lactic acid. Condensation reactions of this 1,2-dione with phenol run exceptionally fast without the need for co-catalysts, when compared to their monoketone analogues. The regioselectivity towards the desired p,p′-isomer is high, especially in the presence of strong acid catalysts. An ionic versus non-ionic mechanism, associated with the acid strength, is proposed to explain the high regioselectivity. Interestingly, the isomer ratios of the bisphenols are very high when compared to those obtained for monoketones, for which the addition of thiol co-catalysts is required.38–41 The new bisphenol isomers show promising plasticizer properties (in PET), comparable to that of bisphenol A, but with a broader temperature processing window. Follow-up studies will focus on the use of the novel bisphenols in the synthesis of polymers like polycarbonates, polyesters and polyether ether ketones with a thorough investigation of their properties.Acknowledgements
This work was performed in the framework of an IAP-PAI network from BELSPO (Federal Agency). W. S., S.-F. K., M. D. and S. V. d. V. acknowledge doctoral and postdoctoral fellowships from the Research Foundation – Flanders (FWO). F. Y. acknowledges a DBOF doctoral fellowship. Also the bilateral grant BIL08/09 is acknowledged for financial support. The authors are grateful to Karel Duerinckx for NMR measurements.References
- A. M. Nelson and T. E. Long, Polym. Int., 2012, 61, 1485–1491 CrossRef CAS.
- H. A. Meylemans, T. J. Groshens and B. G. Harvey, ChemSusChem, 2012, 5, 206–210 CrossRef CAS PubMed.
- P. Czub, in Handbook of Engineering and Specialty Thermoplastics, John Wiley & Sons, Inc., 2011, pp. 221–269 Search PubMed.
- H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, in Industrial Organic Chemicals, John Wiley & Sons, Inc., 2012, pp. 323–373 Search PubMed.
- B. S. Rubin, J. Steroid Biochem. Mol. Biol., 2011, 127, 27–34 CrossRef CAS PubMed.
- S. Van de Vyver, J. Geboers, S. Helsen, F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Chem. Commun., 2012, 48, 3497–3499 RSC.
- S. Van de Vyver, S. Helsen, J. Geboers, F. Yu, J. Thomas, M. Smet, W. Dehaen, Y. Roman-Leshkov, I. Hermans and B. F. Sels, ACS Catal., 2012, 2, 2700–2704 CrossRef CAS.
- Y. Guo, K. Li, X. Yu and J. H. Clark, Appl. Catal., B, 2008, 81, 182–191 CrossRef CAS PubMed.
- Y. Guo, K. Li and J. H. Clark, Green Chem., 2007, 9, 839–841 RSC.
- H.-F. Liu, F.-X. Zeng, L. Deng, B. Liao, H. Pang and Q.-X. Guo, Green Chem., 2013, 15, 81–84 RSC.
- X. Yu, Y. Guo, K. Li, X. Yang, L. Xu, Y. Guo and J. Hu, J. Mol. Catal. A: Chem., 2008, 290, 44–53 CrossRef CAS PubMed.
- H. A. Meylemans, B. G. Harvey, J. T. Reams, A. J. Guenthner, L. R. Cambrea, T. J. Groshens, L. C. Baldwin, M. D. Garrison and J. M. Mabry, Biomacromolecules, 2013, 14, 771–780 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS.
- F. de Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef CAS PubMed.
- L. Li, C. Stroobants, K. Lin, P. A. Jacobs, B. F. Sels and P. P. Pescarmona, Green Chem., 2011, 13, 1175–1181 RSC.
- P. P. Pescarmona, K. P. F. Janssen, C. Delaet, C. Stroobants, K. Houthoofd, A. Philippaerts, C. De Jonghe, J. S. Paul, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1083–1089 RSC.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- P. Y. Dapsens, C. Mondelli and J. Perez-Ramirez, ChemSusChem, 2013, 6, 831–839 CrossRef CAS PubMed.
- Q. Guo, F. T. Fan, E. A. Pidko, W. N. P. van der Graaff, Z. C. Feng, C. Li and E. J. M. Hensen, ChemSusChem, 2013, 6, 1352–1356 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G. Wang and H. Wan, Nat. Commun., 2013, 4 Search PubMed.
- F.-F. Wang, C.-L. Liu and W.-S. Dong, Green Chem., 2013, 15, 2091–2095 RSC.
- G. C. Gunter, D. J. Miller and J. E. Jackson, J. Catal., 1994, 148, 252–260 CrossRef CAS.
- G. C. Gunter, R. H. Langford, J. E. Jackson and D. J. Miller, Ind. Eng. Chem. Res., 1995, 34, 974–980 CrossRef CAS.
- D. C. Wadley, M. S. Tam, P. B. Kokitkar, J. E. Jackson and D. J. Miller, J. Catal., 1997, 165, 162–171 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable & Sustainable Energy Reviews, 2013, 21, 506–523 CAS.
- Q. Bu, H. Lei, S. Ren, L. Wang, Q. Zhang, J. Tang and R. Ruan, Bioresour. Technol., 2012, 108, 274–279 CrossRef CAS PubMed.
