Facile synthesis of 2-methylenecyclobutanones via Ca(OH)2-catalyzed direct condensation of cyclobutanone with aldehydes and (PhSe)2-catalyzed Baeyer–Villiger oxidation to 4-methylenebutanolides†
Lei
Yu
*abc,
Yulan
Wu
a,
Hongen
Cao
a,
Xu
Zhang
ab,
Xinkang
Shi
b,
Jie
Luan
a,
Tian
Chen
a,
Yi
Pan
c and
Qing
Xu
*b
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225002, China. E-mail: yulei@yzu.edu.cn; Fax: (+)-86-514-87975244; Tel: (+)-86-136-65295901
bCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, Zhejiang 325035, China. E-mail: qing-xu@wzu.edu.cn; Fax: (+)-86-577-86689302; Tel: (+)-86-138-57745327
cSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing, Jiangsu 210089, China
First published on 10th October 2013
Abstract
2-Methylenecyclobutanones (2-MCBones), used to be difficult to access, but can now be easily achieved by a green and stereospecific Ca(OH)2-catalyzed direct and simple aldol condensation of cyclobutanone and aldehydes under mild conditions. The obtained (E)-2-MCBones should be a class of potentially useful building blocks in synthesis as they could readily undergo an interesting (PhSe)2-catalyzed Baeyer–Villiger (BV) oxidation with H2O2 at room temperature to give the versatile 4-methylenebutanolides. Mechanistic studies revealed that the BV reaction most possibly proceeded via the initial formation of benzeneseleninoperoxoic anhydride [PhSe(O)O]2O, which then converted to benzeneseleninoperoxoic acid PhSe(O)OOH as the active oxidant, followed by its selective addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of 2-MCBones and then a selective C–C bond cleavage and rearrangement to give 4-methylenebutanolides.
O bond of 2-MCBones and then a selective C–C bond cleavage and rearrangement to give 4-methylenebutanolides.
Introduction
In developing new and efficient methods toward the synthesis of various useful organic skeletons, designing original building blocks (Scheme 1) and exploring their reactivities have become one of the most effective strategies in synthesis.1,2 The recent growing interest in the preparation and applications of methylenecyclopropanes (MCPs) is a typical example in the area,1–5 because the reactive MCPs can be used to construct a wide range of organic skeletons via ring cleavage,3 ring expansion,4 cycloaddition,5 and other types of transformations.1,2 Stimulated by these studies,1–5 methylenecyclobutanes (MCBs), the “sister molecules” of MCPs, were also anticipated to be capable of undergoing analogous transformations such as the ring cleavage and expansion reactions at the cyclobutane moiety. Contrarily, although MCBs are structurally similar to MCPs, literature reports6 and our own results7 revealed that they showed quite different reactivities with each other. Most possibly due to the much lower ring strain and accordingly much lower reactivity of the cyclobutane structure, it usually retained intact in a series of reactions,6,7 making MCBs less useful than the analogous MCPs in synthesis.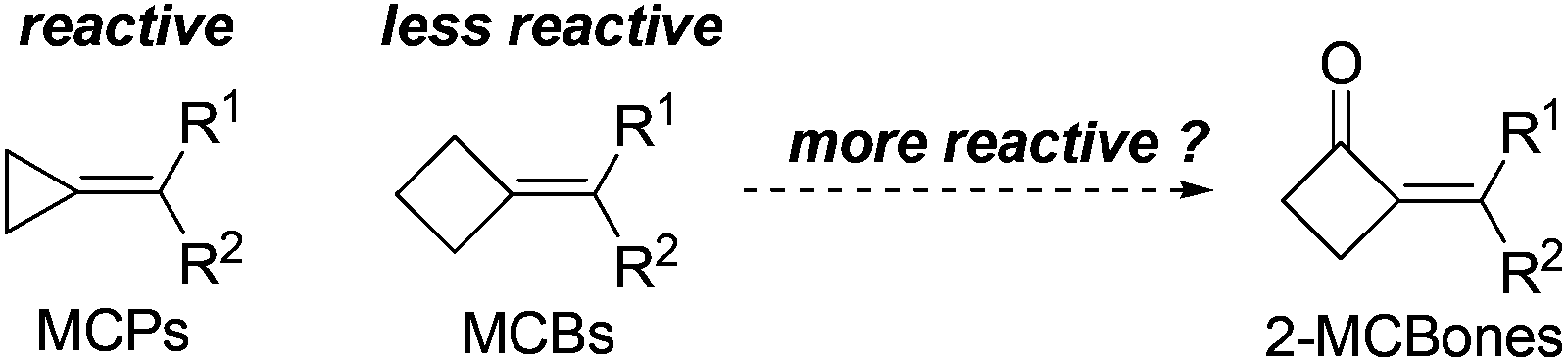 | ||
| Scheme 1 Three structurally-related building blocks. | ||
We envisioned that a direct and simple way to enhance the ring strain and reactivity of MCBs might be converting one of the sp3 carbons in the ring to an sp2 one. Thus, as shown in Scheme 1, 2-methylenecyclobutanones (2-MCBones) may be much more reactive than MCBs for bearing an sp2 ketonic carbon and will possibly provide more synthetic possibilities than MCBs. However, to the best of our knowledge, synthetic applications of 2-MCBones are still very rare to date most possibly because they are still difficult to prepare at present.2,8–12 For example, the groups of Toste and Trost have reported novel methods via Au- or Ru-catalyzed ring expansion reactions of alkynylcyclopropanols to prepare the MCBones,8 but the reported methods for the synthesis of alkynylcyclopropanols have difficulties such as low availability of the substrates, harsh reaction conditions, and multi-step processes.8,9 2-MCBones can also be obtained from other building blocks like MCPs and allenes by cocyclization, cycloaddition, and cycloisomerization reactions,10 but these methods still have their own drawbacks such as low availability and tedious preparation of the substrates, and are thus not practical enough for synthetic purposes. Based on these reports, we initially reckoned that the aldol condensation of cyclobutanone and carbonyl compounds may be a direct and much simpler way to obtain the 2-MCBones. However, although the aldol condensation reactions have been successfully applied in the preparation of various α,β-unsaturated ketones, our literature survey showed that this method was rather unsatisfactory for the preparation of 2-MCBones.11,12 For example, the method initially reported by Conia et al. was low in the selectivities of the products.11 A more recent report described by Ceylan et al. may be a good method for the reactions of a cyclopentene-fused cyclobutanone,12 but we found in our own research that this NaOH-mediated method was ineffective when it was applied to the simple and unsubstituted cyclobutanone, because only <10% GC yield of the target product was detected by employing the reported conditions. All these reports8–12 may also imply that the research on 2-MCBones is most likely bottlenecked by their low availability. Therefore, a convenient and practical preparation method for 2-MCBones is highly desirable in the field. With an ongoing interest in the reactive building blocks,1g,3–5,7 herein we report a green and practical synthesis of (E)-2-MCBones of a Ca(OH)2-catalyzed direct and stereospecific aldol condensation of cyclobutanone and aldehydes, and describe a mild (PhSe)2-catalyzed Baeyer–Villiger oxidation of 2-MCBones to the useful 4-methylenebutanolides.
