PVP-stabilized heteropolyacids as reusable self-assembling catalysts for alcoholysis of cellulosic saccharides†
Jin
Chen
,
Xiaolong
Fang
,
Xinping
Duan
,
Linmin
Ye
,
Haiqiang
Lin
and
Youzhu
Yuan
*
State Key Laboratory of Physical Chemistry of Solid Surfaces, National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P.R. China. E-mail: yzyuan@xmu.edu.cn; Fax: +86-592-2183047; Tel: +86-592-2181659
First published on 8th October 2013
Abstract
Polyvinylpyrrolidone-stabilized heteropolyacids (PVP–HPAs) are synthesized by self-assembling in alcohol. The structure of PVP–HPAs is determined by various characteristic techniques. HPAs can protonate PVP to form polymeric cations. In turn, the protonated PVP interacts strongly with the heteropolyanion by forming an ionic liquid (IL)-like structure. The self-assembling separation and recyclability characteristics are related to the PVP's IL-like structure. The catalyzing performance of PVP–HPAs varies with the species of HPA and the content of PVP. The optimized PVP–H4SiW12O40·5H2O (HSiW) (1/5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3/4) gives more than 60% conversion of cellulose and complete conversion of highly selective cellobiose into butylglucosides. The optimized PVP–HSiW is separated directly by centrifugation and retains the activity without any post-treatment during recycling. The deactivation of PVP–HPAs is related to the loss of the catalyst during recycling. The functional mechanism of the IL-like structure is explored in this control experiment.
:3/4) gives more than 60% conversion of cellulose and complete conversion of highly selective cellobiose into butylglucosides. The optimized PVP–HSiW is separated directly by centrifugation and retains the activity without any post-treatment during recycling. The deactivation of PVP–HPAs is related to the loss of the catalyst during recycling. The functional mechanism of the IL-like structure is explored in this control experiment.
1. Introduction
The global greenhouse effect is increasing, fossil fuel resources are declining, and nuclear energy is still plagued by security issues, so biomass is considered as a promising and secure resource that could meet the requirements for sustainable development.1 Cellulose is one of the most abundant inedible biomasses derived from sustainable photosynthesis. Therefore, the efficient utilization of cellulose can satisfy the requirements of energy and chemical supply without threatening the global food supply. The alcoholysis of cellulose into alkylglucoside is carried out under mild conditions with high selectivity for alkylglucosides; alkylglucosides are highly valued chemicals with wide applications extending to their usage as surfactants and their utilization in the pharmaceutical industry.2,3 Therefore, from the future perspective of green chemistry, the alcoholysis of cellulose is a promising approach.During the catalysis of mineral liquid acids, cellulose undergoes alcoholysis into alkylglucosides under relatively milder conditions. For example, when H2SO4 catalyzes the reaction, 74% conversion of cellulose with 48% total selectivity to α- and β-methylglucosides is achieved at 468 K.4,5 However, neutralization and other multiple post-procedures increase the total cost of using liquid acids. As a better alternative, the application of solid acids has elicited much research interest. However, the transfer barrier leads to a relatively low conversion of cellulose under the catalysis of solid acids. In the last few decades, ionic liquids (ILs) have drawn much interest because of their customizable properties. In 2002, Rogers et al. reported that ILs (e.g., [BMIm]Cl) could be employed as solvents to efficiently dissolve cellulose.6 The application of ILs as reaction media has been extended to the alcoholysis of cellulose by de Vos et al. and Corma et al.7,8 Although ILs clearly promote the degradation of cellulose, the high cost of synthesis and the large amount required increase the overall cost of using ILs. Functionalized ILs (FILs) have been studied with the aim of reducing the required amount of ILs. Heteropolyacids (HPAs), a class of polyoxometalates, have been reported to exhibit outstanding catalyzing performance in the degradation of cellulose.5,9 However, recycling HPAs involves using a large amount of ether for the extraction, which results in the same problem regarding high cost. In 2009, Leng et al. reported that they combined typical FILs with HPAs to synthesize heteropolyanion-based ILs (HILs). HILs exhibited efficient performance in catalyzed esterification and exhibited reaction-induced self-separation.10 Allowing for the risk of losing aromatic imidazole-based or pyridine-based cations, a possibility of damaging the environment during the use of HILs exists.11 To address this issue, the immobilization of ILs onto supporters and the replacement of aromatic ILs with biodegradable compounds have been studied. Apart from the two methods, the application of polymeric ILs (PILs) is opined as an alternative approach.12
Our inspiration for the present work came from PILs. In view of its low toxicity and commercial accessibility, polyvinylpyrrolidone-K30 (PVP) is chosen as a precursor for the in situ synthesis of PVP-stabilized HPAs, called PVP–HPAs. During the catalysis of PVP–HPAs, cellulosic saccharides undergo alcoholysis into alkylglucosides. PVP-based PILs have ever been reported for the application of PVP–HPAs in esterification.13 However, because of the lack of more comprehensive studies, the knowledge about the chemicophysical properties and molecular structure of PVP–HPAs is insufficient. In this work, multiple characteristic techniques were employed to observe the structure–performance correlation.
2. Experimental section
2.1 Materials
PVP, H3PW12O40·nH2O, H4SiW12O40·nH2O, and alcoholic reagents (analytical purity) were purchased from China National Medicines Corporation Ltd. HPAs were purified by extracting from an aqueous solution with ether, evaporating the ether, and drying at 383 K for 12 h to obtain dry white powders with fixed content of crystal water (H3PW12O40·6H2O and H4SiW12O40·5H2O, called HPW and HSiW, respectively). Amberlyst® 15 DRY was purchased from Alfa Company. Cellobiose and cellulose of 85% crystal degree were purchased from Sigma-Aldrich Company. Cellulose required ball milling for 48 h to reduce the crystal degree.2.2 Preparation of PVP–HPAs
Prior to the synthesis of PVP–HPAs, HPAs and PVP were separately dissolved to obtain an alcoholic solution of 20 g L−1 concentration. When dissolving HPAs became difficult, proper heating to 333 K was necessary. The synthesis of PVP–HPA underwent mixing with a calculated volume of alcoholic solution of HPA and PVP in a steel autoclave. PVP–HPA was denoted as PVP–HPA (x:y) according to the mass ratio of PVP and HPA. Taking the synthesis of PVP–HPW (1/5:1) as an example, a 1 mL solution of PVP was added dropwise into a 5 mL solution of HPW with vigorous stirring. After stirring for 6 h, a white alcoholic gel was obtained. The designated name was derived from the input of PVP being equal to one-fifth of the mass of HPW. Since HSiW is a tetrabasic acid and its molecular mass (Mr = 2968 g mol−1) is similar to that of HPW (Mr = 2988 g mol−1), the inputting mass of HSiW was equal to three-fourths of the mass of HPW for the same inputting amount of protons. The gel thus generated was denoted as PVP–HSiW (1/5:3/4). Allowing for the requirement of characterization, the PVP–HPAs were separated by centrifugation and washed with CH2Cl2 five times to remove the alcohol completely. Then, the gel was dried at 313 K for 12 h under a pressure of 1 mmHg column and denoted as D-PVP–HPAs.
