 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow asymmetric cyclopropanation reactions using Cu(I) complexes of Pc-L* ligands supported on silica as catalysts with carbon dioxide as a carrier†
Brunilde
Castano
a,
Emma
Gallo
a,
David J.
Cole-Hamilton
*b,
Vladimiro
Dal Santo
c,
Rinaldo
Psaro
c and
Alessandro
Caselli
*a
aDipartimento di Chimica, Università di Milano, and CNR – Istituto di Scienze e Tecnologie Molecolari, Via Golgi 19, 20133 Milano, Italy. E-mail: alessandro.caselli@unimi.it; Fax: (+39)02 5031 4405; Tel: (+39)02 5031 4372
bEaStCHEM, School of Chemistry, University of St. Andrews, St. Andrews, Fife, KY16 9ST, Scotland, UK. E-mail: djc@st-andrews.ac.uk
cCNR – Istituto di Scienze e Tecnologie Molecolari, Via C. Golgi 19, 20133 Italy
First published on 9th April 2014
Abstract
Continuous flow heterogeneous asymmetric cyclopropanations catalysed by supported hydrogen-bonded (SHB) chiral copper(I) complexes of pyridine containing tetraazamacrocyclic ligands Pc-L* using CO2 as a transport vector are described. The catalytic system showed high stability and good recyclability without loss of activity for at least 24 h in CO2 and catalyst turnover numbers up to 440 were obtained with excellent conversion (up to 99%) and high selectivity (up to 88%). No leaching of copper was observed. Cyclopropane products from both aromatic and aliphatic olefins were obtained in good yields with enantiomeric excesses up to 72%.
Introduction
Homogeneous catalysis presents some inherent problems, compared to its heterogeneous counterpart. Recyclability and separation of the products from the catalyst and any solvent can be difficult, time-consuming and energy intensive; on the other hand, homogeneous catalysts normally show higher chemo- and stereo-selectivities. Extensive effort has been made to develop new methods which combine the ease of catalyst recovery of the heterogeneous systems with the higher performances in terms of activity and selectivity obtained with homogeneous catalysts.1–5A few years ago, our group reported the synthesis and characterisation of copper(I) complexes of pyridine containing tetraazamacrocyclic ligands Pc-L*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and results on their use as catalysts in asymmetric cyclopropanation reactions in the homogeneous phase.7,8 Recently, in order to improve our catalytic system, we have developed new supported hydrogen-bonded (SHB) chiral Pc-L* copper(I) complexes on different ordered and non-ordered silicas. SHB is a method that involves only hydrogen-bonding interactions between the sulfonate group of the triflate counter-anion of the parent homogeneous complex and the free silanol groups of the silica support.9,10 In this way, all the metal centres can be regarded as single-site catalysts dispersed on a very high surface area. They are different from covalent grafting methods which impose structural modification to support the catalyst and that normally reduce the overall stereoselectivities.11 Silica “SHB” chiral Pc-L* copper(I) complexes showed good performances in cyclopropanation reactions, under heterogeneous batch conditions.12 The heterogenised systems showed higher or comparable activities than the homogeneous counterpart8 and good recyclability. Working under batch conditions, we found out that the stereoselective outcome of the reaction was dependent more on the employed solvent (non-polar vs. halogenated) than on the kind of support (ordered or non-ordered).12
6 and results on their use as catalysts in asymmetric cyclopropanation reactions in the homogeneous phase.7,8 Recently, in order to improve our catalytic system, we have developed new supported hydrogen-bonded (SHB) chiral Pc-L* copper(I) complexes on different ordered and non-ordered silicas. SHB is a method that involves only hydrogen-bonding interactions between the sulfonate group of the triflate counter-anion of the parent homogeneous complex and the free silanol groups of the silica support.9,10 In this way, all the metal centres can be regarded as single-site catalysts dispersed on a very high surface area. They are different from covalent grafting methods which impose structural modification to support the catalyst and that normally reduce the overall stereoselectivities.11 Silica “SHB” chiral Pc-L* copper(I) complexes showed good performances in cyclopropanation reactions, under heterogeneous batch conditions.12 The heterogenised systems showed higher or comparable activities than the homogeneous counterpart8 and good recyclability. Working under batch conditions, we found out that the stereoselective outcome of the reaction was dependent more on the employed solvent (non-polar vs. halogenated) than on the kind of support (ordered or non-ordered).12
Immobilised catalysts in general are best used under flow conditions where the reagents and products continuously pass through the catalytic bed. The need for more eco-sustainable and green systems prompted the development of special rigs to conduct catalytic reactions using carbon dioxide as a carrier. In fact CO2 is an attractive solvent as it is safe and an ideal substitute for many hazardous and toxic solvents.13–16 The pressurization of a liquid organic compound with high pressure CO2 will result in an expanded liquid phase of increased CO2 concentration.17 The CO2-dissolved expanded liquid phase has been recognised as an ecologically friendly medium. This kind of catalytic system has been used for a wide range of reactions such as cyclopropanation reactions,18 hydroformylation,2 and alkene methathesis.19 Here we report on the use of SHB copper(I) catalysts in asymmetric cyclopropanations under flow conditions using CO2 as a carrier.