- Z. Ma, E. Troussard and J. A. van Bokhoven, Appl. Catal., A, 2012, 423, 130–136 CrossRef PubMed.
- G. E. Luckachan and C. K. S. Pillai, J. Polym. Environ., 2011, 19, 637–676 CrossRef CAS PubMed.
- M. Ike, M. Y. Chen, E. Danzl, K. Sei and M. Fujita, Water Sci. Technol., 2006, 53, 153–159 CrossRef CAS.
- R. W. Stahlhut, W. V. Welshons and S. H. Swan, Environ. Health Perspect., 2009, 117, 784–789 CAS.
- D. Zalko, C. Jacques, H. Duplan, S. Bruel and E. Perdu, Chemosphere, 2011, 82, 424–430 CrossRef CAS PubMed.
- H. Okada, T. Tokunaga, X. Liu, S. Takayanagi, A. Matsushima and Y. Shimohigashi, Environ. Health Perspect., 2008, 116, 32–38 CrossRef CAS PubMed.
- M. J. Cipullo, General Electric Company, EP0829464A2, 1998 Search PubMed.
- H. H. Szmant, Organic Building Blocks of the Chemical Industry, John Wiley & Sons, Inc., 1989 Search PubMed.
- E. J. Pressman, B. F. Johnson and S. J. Shafer, in Advances in Polycarbonates, American Chemical Society, 2005, ch. 3, vol. 898, pp. 22–38 Search PubMed.
- R. K. Zeidan, V. Dufaud and M. E. Davis, J. Catal., 2006, 239, 299–306 CrossRef CAS PubMed.
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403–9413 CrossRef CAS PubMed.
- E. L. Margelefsky, R. K. Zeidan, V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2007, 129, 13691–13697 CrossRef CAS PubMed.
- E. L. Margelefsky, A. Bendjeriou, R. K. Zeidan, V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2008, 130, 13442–13449 CrossRef CAS PubMed.
- E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118–1126 RSC.
- M. A. Harmer and Q. Sun, Appl. Catal., A, 2001, 221, 45–62 CrossRef CAS.
- A. Chakrabarti and M. M. Sharma, React. Polym., 1993, 20, 1–45 CrossRef CAS.
- R. B. Wesley and B. C. Gates, J. Catal., 1974, 34, 288–293 CrossRef CAS.
- M. Smet, K. Fu, X. Zhang, E. H. Schacht and W. Dehaen, Macromol. Rapid Commun., 2005, 26, 1458–1463 CrossRef CAS.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 CAS.
- E. Raamat, K. Kaupmees, G. Ovsjannikov, A. Trummal, A. Kuett, J. Saame, I. Koppel, I. Kaljurand, L. Lipping, T. Rodima, V. Pihl, I. A. Koppel and I. Leito, J. Phys. Org. Chem., 2013, 26, 162–170 CrossRef CAS.
- A. Kutt, I. Leito, I. Kaljurand, L. Soovali, V. M. Vlasov, L. M. Yagupolskii and I. A. Koppel, J. Org. Chem., 2006, 71, 2829–2838 CrossRef PubMed.
- A. Kuett, T. Rodima, J. Saame, E. Raamat, V. Maeemets, I. Kaljurand, I. A. Koppel, R. Y. Garlyauskayte, Y. L. Yagupolskii, L. M. Yagupolskii, E. Bernhardt, H. Willner and I. Leito, J. Org. Chem., 2011, 76, 391–395 CrossRef PubMed.
- F. Eckert, I. Leito, I. Kaljurand, A. Kuett, A. Klamt and M. Diedenhofen, J. Comput. Chem., 2009, 30, 799–810 CrossRef CAS PubMed.
- S. Saito, S. I. Saito, T. Ohwada and K. Shudo, Chem. Pharm. Bull., 1991, 39, 2718–2720 CrossRef CAS.
- L. Reyes, I. Nicolas-Vazquez, N. Mora-Diez and J. Raul Alvarez-Idaboy, J. Org. Chem., 2013, 78, 2327–2335 CrossRef CAS PubMed.
- T. Sato, G. Sekiguchi, T. Adschiri, R. L. Smith and K. Arai, Green Chem., 2002, 4, 449–451 RSC.
- G. Sartori, F. Bigi, R. Maggi and A. Arienti, J. Chem. Soc., Perkin Trans. 1, 1997, 257–260 RSC.
- J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Inc., New York, 1992 Search PubMed.
- D. Farcasiu, A. Ghenciu, G. Marino and K. D. Rose, J. Am. Chem. Soc., 1997, 119, 11826–11831 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and analytical information, gas chromatograms, mass spectra, 1H, 13C, DEPT, COSY and HSQC NMR spectra, IR spectra and thermograms of BP(2,3-PD) and BPA, the XRD pattern of BP(2,3-PD), DSC analysis and additional catalytic data. See DOI: 10.1039/c4gc00250d |
| This journal is © The Royal Society of Chemistry 2014 |