Results and discussion
Although previous reports11,12 and our preliminary results showed that the aldol condensation reaction may not be a suitable method for the preparation of 2-MCBones, we still chose the strategy for the simplicity, directness, green features, and potential scalability of the method. Thus, an extensive screening on parameters of the reaction conditions was carried out by using cyclobutanone (1) and benzaldehyde (2a) as the model substrates.13 The results showed that, different to previous reports,11,12 the reaction was better to be carried out by refluxing 2a with excess amounts of 114 in EtOH (Table 1), because large amounts of unidentified products could be generated if only stoichiometric amount of 1 was used. By adding excess 1, although the yield of target product 3a was not yet satisfactory, the use of large amounts of base could be avoided. Thus, 10 mol% of NaOH was enough to ensure a smooth reaction to afford 3a in a moderate yield (run 1). More importantly, the obtained 3a was found to be solely the (E)-stereomer as confirmed by NOESY spectroscopic analysis.13 Screening on the loadings of NaOH showed that less NaOH resulted in a lower yield of 3a (run 2) as in the usual cases, but more NaOH surprisingly gave a much lower yield of 3a (run 3), and even no product could be detected when 40 mol% of NaOH was used (run 4). These results showed that NaOH is not a suitable base for the reaction, which may be attributed to the observed NaOH-promoted easy decomposition of 3a.15|
|
||||
|---|---|---|---|---|
| Run | 1 (equiv.) | Base (mol%) | V EtOH (mL) | 3a%b |
| a Excess 1, 2a (1 mmol), and a base in EtOH were heated at 80 °C for 24 h under N2 and then monitored by GC. b GC yields (isolated yields in parenthesis) based on 2a. As determined by NOESY spectroscopic analysis, only the (E)-stereomer of 3a was obtained.13 | ||||
| 1 | 5 | NaOH (10) | 2 | 47 |
| 2 | 5 | NaOH (5) | 2 | 33 |
| 3 | 5 | NaOH (20) | 2 | 3 |
| 4 | 5 | NaOH (40) | 2 | 0 |
| 5 | 5 | Ca(OH)2 (10) | 2 | 68 |
| 6 | 5 | Ca(OH)2 (10) | 3 | 77 (70) |
| 7 | 4 | Ca(OH)2 (10) | 3 | 80 |
| 8 | 3 | Ca(OH)2 (10) | 3 | 83 (75) |
| 9 | 2 | Ca(OH)2 (10) | 3 | 68 |
| 10 | 1 | Ca(OH)2 (10) | 3 | 32 |
| 11 | 3 | Ca(OH)2 (20) | 3 | 75 |
| 12 | 3 | Ca(OH)2 (5) | 3 | 67 |
| 13 | 3 | Ca(OH)2 (1) | 3 | 33 |
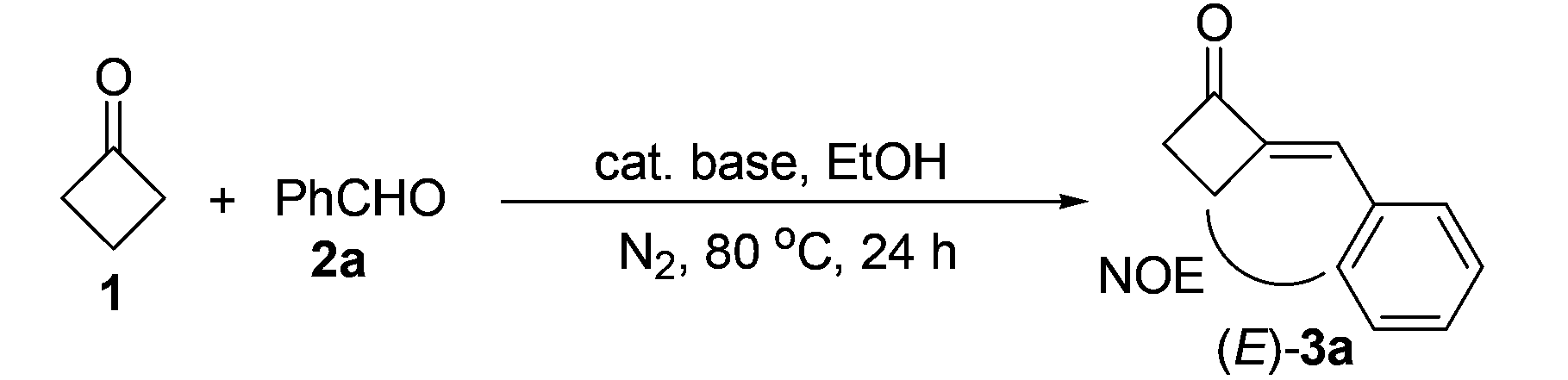
To obtain a better catalyst, a wide range of inorganic and organic bases (LiOH, KOH, CsOH, Mg(OH)2, Ca(OH)2, Sr(OH)2, Ba(OH)2, Fe(OH)3, BeO, MgO, CaO, NaHCO3, Na2CO3, K2CO3, CaCO3, Et3N, DBU, etc.) were then screened.13 To our surprise, the abundant and cheap Ca(OH)2, which is seldom used in organic reactions, was found to be the best base catalyst, giving the highest 68% yield of 3a (run 5) among the tested bases. Solvent and temperature effects were then systematically investigated. The results showed that EtOH is the best solvent and 80 °C the best temperature.13 We also noticed that concentrations of the reactants could also affect the product yield significantly. Thus, either more or less solvent was not beneficial for the reaction,13 and a higher yield of 3a (77%) was achieved by controlling the concentrations of the reactants by using 3 mL of EtOH to a 1 mmol scale reaction (run 6). Further optimization on the ratios of 1/2a showed (runs 6–10) that, although excess amounts of 1 are necessary to achieve a satisfactory reaction with higher yield of 3a and less byproducts,131s loading could be reduced to 3 equiv. without decreasing but slightly enhancing the product's yield (run 8).14 In contrast, less loadings of 1 resulted in a significant drop in the product yield due to the formation of unknown byproducts (runs 9 and 10) as in the case of NaOH.13 In addition, the most appropriate time for the reaction was found to be 24 h, as lower yields of the product were both obtained in shorter or longer reaction times.13 The reaction was also investigated by using more or less amounts of Ca(OH)2 to optimize the best catalyst loading (runs 11–13). Being similar to the trend in the reactions of NaOH (runs 1–4), more Ca(OH)2 resulted in a slight drop in product yield (run 11), and less Ca(OH)2 also led to obviously decreased product yields (runs 12 and 13). As to the reason for this interesting finding, it was observed that 20 mol% bases could lead to decomposition of 3a in varied degrees under the reaction conditions.15 In comparison with other bases such as NaOH that led to total decomposition of 2-MCBones, decomposition can be largely inhibited by using Ca(OH)2 as the catalyst, giving a good recovery yield of 3a.15 All the above results clearly showed that Ca(OH)2 is a much better catalyst than the other bases by acting as both the most effective condensation catalyst and the most inactive decomposition promoter. Since no expensive rare metal catalysts and unavailable substrates are required, this process should be more promising than the known methods,8–12 especially for larger scale preparations. Indeed, a 10 mmol scale reaction of 1 and 2a could readily afford 69% isolated yield of 3a, revealing the synthetic potential of the present Ca(OH)2-catalyzed method.