Experimental
General
NMR spectra were recorded on Bruker Avance 300-DRX or Avance 400-DRX spectrometers. Chemical shifts (ppm) are reported relative to TMS. HPLC analyses were performed on a Hewlett-Packard 1050 instrument equipped with DAI-CEL CHIRALCEL IB, OJ and AD mineralised chiral columns. Infrared spectra were recorded on a BIO-RAD FTS-7 spectrophotometer. CO-DRIFT spectra of the samples were recorded using a FTS-60A spectrophotometer equipped with a homemade reaction chamber. After purging the apparatus with ultra-pure He, spectra of the samples were recorded at RT in He and CO flow, before and after catalysis. Elemental analyses and mass spectra were recorded in the analytical laboratories of Milan University. All starting materials, α-methylstyrene, ethyldiazoacetate (EDA), 2,4-dinitrotoluene, styrene, 4-chlorostyrene, 1,1-diphenylethylene, methyl-2-furoate, 1-octene and 1,2-dichloroethane, were purchased from Aldrich and were used without further treatment. Davisil LC150 (Grace Davison) and Aerosil 380 (Evonik) are commercially available. MCM-4120,21 was synthesized as has already been reported.22 CO2 (99.9995%) was purchased from BOC gases. Unless otherwise specified, all the reactions were carried out under air atmosphere. The synthesis and characterisation of copper(I)(Pc-L*) complexes 1 (ref. 23) and 2 (ref. 24) were done as previously reported.7,8 The water and air sensitive catalysts 1/D, 1/A, 1/M and 2/D (Scheme 1) were synthesized as already reported12 and they were handled in a dry-box, model “Labstar 502”, MBraun, Germany (see Table 1 for details).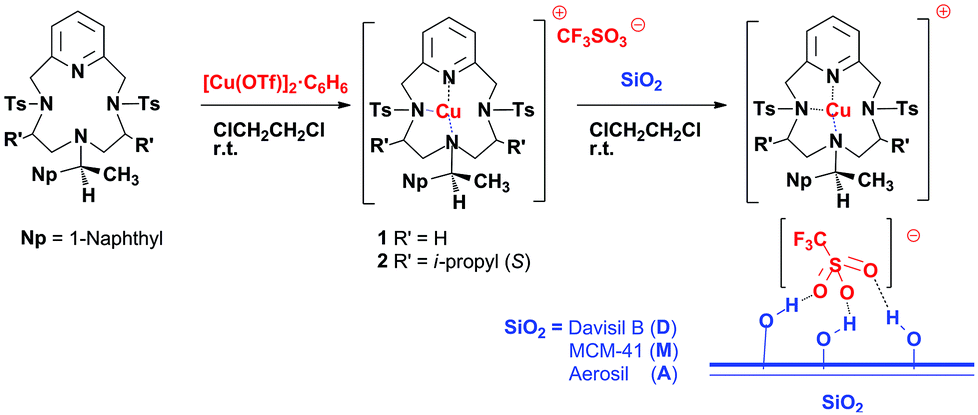 | ||
| Scheme 1 General synthesis of the supported catalysts 1/D-M and 2/D. | ||
| Entry | SiO2 support | Cu loading (wt%) |
|---|---|---|
| a General procedure: metal loadings are determined by ICP-OES using a Thermo iCAP 6300 apparatus. 15 mg of each sample are mineralised by adding 2 mL of concentrated HNO3 63%, 2 mL of H2SO4 98%, and 0.4 g of Se/K2SO4 digesting aid. | ||
| 1/D | Davisil LC150 | 0.84 |
| 1/A | Aerosil 380 | 0.81 |
| 1/M | MCM-41 | 0.85 |
| 2/D | Davisil LC150 | 0.66 |
Cyclopropanation under flow conditions
The supported catalysts were tested in the cyclopropanation reactions. The catalytic reactions were carried out on a specially designed rig, the design of which has previously been reported25,26 using a stainless steel tubular reactor with a volume of 8.8 cm3. The supported catalyst was loaded into a metal tubular reactor in a glove box. The bed was fixed with stoppers of dried glass wool used to prevent it from being flushed from the reactor. Since the used silica has different apparent densities, the eventual void volume of the reactor has been filled by adding glass wool. This tube was then held vertically in an aluminium heating block. All reactions were carried out with the tubular reactor held vertical and with the flow being from bottom to top. Substrate ratios and flow rates are described in table captions. Samples were collected and analysed every 30 min. Some random samples have been analysed in order to determine the amount of copper leaching during the catalysis. For metal analysis, the organic products collected after the cyclopropanation reaction were mineralised and analysed by ICP-OES. All reported yields and diastereoselectivities have been determined by quantitative 1H NMR spectroscopy and are based on EDA, using 2,4-dinitrotoluene as an internal standard. Diethyl-fumarate and maleate, derived from EDA coupling, were the only side products detected and they accounted for the rest of the mass balance of the reaction.General procedure for the catalytic cyclopropanation reactions
Contact times (hereinafter τ), measured in minutes, were calculated as the ratio between the copper(I) amount (measured in mmol) loaded in the reactor and the EDA (the limiting reagent) flow (measured in mmol min−1).