2.3 Characterization
Fourier transform infrared spectra (FTIR) were obtained using Thermo Scientific Nicolet 6700 spectroscopy. The sample was diluted with dry KBr powder, and the mixture was pressed into a sheet for measurement. The H–D isotopic exchange was performed in the in situ liquid absorption cell and was also inspected with Thermo Scientific Nicolet 6700 spectroscopy. The detailed structure of the in situ cell and the procedures are shown in Fig. S1 (ESI†) in detail.X-ray photoelectron spectroscopy (XPS) was carried out using a Qtac-100 LEISS-XPS instrument. The sample was mixed with BaSO4 powder and was pressed into a sheet. Ba2+ in BaSO4 was used as an internal standard for calibrating the spectrum. X-ray powder diffraction measurement (XRD) was carried out using a PANalytical X'pert PRO equipped with Cu-Kα radiation. In contrast to the typical measurement of solid powders, the PVP–HPAs were spread to form a thin film on a glass plate. Firstly, the surface of the glass plate was covered with a piece of plastic tape. Then, the tape was jaded into a shallow pool measuring 1.5 cm × 1.2 cm; 40 μL alcoholic gel was added into the pool and tiled onto the bottom. After absolute volatilization of the solvent in the gel, the residue on the plate formed a uniformly thin white film of PVP–HPA and then the plastic tape was removed. The scanning angle was between 5° and 50°, the step size was 0.0167°, and the scanning time per step was 10 s.
All the solid-state MAS NMR spectra were recorded using a Bruker AVANCE III 400 MHz spectrometer equipped with a 4 mm MAS probe spinning at a rate of 10 kHz. Adamantane was applied as an external standard (1H chemical shift = 1.63 ppm).
Diffuse reflectance UV–vis spectroscopy (DRS UV–vis) was conducted using a Carry 5000 equipped with a mantis arm-type diffuse reflectance room. p-Hydroxyazobenzene (p-HAB) with pKa = −0.9 was used as a probe to compare the acidic intensity of PVP–HPAs. Then, 1.0 mL of a 1.0 mg mL−1 CH2Cl2 solution of p-hydroxyazobenzene (p-HAB) was mixed with 1.0 g D-PVP–HPA, dried under infrared light, and packed into the sample holder for measurement.
2.4 Alcoholysis of cellulose
The alcoholysis of cellulose was performed in a 50 mL Teflon-lined stainless steel autoclave. We take the catalyzed butanolysis of cellulose by PVP–HPW (1/5:1) as a typical example. Ignoring the volume change, an extra 14 mL butanol fixed the total volume of the reaction slurry at 20 mL. After adding 0.5 g cellulose, the autoclave was sealed, the air inside was replaced by Ar three times, and the autoclave was pressured with 1 MPa Ar. Finally, the reaction was carried out at the proposed temperature. The autoclave was cooled to room temperature in a water bath, and the light brown slurry inside it was separated by centrifugation at 4000 r min−1 for 10 min. After separation, the yellow liquid in the upper layer became the alcoholic solution of the products. The liquid was run through a silica gel column that is 3.0 cm in length to remove any insoluble residue that could block the chromatographic column. The products were analyzed by high-performance liquid chromatography (HPLC) (Shimadzu Company) using ethylene glycol as an internal standard. The HPLC was equipped with a Shodex Sugar Column (SH 1011) and a refractive index detector. The equations for calculating selectivity and conversion are shown below.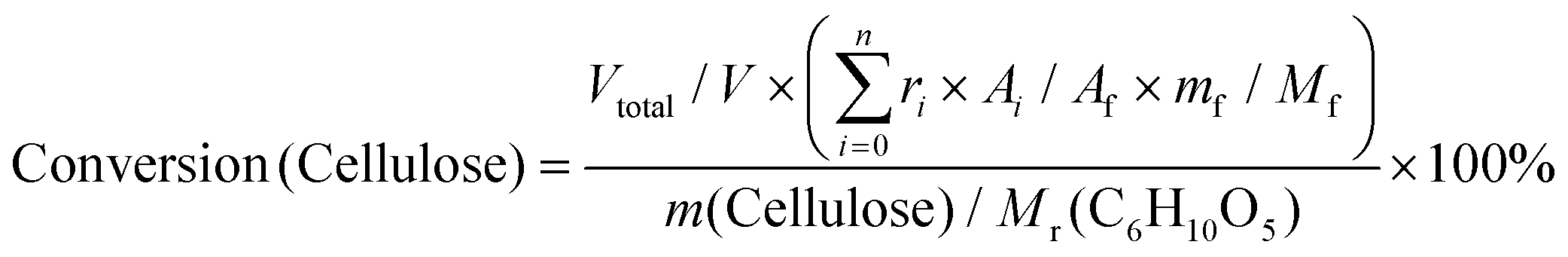 | (1) |
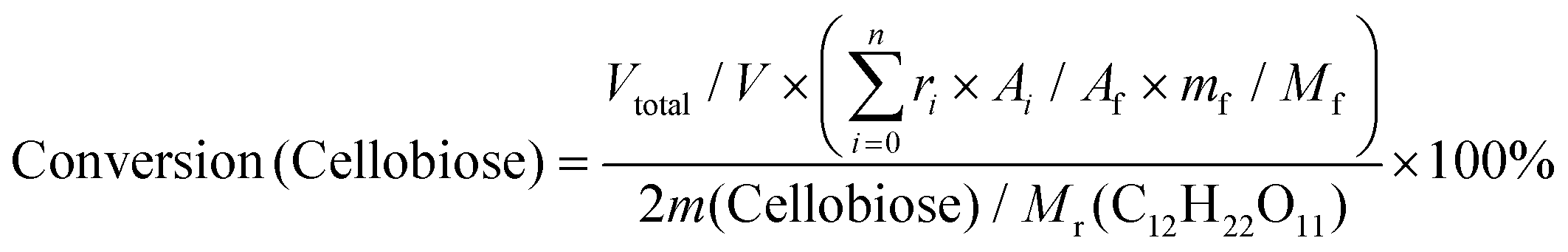 | (2) |
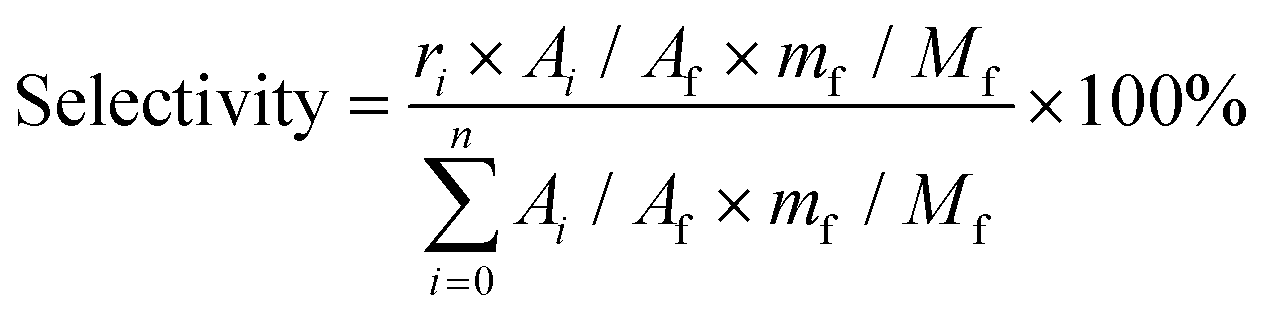 | (3) |
Based on the peak area ratio between products (Ai) and internal standard (Af) and the relative impacts (ri), the conversions of cellulose and cellobiose were calculated. Owing to a few unknown side products, the equation for calculating selectivity was designed according to the area normalization method. By taking the parallel experiments, the standard deviations of the measurement were calculated. In the butanolysis of cellobiose the average standard deviation of conversion was 2.3% and that of selectivity was 1.1%. In the butanolysis of cellulose, the standard deviation of conversion is about 5.0%.