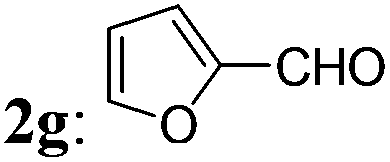
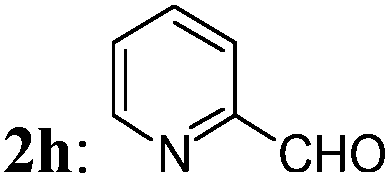
The optimized condition was then applied to prepare various 2-MCBones by using different aldehydes. As shown in Table 2, a series of 2-MCBones could be readily obtained in moderate to good yields via the Ca(OH)2-catalyzed direct aldol condensation of 1 with various aldehydes under similar conditions. Notably, like (E)-3a, all the products obtained were exclusively the (E)-stereomers,13 revealing again the high stereoselectivity of the new method.16 Except 1-naphthaldehyde 2i (run 9), it seemed the reactions of electron-rich aldehydes 2b–2f (runs 1–6), including the sterically more bulky ortho-methyl substituted o-tolualdehyde 2c and mesitaldehyde 2d (runs 3 and 4), and heteroaromatic aldehydes furfural 2g and 2-pyridinecarboxaldehyde 2h (runs 7 and 8), were generally more efficient than those of the electron-deficient ones (runs 10–15), and gave higher yields of the target 2-MCBones. This is most possibly due to the higher reactivities of the electron-deficient aldehydes for the formation of unidentifiable complex byproducts that were observed in the reactions. The reaction of a less electrophilic p-anisaldehdye 2f was not efficient under the standard conditions and therefore had to use more amount of Ca(OH)2 at a higher temperature of 100 °C to give a moderate yield of 3f (run 6). The halogen-substituted electron-deficient 2-MCBones 3j–3n should be potentially useful reagents in synthesis especially in coupling reactions for bearing the reactive halide groups (runs 10–14). In addition, an α,β-unsaturated aldehyde, cinnamaldehyde 2p, could also give the desired 2-MCBone 3p, albeit in a lower yield (run 16). However, although we tried many times, this method is not yet suitable for linear aliphatic aldehydes (such as phenylacetaldehyde, phenylpropyl aldehyde, and n-hexanal) and ketones (such as acetophenone and diphenyl ketone) to prepare the alkyl- or di-substituted 2-MCBones, possibly due to the relatively lower reactivity of the aliphatic aldehydes and the greater steric hindrance of the ketones. Differently, we found hexahydrobenzaldehyde 2q could smoothly afford the corresponding 2-MCBone 3q under the optimized conditions, albeit in a low yield of 32% (run 17). Finally, the simplest aldehyde, formaldehyde 2r, was also employed to prepare the unsubstituted 2-MCBone. Unfortunately, no target product could be obtained under several conditions by using either aqueous formaldehyde solution or paraformaldehyde as the reagent (run 18).
|
|
||
|---|---|---|
| Run | 2: RCHO | 3: Yield%b |
| a Unless otherwise noted, see run 8 of Table 1 for detailed conditions. b Isolated yields based on 2. c 30 mol% of Ca(OH)2, 100 °C. | ||
| 1 | 2a: PhCHO | 3a: 75 |
| 2 | 2b: 4-CH3C6H4CHO | 3b: 73 |
| 3 | 2c: 2-CH3C6H4CHO | 3c: 66 |
| 4 | 2d: 2,4,6-(CH3)3C6H2CHO | 3d: 81 |
| 5 | 2e: 4-t-BuC6H4CHO | 3e: 73 |
| 6c | 2f: 4-CH3OC6H4CHO | 3f: 54 |
| 7 |
|
3g: 63 |
| 8 |
|
3h: 52 |
| 9 | 2i: 1-C10H7CHO | 3i: 44 |
| 10 | 2j: 4-FC6H4CHO | 3j: 50 |
| 11 | 2k: 4-ClC6H4CHO | 3k: 54 |
| 12 | 2l: 4-BrC6H4CHO | 3l: 51 |
| 13 | 2m: 2-BrC6H4CHO | 3m: 44 |
| 14 | 2n: 3-BrC6H4CHO | 3n: 48 |
| 15 | 2o: 4-CF3C6H4CHO | 3o: 52 |
| 16 |
2p: (E)-PhCHCHCHO |
3p: 39 |
| 17 |
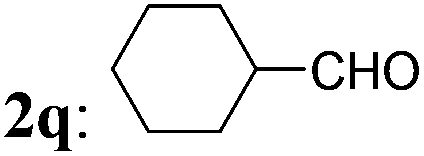
|
3q: 32 |
| 18 | 2r: CH2O | 3r: — |
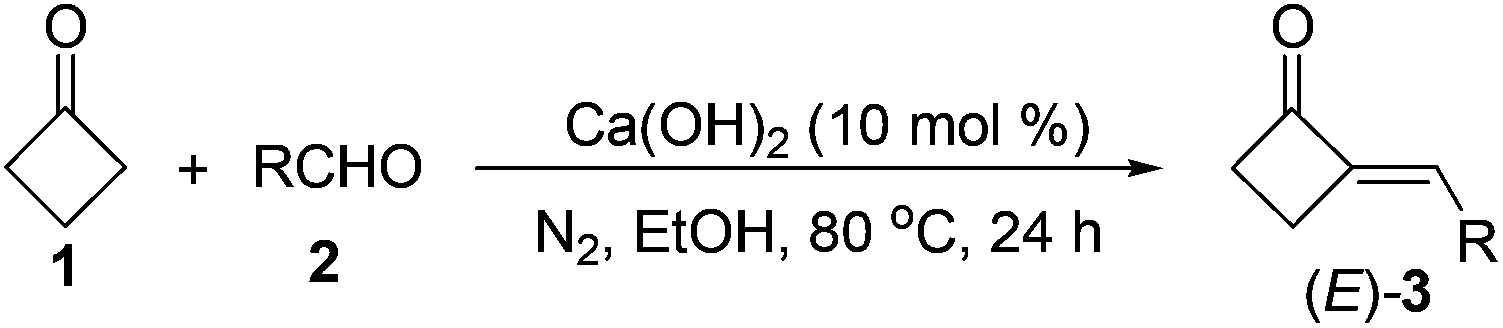
With 2-MCBones in hand, we then turn to investigate their reactivities and potential applications in synthesis. The reaction of (E)-3a with various oxidants such as m-chloroperoxybenzoic acid (MCPBA) in the absence or presence of catalysts was firstly explored. We initially anticipated that, being similar to the ring expansion reactions of MCPs and MCBs17 in both molecular structures and reaction conditions (eqn (1)), 3a may also afford 2-hydroxycyclopentenone 5avia an analogous process under the oxidative conditions (eqn (2)). However, contrary to our expectations and different to the known ring expansion reactions17 and a ring contraction reaction of various cyclic ketones,18 a new compound was obtained under the similar oxidative conditions, which was later determined to be the Baeyer–Villiger (BV) oxidation product of (E)-3a, namely, (E)-4-benzylidenebutanolide 4a19 (eqn (2)). This result may also imply the effect of the carbonyl group in 2-MCBones on the selectivity of the reaction even under similar conditions (vide infra). It is well established that the 4-methylenebutanolide moiety has various biological activities20 and exists widely in natural products such as cytotoxic compounds from the stems of Cinnamomum tenuifolium.20a They are also a class of important synthetic skeleton in many fields such as organic and pharmaceutical synthesis, biochemistry and medicinal chemistry.20 Therefore, conditions of the BV reaction were further screened and optimized.