Results and discussion
At first, we optimised the conditions for the cyclopropanation reaction employing DCE as the carrier. As a benchmark reaction we chose the cyclopropanation of α-methylstyrene, an alkene which is known to give cyclopropanes with low diastereoselectivity27 and which undergoes polymerization less easily with respect to styrene, with ethyldiazoacetate (EDA). The collected data for the cyclopropanation in DCE employing catalysts 1/D are summarised in Table 2, together with typical results for the same reaction conducted in the homogeneous8 and heterogeneous phases under batch conditions.12 The average (conversion, selectivity, cis/trans ratios and ees) values of all collected samples are herein reported; conversion chemo- and diastereo-selectivities in cyclopropanated products were determined by quantitative 1H NMR in CDCl3 based on EDA using 2,4-dinitrotoluene as an internal standard after evaporation of the collected samples. For data for each sample refer to Tables S1–S4 in the ESI.†|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Run | τ (min) | Timeb (min) | Selectivityc (%) | Conversionc (%) |
cis:transc |
eed (%) | |
| cis (1R,2S) | trans (1R,2R) | |||||||
|
a Reactions were performed with [Cu(I)] (5.4 × 10−2 mmol), EDA–α-methylstyrene ratio = 1:5. [EDA] = 0.15 mol L−1 in DCE at room temperature. Reactions were monitored every 30 min and average values for selectivity, conversion, cis/trans ratio and ees are reported.
b Total time of the single flow run.
c Conversion and selectivities determined by 1H NMR in CDCl3 based on EDA using 2,4-dinitrotoluene as an internal standard. Fumarate and maleate accounted for the rest of the mass balance.
d Determined by chiral HPLC equipped with DAICEL CHIRALPAK IB (n-hexane–i-PrOH = 99.25:0.75).
e
Ref. 8.
f
Ref. 12.
g In this run we observed a progressive deactivation of the catalytic system, and instead of the average conversion value, the range is reported. The selectivities and ees reported instead are those averaged.
h The catalyst became completely inactive at the third run. Only traces of cis cyclopropane were detected in the first collected sample.
i We observed a progressive deactivation of the catalytic system, and instead of the average conversion value, the range is reported. The selectivities and ees reported instead are averages.
|
||||||||
| 1e | — | — | 100 | 81 | >99 | 62:38 |
55 | 62 |
| 2f | — | — | 100 | 72 | >99 | 72:28 |
35 | 26 |
| 3 | 1 | 1.8 | 381 | 66.2 | >99 | 56:44 |
34 | 15 |
| 2g | 399 | 64.6 | 39–4 | 46:54 |
25 | 14 | ||
| 3h | 385 | n.d. | <5 | — | — | — | ||
| 4 | 1 | 3.7 | 668 | 56.9 | >99 | 61:39 |
25 | 26 |
| 5 | 1 | 0.7 | 346 | 60.5 | >99 | 63:37 |
58 | 29 |
| 2i | 121 | 55.6 | 99–11 | 54:46 |
38 | 30 | ||
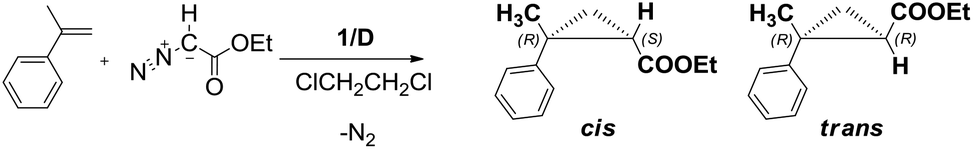
The ratio between EDA and α-methylstyrene was at first maintained at 1:5 with [EDA] = 0.15 M (global volume of added reagents in DCE). In fact a fivefold excess of the alkene has to be used to minimise the side products derived from EDA self-condensation, fumarate and maleate. Interestingly, we always observed complete conversion of the starting EDA in the first reaction runs, with a good chemoselectivity in cyclopropane products. A 0.2 mL min−1 flow (τ = 1.8 min) was first tested and complete conversion with good chemoselectivity (66%) was obtained with the selectivity remaining constant during all the time needed for the reagent solution to be eluted from the reactor (381 min, entry 3, run 1, Table 2). The EDA conversion only decreased slightly (91%) in the last collected sample (see Table S1a†).
The catalyst was then recycled for a second run by flowing a fresh mixture of α-methylstyrene and EDA in DCE (the same amounts as those used for the first run). In this case we noticed that the catalyst had lost its initial activity (39% conversion of the added EDA). Progressively the conversion decreased to <4% at the end of the reaction (overall 780 minutes, entry 3, run 2, Table 2; see Table S1b† for details). In order to better understand the nature of the deactivation process (i.e. poisoning of the catalyst by the cyclopropane products), after a DCE washing, new fresh reagents were introduced and a third run was performed (entry 3, run 3, Table 2). No conversion of EDA was observed meaning that a complete deactivation of the catalyst occurred after prolonged reaction times. ICP-OES analysis of the silica post-catalysis showed a copper content lower than that in the starting material (determined copper loading = 0.60%), a fact that can explain the progressive decrease in catalytic activity. Moreover, the silica material after catalysis has some adsorbed organic compounds (see below) that can also explain the very low copper content found. A big difference from our previous work in the homogeneous phase and under heterogeneous batch conditions was that we did not run the catalytic reaction under a protective atmosphere and we employed commercially available solvents and reagents without any further distillation.