3. Results and discussion
3.1 Structure and properties of PVP–HPAs
The FTIR results of PVP, HPAs, and PVP–HPAs (Fig. 1) provide insight into the structural changes that occur. HPW contains four characteristic bands at the finger range, including 1080, 987, 890, and 816 cm−1, assigned to the stretching modes of P–O, W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, W–Oe–W (edge-sharing), and W–Oc–W (corner-sharing), respectively. Within the spectrum of HSiW, the bands at 1020, 982, 921, and 798 cm−1 are separately assigned to the stretching modes of Si–O, WO, W–Oe–W, and W–Oc–W. According to a published report, protons stay in WO.14 When PVP is added to form PVP–HPW (1/5:1) and PVP–HSiW (1/5:3/4), the bands assigned to WO move to 979 and 974 cm−1, respectively, indicative of a possible migration of protons. Apart from the shift from WO, an obvious change in the band takes place at about 1650 cm−1, which is assigned to >CO. Within the spectrum of PVP, the band of >CO extends to a higher wavenumber owing to the tautomerism between >CO and >N+C–O−. Protonating PVP in PVP–HPW (1/5:1) eliminates tautomerism, which leads to the absence of a band extension of >CO. A similar phenomenon occurs in the spectrum of PVP–HSiW (1/5:3/4). The addition of PVP results in the band at 890 cm−1, assigned to W–Oe–W of HPW, undergoing a blue shift to 897 cm−1 within the spectrum of PVP–HPW (1/5:1); the band at 921 cm−1, assigned to W–Oe–W of HSiW, moves to 915 cm−1 within the spectrum of PVP–HSiW (1/5:3/4). PVP withdraws the protons from HPAs, resulting in an averaging effect on the charge dispersion of the heteropolyanion. Therefore, the bond strength of W–Oe–W trends toward the similarity. As a result, the band appears at a closer position. The results of FTIR and the discussions point to the protons migrating from HPAs to protonate PVP.
O, W–Oe–W (edge-sharing), and W–Oc–W (corner-sharing), respectively. Within the spectrum of HSiW, the bands at 1020, 982, 921, and 798 cm−1 are separately assigned to the stretching modes of Si–O, WO, W–Oe–W, and W–Oc–W. According to a published report, protons stay in WO.14 When PVP is added to form PVP–HPW (1/5:1) and PVP–HSiW (1/5:3/4), the bands assigned to WO move to 979 and 974 cm−1, respectively, indicative of a possible migration of protons. Apart from the shift from WO, an obvious change in the band takes place at about 1650 cm−1, which is assigned to >CO. Within the spectrum of PVP, the band of >CO extends to a higher wavenumber owing to the tautomerism between >CO and >N+C–O−. Protonating PVP in PVP–HPW (1/5:1) eliminates tautomerism, which leads to the absence of a band extension of >CO. A similar phenomenon occurs in the spectrum of PVP–HSiW (1/5:3/4). The addition of PVP results in the band at 890 cm−1, assigned to W–Oe–W of HPW, undergoing a blue shift to 897 cm−1 within the spectrum of PVP–HPW (1/5:1); the band at 921 cm−1, assigned to W–Oe–W of HSiW, moves to 915 cm−1 within the spectrum of PVP–HSiW (1/5:3/4). PVP withdraws the protons from HPAs, resulting in an averaging effect on the charge dispersion of the heteropolyanion. Therefore, the bond strength of W–Oe–W trends toward the similarity. As a result, the band appears at a closer position. The results of FTIR and the discussions point to the protons migrating from HPAs to protonate PVP.
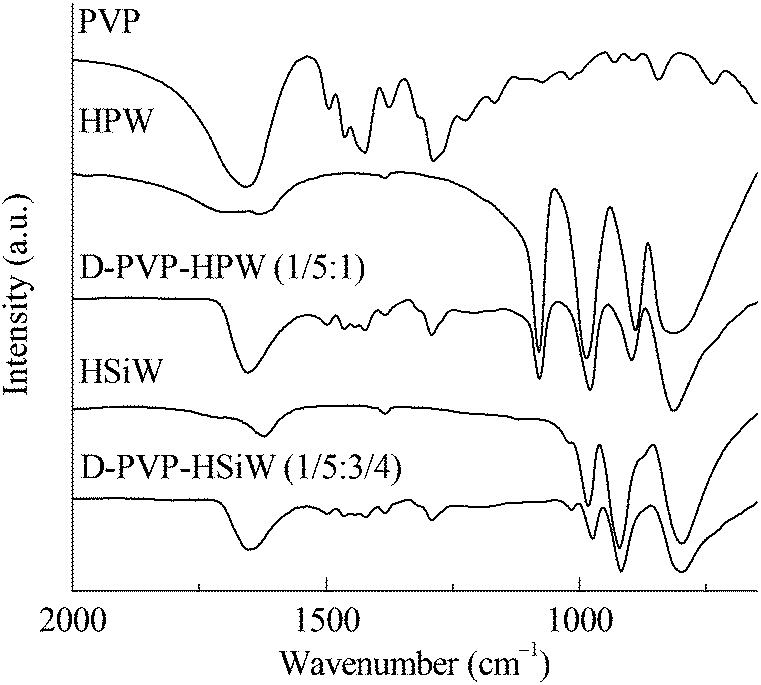 | ||
| Fig. 1 FTIR spectra of PVP, HPAs, and PVP–HPAs. | ||
There still exists a puzzling problem concerning which atom of PVP is protonated during the synthesis of PVP–HPAs. Illuminated by the structure of amide-based ILs, the N-atom in PVP is speculated to be protonated, but from the viewpoint of the FTIR spectra no direct evidence is released, because the band assigned to N–H is close to that of water in air; hence, H–D isotopic exchange is taken. As shown in Fig. S2 (ESI†), after deuteration a new band appearing at about 2400 cm−1 indicates deuteration of protons by D2O. For convenience, the samples are renamed as DPW, DSiW, PVP–DPW (1/5:1) and PVP–DSiW (1/5:3/4). In the spectra of DPW and DSiW the bands at 2370 cm−1 are assigned to the stretching model of O–D of crystal D2O and the H-bond between D+ and D2O. In the spectra of PVP–DPW (1/5:1) and PVP–DSiW (1/5:3/4) the bands at 2420 cm−1 are probably assigned to N–D of protonated PVP and H-bond between D+ and >CO of PVP according to the published paper.15 From the viewpoint of the spectra, the bands of PVP–DPAs are broader than those in the spectra of DPAs. As we know, the width of the band in FTIR is related to the degree of freedom of the group. Therefore, it is speculated that because of the identity property of each pyrrolidone group of PVP, the proton can move freely on PVP and interacts with each pyrrolidone group; namely, the protonated groups are gifted with a higher degree of motion freedom than the degree of vibration and rotation freedom of those in HPAs, so the corresponding bands of PVP–DPAs are broader. This phenomenon shows the migration of protons from HPA to PVP during the synthesis of PVP–HPA again.