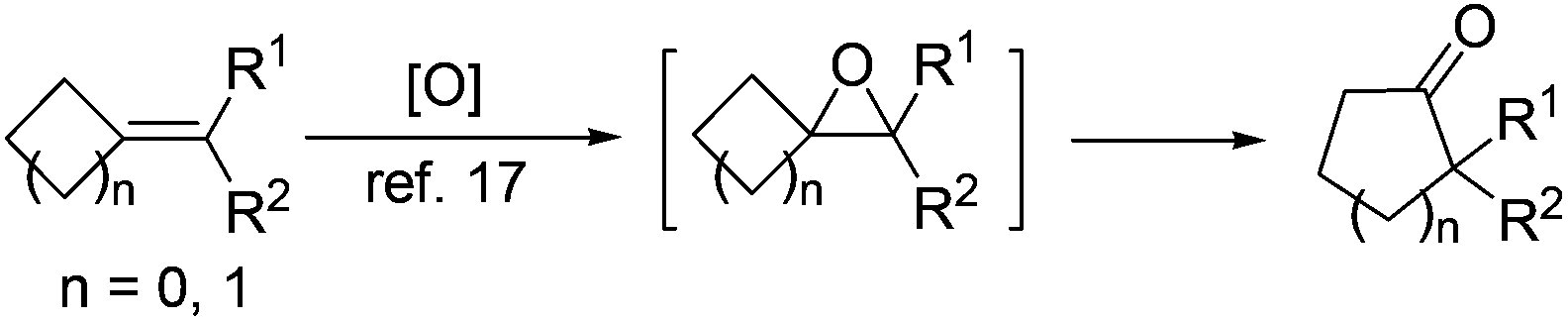 | (1) |
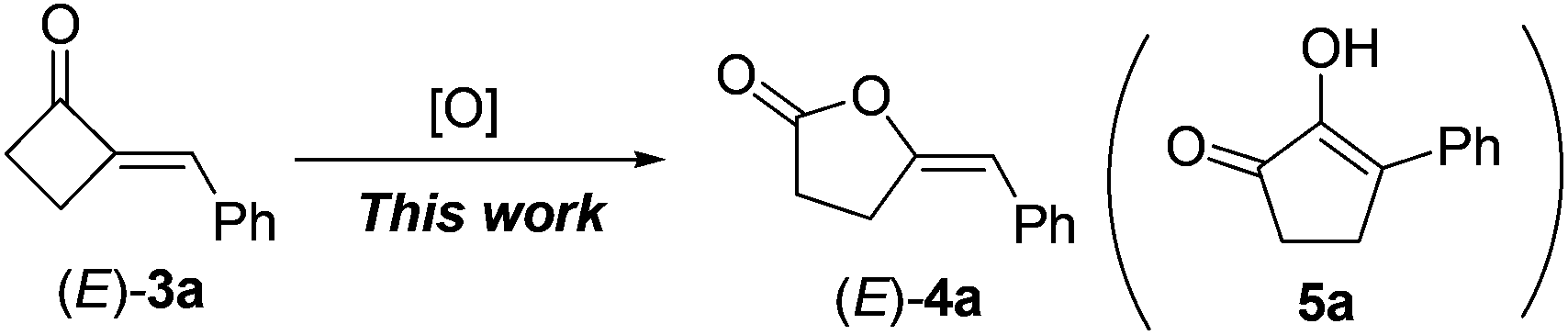 | (2) |
As shown in Table 3, (E)-3a was initially treated with MCPBA at room temperature, affording (E)-4a in only a low yield (run 1). Other oxidants such as H2O2, benzoyl peroxide (BPO), PhI(OAc)2·H2O, and SeO2 were also tested to achieve better results. However, without any catalyst, conversion of 3a to 4a by using these oxidants was even less effective than MCPBA (runs 2–5). Interestingly, when selenium powder was added as the catalyst in the reaction of H2O2, it dramatically promoted the totally ineffective blank reaction (run 2) to occur to afford 24% yield of 4a (run 6). Similarly, although SeO2 was not effective at all by itself as the oxidant (run 5), it could catalyze the reaction by using H2O2 as the oxidant to give 21% yield of 4a (run 7). These results indicated that, as in the reported reactions of cyclic ketones,18,23 certain selenium species are most possibly generated to ensure the BV oxidation of 2-MCBones. Therefore, several chalcogen catalysts were screened by using the greener H2O2 as the oxidant. As shown in the table, (PhS)2 was found to be totally ineffective under the same condition (run 8) and (PhSe)2 is contrarily the best catalyst for the reaction, giving 4a in 82% isolated yield (run 9). Further condition screening on reaction temperature (runs 10 and 11), catalyst loadings (runs 12–14), and H2O2 loadings (runs 15 and 16) revealed that the reaction was best carried out by using 5 mol% of (PhSe)2 and 5 equiv. of H2O2 at room temperature (run 9).
|
|
||||
|---|---|---|---|---|
| Run | Cat. (mol%) | [O] (equiv.)b | t | 4a%c |
| a The mixture of 3a (0.3 mmol), catalyst, and oxidant was stirred in CH3CN (1 mL) for 24 h and then monitored by TLC. b H2O2 of 30% (w/w) was used. c Isolated yields based on 3a. d Ca. 30 °C. | ||||
| 1 | — | MCPBA (1.5) | r.t.d | 35 |
| 2 | — | H2O2 (5) | r.t. | 0 |
| 3 | — | BPO (1.5) | r.t. | 26 |
| 4 | — | PhI(OAc)2·H2O (1.5) | r.t. | 12 |
| 5 | — | SeO2 (1.5) | r.t. | 0 |
| 6 | Se (5) | H2O2 (5) | r.t. | 24 |
| 7 | SeO2 (5) | H2O2 (5) | r.t. | 21 |
| 8 | (PhS)2 (5) | H2O2 (5) | r.t. | 0 |
| 9 | (PhSe)2 (5) | H 2 O 2 (5) | r.t. | 82 |
| 10 | (PhSe)2 (5) | H2O2 (5) | 40 °C | 47 |
| 11 | (PhSe)2 (5) | H2O2 (5) | 60 °C | 33 |
| 12 | (PhSe)2 (1) | H2O2 (5) | r.t. | Trace |
| 13 | (PhSe)2 (3) | H2O2 (5) | r.t. | 42 |
| 14 | (PhSe)2 (10) | H2O2 (5) | r.t. | 58 |
| 15 | (PhSe)2 (5) | H2O2 (3) | r.t. | 40 |
| 16 | (PhSe)2 (5) | H2O2 (8) | r.t. | 67 |
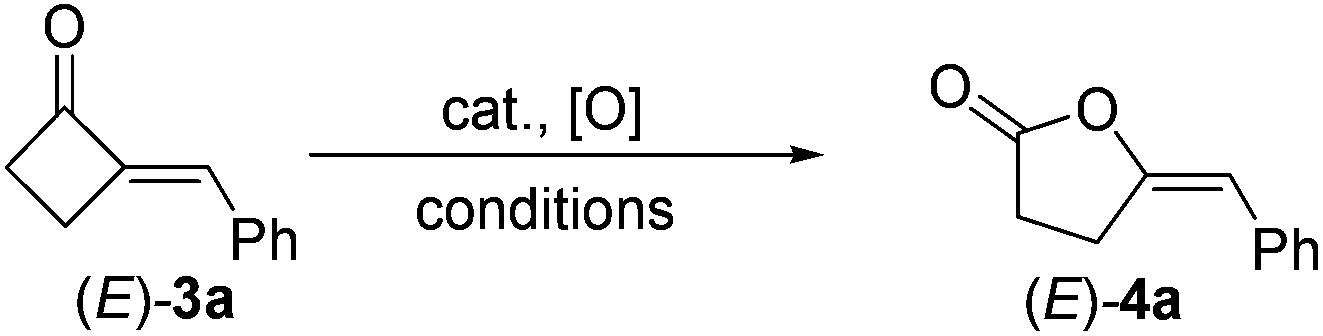
This optimal condition was then applied in the synthesis of various (E)-4-methylenebutanolides 4 from the corresponding 2-MCBones 3 (Table 4).19 Thus, by treating with H2O2, the (PhSe)2-catalyzed BV oxidation of (E)-3 readily occurred at room temperature to give (E)-4 in moderate to good yields.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,19
a,19
|
|
||
|---|---|---|
| Run | 3: R | 4: Yield%b |
| a See run 9 of Table 3 for reaction conditions. b Isolated yields based on 3. | ||
| 1 | 3a: Ph | 4a: 82 |
| 2 | 3b: 4-CH3C6H4 | 4b: 73 |
| 3 | 3c: 3-CH3C6H4 | 4c: 61 |
| 4 | 3d: 2,4,6-(CH3)3C6H2 | 4d: 65 |
| 5 | 3e: 4-t-BuC6H4 | 4e: 67 |
| 6 | 3f: 4-CH3OC6H4 | 4f: 54 |
| 7 | 3i: 1-C10H7 | 4i: 53 |
| 8 | 3j: 4-FC6H4 | 4j: 58 |
| 9 | 3l: 4-BrC6H4 | 4l: 50 |
| 10 | 3m: 2-BrC6H4 | 4m: 40 |
| 11 | 3n: 3-BrC6H4 | 4n: 67 |
| 12 | 3q: cyclo-C6H11 | 4q: 42 |
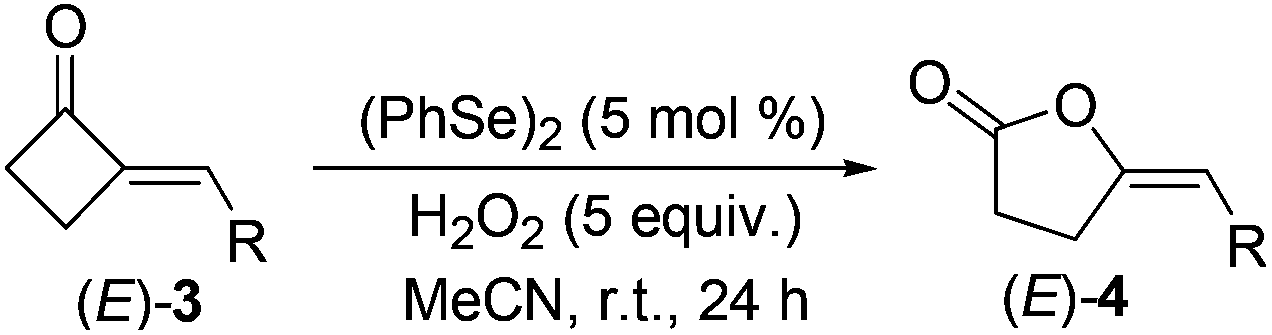
It is interesting that even under similar oxidative conditions 2-MCBones behaved differently to the usual strained ring compounds MCPs or MCBs.17 The mechanism of the BV oxidation of 2-MCBones 3 is thus our next concern.
