 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sustainable route to produce the scytonemin precursor using Escherichia coli†
Sailesh
Malla
a and
Morten O. A.
Sommer
*ab
aThe Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, DK-2970 Hørsholm, Denmark
bDepartment of Systems Biology, Technical University of Denmark, DK-2800 Lyngby, Denmark. E-mail: msom@bio.dtu.dk
First published on 1st April 2014
Abstract
Scytonemin is an indolic–phenolic natural product with potent pharmaceutical activities and possible application as a sunscreen. However, the productivity of the existing synthesis systems restrains its applications in medicine and cosmetics. In this paper, we report the generation of the monomer moiety of scytonemin from tryptophan and tyrosine in Escherichia coli. We heterologously expressed the biosynthetic pathway from Nostoc punctiforme and discovered that only three enzymes from N. punctiforme are required for the in vivo production of the monomer moiety of scytonemin in E. coli. We also found that the constructed recombinant E. coli strains are capable of producing novel alkaloids as shunt products. The recombinant E. coli strain expressing the putative scytonemin biosynthetic gene cluster produced 4.2 mg L−1 (2.46 μg mg−1 dry cell weight) of the monomer moiety of scytonemin without supplementation of extracellular substrates whereas upon supplementation with 1 mM of the substrates to the E. coli strain harboring scyABC genes, 8.9 mg L−1 (4.56 μg mg−1 dry cell weight) of the monomer moiety of scytonemin was produced in 5 days. Combining this cell factory with the previously described chemical dimerization process will contribute to a sustainable production of semi-synthetic scytonemin.
Introduction
Alkaloids, a diverse group of nitrogen-containing natural products, are produced by a large variety of organisms including bacteria, fungi, insects, plants and animals. Numerous alkaloids are pharmacologically well characterized and are used as clinical drugs, ranging from chemotherapeutics to analgesic agents.1 Studies on plant alkaloids suggest that they are involved in the defense mechanism against herbivores, insects and pathogens.2 Since alkaloids are toxic, they are usually produced in small quantities by their native producer organisms. Scytonemin is an alkaloid pigment consisting of a symmetrical dimeric carbon skeleton composed of fused heterocyclic units with conjugated double-bond distribution (Fig. 1) synthesized by numerous cyanobacteria.3 Scytonemin is the first described small molecule that inhibits human polo-like kinase 1 (PLK1).4 PLK1 has multiple functions during mitosis and plays a significant role in maintaining the genomic stability.5 Furthermore, PLK1 is highly expressed in a broad spectrum of cancer cells, indicating its possibility of being involved in carcinogenesis.6 Scytonemin (at 3–4 μM concentration) can inhibit cell growth by cell cycle arrest in multiple myeloma cells and renal cancer cells through specific down-regulation of the PLK1 activity.7,8 Scytonemin is not cytotoxic (up to 10 μM) to non-proliferating cells, highlighting its possible application in medicine.9,10 In addition to kinase inhibitory activities, scytonemin also acts as a natural microbial sunscreen by effectively minimizing cellular damage caused by UV (315–400 nm) exposure.11 Scytonemin also exhibits a radical-scavenging activity12 and its synthesis was enhanced by oxidative stress in cyanobacteria.13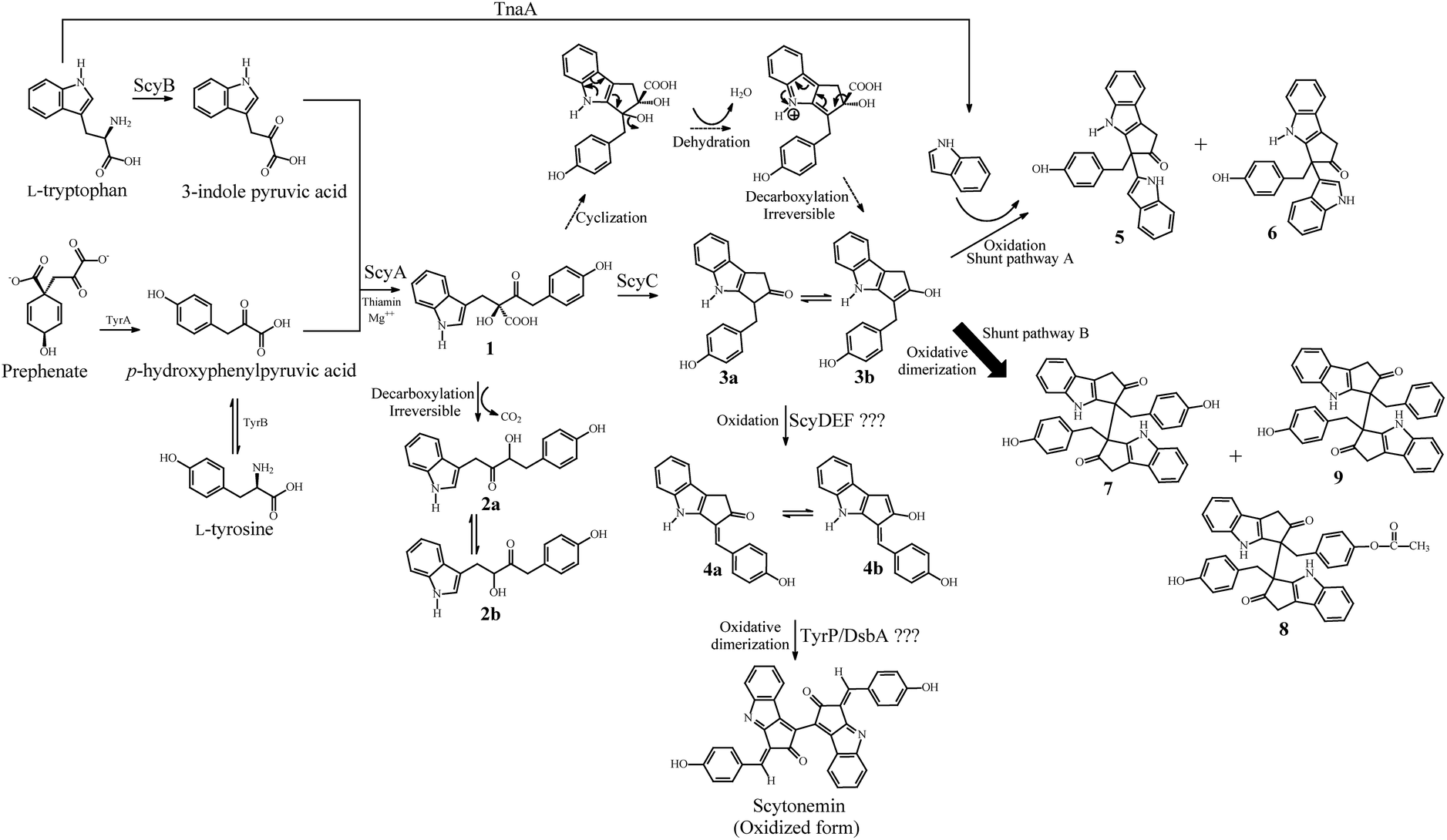 | ||
| Fig. 1 Proposed biosynthetic pathway for scytonemin and the competing shunt pathways A and B in E. coli. The shunt pathways A and B produced new alkaloid derivatives. | ||
The putative scytonemin biosynthetic gene cluster from Nostoc punctiforme ATCC 29133 consists of 18 unidirectional open reading frames (orfs) (Fig. 2). Native expression of this gene cluster is triggered by exposure to UV light, resulting in extracellular pigment accumulation. Once scytonemin has reached sufficient quantities in the extracellular slime layer to block the incoming UVA, the gene expression returns to background levels and halts further scytonemin synthesis.14,15 Due to the potent UV light absorption of scytonemin, the accumulated scytonemin concentration is low (∼1.3 μg mg−1 of dry cell weight (DCW)) in currently characterized cyanobacterial strains under laboratory culture conditions16 whereas naturally growing colonies of a terrestrial cyanobacterium N. commune contained only 0.4 μg mg−1 of DCW of scytonemin.17 Consequently, direct extraction from natural producers is unfeasible on a large scale. Another route to produce scytonemin is through chemical synthesis. The total synthesis of scytonemin has been reported from 3-indole acetic acid through a process comprising nine chemical steps resulting in approximately 4% conversion to the final product.18 Accordingly, more effective approaches are desired for the continuous, rapid and cost effective production of scytonemin.