Compared with H–D isotopic exchange, XPS gives more direct evidence for pointing out the protonated site of PVP. In Fig. S3 (ESI†), the peak assigned to N1s of PVP at 398.9 eV is a symmetric curve. Within the spectra of PVP–HPW (1/5:1) and PVP–HSiW (1/5:3/4), the peaks of N1s undergo a blue shift to 399.2 eV and 390.0 eV respectively. In addition, the peaks are asymmetric, namely, a shoulder peak extends to higher binding energy. The shift of N1s and the presence of a shoulder peak indicate the existence of new species of N-atoms in PVP–HPW (1/5:1) and PVP–HSiW (1/5:3/4). In view of the species with more electronegativity than that in PVP, it is identified to be a protonated N-atom according to the published report.16 Combining the results of H–D isotopic exchange and XPS, the N-atom of PVP is pointed out as the protonated site, but the existence of H-bonds between protons and >CO of PVP should not be eliminated.
XRD was employed to investigate the effect of the content of PVP on the micro/nanostructure of PVP–HPAs. The absorption of water from air during the preparation of the thin film at room temperature resulted in some differences between the spectra of Wet HPW and Dry HPW (dried at 383 K for 12 h). However, according to the published report,17 the Wet HPW retains the Keggin-type structure of Dry HPW. The sharp peaks of Wet HPW at 2θ = 7.93°, 8.76°, 17.57°, and 26.36° are referred to as symptoms of dispersion (Fig. S4 in ESI†). At the initial range of mass ratios between 1/39:1 and 3/37:1, the presence of the four characteristic peaks indicates that the bulk of HPW stays in the microcrystal state; this portion of HPW does not or only weakly interact with PVP. However, the intensity of these peaks decreases regularly with increasing content of PVP, indicative of the positive effect of PVP on the dispersion of HPW. When the mass ratio increases to 1/9:1, the four peaks disappear and no characteristic peak appears as the mass ratio increases sequentially. This result indicates that HPW is well dispersed, which is also supported by the results of SEM (Fig. S5 in ESI†). The SEM images show that HPW remains a block crystal, but F-PVP–HPW (1/5:1) exists in the amorphous state. Interestingly, HPW is a tribasic acid, so three molar equivalents of monomers of PVP can be protonated by one equivalent of HPW (n(monomer)/n(HPW) = 3/1). Therefore, based on eqn (4) shown below:
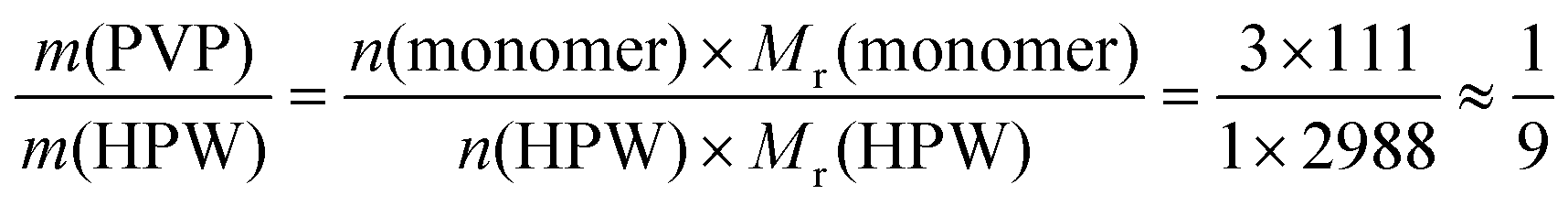 | (4) |
:1, HPW interacts with PVP fully. In the experiment, 1/9 is the point where the structure of PVP–HPW changes. The coincidence between experimental and theoretical results demonstrates a chemical reaction that occurs during the mixing of PVP and HPW, which agrees with the conclusion regarding the previous results.
Through the use of solid MAS-NMR, the details of the mechanism of the migration of protons from HPA to protonated PVP were clearly elucidated. In Fig. 2, according to the published reports,14,18 the single sharp peak in the spectrum of HPW belongs to the protons. In our experiment, the chemical shift is 6.72 ppm instead of 9 ppm, possibly due to the high hydration level of 6. In the spectra of PVP–HPW, a series of bands assigned to the covalent hydrogen of PVP is found at a chemical shift range between 4 ppm and 0 ppm, and the values do not vary with the change in the content of PVP. Unlike covalent hydrogen, the shift of protons is sensitive to the content of PVP. When a small amount of PVP is added, a sharp peak of D-PVP–HPW (1/39:1) appears at 6.70 ppm, close to the 6.72 ppm of HPW. We speculate that a majority of the protons are still occupied by heteropolyanions. However, when the content of PVP increases sequentially at a certain scale, the chemical shifts of the sharp peaks of D-PVP–HPW (1/9:1), (1/5:1), and (1/3:1) are 7.49, 7.50, and 7.53 ppm, respectively, and these values are very close. If associated with the results of XRD, this phenomenon serves as another proof of the existence of a chemical interaction between PVP and HPAs. A mass ratio above 1/9:1 reportedly indicates that all the protons have migrated from HPAs to PVP, and the chemical conditions of the protons do not differ from the increasing content of PVP. Moreover, the shielding effect from PVP is omitted. However, in the case of a high content of PVP, the effect becomes remarkable. Therefore, in the spectrum of PVP–HPW (2/3:1), the sharp peak moves back to 6.44 ppm. Summarizing the results of XRD, FTIR, XPS, and NMR, a proposed model for the synthesis of PVP–HPAs is described in Scheme 1. Taking the synthesis of PVP–HPW as a model, HPW protonates PVP to form a polymeric cation; the polymeric cation interacts with the heteropolyanion by Coulomb force ignoring the effect of H-bonds. ILs are composed of large organic cations and inorganic anions that interact by Coulomb force, which is analogous to the molecular structure of PVP–HPAs. Thus, a definition of the IL-like structure is presented to describe the structure of PVP–HPAs.