In fact, we have already observed that yields and selectivities were not severely affected by the use of non-distilled solvents.7
Different flow rates (0.1 and 0.5 mL min−1, τ = 3.7 and 0.7 min, respectively) were subsequently tested. The best results in terms of diastereo- and enantio-selection were achieved with 0.5 mL min−1 flow (entry 5, run 1, Table 2), meaning that under those flow conditions the catalytic system worked even better than in batch. Unfortunately, during the second run again a decrease of chemoselectivity was observed together with a decrease in activity (entry 5, run 2, Table 2). Notably in these catalytic systems starting materials were mixed together and the reactor was fed straight with the mixture and reactions were performed at R.T. Conversely, in the homogeneous phase EDA was slowly added by a syringe pump over 100 minutes at 0 °C in order to avoid the formation of the homocoupled products. Despite the higher temperature, however, the ees obtained were close to or even better than those recorded under batch conditions. The leaching of the catalyst employing DCE as a vector, however, is still a problem to be faced. Random samples have been analysed in order to determine the amount of copper leached during the catalysis (see ESI†). In DCE, a copper content up to 3788 ppb (1.9% of total copper) was found.
Our intention was to shift to a more sustainable and greener system using carbon dioxide in place of DCE as the flowing solvent. CO2 was chosen because it can reach the supercritical state under moderate conditions of pressure and temperature, avoiding the degradation of thermolabile and volatile substances and providing simultaneously an inert medium suitable for use as a vector.16 The critical point of pure CO2 is 73.8 bar and 31.1 °C. However, with the substrates and flow rates involved it is likely that the system should be considered as operating in an expanded liquid phase. Previous studies have shown that such expanded liquids can give superior performance under flow conditions.26 Maintaining the ratio 1:5 with EDA–α-methylstyrene, complex 1, grafted on the three different types of silica, was tested under flow conditions with carbon dioxide (Table 3). Reactions in CO2 were carried out at 40 °C (while the best ees in the homogeneous phase reactions were obtained at 0 °C).
| Entry | Run | Catalyst | Timeb (min) | Selectivityc (%) | Conversionc (%) |
cis:transc |
eed (%) | |
|---|---|---|---|---|---|---|---|---|
| cis (1R,2S) | trans (1R,2R) | |||||||
|
a Reactions were performed with [Cu(I)] (5.2 × 10−2 mmol), EDA–α-methylstyrene ratio = 1:5, T = 40 °C, PCO2 = 130 bar, flow CO2 = 0.5 mL min−1, flow HPLC = 0.02 mL min−1, τ = 2.0 min. Reactions were monitored every 30 min and average values for selectivity, conversion, cis/trans ratio and ees are reported.
b
,
c
,
d Same as in Table 2.
e Reaction was performed with EDA–α-methylstyrene 1:2.
f Reaction was performed with EDA–α-methylstyrene 1:10.
|
||||||||
| 1 | 1 | 1/D | 468 | 65.8 | >99 | 67:33 |
36 | 34 |
| 2 | 159 | 65.5 | >99 | 68:32 |
44 | 26 | ||
| 2e | 1 | 1/D | 543 | 46.3 | >99 | 62:38 |
38 | 25 |
| 3f | 1 | 1/D | 603 | 72.7 | >99 | 72:28 |
36 | 27 |
| 4 | 1 | 1/M | 470 | 87.9 | >99 | 59:41 |
40 | 29 |
| 5 | 1 | 1/A | 472 | 72.2 | >99 | 57:43 |
33 | 21 |
Initially, the catalyst 1/D, used for screening catalysis in DCE flow, was also tested under CO2. The CO2 pressure was adjusted to 130 bar, with a flow rate of 0.5 mL min−1. The reagents were fed at 0.02 mL min−1via an HPLC pump. Again, complete conversion of EDA was observed and, in this case, the reaction products were just collected and weighed every half hour and analysed by quantitative 1H NMR after the addition of 2,4-dinitrotoluene as an internal standard. The chemoselectivity of the reaction towards cyclopropanes was similar to that observed in DCE, but interestingly, in this case the cis/trans ratio was closer to the value observed under optimised batch conditions employing n-hexane as a solvent (compare entry 1, Table 3, with entry 2, Table 2). Moreover, in this case no deactivation of the catalyst was observed and quantitative conversion of the EDA was also obtained in the second run.
We next monitored the effect of different ratios between EDA–α-methylstyrene on the reaction outcome. A decrease of the fivefold excess of alkene to a ratio 1:2 of EDA–α-methylstyrene led, as expected, to a lower chemoselectivity, while the ee values for both isomers were almost unaffected (entry 3, Table 3). When a 10-fold excess of α-methylstyrene was used, a better chemoselectivity and a higher diastereoselectivity in favour of the cis isomer were observed (entry 3, Table 3). On the other hand, the enantioselectivity was unaffected by a tenfold excess of alkene. A five-fold excess of the alkene seems to be the better choice to maximise the reaction yield. At the end of the reaction, the unreacted alkene can be easily recovered from the cyclopropane products and eventually recycled in the next run.