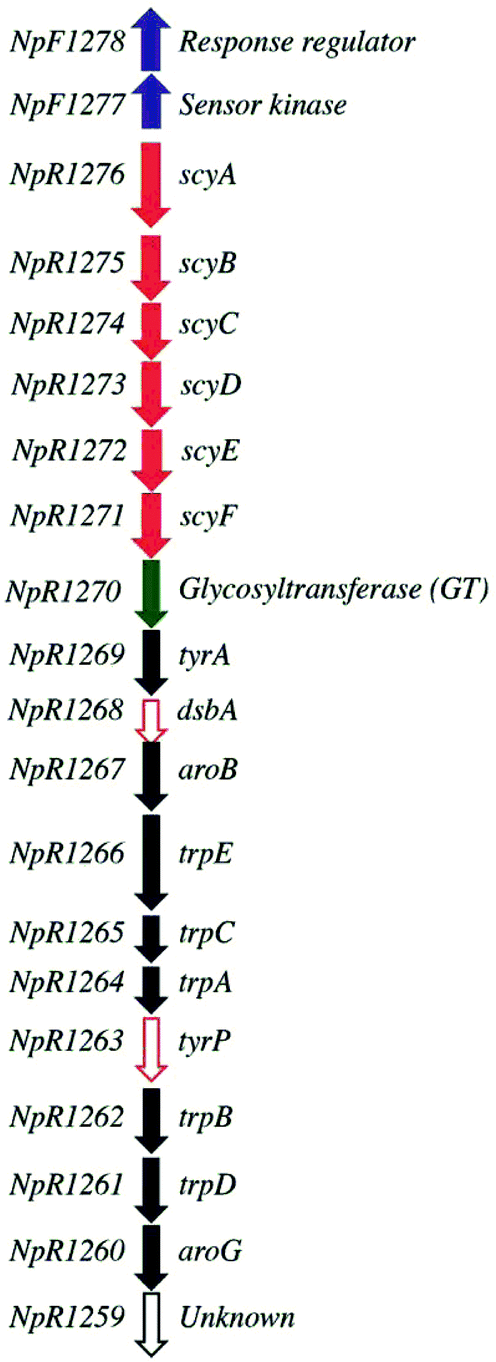 | ||
| Fig. 2 Putative scytonemin biosynthetic gene cluster in ATCC 29133 (adapted from Soule et al., 2009). Arrows represent genes and their transcriptional orientation. Blue filled arrow, regulatory gene; red filled arrow, core structural biosynthetic gene; red open arrow, anticipated core biosynthetic gene for final dimerization step; green filled arrow, glycosyltransferase; black filled arrow, aromatic amino acid biosynthetic gene; and black open arrow, unknown function. | ||
Microbial cell factories offer extensive opportunities for the industrial production of complex biomolecules for cost effective biological synthesis.19–22 Furthermore, microbial fermentation often reduces the need for energy intensive reaction conditions, toxic organic solvents, heavy metal catalysts, and strong acids/bases, which are widely utilized in chemical synthesis routes.23 Among the microbial cell factories designed, the Gram-negative bacterium Escherichia coli has become one of the most promising hosts, with a highly tractable genetic system and favorable fermentation conditions for production purposes.24–26 Indeed, plant based alkaloid compounds have been successfully produced from the engineered E. coli strains. For example, 46 mg L−1 of the plant benzylisoquinoline alkaloid, (S)-reticuline, is produced from fermentation of metabolically engineered E. coli by utilizing simple carbon sources such as glucose or glycerol.19 Similarly, production of indole, a signaling molecule, from exogenous tryptophan in E. coli has been extensively studied.27 Yields up to 6 mM of indole have been achieved from E. coli by supplementation of enough tryptophan in culture media.28 In the present study, we described the construction of an E. coli cell factory for bio-based production of the key pharmaceutical intermediate, the monomer moiety of scytonemin (compound 4 in Fig. 1).
Materials and methods
Bacterial strains, plasmids, cultured conditions and chemicals
All strains, vectors and plasmids used in this study are listed in Table 1. All DNA manipulations were carried out by following standard protocols.29E. coli strains were routinely cultured in Luria–Bertani (LB) broth or on agar supplemented with the appropriate amount of antibiotics (ampicillin 100 μg mL−1, chloramphenicol 25 μg mL−1, streptomycin–spectinomycin 50 μg mL−1 and kanamycin 35 μg mL−1) when necessary. M9 minimal medium was used for production of intermediates and derivatives of scytonemin. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (Hertfordshire, UK) and Fermentas (Denmark). The DNA sequence was determined on an automated DNA sequence analyzer. The authentic scytonemin standard was kindly provided by Professor Jerker Mårtensson (Chalmer University of Technology, Sweden).| Strains/plasmids | Description | Source/reference |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| DH5α | General cloning host | Invitrogen |
| BL21(DE3) | ompT hsdT hsdS (rB− mB−) gal (DE3) | Novagen |
| SM1 | BL21(DE3) carrying pCDF-ScyA and pACYC-ScyB | This study |
| SM2 | BL21(DE3) carrying pCDF-ScyAC and pACYC-ScyB | This study |
| SM3 | BL21(DE3) carrying pCDF-ScyACD, pACYC-ScyB and pET-ScyEF | This study |
| SM4 | BL21(DE3) carrying pCDF-ScyACD, pACYC-ScyB, pET-ScyEF and pRSF-tyrP-dsbA | This study |
| STN | BL21(DE3) carrying pC-ScyABC-ScyDEF, pE-GtAroB-TrpEC and pA-TrpAB-TrpDU | This study |
| Plasmids and vectors | ||
| pET-Duet-1 | Double T7 promoters, ColE1 ori, Ampr | Novagen |
| pCDF-Duet-1 | Double T7 promoters, CloDF13 ori, Smr | Novagen |
| pRSF-Duet-1 | Double T7 promoters, RSF ori, Kmr | Novagen |
| pACYC-Duet-1 | Double T7 promoters, P15A ori, Cmr | Novagen |
| pCDF-ScyA | pCDF-Duet-1 carrying scyA from Nostoc punctiforme | This study |
| pCDF-ScyAC | pCDF-Duet-1 carrying scyA and scyC from N. punctiforme | This study |
| pCDF-ScyACD | pCDF-Duet-1 carrying scyA, scyC and scyD from N. punctiforme | This study |
| pACYC-ScyB | pACYC-Duet-1 carrying scyB from N. punctiforme | This study |
| pET-ScyEF | pET-Duet-1 carrying scyE and scyF from N. punctiforme | This study |
| pRSF-TyrP-DsbA | pRSF-Duet-1 carrying tyrP and dsbA from N. punctiforme | This study |
| pC-ScyABC-ScyDEF | pCDF-Duet-1 carryng scyABC and scyDEF from N. punctiforme | This study |
| pE-GtAroB-TrpEC | pET-Duet-1 carrying Gt-tyrA-dsbA-aroB and trpE-trpC from N. punctiforme | This study |
| pA-TrpAB-TrpDU | pACYC-Duet-1 carrying trpA-tyrP-trpB and trpD-aroG-Npr1259 from N. punctiforme | This study |
Plasmid construction
The construction of recombinant plasmids pCDF-ScyA, pCDF-ScyAC, pCDF-ScyACD, pACYC-ScyB, pET-ScyEF, pRSF-TyrP-DsbA, pC-ScyABC-ScyDEF, pE-GtAroB-TrpEC and pA-TrpAB-TrpDU is described below. All PCR primers used in this study are described in Table 2.| Primers | Oligonucleotide sequences (5′–3’) | Restriction site |
|---|---|---|
| a Restriction sites are indicated by underline and italics. | ||
| ScyA_F | TA![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) ![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/i_char_0047_0332.gif) GCATGAGTCAAAACTATACTGGT GCATGAGTCAAAACTATACTGGT |
NcoI |
| ScyA_R | TTCTCAAACCATTGGAAATGAAAC |
BamHI |
| ScyB_F | TAGCATGCTGCTATTTGAAACTGTT |
NcoI |
| ScyB_R | TTCTTAAGCTGCGATCGCTTTAG |
BamHI |
| ScyC_F | ATAGAAAAAAATACTTTTGCAACA |
NdeI |
| ScyC_R | TTGTTAGTTGGGAACTAGGGATTC |
BglII |
| ScyC_R_BamHI | TTGTTAGTTGGGAACTAGGGATTC |
BamHI |
| ScyD_F | ATAAAACTGAAGCCATTCACTATT |
NdeI |
| ScyD_R | GAGTTAGTTGAGATTTATGGGAGGTG |
KpnI |
| ScyD_F_BglII | GTATTGTACACGGCCGCATAAT |
BglII |
| ScyE_F | TAGCATGAAACTCAAATCACTTACT |
NcoI |
| ScyE_R | TTCTTAGACAGTCTCTGCTTTCAC |
BamHI |
| ScyF_F | ATAGGATTAGTCAAAAATTTGTCAA |
NdeI |
| ScyF_R | TTGTCAGCATTGCTTTTGCAGTTC |
BglII |
| TyrP_F | TAGCATGAAACTCCTGCTAAAATC |
NcoI |
| TyrP_R | TTCTCATCTTTGCGTTTTTCTTTC |
BamHI |
| DsbA_F | ATACTAATAGATATCTTTCATGATA |
NdeI |
| DsbA_R | TTGTCATATTTTTGCGGGTATATC |
BglII |
| GT-AroB_F | TGCATGCAAATTCTGATTTATTCAT |
NcoI |
| GT-AroB_R | CCTCTAAAATTCCTGCAATAGTGA |
BamHI |