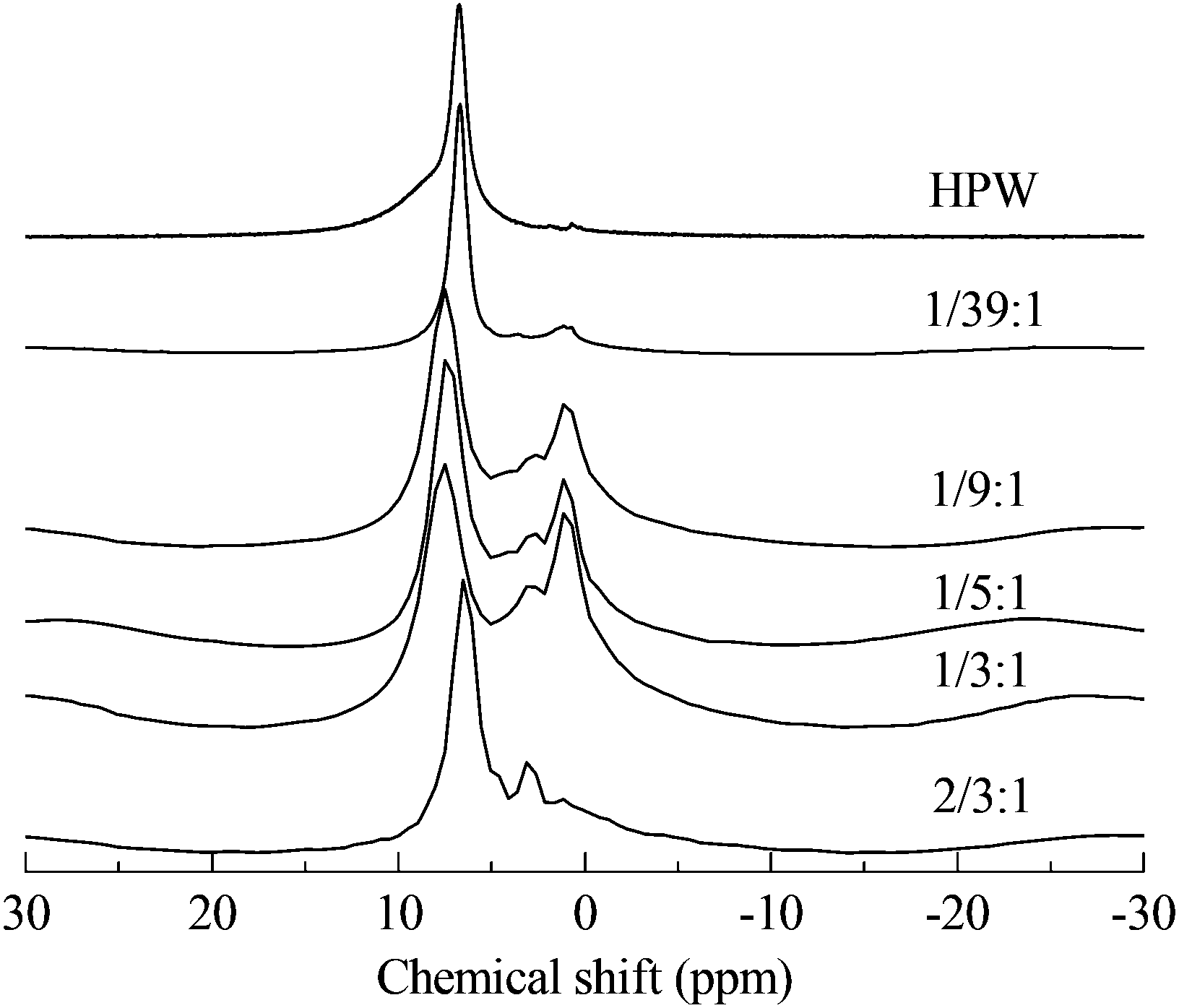 | ||
| Fig. 2 Results of solid MAS 1H-NMR of PVP–HPW (x:y). | ||
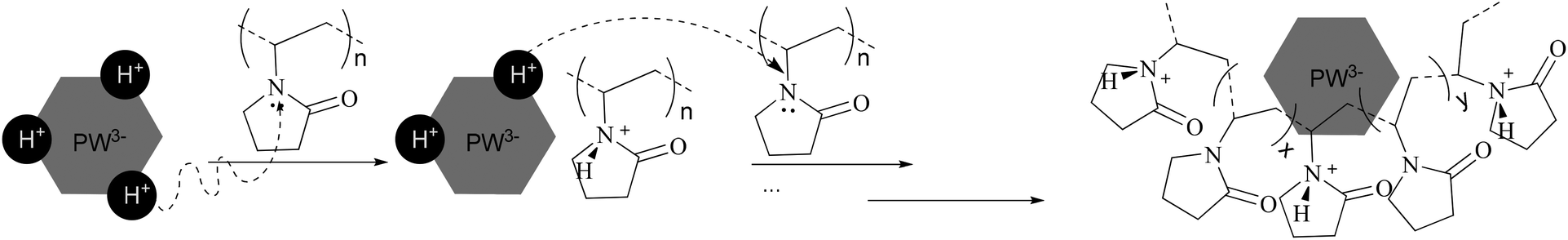 | ||
| Scheme 1 Diagram of the synthesis route of PVP–HPW. | ||
Apart from the IL-like structure, the effect of the content of PVP on the Brønsted acidic intensity of PVP–HPAs has also drawn some interest. The UV–vis absorption of dye is a commonly accepted means of detecting the acidic intensity of solid acids.19–21 Thus, a combination of dye absorption and DRS UV–vis was applied. Fig. 3 shows that the absorption band of p-HAB is at 355 nm when absorbed on BaSO4, a neutral salt. Given the full protonation on HPW, the band at 485 nm assigned to the protonated p-HAB appears without the presence of the band at 355 nm. When absorbed on PVP–HPW, the intensity of the band at 485 nm is obviously lower. Meanwhile, the intensity decreases as a function of the increasing content of PVP. The amount of p-HAB added is much less than the content of protons in the sample. Therefore, when the effect of the granularity of the powder samples on measurement is ignored, the level of protonation of p-HAB reflects the relative activity of the protons. The intensity of the band at 485 nm is related to the acidic intensity of the sample. Therefore, the addition of PVP reduces the acidic intensity of HPW, and this effect can be tuned by changing the mass ratio. The results of HSiW and PVP–HSiW (1/5:3/4) (Fig. S6 in ESI†) also show the effect of PVP.