In the last two entries (entries 4 and 5, Table 3) complex 1 grafted on MCM-41 (1/M) and on Aerosil (1/A), respectively, was used as the catalyst, in order to test a possible confinement effect provided by the support. Under batch conditions, experimental data showed only a very weak dependence of the reaction efficiency upon the surface area characteristics of the silica and almost indistinguishable diastereo- and enantio-selectivities using commercial Davisil, Aerosil, or ordered mesoporous MCM-41 silicas were obtained.12 Under CO2 flow, we observed a slight decrease in the cis selectivity with both MCM-41 and Aerosil as a support, but a better yield of cyclopropanes was obtained, especially with 1/M as the catalyst. Again, random samples have been analysed in order to determine the amount of copper leached into the flowing medium. A maximum of 10.8 ppb and 4.3 ppb for compounds 1/M and 1/D, respectively, of copper were found by ICP-OES analysis of the reaction products, meaning that under those conditions the copper complex is stable and strongly bound to the silica.
Finally, since the chiral Cu(I) complex 2 gave the best results in terms of enantioselectivity in the homogeneous phase,8 and subsequently its reactivity was studied when supported on Davisil under batch conditions,12 we tested the catalytic performances of 2/D (Scheme 1) in asymmetric cyclopropanation reactions under CO2.
The less expensive commercial Davisil silica was chosen as the support. Davisil was also used as our benchmark silica in heterogeneous phase batch reactions,12 since the possibility of using commercially available silica as a support in the system was very interesting and could pave the way for the employment of this immobilization technique in laboratories not equipped for the synthesis of mesoporous materials. For the sake of clarity, the results obtained using CO2 under optimised conditions employing the catalyst 2/D are reported in Table 4 and are compared to those obtained in the homogeneous phase8 (DCE as a solvent, entry 2, Table 4) and in heterogenised batch reactions12 (n-hexane as a solvent, entry 3, Table 4) with the same copper(I) complex 2. Complex 2 has the opposite configuration at the α-naphthylic stereogenic carbon (see Experimental, ref. 23 and 24), thus affording the opposite enantiomeric excesses in the cyclopropanation reaction.8
| Entry | Run | Timeb (min) | Selectivityc (%) |
cis:transc |
eed (%) | |
|---|---|---|---|---|---|---|
| cis (1S,2R) | trans (1S,2S) | |||||
|
a In all cases a complete conversion of the starting EDA was observed.
b
,
c
,
d Same as in Table 2.
e Reactions were performed with [Cu(I)] (4.0 × 10−2 mmol), EDA–α-methylstyrene ratio = 1:5, T = 40 °C, PCO2 = 130 bar, flow CO2 = 0.5 mL min−1, flow HPLC 0.02 mL min−1, τ = 1.5 min. Reactions were monitored every 30 min and average values for selectivity, conversion, cis/trans ratio and ees are reported. For the second run, fresh reagents were added and the reaction was left overnight. The reaction products were analysed after the end of the single run.
f
Ref. 8.
g
Ref. 12.
|
||||||
| 1e | 1 | 518 | 68.6 | 57:43 |
58 | 68 |
| 2 | 830 | 74.0 | 50:50 |
59 | 70 | |
| 2f | — | 100 | 98 | 50:50 |
88 | 99 |
| 3g | 1 | 100 | 63 | 68:32 |
60 | 59 |
| 2 | 100 | 71 | 65:35 |
56 | 65 | |
| 3 | 100 | 65 | 63:37 |
60 | 67 | |
Under CO2 with the catalyst 2/D, again complete conversion of EDA was obtained (entry 1, Table 4). After the first run, a second consecutive reaction was performed, by charging the reactor with fresh α-methylstyrene and EDA in the same concentrations and relative ratios, and the reactor was fed overnight (entry 1, run 2, Table 4). For that reason, in this case, products were collected only at the end of the run. No deactivation of the catalyst was observed, but instead the chemoselectivity of the catalytic system seemed to improve with time (see later). Again, we did not observe any metal leaching and a maximum of only 3.2 ppb of copper was found in the collected samples. The catalyst was stable over at least 23 hours, as illustrated in Fig. 1, with constant quantitative conversion of added EDA. Again ees under CO2 were similar to those obtained in heterogeneous phase batch reactions, despite the fact that higher temperatures are normally detrimental for enantioselectivity.28 In CO2 the reactions were carried out at 40 °C while homogeneous phase and heterogeneous phase batch reactions were run at 0 °C. The turnover number was improved up to 440 with respect to homogeneous phase (30) and heterogeneous phase batch (90) reactions. In the last two catalytic systems the TON was limited by the slow addition of the EDA solution to the alkene and the high concentration of the catalyst needed for the reaction to proceed to complete conversion.