| TrpEC_F | TTAATTTTTAATTCCCGTTCCTAC |
NdeI |
| TrpEC_R | GTCCTAAGAAAGCCTTAAAAGACT |
BglII |
| TrpAB_F | TGCATGACCTCTATCTCCAATTCC |
NcoI |
| TrpAB_R | ACATTAAGGAATCAGGACTTTGGC |
BamHI |
| TrpDU_F | CTAATAGCTGTAACTCAAACTCCA |
NdeI |
| TrpDU_R | TATTCAAGAACGGATTAACATCGG |
BglII |
Based on pCDF-Duet-1, the expression recombinant plasmid pCDF-ScyACD was constructed, which allowed the simultaneous expression of the thiamin diphosphate (ThDP) dependent enzyme acetolactate synthase homologue, ScyA, (NpR1276, Genbank accession no. YP_001864940), ScyC (NpR1274, Genbank accession no. YP_001864938), and ScyD (NpR1273, Genbank accession no. YP_001864937) from Nostoc punctiforme ATCC 29133 in E. coli. Primer pairs ScyA_F/ScyA_R, ScyC_F/ScyC_R and ScyD_F/ScyD_R were used for the amplification of nucleotide sequences of scyA (1875 bp), scyC (969 bp) and scyD (1272 bp), respectively, from the genomic DNA of N. punctiforme. The PCR product of scyA was cloned into the NcoI/BamHI (MCS1) sites of pCDFDuet-1 to construct the pCDF-ScyA recombinant expression plasmid. Similarly, the PCR product of scyC was cloned into the NdeI/BglII (MCS2) of the pCDF-ScyA plasmid to get the pCDF-ScyAC recombinant plasmid. The PCR product of scyD was cloned into the NdeI/KpnI (MCS2) of pCDF-Duet-1 vector to construct the pCDF-ScyD recombinant plasmid. Finally, using the primer pair ScyD_F_BglII/ScyD_R and pCDF-ScyD as a template, PCR was performed which allowed the amplification of the T7lac sequence along with the scyD structural gene. The PCR product, T7-rbs-ScyD, was then cloned into the BglII/KpnI sites of pCDF-ScyAC to create the pCDF-ScyACD recombinant plasmid.
The primer pair ScyB_F/ScyB_R was used for the amplification of the leucine dehydrogenase homologue, ScyB (NpR1275, Genbank accession no YP_001864939), from N. punctiforme ATCC 29133 and the PCR product was cloned into pACYC-Duet-1 in NcoI/BamHI sites to construct the pACYC-ScyB expression recombinant plasmid.
Similarly, the primer pairs ScyE_F/ScyE_R and ScyF_F/ScyF_R were used to amplify scyE (NpR1272, Genbank accession no. YP_001864936) and scyF (NpR1271, Genbank accession no. YP_001864935) from the genomic DNA of N. punctiforme, respectively. The PCR product of scyE was cloned into pET-Duet-1 in NcoI/BamHI sites to construct the pET-ScyE expression recombinant plasmid. Furthermore, the PCR product of scyF was cloned into pRSF-ScyE excised with NdeI/BglII sites to construct the pET-ScyEF expression recombinant plasmid.
Likewise, the primer pairs TyrP_F/TyrP_R and DsbA_F/DsbA_R were used to amplify TyrP (NpR1263, Genbank accession no. YP_001864927) and DsbA (NpR1268, Genbank accession no. YP_001864932) from the genomic DNA of N. punctiforme, respectively. The PCR products of tyrP and dsbA were consecutively cloned into NcoI/BamHI and NdeI/BglII sites of pRSF-Duet-1 vector to construct the pRSF-TyrP-DsbA expression recombinant plasmid.
To express the putative scytonemin gene cluster (Fig. 2), recombinant plasmids pC-ScyABC-ScyDEF, pE-GTAroB-TrpEC and pA-TrpAB-TrpDU were constructed based upon pCDF-Duet-1, pET-Duet-1 and pACYC-Duet-1 expression vectors, respectively. The primer pairs ScyA_F/ScyC_R_BamHI, ScyD_F/ScyF_R, GT-AroB_F/GT-AroB_R, TrpEC_F/TrpEC_R, TrpAB_F/TrpAB_R, and TrpDU_F/TrpDU_R were used for amplification of scyABC, scyDEF, Gt-tyrA-dsbA-aroB, trpE-trpC, trpA-tyrP-trpB, and trpD-aroG-NpR1259 regions of the putative scytonemin gene cluster from the genomic DNA of N. punctiforme, respectively. The PCR product of scyABC was cloned into the NcoI/BamHI (MCS1) sites of pCDF-Duet-1 to construct the pC-ScyABC recombinant expression plasmid. Further, the PCR product of scyDEF was cloned into the NdeI/BglII (MCS2) of the pC-ScyABC plasmid to construct pC-ScyABC-ScyDEF. Similarly, the PCR products of Gt-tyrA-dsbA-aroB and trpE-trpC were cloned into NcoI/BamHI (MCS1) and NdeI/BglII (MCS2) of pET-Duet-1 vector, respectively, to construct the pE-GtAroB-TrpEC recombinant plasmid. Finally, the PCR products of trpA-tyrP-trpB and trpD-aroG-Npr1259 were cloned into NcoI/BamHI (MCS1) and NdeI/BglII (MCS2) of the pACYC-Duet-1 vector, respectively, to construct the pA-TrpAB-TrpDU recombinant plasmid.
In all cases, construction of recombinant plasmids was verified by both restriction mapping and direct nucleotide sequencing of respective genes in the recombinant plasmids.
Recombinant protein expression, whole-cell biotransformation, product isolation and determination of biomass
E. coli BL21(DE3) harboring recombinant plasmids were precultured into 3 mL of LB liquid media with appropriate antibiotics and incubated at 37 °C with 220 rpm overnight. The following day 200 μL of preinoculum was transferred into 4 mL of LB liquid media (with antibiotics) and cultured at 37 °C until the optical density at 600 nm (OD600 nm) reached approximately 0.6. Then isopropyl-β-D-thiogalactopyranoside (IPTG) was added at a final concentration of 1 mM and the culture was incubated at 30 °C for 20 h. The cells (∼5 × 108 cells) were harvested by centrifugation, washed with 1 mL of phosphate buffer (pH 7.0) and then resuspended in 100 μL of phosphate buffer. The recombinant protein was released by following six cycles of the freeze–thaw method and checked by SDS-PAGE (ESI Fig. S1†). For freeze–thaw cycles, the cell suspension in phosphate buffer was frozen in a dry ice and isopropanol bath for 5 min and thawed in a 37 °C water bath.For whole-cell biotransformation, after IPTG induction the culture was incubated at 30 °C for 5 h to increase biomass. The cell pellet was collected by centrifugation and resuspended in M9 minimal medium (resulting in an OD600 nm of ∼1.5) with 1 mM of IPTG. The culture broth was aliquoted (500 μL in each well) in the 96-deep-well plate (VWR, Denmark) and supplemented with tryptophan and tyrosine (0.5 mM or 1 mM of each). The plate was then incubated at 30 °C and 300 rpm for 5 days. The culture broth was extracted with an equal volume of methanol for high performance liquid chromatography (HPLC) and electrospray ionization mass analysis.