Firstly, the reactive organoselenium species in the reaction was investigated. It has been documented that (PhSe)2 can be readily oxidized to benzeneseleninic acid (A)21 in the presence of oxidants; whereas A may afford benzeneseleninoperoxoic acid (B) upon further oxidation with excess amounts of oxidants (Scheme 2, path a).22–24 Thus, whether A or B is the key oxidative catalyst in the present reaction was investigated. As shown in eqn (3), 3a was firstly mixed with 1 equiv. of A under the standard condition, but no target reaction occurred at all with most of 3a recovered. This result ruled out the possibility of A to oxidize 3a in the reaction mechanism. Since B is a highly oxidative oxidant unstable under usual conditions, we next investigated the reaction of 3a with excess H2O2 in the presence of a catalytic amount of A (eqn (4)). In contrast to the previous negative result (eqn (3)), in this case the BV oxidation readily occurred and gave 76% isolated yield of 4a (eqn (4)). These results indicated that B, the oxidation product of A in the presence of oxidants, is most likely the active oxidation catalyst in the reaction.
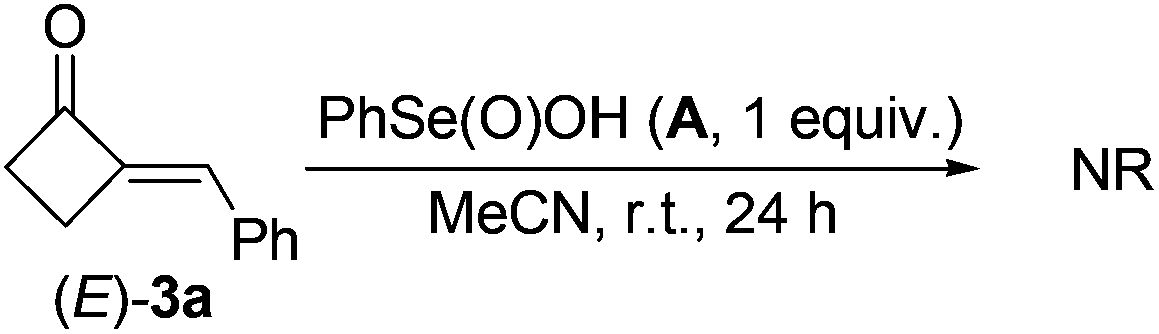 | (3) |
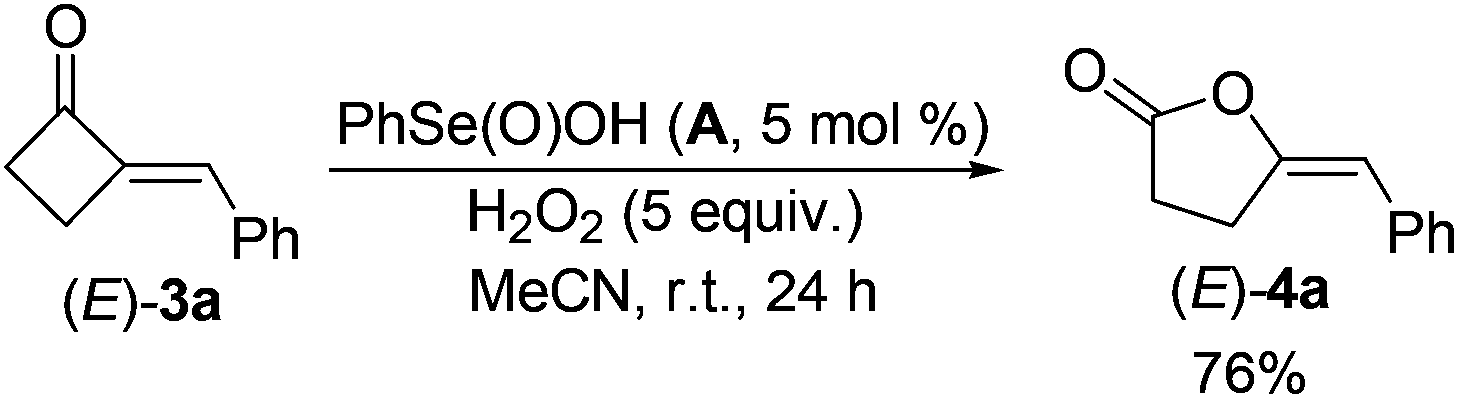 | (4) |
 | ||
| Scheme 2 77Se NMR studies on reactive organoselenium species. | ||
However, 77Se NMR spectroscopic analysis revealed that a new peak at 1248 ppm that is consistent with the literature data of benzeneseleninoperoxoic anhydride [PhSe(O)O]2O (C, 1241 ppm)25 was only observed when (PhSe)2 was treated with H2O2.13 This result indicated that, different to the generally-accepted view that A was the first oxidation intermediate in the reaction of (PhSe)2 and oxidants (Scheme 2, path a),22–24 a new organoselenium species, [PhSe(O)O]2O (C), was most possibly the first generated oxidation intermediate in the reaction (Scheme 2, path b). Further 77Se NMR spectroscopic analysis also revealed that C could convert to B and then A slowly by standing or quickly by vigorous stirring.13 Clearly, the latter results are consistent with the viewpoint of the literature,22–24 especially when vigorous stirring was applied to the reaction.
On the other hand, due to the presence of a carbonyl group adjacent to the exocyclic CC double bond in 2-MCBones that may control the chemoselectivity of the reaction, the present reaction should proceed via a different mechanism to the known ring expansion or ring contraction reactions of MCPs, MCBs, and cyclic ketones,17,18 but similar to those of the oxidative BV oxidations of cyclic ketones.23
Therefore, based on our above findings including the 77Se NMR spectroscopic analysis of the involved organoselenium species (Scheme 2) and the literature on organoselenium-catalyzed oxidation reactions,22–24 a possible mechanism was proposed for the present organoselenium-catalyzed BV oxidation of 2-MCBones. As shown in Scheme 3, (PhSe)2 might firstly be oxidized by H2O2 to give benzeneseleninoperoxoic anhydride [PhSe(O)O]2O (C), which upon hydrolysis may give the active oxidant B. Then, most probably due to the higher reactivity of the carbonyl moiety, nucleophilic addition of B to the CO bond23 should be more preferable to take place than the epoxidation of the exocyclic CC bond,17 which then leads to the formation of intermediate 6 rather than 7. In the next step, although both path c and d are possible routes for the C–C bond cleavage of 6, a regiospecific reaction (path c) and rearrangement of 6 should take place to give the final product (E)-4-methylenebutanolides 4 and release free acid A. In the presence of excess H2O2, A should then be oxidized to regenerate the active oxidant B.22–24 Moreover, the selectivity of the C–C bond cleavage, path c but not d, is most possibly attributed to the activation by the adjacent CC bond, which could also be supported by the findings in ring cleavage reactions of MCPs and MCBs.3–5,6g,17 In addition, different to the known BV oxidations of the usual cyclic ketones including cyclobutanones that require not readily available substituted diaryl diselenides as the effective precatalysts,23 the present reaction can readily use the simplest, cheapest, and most available (PhSe)2 as the catalyst precursor. This might be attributed to the presence of an adjacent exocyclic CC double bond that contributes to activating the CO bond in 2-MCBones 3.