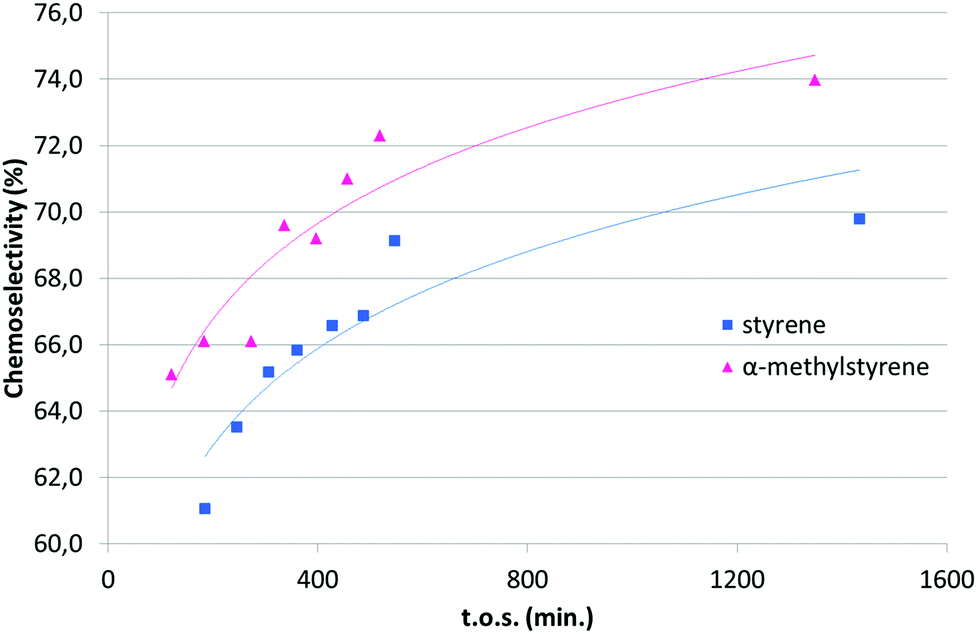 | ||
| Fig. 1 Chemoselectivity (%) vs. t.o.s. (minutes) of styrene and α-methylstyrene catalysed by 2/D. | ||
To explore the synthetic scope, different alkenes were tested under the optimised conditions using the supported catalyst 2/D (Table 5).
a
| Entry | Run | Alkene | Timeb (min) | Selectivityc (%) | Conversionc (%) |
cis:transc |
eed (%) | |
|---|---|---|---|---|---|---|---|---|
| cis | trans | |||||||
|
a Reactions were performed with [Cu(I)] (4.2 × 10−2 mmol), under otherwise identical conditions as those reported in Table 3.
b
,
c Same as in Table 2.
d Determined by chiral HPLC; absolute configurations: for entries 1 and 2 cis-cyclopropanes were (1S,2R), trans-cyclopropanes were (1S,2S); for entry 3 the absolute configuration was (1S). For entry 4 trans-cyclopropane was (1R,5R,6R); for entry 5 the absolute configurations were not determined (see ESI for details).
e Reaction was performed overnight and checked only at the end.
f Reaction was performed with EDA–alkene 1:10.
|
||||||||
| 1 | 1 |
|
547 | 65.5 | >99 | 32:68 |
62 | 55 |
| 2e | 886 | 69.8 | >99 | 31:69 |
56 | 55 | ||
| 2 | 1 |
|
582 | 77.7 | >99 | 48:52 |
43 | 72 |
| 3 | 1 |
|
565 | 51.7 | >99 | — | 65 | |
| 4 | 1 |
|
542 | 33.1 | >99 | <1:>99 |
— | 67 |
| 5f | 1 | 1-Octene | 477 | 71.2 | >99 | 48:52 |
68 | 40 |
At an EDA–alkene ratio of 1:5 with [Cu(I)] (4.0 × 10−2 mmol), 2/D catalysed the reaction of all the tested substrates and in all cases complete conversion of EDA was observed. Reported conversion and selectivities have been determined by quantitative 1H NMR and are based on EDA. Fumarate and maleate were again the only detected side products. Catalysis was at first performed with styrene in order to study the influence of the absence of α-substituents on the styrenic double bond (entry 1, Table 5). As expected, the diastereoselectivity of the reaction was strongly affected, in favour of the less hindered trans isomer. A similar effect was observed also in the homogeneous phase.8 Again the second run for that alkene was performed overnight (25 hours of continuous reaction).
Reporting the chemoselectivity (%) vs. time of stream (t.o.s.) obtained with α-methylstyrene (Table 4, entry 1) and styrene (Table 5, entry 1), we observed that in both cases selectivity increased during the catalysis and it reached a maximum after about 550 minutes (Fig. 1). No catalyst deactivation was observed in both cases, indicating that the catalyst could perform catalytic reactions for prolonged times. The observed increase of the chemoselectivity upon prolonged t.o.s. is difficult to rationalise at the present stage, since we could not perform any kinetic study under the employed reaction conditions. In fact, reactants are fed with the same concentration and at the same rate through the catalytic run. A blank experiment performed charging the reactor with activated Davisil without any copper grafted showed that the silica has some activity in the EDA conversion (40% under the typical reaction conditions) to yield fumarate and maleate as major products. Moreover, the infrared spectra of 1/D at the end of the catalytic runs (vide infra) confirm the presence of adsorbed organic products. These observations suggest the progressive poisoning of active sites (acidic silanols) on the silica surface due to the deposition of by-products derived from diazoacetate polymerization (see the “Post-catalysis characterization” section).29 This results in a decrease of parasitic reactions thus leading to the emergence of the desired chemoselective reactions on copper(I) single sites. Notably, this behaviour of silica, acting like a non-innocent support, was already reported for the hydrogenation of indoles with SHB Ru supported catalysts.30
The best yields of cyclopropanes were obtained in the case of 4-chlorostyrene (entry 2, Table 5), a result that was observed also in the homogeneous phase. Also the diastereoselectivity and the enantioselectivity observed for the trans isomer were very close to those obtained employing complex 2 in DCE. Even in the presence of a bulky substrate such as 1,1-diphenylethylene (entry 3, Table 5) a conversion up to 99% was achieved, with a selectivity higher than those observed in the homogeneous reactions.8 The reaction was not limited to aromatic alkenes. Good results were achieved with methyl-2-furoate with excellent diastereoselectivity where only the desired trans isomer (attack at the non-substituted double bond) was obtained (entry 4, Table 5). This result is of particular interest, since the obtained cyclopropane is an important building block in the synthesis of bioactive compounds such as rocellaric acid.31
Also aliphatic alkenes gave very good results. For instance, the cyclopropanation occurred even with the non-activated double bond of 1-octene with good enantioselectivity, although in this case the ratio of alkene–EDA was increased to 10:1 (entry 5, Table 5). A very good chemoselectivity (71%) was achieved and this result is comparable with that for the homogeneous counterpart (73%).8 Moreover, enantioselectivities (ee up to 72% for the cis isomer and up to 42% for the trans isomer, see Table S4e in ESI†) very close to those obtained in the homogeneous case (75% and 55%) were observed.