To calculate dry cell weight (DCW) of the E. coli recombinant strains, the cell pellets were collected in a pre-weighed Eppendorf tube by centrifuging 1 ml of culture broth (combining samples from two wells) at 6000g for 10 min. Then the cell pellets were dried at 60 °C in a vacuum oven until a constant weight was obtained. The cell pellets were used to determine the DCW as the biomass. Triplicate reading was carried out.
Product analysis and quantification
The bioconversion products from E. coli recombinant strains were analyzed and quantified by HPLC (Ultimate 3000, Thermo Scientific, USA) equipped with a Discovery® HS F5 column (4.6 × 150 mm, 5.0 μM particle size, Supelco, Sigma-Aldrich) connected to a UV detector (260 nm, 290 nm, 360 nm and 370 nm). A flow rate of 0.5 mL min−1 was used with a linear gradient of 10 mM ammonium formate buffer (pH 3 adjusted with formic acid) (Phase A) and acetonitrile (Phase B) by the following method: 0–3 min (25% B), 3–15 min (25–75% B), 15–25 min (75% B), and 25–29 min (75–25% B) and 29–30 min (25% B). For quantification of metabolites, calibration curves of purified compounds were drawn using 6.25, 12.5, 25, and 50 μg mL−1 concentrations. The exact mass of the compounds were analyzed by using Oribtrap Fusion (Thermo Scientific, USA) with a Dionex 3000 RX HPLC system (Thermo Scientific, USA) in the positive and negative ion modes.Structural elucidation
The recombinant strain E. coli SM4 (E. coli BL21(DE3) harboring scyA, scyB, scyC, scyD, scyE, scyF, tyrP and dsbA) was cultured in 1 L of M9 minimal media. During induction by IPTG, 500 μM of tryptophan and tyrosine were also supplemented and after 5 days of incubation, the isolation process of the biotransformation product was undertaken. The culture broth was centrifuged at 6800g for 12 min to separate supernatant and cell pellet. Then the supernatant was extracted with an equal volume of ethyl acetate whereas the cell pellet was extracted with 100 mL of an ethyl acetate and acetone (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. The organic phase was collected and concentrated to dryness by evaporation of excess solvent. The remaining products were dissolved in methanol and the isolated crude extracts from supernatant and cell pellet were combined. The extracted crude compound was chromatographed on PREP-HPLC (Ultimate 3000, Thermo Scientific, USA) under the following conditions: column, Discovery® HS F5 (4.6 × 150 mm, 5.0 μM particle size, Supelco, Sigma-Aldrich); UV detection, 290 nm; flow rate, 1.0 mL min−1; under similar gradient conditions of solvents as mentioned above. The fractions were collected and the purified fractions were completely dried in a SpeedVac concentrator (SAVANT SC210A, Thermo Scientific, USA). The structural elucidation of the purified compounds was done by NMR analysis (1H, 13C, Correlation Spectroscopy (COSY), Heteronuclear Single Quantum Coherence (HSQC), Heteronuclear multiple-bond correlation spectroscopy (HMBC)) and the relative stereochemistry for compound 4 was assigned from 1D-Nuclear Overhauser effect (NOE) experiment. The NMR analysis for structural elucidation of compounds 5, 6, 7, 8 and 9 are described in ESI.† NMR spectra were obtained in DMSO-d6 (Aldrich, Chicago, IL, USA) using a Bruker Advance 600 instrument (600 MHz). For the 1H-NMR experiment, 32 transients spectra were acquired with a spectral width of 8000 Hz. All NMR data were processed using XWINNMR (Bruker).
:1) mixture. The organic phase was collected and concentrated to dryness by evaporation of excess solvent. The remaining products were dissolved in methanol and the isolated crude extracts from supernatant and cell pellet were combined. The extracted crude compound was chromatographed on PREP-HPLC (Ultimate 3000, Thermo Scientific, USA) under the following conditions: column, Discovery® HS F5 (4.6 × 150 mm, 5.0 μM particle size, Supelco, Sigma-Aldrich); UV detection, 290 nm; flow rate, 1.0 mL min−1; under similar gradient conditions of solvents as mentioned above. The fractions were collected and the purified fractions were completely dried in a SpeedVac concentrator (SAVANT SC210A, Thermo Scientific, USA). The structural elucidation of the purified compounds was done by NMR analysis (1H, 13C, Correlation Spectroscopy (COSY), Heteronuclear Single Quantum Coherence (HSQC), Heteronuclear multiple-bond correlation spectroscopy (HMBC)) and the relative stereochemistry for compound 4 was assigned from 1D-Nuclear Overhauser effect (NOE) experiment. The NMR analysis for structural elucidation of compounds 5, 6, 7, 8 and 9 are described in ESI.† NMR spectra were obtained in DMSO-d6 (Aldrich, Chicago, IL, USA) using a Bruker Advance 600 instrument (600 MHz). For the 1H-NMR experiment, 32 transients spectra were acquired with a spectral width of 8000 Hz. All NMR data were processed using XWINNMR (Bruker).
Results and discussion
Heterologous expression of the putative scytonemin gene cluster
Recent studies have shown that N. punctiforme genes have been well expressed and functional in E. coli.30,31 Accordingly, we chose to construct our recombinant pathway in E. coli using the native genes of N. punctiforme. For the expression of the putative scytonemin biosynthetic gene cluster, the recombinant plasmids pC-ScyABC-ScyDEF, pE-GtAroB-TrpEC and pA-TrpAB-TrpDU were constructed, and they were transformed into E. coli BL21 to create the strain E. coli STN. Upon IPTG induction, the cultures of E. coli expressing the putative scytonemin biosynthetic gene cluster (E. coli STN strain) in M9 minimal media turned yellow whereas the uninduced cultures did not have any color (data not shown). The metabolites produced by E. coli STN strain were analyzed by HPLC and mass analysis. The E. coli STN strain did not produce scytonemin upon IPTG induction. However, the monomer of scytonemin (compounds 4) and a new alkaloid derivative (compound 7) were produced as the dominant products from the endogenous amino acids (Fig. 3). | ||
| Fig. 3 (A) HPLC analysis of bioconversion products from E. coli SM4 and STN strains. (i) metabolites from E. coli SM4 supplemented with 1 mM of L-tryptophan and 1 mM of L-tyrosine, (ii) metabolites from E. coli SM4 without supplementation of substrates (control), (iii) metabolites from E. coli STN with IPTG induction, and (iv) metabolites from E. coli STN without IPTG induction (control). Compounds 2, 3, 4, 5, 6, 7, 8, and 9 have a retention time of 14.2, 17.3, 18.8, 16.1, 17.8, 18.6, 20.3 and 21.1 min, respectively. (B) LC/ESI-MS analysis of metabolites from E. coli SM4 in the positive mode: (i) exact mass of compound 3 [M + H]+ [m/z] (278.11783), (ii) exact mass of compound 4 [M + H]+ [m/z] (276.10229), (iii) exact mass of compound 5 [M + H]+ [m/z] (393.16022), (iv) exact mass of compound 6 [M + H]+ [m/z] (393.16049), (v) exact mass of compound 7 [M + H]+ [m/z] (553.21387), (vi) exact mass of compound 8 [M + H]+ [m/z] (595.22323), and (vii) exact mass of compound 9 [M + H]+ [m/z] (537.21747). | ||
Despite the production of compound 4, the absence of scytonemin in the metabolites from STN was either due to the lack of dimerization enzyme(s) in the putative scytonemin gene cluster or inactive putative dimerization enzyme(s) during heterologous expression in E. coli. Genome analysis and comparison among several cyanobacterial strains for the conserved localization in the scytonemin clusters revealed a five-gene satellite cluster, oriented in the same transcriptional direction in N. punctiforme.15 Of five genes in the cluster, two genes are annotated as unknown hypothetical proteins, and three genes are annotated as putative metal-dependent hydrolase, putative prenyltransferase and putative type I phosphodiesterase. In addition, the transcriptional studies showed that all five genes in this cluster were upregulated under UV irradiation.16 Hence, it was predicted that besides the putative gene cluster shown in Fig. 2, this satellite five-gene cluster might be involved during scytonemin biosynthesis. However, due to unclear annotations and lack of biochemical characterization, the role of this satellite cluster is still ambiguous.