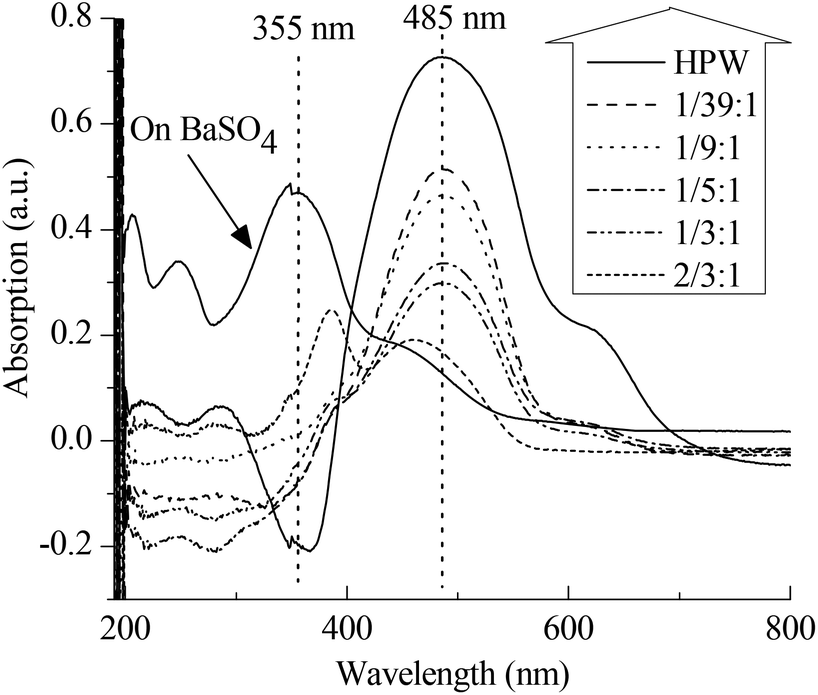 | ||
| Fig. 3 Results of DRS UV–vis spectra of p-HAB absorbed on PVP–HPW (x:y), HPW, and BaSO4. | ||
3.2 Alcoholysis of cellulosic saccharides
Cellulose, oligosaccharides, and glucose are cellulosic saccharides. Oligosaccharides are derived from the partial hydrolysis of cellulose; further degradation is considered an indirect method of transforming cellulose. Cellobiose is a kind of disaccharide that contains the basic structure of cellulose, so it was employed as a representation of oligosaccharides and as a molecular model for the alcoholysis of cellulose (Scheme 2).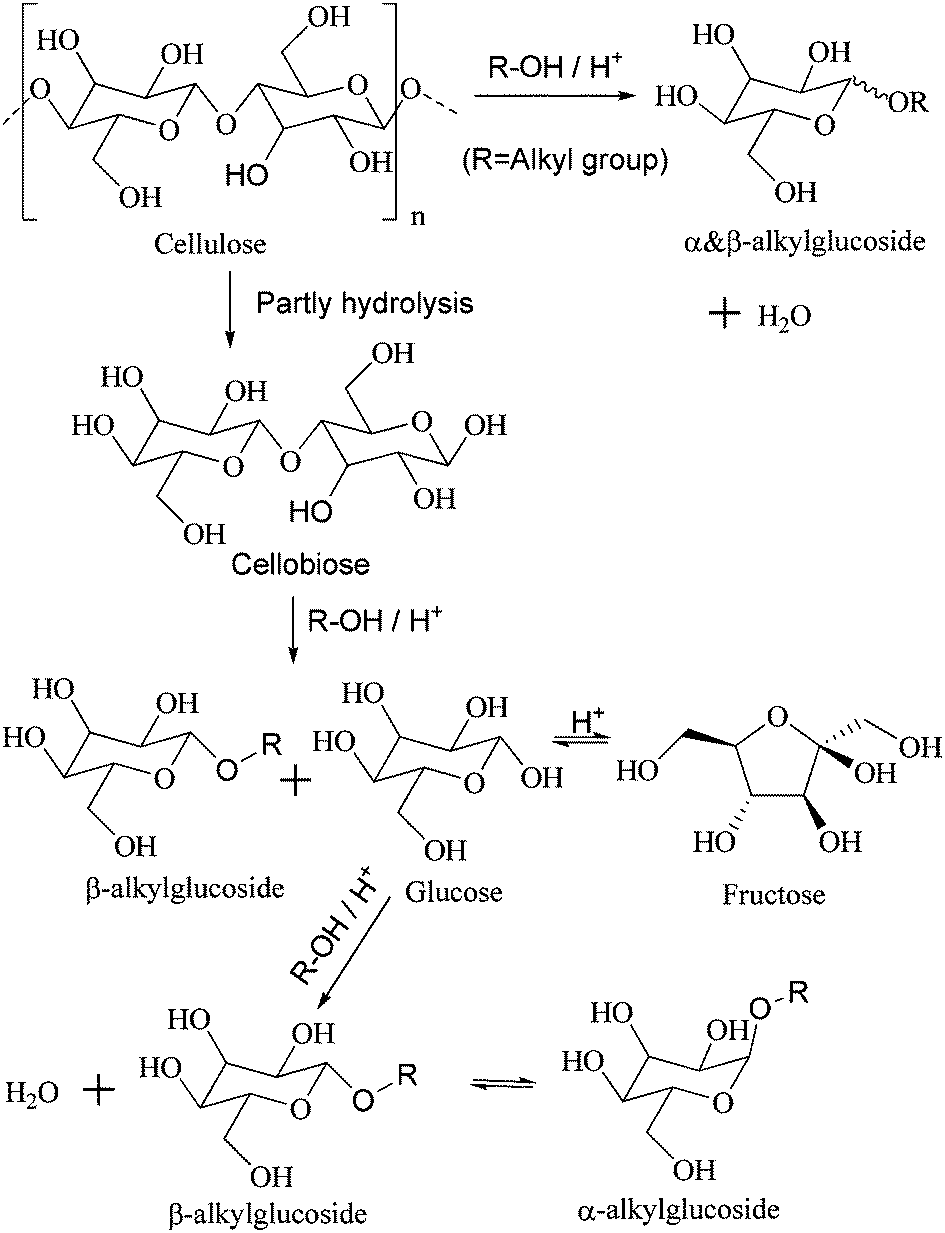 | ||
| Scheme 2 Direct and indirect routes for the alcoholysis of cellulose and the reaction mechanism. | ||
Various catalysts were tested and the results are listed in Table 1. Among them, H2SO4 (entries 1A and B) exhibits the most outstanding catalytic activity in the butanolysis of cellobiose and cellulose due to its strong acidity and homogeneous catalysis in alcohol. Meanwhile, the total selectivity of butylglucosides reaches more than 90%. However, corrosion in the reactor and multiple post-pretreatments for separation increase the cost of using H2SO4. In contrast to inorganic liquid acids, solid acids can be separated directly, but the transfer barrier is viewed as a great disadvantage. Therefore, HZSM-5 (entries 2A and B) displays very low catalytic activity, with only 11.3% conversion of cellobiose and rare conversion of cellulose. Compared with HZSM-5, Amberlyst® 15 DRY, a kind of solid acidic resin with macropores, has a lower transfer barrier. As such, 74.6% and 32.0% conversions of cellobiose and cellulose are achieved (entries 3A and B), respectively. However, –SO3H groups of Amberlyst® 15 DRY reportedly leach easily, so low thermal stability limits its use.5,22 HPAs are stable solid acids. However, compared with HZSM-5 and Amberlyst® 15 DRY, HPAs are dissolved in most polar solvent. Therefore, the alcoholysis catalyzed by HPAs is a homogeneous reaction. Congruent to published results,5 during the catalysis of HPW and HSiW, 92.0% and 93.6% of cellulose respectively undergo alcoholysis into butylglucosides of above 90% total selectivity (entries 4B and 5B). However, after the reaction, vaporizing the alcohol and extracting the HPAs with a large amount of ether are necessary.