Reporting the chemoselectivity (%) vs. t.o.s. for all the tested alkenes, selectivity generally increased during the catalytic run (Fig. S1†). Only in the case of methyl-2-furoate as a substrate was a progressive decrease of this value over time observed.
In order to check the effect of different substrates on copper leaching employing CO2, random samples of all the performed catalytic tests were also analysed by ICP-OES (for the complete series of collected data see ESI†). Again, negligible copper leaching was observed in all cases. Slightly higher contents were found in the case of methyl-2-furoate, where concentrations up to 122.5 ppb of copper were obtained. This last observation demonstrates that, effectively, the presence of an oxygen donor atom in the substrate might be detrimental to the catalyst stability.
Post-catalysis characterisation
Consistent with our previous report12 on Pc-L* (pyridine containing macrocyclic ligands) copper(I) complexes, DRIFT spectra showed that the Cu complexes are grafted without any modification of the ligand structure, since the IR absorption bands, detected for 1/D, A, M and 2/D, did not show any appreciable modification with respect to solid 1 or 2, either in location or in intensity (see ESI, Fig. S2,† for a comparison of DRIFT spectra of complex 2 and the supported catalyst 2/D). Grafted complexes were investigated also after the catalytic runs. DRIFT spectroscopy, again, confirmed that the structures of Cu complexes are stable under the reaction conditions, showing the presence of all the bands in the skeletal range of the spectrum typical of 1 and 2 (see ESI, Fig. S4†).However, in most cases the occurrence of an intense absorption band, located around 1750 cm−1, in samples collected at the end of catalytic runs suggests the presence of adsorbed reaction intermediates. Experiments, conducted in dichloroethane solutions of the unsupported Cu complex, revealed the occurrence of an interaction between maleate and fumarate and the copper complex, which gave rise to a band at 1713 cm−1 (see ESI, Fig. S5 and S6†). Thus, products adsorbed on the samples after reaction seem to be of a different nature, and can be ascribed to diazoacetate polymers as already reported for bis(oxazoline) copper complexes.29
Conclusions
In summary, we have developed a new catalytic system based on supported hydrogen-bonded (SHB) chiral copper(I) complexes. These are competent catalysts for asymmetric cyclopropanation reactions allowing the use of more eco-sustainable CO2 as a vector instead of organic solvents normally used for these reactions. The heterogenised systems under flowing CO2 showed comparable or even higher chemoselectivities than the homogeneous counterpart. In terms of enantioselection excellent results were obtained also with non-activated alkenes like 1-octene where ees are comparable to those observed in the homogeneous phase. Interestingly, all the data in CO2 were obtained at 40 °C, while the best temperature for the cyclopropanation reaction in the homogeneous phase was 0 °C. Moreover, supported catalysts showed a good recyclability and the turnover number has been increased (up to 440). The catalysts remained active up to 25 h without any loss in activity and chemoselectivity improved upon prolonged reaction times. Catalysts were stable and robust with negligible copper leaching (only 0.007% of total copper) and can be easily recycled by just re-feeding the reactor with the reagents. The alkene excess can be easily separated from the reaction products and eventually re-used. Future studies will be devoted to improving the ligand design in order to enhance the stereo-selective outcome of the reaction. In principle, in fact, if we can obtain a single diastereoisomer while still maintaining high chemoselectivity under the present conditions, no further purification will be needed to yield a pure product of high added synthetic value containing a cyclopropane unit.Acknowledgements
One of the authors, B. C., is grateful to Erasmus Life-Learning Program for a fellowship of three months at the University of St. Andrews. V. D. S. and R. P. thank the Italian Ministry of Education, University and Research for financial support through the project “ItalNanoNet” (Rete Nazionale di Ricerca sulle Nanoscenze; prot. no. RBPR05JH2P).Notes and references
- V. Dal Santo, F. Liguori, C. Pirovano and M. Guidotti, Molecules, 2010, 15, 3829–3856 CrossRef CAS PubMed.
- P. B. Webb, M. F. Sellin, T. E. Kunene, S. Williamson, A. M. Z. Slawin and D. J. Cole-Hamilton, J. Am. Chem. Soc., 2003, 125, 15577–15588 CrossRef CAS PubMed.