Expression of structural core biosynthetic genes
Comparative genomic analysis of the scytonemin gene cluster from various cyanobacterial strains revealed that six gene products of ScyA-F are anticipated to produce the monomer moiety of scytonemin, and the final reaction, i.e., dimerization step was predicted to catalyze by tyrosinase (TyrP) and/or oxidoreductase (DsbA) (Fig. 1).15 Accordingly, we constructed the recombinant plasmids pACYC-ScyB, pCDF-ScyAB, pRSF-ScyEF and pET-TyrP-DsbA and introduced them into E. coli BL21 (DE3). The resulting strain was designated as E. coli SM4. The in vivo isotope labeling studies in cyanobacterial strains showed that both labeled tryptophan and tyrosine were incorporated into the scytonemin structure during its biosynthesis.32 So, we supply tryptophan and tyrosine as precursor substrates during the biotransformation of E. coli SM4. The culture broth of E. coli SM4 strain supplemented with these precursors turned yellow and the yellowish product was primarily accumulated in the cell pellet (ESI Fig. S2†). HPLC and mass analysis of the bioconversion products of SM4 strain upon supplementation of 500 μM of tryptophan and tyrosine accumulated compound 2 (C18H17NO3 calculated [M + H]+: 296.12866, found: 296.12876 and calculated [M − H]−: 294.11301, found 294.11344), compound 3 (C18H15NO2 calculated [M + H]+: 278.11810, found: 278.11783), compound 4 (C18H13NO2 calculated [M + H]+: 276.10245, found: 276.10229) along with new alkaloid derivatives compound 5 (C26H20N2O2 calculated [M + H]+: 393.16030, found: 393.16022), compound 6 (C26H20N2O2 calculated [M + H]+: 393.16030, found: 393.16049), compound 7 (C36H28N2O4 calculated [M + H]+: 553.21273, found: 553.21387), compound 8 (C38H30N2O5 calculated [M + H]+: 595.22329, found: 595.22323) and compound 9 (C36H28N2O3 calculated [M + H]+: 537.21781, found: 537.21747) (Fig. 3A and 3B and ESI Fig. S4†). All of these five new alkaloid derivatives have very similar UV-absorption spectra to that of compound 3 (ESI Fig. S3†). The structure of compounds 4, 6, 7, 8 and 9 was confirmed by NMR analysis (1H, 13C, HSQC, HMBC) (Table 3, S1–S4 and ESI Fig. S5–S10†).| Position | Chemical shift (ppm) | |
|---|---|---|
| 13C | 1H | |
| a Assignments of carbon 3 and 11 may be switched. | ||
| 1 | 204.85 | |
| 2 | 36.33 | 3.51, s |
| 3a | 119.37 | |
| 4 | 139.98 | |
| 5 | 119.10 | 7.50, d (J = 7.79 Hz) |
| 6 | 120.01 | 7.07, t (J = 7.48 Hz) |
| 7 | 123.28 | 7.19, t (J = 7.25 Hz) |
| 8 | 112.93 | 7.53, d (J = 8.20 Hz) |
| 9 | 123.56 | |
| 10 | 10.93, s | |
| 11a | 139.69 | |
| 12 | 125.54 | |
| 13 | 124.34 | 6.98, s |
| 14 | 125.78 | |
| 15 | 130.64 | 7.63, d (J = 8.51 Hz) |
| 16 | 116.21 | 6.93, d (J = 8.52 Hz) |
| 17 | 158.60 | |
| 18 | 10.03, s | |
The absence of scytonemin in the bioconversion products of both SM4 and STN strains indicates that the final dimerization step is the major bottleneck in E. coli. Structural elucidation of the new alkaloid derivatives (shunt products) revealed that all five compounds were produced from the oxidation of intermediate compound 3, i.e., either by the formation of C–C bond with indole or dimerization of compound 3. To gain more information about these new derivatives such as their synthetic origin and plausible bioactivities, we searched the literature to ascertain whether any of these compounds have been previously reported. An anti-inflammatory drug target IκB kinase inhibitor, PS1145, and a proteasome inhibitor, Nostodione A, are structurally similar to the monomer moiety of scytonemin.33 Nostodione A is generated upon ozonolysis of the reduced form of scytonemin,34 and this compound has been isolated from N. commune35 and a fresh water cyanobacterium, Scytonema hofmanni.36 Similarly, the three new scytonemin derivatives, dimethoxyscytonemin, tetramethoxyscytonemin and scytonin, have been identified from the organic extracts of Scytonema sp. These compounds do not possesses cytotoxic effects even at 10 μM and also did not inhibit the growth of Gram positive, Gram negative and fungi at the concentration of 1 μM.37 All of these previously reported derivatives are derived from the scytoneman skeleton of scytonemin. To the best of our knowledge, all the shunt products we found in this study have not been reported yet from any cyanobacterial strains including N. punctiforme. So, it is plausible that these shunt oxidation pathways are catalyzed by E. coli endogeneous enzyme(s) consuming the accumulated compound 3 in the cell.
Structural elucidation of compound 4
The 1H and 13C-NMR signals of compound 4 are given in Table 3 whereas the COSY, HMBC, HSQC and NEO spectrum are given in ESI (ESI Fig. S5†). In the 1H-NMR spectrum, the low field singlet signals at 10.03 ppm and 10.93 ppm correspond to phenyl hydroxyl and indole amide groups, respectively, the signals between 7 ppm and 8 ppm correspond to typical aromatic phenyl and indole rings and the signal at 3.51 ppm corresponds to an aliphatic signal. The COSY spectrum confirmed the proton observations and revealed a correlation between the amide and one of the terminal protons of the indole proton system (4 bonds apart), allowing a sequential assignment of the proton spectrum (in fact this seems to be a 5 bond correlation from the NH to the opposite side of the indole proton network). The HSQC correlated these proton signals to their respective carbons, permitting the firm assignment of all non-quaternary carbons. The HMBC allowed the assignment of some quaternary signals and the observation of a correlation between the aliphatic signal and a resonance at 204.85 ppm (only ketones resonate at this frequency). Due to the scarcity of protons in this molecule, the fact that HMBC signals can correlate to 2, 3 or 4 bonds apart and the cyclic nature of the molecule, sequential assignment and structural confirmation of the 5 membered ring becomes virtually impossible. The presence of an indole, a phenyl and a ketone group is indisputable, however their position could not be ascertained so six structures as shown in Fig. 4A were possible.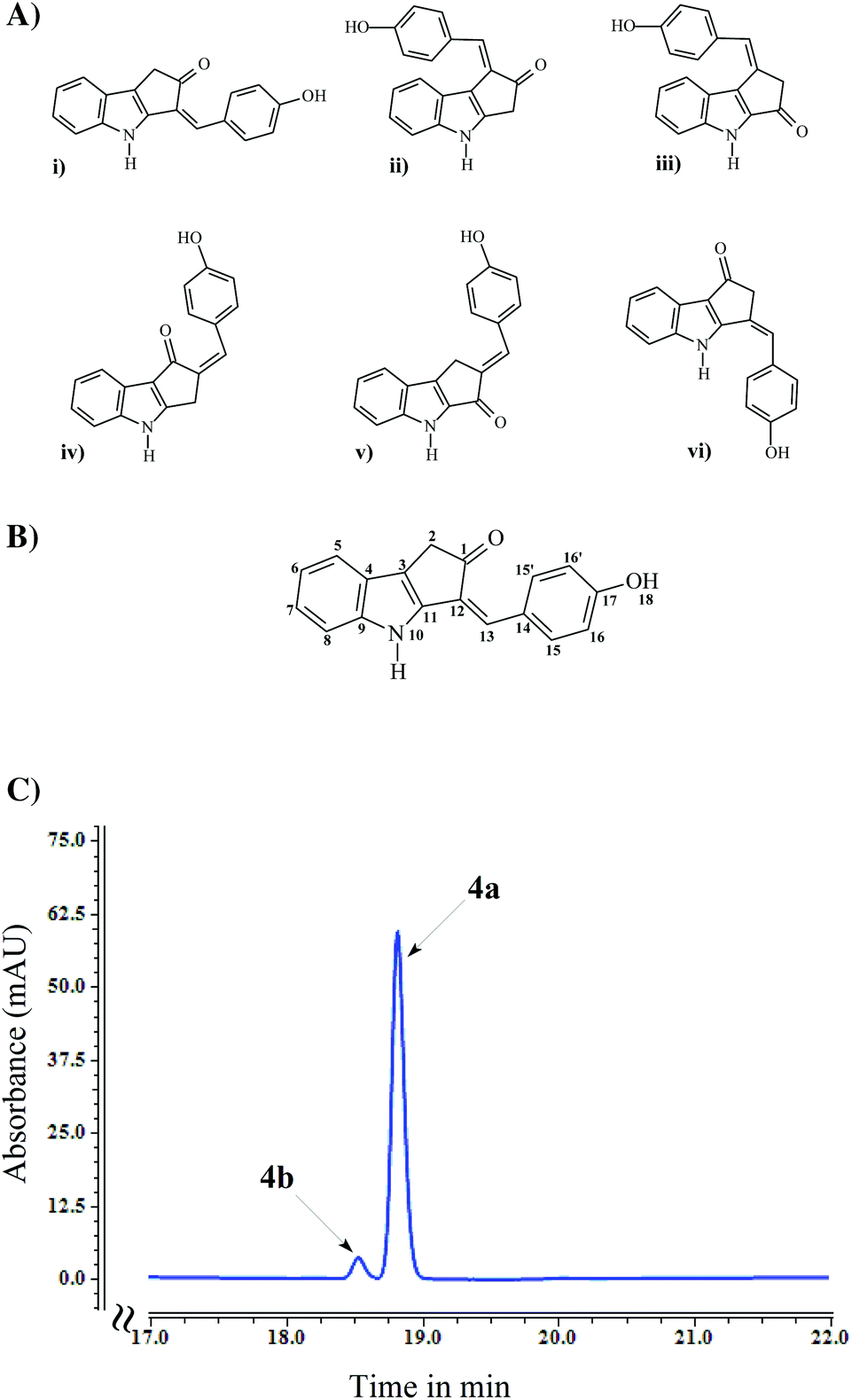 | ||