| Entry | Catalyst | Conversion (%) (recycling number)b | Selectivity (%) | ||
|---|---|---|---|---|---|
| TRS | β-BGS | α-BGS | |||
| a Butanolysis of cellobiose and cellulose is denoted by A and B, respectively, and ethanolysis of cellobiose is denoted by C. Reaction conditions: m(cellobiose or cellulose) = 0.5 g, V(butanol or ethanol) = 20 mL, n(H+) = 0.1 mmol or m(solid acid) = 0.1 g (entries 2 and 3), P(Ar) = 1 MPa, T = 428 K for alcoholysis of cellobiose or 433 K for alcoholysis of cellulose, and t = 4 h. b Separate catalysts by 4000 r min−1 centrifugation for 10 min; recycle the catalysts unless the conversion of cellobiose falls to 85%. c m(PVP) = 0.2 g. Abbreviation list: BGS (butylglucosides), EGS (methylglucosides), TRS (total reduced saccharides, including glucose, fructose and derivatives from hydrolysis). | |||||
| 1A | H2SO4 | >99 | 9.5 | 29.2 | 61.3 |
| 1B | 93.9 | 9.2 | 29.6 | 61.2 | |
| 2A | HZSM-5 (Si/Al = 38) | 11.3 | 74.1 | ND | 25.9 |
| 2B | 0 | — | — | — | |
| 3A | Amberlyst® 15 Dry | 74.6 | 7.9 | 29.2 | 60.5 |
| 3B | 32.0 | 8.9 | 24.0 | 53.8 | |
| 4B | HPW | 92.0 | 6.1 | 30.4 | 63.5 |
| 5B | HSiW | 93.6 | 6.3 | 30.4 | 63.3 |
| 6A | PVPc | 0 | — | — | — |
| 7A | PVP–HPW (1/5:1) |
>99 (7) | 7.6 | 29.6 | 62.8 |
| 7B | 73.7 | 10.2 | 28.2 | 61.6 | |
| 7C | >99 (5) | 5.9 | 28.6 (β-EGS) | 56.9 (α-EGS) | |
| 8A | PVP–HPW (2/3:1) |
64.4 | 34.2 | 25.3 | 40.4 |
| 9A | PVP–HPW (1/39:1) |
>99 (3) | 9.0 | 29.0 | 62.0 |
| 10A | PVP–HSiW (1/5:3/4) |
>99 (20) | 9.9 | 29.0 | 61.1 |
| 10B | 87.0 | 9.7 | 29.5 | 60.8 | |
PVP–HPAs provide a promising approach to overcoming these challenges. According to the definition of gel,23,24 it is a soft material that can absorb a great amount of solvents and substrates like a piece of sponge. This property allows PVP–HPAs to address the problem of the transfer barrier. PVP has no intrinsic activity (entry 6A), so the activity of PVP–HPAs benefits from HPAs. PVP–HPW (1/5:1) catalyzes a complete conversion of cellobiose and 73.7% conversion of cellulose alcoholysis into butylglucosides (entries 7A and B), which is comparable to that of H2SO4. Additionally, PVP–HPW (1/5:1) is separated directly by centrifugation and reused without any regeneration after the reaction. In the butanolysis of cellobiose PVP–HPW (1/5:1), high activity is maintained for six recycles before beginning to decline (Fig. 4(a)). In the butanolysis of cellulose, the activity declines tremendously during recycling (Fig. S7 in ESI†). When PVP–HPW (1/5:1) is applied during the alcoholysis of cellobiose in ethanol (entry 7C), it also displays good catalyzing performance. Although PVP–HPW (1/5:1) exhibits reusable catalyzing performance, it still falls short of our objectives. According to the published report, the SiW anion has better stability than the PW anion in the liquid phase,25 so HPW is substituted with HSiW to synthesize PVP–HSiW (1/5:3/4). Similar to PVP–HPW (1/5:1), PVP–HSiW (1/5:3/4) exhibits remarkable activity in the alcoholysis of cellobiose and cellulose (entries 10A and B). However, PVP–HSiW (1/5:3/4) exhibits a more stable recyclability. In the butanolysis of cellobiose, the activity declines to about 60% conversion at the 20th cycle (Fig. 4(b)). During the recycling of PVP–HSiW (1/5:3/4) in the butanolysis of cellobiose, the accumulation of water from each cycle leads to a gradual increase in the selectivity for TRS. In the butanolysis of cellulose, the deactivation tendency of PVP–HSiW (1/5:3/4) seems mild; after the first two recycles, the conversion of the cellulose tends toward 60%, with a constant selectivity of about 90% for butylglucosides (Fig. 4(c)).
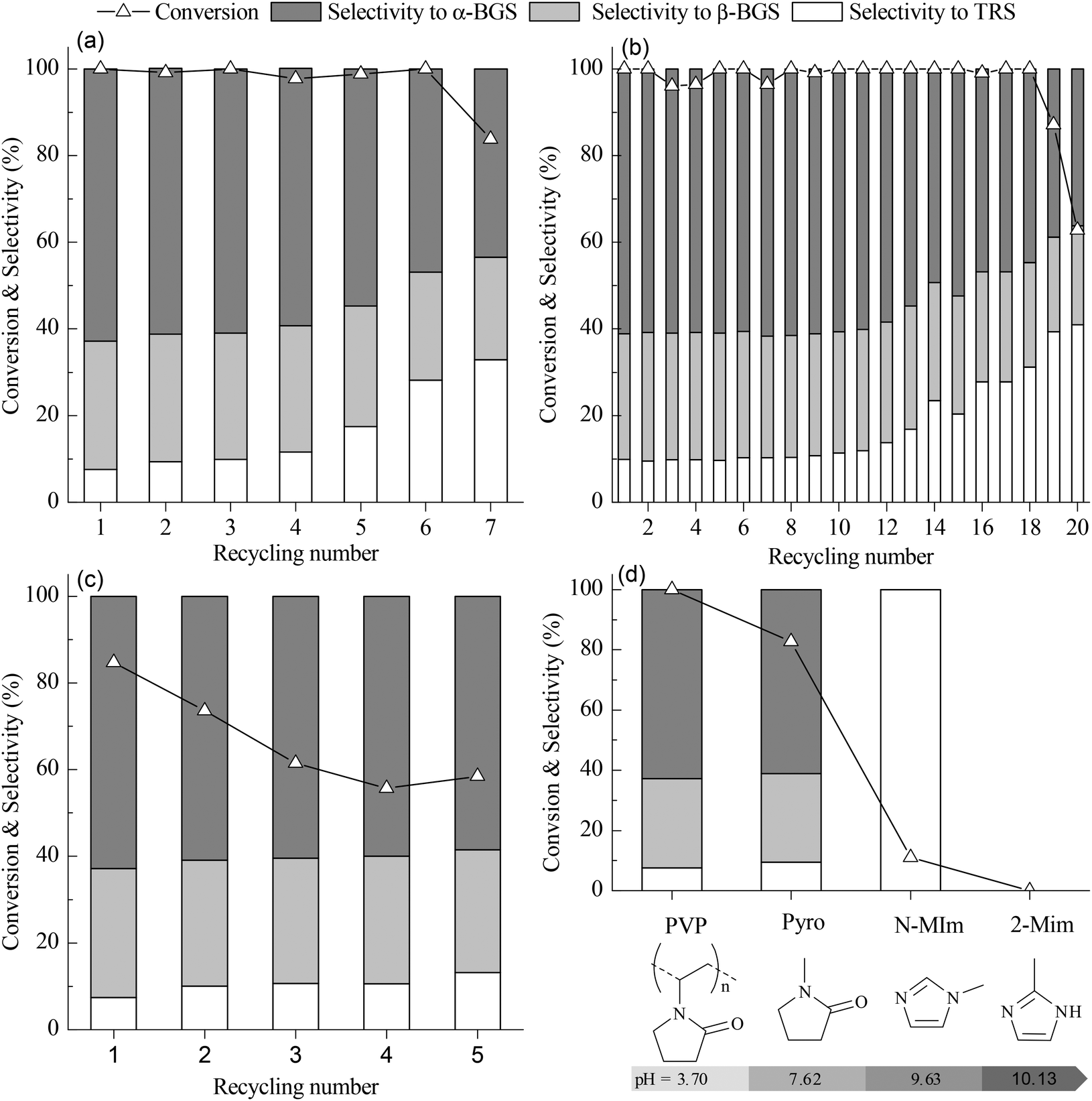 | ||
| Fig. 4 (a) Recycling performance of PVP–HPW (1/5:1) in catalyzed butanolysis of cellobiose. (b) Recycling performance of PVP–HPW (1/5:3/4) in catalyzed butanolysis of cellobiose. (c) Recycling performance of PVP–HPW (1/5:3/4) in catalyzed butanolysis of cellulose. (d) The relationship between the catalytic performance for butanolysis of cellobiose and pH values of 0.1 mol L−1 aqueous solution of cation precursors. The reaction conditions of (a), (b), (c), and (d) are the same as those indicated in Table 1. | ||