- V. Dal Santo, M. Guidotti, R. Psaro, L. Marchese, F. Carniato and C. Bisio, Proc. R. Soc. London, Ser. A, 2012, 468, 1904–1926 CrossRef CAS.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- C. Aprile, H. Garcia and P. P. Pescarmona, in Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques, and Applications, ed. M. Gruttadauria and F. Giacalone, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 177–208 Search PubMed.
- B. Castano, T. Pedrazzini, M. Sisti, E. Gallo, F. Ragaini, N. Casati and A. Caselli, Appl. Organomet. Chem., 2011, 25, 824–829 CrossRef CAS.
- A. Caselli, F. Cesana, E. Gallo, N. Casati, P. Macchi, M. Sisti, G. Celentano and S. Cenini, Dalton Trans., 2008, 4202–4205 RSC.
- B. Castano, S. Guidone, E. Gallo, F. Ragaini, N. Casati, P. Macchi, M. Sisti and A. Caselli, Dalton Trans., 2013, 42, 2451–2462 RSC.
- C. Bianchini, V. Dal Santo, A. Meli, W. Oberhauser, R. Psaro and F. Vizza, Organometallics, 2000, 19, 2433–2444 CrossRef CAS.
- P. Barbaro, C. Bianchini, G. Giambastiani, W. Oberhauser, L. M. Bonzi, F. Rossi and V. Dal Santo, Dalton Trans., 2004, 1783–1784 RSC.
- D. Rechavi and M. Lemaire, Chem. Rev., 2002, 102, 3467–3494 CrossRef CAS PubMed.
- B. Castano, P. Zardi, Y. C. Honemann, A. Galarneau, E. Gallo, R. Psaro, A. Caselli and V. Dal Santo, RSC Adv., 2013, 3, 22199–22205 RSC.
- P. G. Jessop and W. Leitner, Chemical Synthesis Using Supercritical Fluids, Wiley-VCH Verlag GmbH, Weinheim, Germany, 1999 Search PubMed.
- W. Leitner, in Modern Solvents in Organic Synthesis, ed. P. Knochel, Springer, Berlin, Heidelberg, 1999, vol. 206, ch. 5, pp. 107–132 Search PubMed.
- D. J. Cole-Hamilton, Adv. Synth. Catal., 2006, 348, 1341–1351 CrossRef CAS.
- B. S. Sekhon, Int. J. Pharm. Technol. Res., 2010, 2, 810–826 CAS.
- R. Liu, H. Yoshida, S. Fujita, N. Lu, W. H. Tu and M. Arai, Appl. Catal., A, 2013, 455, 32–38 CrossRef CAS PubMed.
- M. I. Burguete, A. Cornejo, E. García-Verdugo, M. J. Gil, S. V. Luis, J. A. Mayoral, V. Martínez-Merino and M. Sokolova, J. Org. Chem., 2007, 72, 4344–4350 CrossRef CAS PubMed.
- R. Duque, E. Ochsner, H. Clavier, F. Caijo, S. P. Nolan, M. Mauduit and D. J. Cole-Hamilton, Green Chem., 2011, 13, 1187–1195 RSC.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlencker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- T. Martin, A. Galarneau, F. Di Renzo, F. Fajula and D. Plee, Angew. Chem., Int. Ed., 2002, 41, 2590–2592 CrossRef CAS.
- Complex 1 refers to ligand 6-[(S)-1-(1-naphthyl)ethyl]-3,9-ditosyl-3,6,9,15-tetraazabicyclo[9,3,1]pentadeca-1(15),11,13-triene.
- Complex 2 refers to ligand (6-[(R)-1-naphthylethyl]-3,9-ditosyl-3,8-[(S,S)-iso-propyl]-3,6,9,15-tetraazabicyclo[9,3,1]pentadeca-1(15),11,13-triene).
- U. Hintermair, G. Zhao, C. C. Santini, M. J. Muldoon and D. J. Cole-Hamilton, Chem. Commun., 2007, 1462–1464 RSC.
- U. Hintermair, Z. Gong, A. Serbanovic, M. J. Muldoon, C. C. Santini and D. J. Cole-Hamilton, Dalton Trans., 2010, 39, 8501–8510 RSC.
- T. Niimi, T. Uchida, R. Irie and T. Katsuki, Adv. Synth. Catal., 2001, 343, 79–88 CrossRef CAS.
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Supplement 2, Springer-Verlag, Berlin, Heidelberg, New York, 2004 Search PubMed.
- L. J. Liu, Y. Song and H. Li, Polym. Int., 2002, 51, 1047–1049 CrossRef CAS.
- C. Bianchini, V. Dal Santo, A. Meli, S. Moneti, M. Moreno, W. Oberhauser, R. Psaro, L. Sordelli and F. Vizza, J. Catal., 2003, 213, 47–62 CrossRef CAS.
- C. Bohm and O. Reiser, Org. Lett., 2001, 3, 1315–1318 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and tables of all the catalytic runs, DRIFT spectra of samples pre- and post-catalysis and selected 1H NMR spectra and HPLC chromatograms. See DOI: 10.1039/c4gc00119b |
| This journal is © The Royal Society of Chemistry 2014 |