| Fig. 4 (A) Six possible structures for compound 4 compatible with proton, carbon, COSY, HSQC and HMBC NMR analysis. (B) Structure of the compound 4 with atom numbering. (C) HPLC analysis of purified compound 4 from biotransformation of E. coli SM4 at 360 nm. | ||
At this stage a NOE spectrum was acquired. The NOE spectrum revealed a correlation between the amide proton and signals of the phenyl group suggesting only possible structures (i) and (vi) in Fig. 4A. Also, a signal was observed between the aliphatic group and a proton on the indole ring but not with the phenyl ring and the amide group, which strongly suggests that the possible structure for compound 4 is structure (i) in Fig. 4A. The confirmed structure of compound 4 along with atom numbering is given in Fig. 4B.
Although the NMR analysis confirmed structure 4a, two isomeric forms i.e., keto (4a) and enol (4b) forms are feasible structures for compound 4 as a result of keto–enol tautomerization. Owing to the lower energy, the keto form is thermodynamically more stable than the enol form, so the equilibrium heavily favors the formation of the keto form at room temperature.38,39 In addition, the equilibrium shifts toward the keto form in polar solvents mainly due to the involvement of lone pairs (present in oxygen of the keto group) in hydrogen bond formation with the solvent, making them less available to form hydrogen bonds with the enol form.40,41 HPLC chromatogram of the purified compound 4 contained two peaks: a major peak at a retention time of 18.8 min and a minor peak at 18.5 min retention time (Fig. 4C). Regardless of an absorbance maxima shifting (from 408 nm for major peak to 429 nm for minor peak), both of these compounds had very much similar UV absorbance spectra (ESI Fig. S3†). Hence, despite the formation of both keto and enol forms of compound 4, only keto form (4a) was detected in NMR analysis.
Minimal genes for the production of the scytonemin monomer
To identify the minimal set of genes required for the production of the monomer moiety of scytonemin, a number of E. coli recombinant strains were constructed and their metabolites were analyzed following whole-cell biotransformation supplemented with tryptophan and tyrosine. First, the recombinant strain E. coli SM1 was constructed by introducing the plasmids pACYC-ScyB and pCDF-ScyA into E. coli BL21. Upon supplementation of tryptophan and tyrosine, this strain predominantly accumulated a decarboxylated product of intermediate 1 (i.e., compounds 2a or 2b), which was detected by HPLC at 14.2 min retention time and identified by mass analysis (ESI Fig. S4†). Unlike the yellowish culture broth of SM4, the culture broth of SM1 supplemented with tryptophan and tyrosine was similar to the control strain (ESI Fig. S2†).We then constructed the recombinant E. coli strains SM2 (E. coli BL21 harboring pACYC-ScyB and pCDF-ScyAC) and SM3 (E. coli BL21 harboring pACYC-ScyB, pCDF-ScyACD and pRSF-ScyEF). The biotransformation products of these strains were analyzed by exogenously supplying tryptophan and tyrosine. The culture broth of SM2 and SM3 strains is similar to that of the SM4, and both of these strains accumulated compound 4, along with all five shunt products (compounds 5, 6, 7, 8, and 9).
The in vitro characterization of the early biosynthetic enzymes of the scytonemin gene cluster proved that ScyB converts L-tryptophan to indole-3-pyruvic acid, which is coupled with p-hydroxyphenylpyruvic acid in the presence of ScyA to produce a labile β-keto acid adduct 1.42 The endogeneous E. coli enzyme, TyrB, catalyzes deamination of tyrosine providing one of the substrates, p-hydroxyphenylpyruvic acid, for ScyA.43 However, in the absence of ScyC, the adduct 1 undergoes a facile, non-enzymatic decarboxylation to produce the regioisomers 2a and 2b.44 On the other hand, in the presence of ScyC, this non-enzymatic decarboxylation reaction is suppressed in favor of an intramolecular cyclization followed by dehydration and irreversible decarboxylation to produce compound 3a.44 Although the in vitro studies on scyC only accumulated 3a,44 we found that in vivo production of a monomer moiety of scytonemin (compound 4) in E. coli can be achieved by expression of only three genes, scyABC, from N. punctiforme. This indicates that the endogenous enzyme(s) from the E. coli host catalyze the oxidation reaction to convert compound 3 into compound 4. Furthermore, the dimerization reaction for the generation of compounds 7, 8 and 9 are also likely catalyzed by the E. coli endogeneous enzyme(s) instead of TyrP/DsbA from N. punctiforme as all five shunt products were also accumulated in the SM2 strain harboring only scyABC genes.