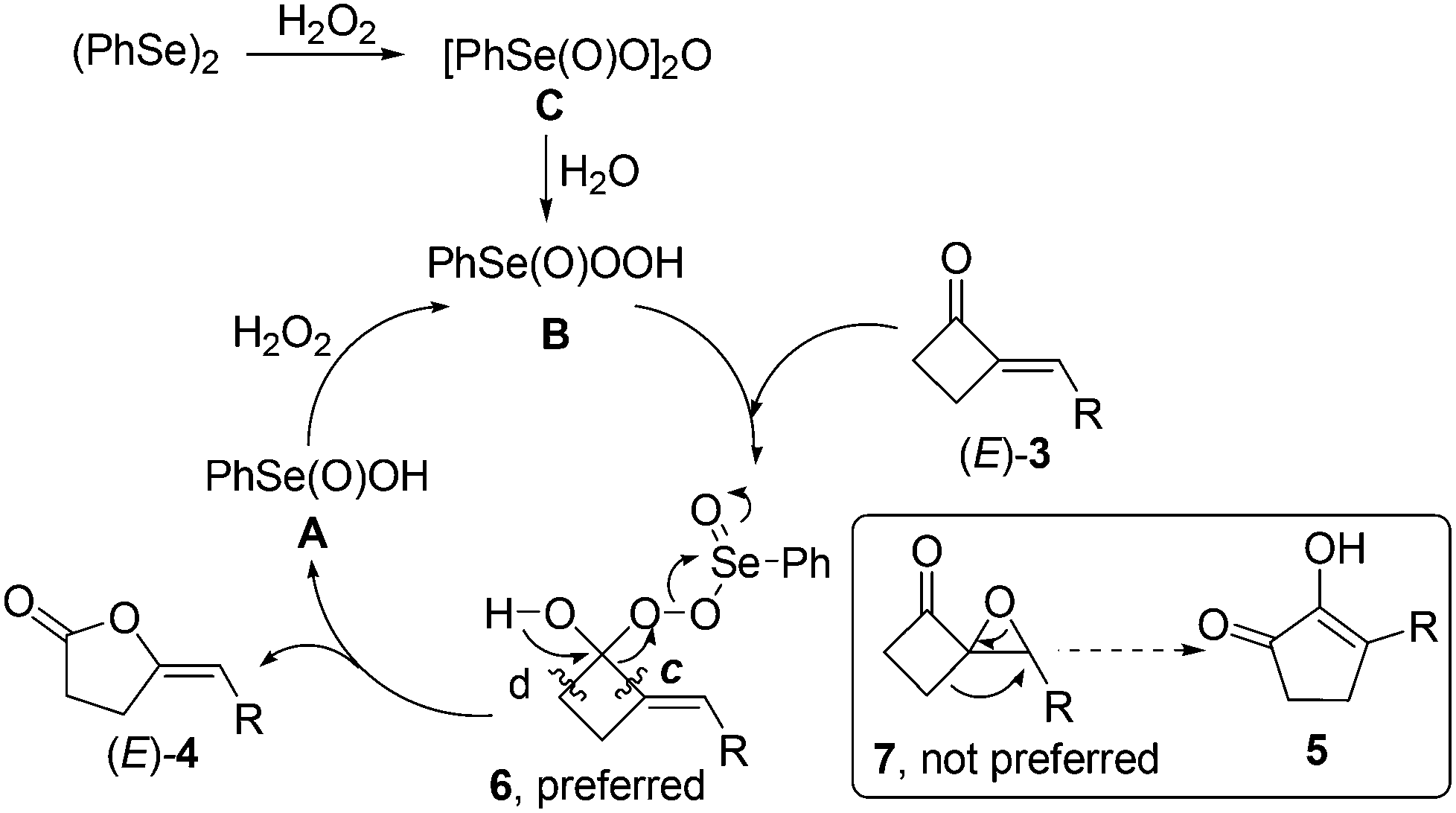 | ||
| Scheme 3 Proposed mechanism for (PhSe)2-catalyzed Baeyer–Villiger oxidation of 2-MCBones 3 to 4-methylenebutanolides 4. | ||
Conclusion
In conclusion, we have developed a green and concise method for the preparation of 2-methylenecyclobutanones (2-MCBones) via a stereospecific and (E)-selective Ca(OH)2-catalyzed direct aldol condensation of cyclobutanone and aldehydes, which is to the best of our knowledge a comparatively simpler and more practical method to obtain the difficult-to-be-accessed 2-MCBones. 2-MCBones should be more reactive and potentially more useful building blocks than analogous MCBs as they could be readily converted to the useful (E)-4-methylenebutanolides under mild conditions by a (PhSe)2-catalyzed Baeyer–Villiger oxidation by using H2O2 as the green oxidant. Mechanistic studies revealed that the reaction most possibly proceeded via the initial formation of [PhSe(O)O]2O from (PhSe)2 by oxidation, followed by conversion to PhSe(O)OOH as the active oxidant, which was then selectively added to the CO bond of 2-MCBones to undergo a selective C–C bond cleavage and rearrangement to give the 4-methylenebutanolides. More methods for the preparation of different MCBones and their potential utilities in synthesis via ring cleavage, ring expansion, and cycloaddition reactions are underway.
Experimental section
General methods
The chemicals, cyclobutanone, aldehydes, bases, solvents, and organoselenium catalysts were all purchased. Liquid aldehydes were redistilled under vacuum before use. Solid aldehydes were recrystallized in EtOH–H2O under N2 before use. All the reactions were monitored by TLC and/or GC analysis. GC yields were calculated according to the internal standard curve. (E)-2-methylenecyclobutanones 3 and (E)-4-methylenebutanolides 4 were all purified by column chromatogram. 1H and 13C NMR and NOESY spectra were recorded on a Bruker Avance 600 instrument (600 MHz for 1H and 150 MHz for 13C NMR spectroscopy) by using CDCl3 as the solvent and Me4Si as the internal standard. Chemical shifts for 1H and 13C NMR were referred to internal Me4Si (0 ppm) and J-values were shown in Hz. 77Se NMR were recorded on an Agilent DD2 600 instrument (114 MHz) by using D2O as the solvent. Melting points were measured using a WRS-2A digital instrument. Mass spectra were measured on a Thermo Trace DSQ II or a Shimadzu GCMS-QP2010 Ultra spectrometer (EI). Elemental analysis was performed on an Elementar Vario EL cube instrument. HRMS (ESI) analysis was performed using a Bruker microTOF-Q II instrument.General procedure for preparation of (E)-2-MCBones 3
To a 10 mL round-bottomed flask was added 0.1 mmol of Ca(OH)2. A solution of aldehyde 2 (1.0 mmol) and cyclobutanone 1 (3.0 mmol) in 3 mL of anhydrous ethanol was then injected via a syringe under N2. The mixture was then stirred at 80 °C for 24 hours under N2. The solvent was evaporated under vacuum and the residue was purified through flash column chromatogram (eluent: petroleum ether–EtOAc 9:1) to give (E)-3.