Comparison of compounds 4 and 7 yields
The production of the monomer moiety of scytonemin 4 and shunt dimer compound 7 from E. coli strains SM2 and SM4 was analyzed by supplementing tryptophan and tyrosine in M9 minimal medium at 5 days of reaction time. Utilizing endogenous tryptophan and tyrosine, the strains can produce compounds 4 and 7 upon IPTG induction. However, the yields of these compounds are higher upon supplementation of tryptophan and tyrosine. The biotransformation of strain SM2 supplemented with 500 μM of substrates produced 5.0 mg L−1 of compound 4 and 46.9 mg L−1 of compound 7 whereas at 1 mM of substrate supplementation 7.3 mg L−1 of compound 4 and 77.0 mg L−1 of compound 7 were produced. Likewise, the strain SM4 produced 6.1 mg L−1 of compound 4 and 46.3 mg L−1 of compound 7 at 500 μM substrate supplementation whereas at 1 mM of substrate supplementation 8.9 mg L−1 of compound 4 and 87.1 mg L−1 of compound 7 were produced (Fig. 5). On the other hand, upon IPTG induction the strain STN produced 4.2 mg L−1 of compound 4 and 39.2 mg L−1 of compound 7, respectively, in M9 minimal media at 5 days.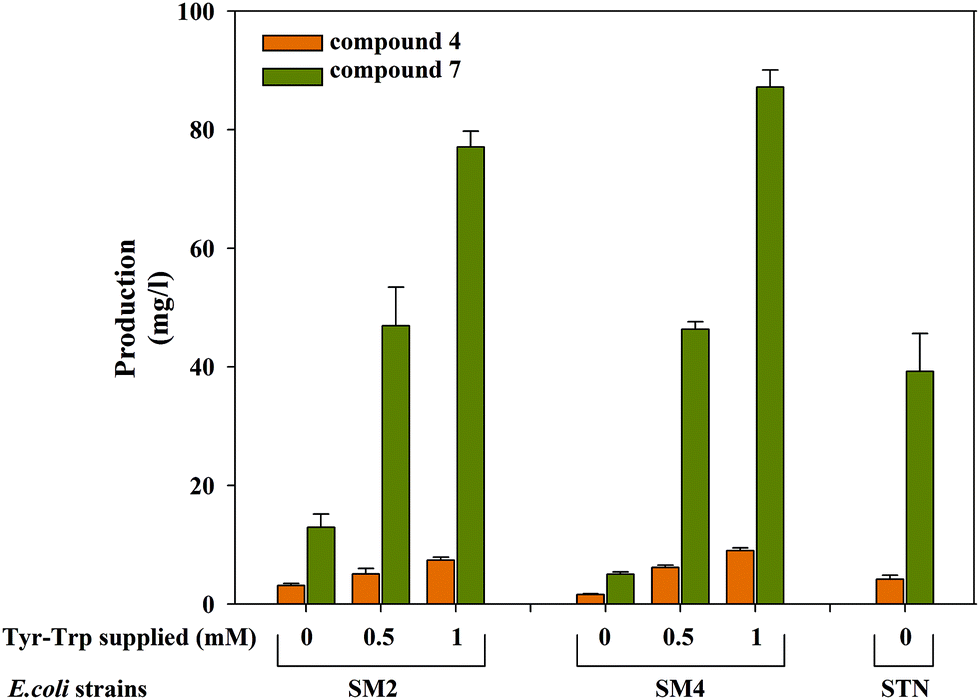 | ||
| Fig. 5 Production of compounds 4 and 7 by E. coli recombinant strains SM2 and SM4 with/without supplementation of tryptophan and tyrosine and strain STN with/without IPTG induction. | ||
The biomass (DCW) of IPTG induced substrate supplemented (1 mM of each) SM2 and SM4 strains were 1.84 g L−1 and 1.94 g L−1 at 5 days whereas those of the control strains were 1.87 g L−1 and 1.81 g L−1, respectively. Similarly, upon IPTG induction STN strain had 1.70 g L−1 of DCW, whereas in the absence of induction this strain had 1.86 g L−1 of DCW at 5 days. This showed the yield of 2.46 μg mg−1 DCW, 3.96 μg mg−1 DCW, and 4.56 μg mg−1 DCW of the compound 4 by STN, SM2 and SM4 strains, respectively.
Conclusions
Following our work, the final dimerization step remains a major hurdle for the complete production of scytonemin in E. coli. Yet commercially, many drugs such as an anticancer drug, paclitaxel (Taxol),45 and an antimalarial drug, artemisinin46 have been produced by combining the biosynthetic and chemical synthetic approaches, highlighting the advantageous features of the bio-chemical approach for production of complex compounds. Our construction of a cell factory producing the monomer moiety of scytonemin could facilitate such production when combined with the already described chemo-synthetic dimerization step.Upon supplementation of 1 mM of tryptophan and tyrosine, ca. 158 μM of compound 7 (i.e., 316 μM of the equivalent substrate concentration), ca. 32 μM of the monomer moiety of scytonemin, and comparable amounts of other derivatives (compounds 2, 3, 5, 6, 8, and 9) to that of compound 4 were produced. This indicates that nearly half of the supplemented substrates were utilized by the heterologously expressed scytonemin pathway in the constructed E. coli strain. This E. coli cell factory has a 3.5 fold higher yield of the scytonemin monomer moiety as compared to the scytonemin produced by the native producer N. punctiforme. Accordingly, our work represents an important milestone towards a green scytonemin process. However, the industrial applicability of this system requires a maximal conversion of substrates into the targeted product without (or low) the production of side products. Several techniques could possibly be applied for further optimization of this strain and biotransformation systems to enhance production. For example, inactivation of the targeted gene(s) could facilitate the production yields by preventing metabolic flux through undesired branch pathways.47,48 Furthermore, expression level optimizations of heterologous pathway enzymes could be achieved by altering the plasmid copy number49 and promoter strength50 and engineering the ribosome binding sites (RBS).51 Similarly, adaptive laboratory evolution (ALE) strategies have been broadly applied in metabolic engineering of E. coli for improving fitness, yield, production rate and cost-effectiveness. The ALE techniques are greatly effective for non-native pathway optimization which allows the selection of beneficial mutations in the production strains in an unbiased fashion.52 Likewise, immobilization of enzymes or whole cells has been successfully applied in numerous scientific and industrial processes.53 Enzyme properties such as stability, activity, specificity, selectivity, etc. have been greatly improved by enzyme immobilization and multi-enzyme co-localization.54,55 During biotransformation, supplementation of high substrate concentration may have a tendency to change the pH, osmotic pressure, etc. of culture media (or reaction conditions), thus limiting the bioconversion process. However, immobilization of the enzyme could increase resistance to such changes and it may also increase the enzyme concentration, which favors supplementation of higher substrate concentrations and hence increase the product yield. Immobilized technology has been extensively used in bioreactors for significant improvement of the yields in fermentation.56 In addition, systematic and careful design in bioreactor and optimization of physical parameters such as cultivation conditions (temperature, dissolved oxygen and RPM), pH condition, media composition, etc. has a great impact in the bioconversion process.57
Further in-depth studies to better understand the shunt pathway B is essential as a majority of compound 3 was consumed by this pathway. Likewise, compound 3 was also consumed by forming an adduct with the indole moiety through a shunt pathway A. Since tryptophanase is responsible for degradation of L-tryptophan into indole, pyruvate and ammonia,58 the prevention of tryptophan degradation as well as the effect of shunt pathway A could be abolished by inactivation of chromosomal tryptophanase (tnaA) in E. coli. These strains could be further metabolically engineered for the overproduction of endogenous tryptophan and the tyrosine pool.59,60 For example, overexpression of branch pathway genes from chorismate to L-tyrosine and L-tryptophan can overproduce these amino acids.61 Hence, studies on the dimerization reaction for the complete synthesis of scytonemin in E. coli along with pathway optimizations to improve the yield of compound 4 will be the focus of future investigations.
Acknowledgements
This work was supported by Novo Nordisk Foundation. We are grateful to Prof. Søren Molin. We thank Dr Pedro Lamosa (ITQB, Portugal) for assistance with the NMR spectroscopic analyses and Dr Scott James Harrison for the MS analysis. We also thank Dr Hao Luo, Dr Jiangfeng Zhu and Dr Ariane Zutz for discussion during the manuscript preparation.References
- E. Leonard, W. Runguphan, S. O'Connor and K. J. Prather, Nat. Chem. Biol., 2009, 5, 292–300 CrossRef CAS PubMed.
- T. Hartmann, Planta, 2004, 219, 1–4 CrossRef CAS PubMed.
- P. J. Proteau, W. H. Gerwick, F. Garcia-Pichel and R. Castenholz, Experientia, 1993, 49, 825–829 CrossRef CAS.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2006, 6, 321–330 CrossRef CAS PubMed.
- F. A. Barr, H. H. Silljé and E. A. Nigg, Nat. Rev. Mol. Cell Biol., 2004, 5, 429–440 CrossRef CAS PubMed.
- Y. Ito, H. Yoshida, F. Matsuzuka, N. Matsuura, Y. Nakamura, H. Nakamine, K. Kakudo, K. Kuma and A. Miyauchi, Anticancer Res., 2004, 24, 259–263 CAS.
- G. Zhang, Z. Zhang and Z. Liu, Tumour Biol., 2013, 34, 1887–1894 CrossRef CAS PubMed.
- G. Zhang, Z. Zhang and Z. Liu, Tumour Biol., 2013, 34, 2241–2247 CrossRef CAS PubMed.
- C. S. Stevenson, E. A. Capper, A. K. Roshak, B. Marquez, C. Eichman, J. R. Jackson, M. Mattern, W. H. Gerwick, R. S. Jacobs and L. A. Marshall, J. Pharmacol. Exp. Ther., 2002, 303, 858–866 CrossRef CAS PubMed.
- C. S. Stevenson, E. A. Capper, A. K. Roshak, B. Marquez, K. Grace, W. H. Gerwick, R. S. Jacobs and L. A. Marshall, Inflamm. Res., 2002, 51, 112–114 CrossRef CAS.
- F. Garcia-Pichel, N. D. Sherry and R. W. Castenholz, Photochem. Photobiol., 1992, 56, 17–23 CrossRef CAS.
- K. Matsui, E. Nazifi, Y. Hirai, N. Wada, S. Matsugo and T. Sakamoto, J. Gen. Appl. Microbiol., 2012, 58, 137–144 CrossRef CAS.
- J. G. Dillon, C. M. Tatsumi, P. G. Tandingan and R. W. Castenholz, Arch. Microbiol., 2002, 177, 322–331 CrossRef CAS PubMed.
- C. M. Sorrels, P. J. Proteau and W. H. Gerwick, Appl. Environ. Microbiol., 2009, 75, 4861–4869 CrossRef CAS PubMed.
- T. Soule, K. Palmer, Q. Gao, R. M. Potrafka, V. Stout and F. Garcia-Pichel, BMC Genomics, 2009, 10, 336–345 CrossRef PubMed.
- T. Soule, F. Garcia-Pichel and V. Stout, J. Bacteriol., 2009, 191, 4639–4646 CrossRef CAS PubMed.
- T. Sakamoto, K. Kumihashi, S. Kunita, T. Masaura, K. Inoue-Sakamoto and M. Yamaguchi, FEMS Microbiol. Ecol., 2011, 77, 385–394 CrossRef CAS PubMed.
- A. Ekebergh, I. Karlsson, R. Mete, Y. Pan, A. Börje and J. M årtensson, Org. Lett., 2011, 13, 4458–4461 CrossRef CAS PubMed.
- A. Nakagawa, H. Minami, J. S. Kim, T. Koyanagi, T. Katayama, F. Sato and H. Kumagai, Nat. Commun., 2011, 2, 326 CrossRef PubMed.
- J. Du, Z. Shao and H. Zhao, J. Ind. Microbiol. Biotechnol., 2011, 38, 873–890 CrossRef CAS PubMed.
- S. Y. Lee, D. Mattanovich and A. Villaverde, Microb. Cell Fact., 2012, 11, 156 CrossRef CAS PubMed.
- C. D. Murphy, Org. Biomol. Chem., 2012, 10, 1949–1957 CAS.
- E. Matsumura, M. Matsuda, F. Sato and H. Minami, Natural Products, Springer, Berlin Heidelberg, 2013 Search PubMed.
- D. P. Clark, FEMS Microbiol. Rev., 1989, 5, 223–234 CAS.
- S. Malla, M. A. Koffas, R. J. Kazlauskas and B. G. Kim, Appl. Environ. Microbiol., 2012, 78, 684–694 CrossRef CAS PubMed.
- B. Kim, H. Park, D. Na and S. Y. Lee, Biotechnol. J., 2013 DOI:10.1002/biot.201300263.
- W. Chu, T. R. Zere, M. M. Weber, T. K. Wood, M. Whiteley, B. Hidalgo-Romano, E. Valenzuela Jr. and R. J. McLean, Appl. Environ. Microbiol., 2012, 78, 411–419 CrossRef CAS PubMed.
- G. Li and K. D. Young, Microbiology, 2013, 159, 402–410 CrossRef CAS PubMed.
- J. Sambrook and D. W. Russell, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 3rd edn, 2001 Search PubMed.
- K. Koketsu, S. Mitsuhashi and K. Tabata, Appl. Environ. Microbiol., 2013, 79, 2201–2208 CrossRef CAS PubMed.
- Q. Gao and F. Garcia-Pichel, J. Bacteriol., 2011, 193, 5923–5928 CrossRef CAS PubMed.
- C. S. Jones, E. Esquenazi, P. C. Dorrestein and W. H. Gerwick, Bioorg. Med. Chem., 2011, 19, 6620–6627 CrossRef CAS PubMed.
- B. Haefner, Drug Discovery Today, 2003, 8, 536–544 CrossRef CAS.
- P. J. Proteau, W. H. Gerwick, F. Garcia-Pichel and R. Castenholz, Experientia, 1993, 49, 825–829 CrossRef CAS.
- A. Kobayashi, S. I. Kajiyama, K. Inawaka, H. Kanzaki and K. Z. Kawazu, Z. Naturforsch., C: Biosci., 1994, 49, 464–470 CAS.
- S. H. Shim, G. Chlipala and J. Orjala, J. Microbiol. Biotechnol., 2008, 18, 1655–1658 CAS.
- V. Bultel-Poncé, F. Felix-Theodose, C. Sarthou, J. F. Ponge and B. Bodo, J. Nat. Prod., 2004, 67, 678–681 CrossRef PubMed.
- A. J. Kresge, Pure Appl. Chem., 1991, 63, 213–221 CrossRef CAS.
- B. Capon, The Chemistry of Enols, ed. Z. Rappoport, Wiley, NY, 1990 Search PubMed.
- S. G. Mills and P. J. Beak, Org. Chem., 1995, 50, 1216–1224 CrossRef.
- W. Blokzijl, J. B. F. N. Engbert and M. J. Blandamer, J. Chem. Soc., Perkin Trans. 2, 1994, 455–458 RSC.
- E. P. Balskus and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 15260–15261 CrossRef CAS PubMed.
- I. G. Fotheringham, S. A. Dacey, P. P. Taylor, T. J. Smith, M. G. Hunter, M. E. Finlay, S. B. Primrose, D. M. Parker and R. M. Edwards, Biochem. J., 1986, 234, 593–604 CAS.
- E. P. Balskus and C. T. Walsh, J. Am. Chem. Soc., 2009, 131, 14648–14649 CrossRef CAS PubMed.
- C. McNeil, J. Natl. Cancer Inst., 1995, 87, 1106–1108 CrossRef CAS PubMed.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, H. Jiang, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, Nature, 2013, 496, 528–532 CrossRef CAS PubMed.
- S. Malla, R. P. Pandey, B. G. Kim and J. K. Sohng, Biotechnol. Bioeng., 2013, 110, 2525–2535 CrossRef CAS PubMed.
- Z. L. Fowler, W. W. Gikandi and M. A. Koffas, Appl. Environ. Microbiol., 2009, 75, 5831–5839 CrossRef CAS PubMed.
- E. Chaignat, E. A. Yahya-Graison, C. N. Henrichsen, J. Chrast, F. Schütz, S. Pradervand and A. Reymond, Genome Res., 2011, 21, 106–113 CrossRef CAS PubMed.
- Z. Shao, G. Rao, C. Li, Z. Abil, Y. Luo and H. Zhao, ACS Synth. Biol., 2013, 15, 662–669 CrossRef PubMed.
- N. R. Sandova, J. Y. Kim, T. Y. Glebes, P. J. Reeder, H. R. Aucoin, J. R. Warner and R. T. Gill, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10540–10545 CrossRef PubMed.
- V. A. Portnoy, D. Bezdan and K. Zengler, Curr. Opin. Biotechnol., 2011, 22, 590–594 CrossRef CAS PubMed.
- M. B. Cassidy, H. Lee and J. T. Trevors, J. Ind. Microbiol., 1996, 16, 79–101 CrossRef CAS.
- F. Jia, B. Narasimhan and S. Mallapragada, Biotechnol. Bioeng., 2013 DOI:10.1002/bit.25136.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed.
- P. Brodelius and J. Vandamme, Biotechnology: A Comprehensive Treatise in Eight Volumes, ed. J. F. Kennedy, VCH Verlagsgesellschaft mbH, Germany, 1987, vol. 7a Search PubMed.
- F. R. Schmidt, Appl. Microbiol. Biotechnol., 2005, 68, 425–435 CrossRef CAS PubMed.
- M. N. Kazarinoff and E. E. Snell, J. Biol. Chem., 1977, 252, 7598–7602 CAS.
- D. Juminaga, E. E. Baidoo, A. M. Redding-Johanson, T. S. Batth, H. Burd, A. Mukhopadhyay, C. J. Petzold and J. D. Keasling, Appl. Environ. Microbiol., 2012, 78, 89–98 CrossRef CAS PubMed.
- M. I. Chávez-Béjar, A. R. Lara, H. López, G. Hernández-Chávez, A. Martinez, O. T. Ramírez, F. Bolívar and G. Gosset, Appl. Environ. Microbiol., 2008, 74, 3284–3290 CrossRef PubMed.
- M. Ikeda, Appl. Microbiol. Biotechnol., 2006, 69, 615–626 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc00118d |
| This journal is © The Royal Society of Chemistry 2014 |