General procedure for (PhSe)2-catalyzed Baeyer–Villiger oxidation of (E)-2-MCBones 3 to (E)-4-methylenebutanolides with H2O2
(E)-2-MCBones 3 (0.3 mmol) and (PhSe)2 (0.015 mmol) were added to a reaction tube. A solution of H2O2 (1.5 mmol) in 1 mL of CH3CN was then injected via a syringe. The mixture was then stirred at room temperature (ca. 30 °C) for 24 h. After the reaction was complete as monitored by TLC, 2 mL of water was added and the mixture was extracted with EtOAc (2 mL × 3). The combined organic layer was dried over Na2SO4 and the solvent evaporated under vacuum. The residue was purified by flash column chromatogram (eluent: petroleum ether–EtOAc 8:1) to give (E)-4.
Acknowledgements
We thank NNSFC (21202141, 20902070), Priority Academic Program Development of Jiangsu Higher Education Institutions, Opening Foundation of the Key Laboratory of Environmental Materials and Engineering of Jiangsu Province (K11024), and Opening Foundation of the Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University (2010GDGP0106) for financial support. We also thank Prof. Mark Lautens and Dr Joel Tang (University of Toronto) for direction and assistance in 77Se NMR measurement, the anonymous reviewers for their invaluable advices, and the Analysis Center of Yangzhou University.Notes and references
- For reviews on MCPs: (a) A. Brandi and A. Goti, Chem. Rev., 1998, 98, 589 CrossRef CAS PubMed; (b) E. Nakamura and S. Yamago, Acc. Chem. Res., 2002, 35, 867 CrossRef CAS PubMed; (c) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef CAS; (d) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS PubMed; (e) H. Pellissier, Tetrahedron, 2010, 66, 8341 CrossRef CAS; (f) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883 CrossRef CAS PubMed; (g) L. Yu and R. Guo, Org. Prep. Proced. Int., 2011, 43, 209 CrossRef CAS.
- D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272 CrossRef CAS.
- (a) T. Kippo, K. Hamaoka and I. Ryu, J. Am. Chem. Soc., 2013, 135, 632 CrossRef CAS PubMed; (b) K. Ogata, D. Shimada, S. Furuya and S.-I. Fukuzawa, Org. Lett., 2013, 15, 1182 CrossRef CAS PubMed; (c) L.-X. Shao, J. Li, B.-Y. Wang and M. Shi, Eur. J. Org. Chem., 2010, 6448 CrossRef CAS; (d) D.-H. Zhang, L.-Z. Dai and M. Shi, Eur. J. Org. Chem., 2010, 5454 CrossRef CAS; (e) L. Yu and X. Huang, Synlett, 2007, 1371 CAS; (f) L. Yu and X. Huang, Synlett, 2006, 2136 CAS; (g) X. Huang and L. Yu, Synlett, 2005, 2953 CrossRef CAS.
- (a) M.-Z. Miao, J. Cao, J.-J. Zhang, X. Huang and L.-L. Wu, Org. Lett., 2012, 14, 2718 CrossRef CAS PubMed; (b) Z. Zhang and M. Shi, Tetrahedron Lett., 2011, 52, 6541 CrossRef CAS; (c) L. Yu, L.-F. Ren, R. Yi, Y.-L. Wu, T. Chen and R. Guo, J. Organomet. Chem., 2011, 696, 2228 CrossRef CAS; (d) L. Yu, J.-D. Meng, L. Xia and R. Guo, J. Org. Chem., 2009, 74, 5087 CrossRef CAS PubMed; (e) M. Shi, L.-P. Liu and J. Tang, J. Am. Chem. Soc., 2006, 128, 7430 CrossRef CAS PubMed; (f) A. Fürstner and C. Aïssa, J. Am. Chem. Soc., 2006, 128, 6306 CrossRef PubMed; (g) Ref. 3d and 3e .
- (a) L. Yu, Y.-L. Wu, T. Chen, Y. Pan and Q. Xu, Org. Lett., 2013, 15, 144 CrossRef CAS PubMed; (b) K. Chen, M. Jiang, Z. Zhang, Y. Wei and M. Shi, Eur. J. Org. Chem., 2011, 7189 CrossRef CAS; (c) S.-Y. Li, Y. Luo and J. Wu, Org. Lett., 2011, 13, 3190 CrossRef CAS PubMed; (d) B. Hu, J.-L. Zhu, S.-Y. Xing, J. Fang, D. Du and Z.-W. Wang, Chem.–Eur. J., 2009, 15, 324 CrossRef CAS PubMed; (e) M. Gulías, J. Durán, F. López, L. Castedo and J. L. Mascareñas, J. Am. Chem. Soc., 2007, 129, 11026 CrossRef PubMed; (f) P. A. Wender, L. O. Haustedt, J. Lim, J. A. Love, T. J. Williams and J.-Y. Yoon, J. Am. Chem. Soc., 2006, 128, 6302 CrossRef CAS PubMed; (g) T. Kurahashi and A. de Meijere, Angew. Chem., Int. Ed., 2005, 44, 7881 CrossRef CAS PubMed; (h) A. I. Siriwardana, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2004, 69, 3202 CrossRef CAS PubMed.
- (a) F. M. Cordero, C. Vurchio, A. Brandi and R. Gandolfi, Eur. J. Org. Chem., 2011, 5608 CrossRef CAS; (b) M. Jiang and M. Shi, Tetrahedron, 2009, 65, 798 CrossRef CAS; (c) M. Jiang and M. Shi, J. Org. Chem., 2009, 74, 2516 CrossRef CAS PubMed; (d) M. Jiang, L.-P. Liu and M. Shi, Tetrahedron, 2007, 63, 9599 CrossRef CAS; (e) L.-P. Liu, J.-M. Lu and M. Shi, Org. Lett., 2007, 9, 1303 CrossRef CAS PubMed . For limited examples on ring cleavage reactions of MCBs: ; (f) C. Aissa, D. Crépin, D. J. Tetlow and K. Y. T. Ho, Org. Lett., 2013, 15, 1322 CrossRef CAS PubMed; (g) W. Li and M. Shi, Tetrahedron, 2007, 63, 11016 CrossRef CAS; (h) M. Jiang and M. Shi, Org. Lett., 2008, 10, 2239 CrossRef CAS PubMed.
- (a) L. Yu, L.-F. Ren, R. Yi, Y.-L. Wu, T. Chen and R. Guo, Synlett, 2011, 579 CrossRef CAS; (b) L. Yu, L.-F. Ren, Y. Rong and R. Guo, Synth. Commun., 2011, 41, 2530 CrossRef CAS; (c) L. Yu, L.-F. Ren, B. Xu and R. Guo, Synth. Commun., 2011, 41, 3237 CrossRef CAS.
- (a) J. P. Markham, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 9708 CrossRef CAS PubMed; (b) B. M. Trost, J. Xie and N. Maulide, J. Am. Chem. Soc., 2008, 130, 17258 CrossRef CAS PubMed.
- (a) J. Salaün, J. Org. Chem., 1976, 41, 1237 CrossRef; (b) J. Salaün, F. Bennani, J.-C. Compain, A. Fadel and J. Oliver, J. Org. Chem., 1980, 45, 4129 CrossRef; (c) N. Iwasawa, T. Matsuo, M. Iwamoto and T. Ikeno, J. Am. Chem. Soc., 1998, 120, 3903 CrossRef CAS.
- (a) M. Toyofuku, S. Fujiwara, T. Shin-ike, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2005, 127, 9706 CrossRef CAS PubMed; (b) M. Bertrand, R. Maurin, J. L. Gras and G. Gil, Tetrahedron, 1975, 31, 849 CrossRef CAS; (c) M. Bertrand, J.-L. Gras and J. Gore, Tetrahedron, 1975, 31, 857 CrossRef CAS; (d) Ref. 4a and 5g .
- (a) J. M. Conia and J. P. Sandre, Bull. Soc. Chim. Fr., 1963, 744 CAS; (b) P. Thieme, Chem. Ber., 1968, 101, 378 CrossRef CAS.
- M. Ceylan and E. Fındık, Synth. Commun., 2009, 39, 1046 CrossRef CAS.
- See ESI† for details.
- In our hands, cyclobutanone 1 can be purchased from Sichuan Hongsheng Bio-tech Co. Ltd at a lower price (∼$611 per kg) compared to the usual commercial sources (∼$116 per g). Besides, excess amounts of 1 used can be readily recovered from the reaction residue by distillation and reused.
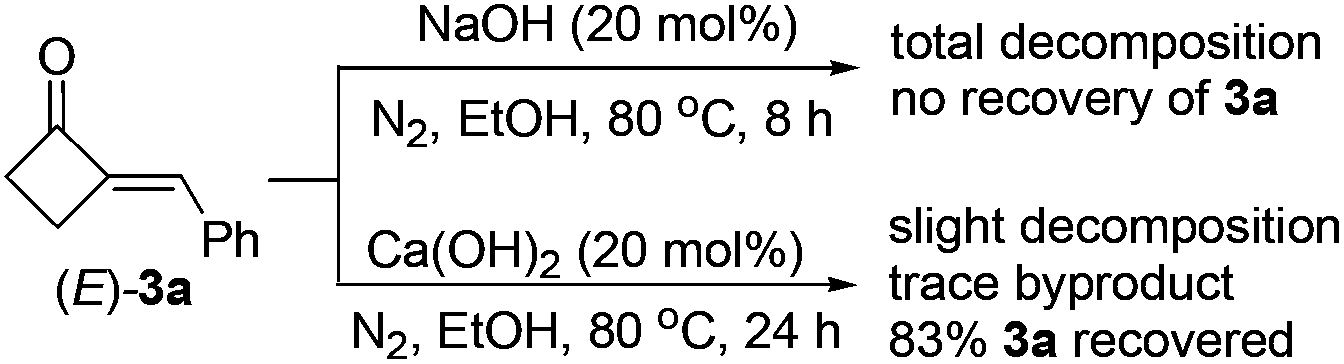
- We later observed that, in the presence of 20 mol% of NaOH, MCBone 3a totally decomposed under the reaction conditions in only 8 h (see below). This result can give the reason why NaOH is not a suitable base catalyst for the aldol condensation of 1 and aldehydes. In contrast, only slight decomposition of 3a was observed and a high yield of 3a could be recovered when 20 mol% of Ca(OH)2 was
used. These results also revealed that Ca(OH)2 is a much better catalyst than other bases for the preparation of 2-MCBones, because decomposition of the in situ generated 2-MCBones can be largely inhibited in the presence of Ca(OH)2.
. - The (E)-stereoselectivity in the present reactions is contrary to the (Z)-stereoselectivity in some of the known methods (ref. 8, 10), being a simple and good complement for the preparation of sterically different 2-MCBones.
- For a review and reports on ring expansion reactions of the related building blocks: (a) E. Leemans, M. D'hooghe and N. De Kimpe, Chem. Rev., 2011, 111, 3268 CrossRef CAS PubMed; (b) W. Yuan, X. Dong, Y. Wei and M. Shi, Chem.–Eur. J., 2012, 18, 10501 CrossRef CAS PubMed; (c) T. O. Painter, P. D. Thornton, M. Orestano, C. Santini, M. G. Organ and J. Aube, Chem.–Eur. J., 2011, 17, 9595 CrossRef CAS PubMed; (d) Y.-M. Shen, B. Wang and Y. Shi, Angew. Chem., Int. Ed., 2006, 45, 1429 CrossRef CAS PubMed; (e) M. C. Pirrung and S. A. Thomson, J. Org. Chem., 1988, 53, 227 CrossRef CAS; (f) R. G. Gadwood, L. M. Mallick and A. J. DeWinter, J. Org. Chem., 1987, 52, 774 CrossRef CAS; (g) J. K. Crandall and W. W. Conover, J. Org. Chem., 1978, 43, 3533 CrossRef CAS; (h) B. M. Trost and L. H. Latimer, J. Org. Chem., 1978, 43, 1031 CrossRef CAS; (i) Ref. 6f .
- M. Giurg and J. Mlochowski, Synth. Commun., 1999, 29, 2281 CrossRef CAS.
- The stereochemistry of (E)-4 was determined by comparing the NMR spectra with those of (E)-3 (ref. 13), known (Z)- and (E)-4 described in the literature. See: (a) C. Sun, Y. Fang, S. Li, Y. Zhang, Q. Zhao, S. Zhu and C. Li, Org. Lett., 2009, 11, 4084 CrossRef CAS PubMed; (b) G. Tsolomiti and A. Tsolomitis, Heterocycl. Commun., 2006, 12, 93 CAS.
- (a) R.-J. Lin, M.-J. Cheng, J.-C. Huang, W.-L. Lo, Y.-T. Yeh, C.-M. Yen, C.-M. Lu and C.-Y. Chen, J. Nat. Prod., 2009, 72, 1816 CrossRef CAS PubMed; (b) X. Li, F. Wang, N. Dong and J.-P. Cheng, Org. Biomol. Chem., 2013, 11, 1451 RSC; (c) X. Ji, Y. Zhou, J.-F. Wang, L.-X. Zhao, H.-L. Jiang and H. Liu, J. Org. Chem., 2013, 78, 4312 CrossRef CAS PubMed; (d) C. R. Johnson, S. K. Gorla, M. Kavitha, M.-J. Zhang, X.-P. Liu, B. Striepen, J. R. Mead, G. D. Cuny and L. Hedstrom, Bioorg. Med. Chem. Lett., 2013, 23, 1004 CrossRef CAS PubMed; (e) P. A. Procopiou, C. Browning, P. M. Gore, S. M. Lynn, S. A. Richards, R. J. Slack and S. L. Sollis, Bioorg. Med. Chem., 2012, 20, 6097 CrossRef CAS PubMed; (f) G. Tsolomiti and A. Tsolomitis, Heterocycl. Commun., 2006, 12, 93 CAS; (g) I. Regla, A. Reyes, C. Korber, P. Demare, O. Estrada and E. Juaristi, Synth. Commun., 1997, 27, 817 CrossRef CAS; (h) W. Kirmse and K. Kund, J. Am. Chem. Soc., 1989, 111, 465 Search PubMed; (i) E. Guntrum, W. Kuhn, W. Spoenlein and V. Jaeger, Synthesis, 1986, 921 CrossRef CAS; (j) M. Yamamoto, J. Chem. Soc., Chem. Commun., 1978, 649 RSC.
- D. Dowd and P. Gettins, Magn. Reson. Chem., 1988, 26, 1 CrossRef CAS.
- L. Syper and J. Mlochowski, Tetrahedron, 1987, 43, 207 CrossRef CAS.
- (a) G.-J. ten Brink, J.-M. Vis, I. W. C. E. Arends and R. A. Sheldon, J. Org. Chem., 2001, 66, 2429 CrossRef CAS PubMed; (b) G.-J. ten Brink, J.-M. Vis, I. W. C. E. Arends and R. A. Sheldon, Tetrahedron, 2002, 58, 3977 CrossRef CAS; (c) H. Ichikawa, Y. Usami and M. Arimoto, Tetrahedron Lett., 2005, 46, 8665 CrossRef CAS; (d) Y. Miyake, Y. Nishibayashi and S. Uemura, Bull. Chem. Soc. Jpn., 2002, 75, 2233 CrossRef CAS.
- (a) D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409 CrossRef CAS PubMed; (b) S. Santoro, C. Santi, M. Sabatini, L. Testaferri and M. Tiecco, Adv. Synth. Catal., 2008, 350, 2881 CrossRef CAS.
- The determined chemical shift of C (1248 ppm) in 77Se NMR is consistent with its literature data of 1241 ppm. See: http://www.chem.wisc.edu/areas/reich/handouts/nmr/se-data.htm.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, condition screening tables, characterization data, 1H, 13C NMR and NOESY spectra of the products, and 77Se NMR spectroscopic studies on the involved organoselenium species. See DOI: 10.1039/c3gc41562g |
| This journal is © The Royal Society of Chemistry 2014 |