After the first cycle, PVP–HPAs were separated and washed with CH2Cl2 for FTIR characterization. Based on the results of spent PVP–HPW (1/5:1) and spent PVP–HSiW (1/5:3/4) (Fig. S8 in ESI†), no obvious structural change takes place during the reaction. The results of thermogravimetry (TG) also show that PVP–HPAs possess reliable stability for the alcoholysis reaction because the temperature at the beginning of the weight loss is about 530 K much higher than the reaction temperature (Fig. S9 in ESI†). Therefore, the possibility of structural decomposition during the reaction is excluded. With the help of inductively coupled plasma optical emission spectroscopy (ICP-OES), a portion of the PVP–HPAs is found to have leached into the solution during recycling (Table S1 in ESI†). According to the results, 10.6 and 5.3 wt% of PVP–HPW (1/5:1) and PVP–HSiW (1/5:3/4), respectively, are leached in the first cycle. However, in the following cycles, the leached amount decreases noticeably. The leached amount of PVP–HSiW (1/5:3/4) is obviously less than that of PVP–HPW (1/5:1) in each cycle. The foregoing provides a reasonable explanation for the activity of PVP–HPAs toward the end of the cycle. The findings indicate that PVP–HSiW (1/5:3/4) has a longer lifespan. At the blank experiment, the FTIR spectra of leached compounds collected from the solution (Fig. S10 in ESI†) are similar to those of fresh PVP–HPW (1/5:1) and PVP–HSiW (1/5:3/4) ignoring the interference from residue butanol, indicating that the catalyst itself is leached during recycling.
Aiming to explore the reason for PVP–HPAs leaching, the butanolysis of cellobiose was conducted at a low conversion. As shown in Fig. S11 (ESI†), during recycling PVP–HPW (1/5:1) the conversion decreases gradually with increasing recycling number, but no obvious change of selectivity is found, which indicates that the loss of the catalyst has no influence on the catalytic active site. However, this is different from the previous results shown in Fig. 4(a). At higher conversion the selectivity to TRS increases faster during recycling. TRS are the products from the hydrolysis of cellobiose, so the water in the reaction may play a key role to affect the recyclability and the catalytic selectivity of PVP–HPAs. Replacing butanol by water, PVP–HPW (1/5:1) and PVP–HSiW (1/5:3/4) can catalyze hydrolysis of cellobiose into TRS with about 95% selectivity to glucose shown in Table S2 (ESI†); and it is found that PVP–HPAs are solute in water after reaction, so water is suspected to steal the catalyst and lead to increasing selectivity to TRS during recycling. In order to get more evidence, in the control experiment the deliberate addition of water leads to less recycling number of PVP–HPW (1/5:1) accompanied by the increasing selectivity to TRS (Fig. S12 in ESI†). Because of the gel property of hydrophilic PVP–HPAs, PVP–HPAs can swallow and store a great amount of water from reaction, which is the reason for the performance change of PVP–HPAs during recycling.
However, there arises a new question of where water comes from. GC-MS analysis detects a small amount of butyl ether under the present reaction conditions, so dehydrolysis of butanol is viewed as a source of water. In general, the amount of water derived from dehydrolysis increases at high reaction temperature, so it is speculated that the selectivity to TRS is promoted by increasing temperature. That is, contrary to the speculation, the results (Fig. S13 in ESI†) show that the selectivity to TRS decreases as the temperature increases. It is necessary to get knowledge about the mechanism of the alcoholysis reaction. Taking the alcoholysis of cellobiose as an example (Scheme 2), the first stage is alcoholysis of cellobiose into one β-alkylglucoside and one glucose molecule; then dehydrolysis between one glucose and one alcohol molecule produces one β-alkylglucoside and one water molecule; finally, a portion of β-alkylglucoside is transformed into α-alkylglucoside. In the mechanism, the first and the third stage can proceed under relatively milder conditions, but dehydrolysis is viewed as a rate-determining step requiring a relatively higher reaction temperature, so the selectivity to alkylglucosides is better at higher temperature. Meanwhile, the alcoholysis reaction is recognized as another source of water. The catalytic performance of different catalysts at low conversion is shown in Table S2 (ESI†). From the viewpoint of the TOF values, there are some changes taking place to the catalytic active site between HPAs and PVP–HPAs, which has been given an explanation by the previous characterization.
According to the results of characterization, the content of PVP has a significant influence on the structure and acidic intensity of PVP–HPAs. This influence is also reflected in the catalytic performance of PVP–HPAs. When the mass ratio of PVP and HPW is increased to 2/3:1, the conversion of cellobiose is only 64.4% (entry 8A). The results of DRS UV–vis demonstrate that excessive PVP suppresses the acidity of PVP–HPAs. Thus, the catalytic activity of PVP–HPW (2/3:1) is poor. In contrast to PVP–HPW (2/3:1), the catalytic activity of fresh PVP–HPW (1/39:1) is remarkable, but the conversion of cellobiose decreases remarkably after just three cycles. Therefore, its recycling number is only 4 (entry 9A). This phenomenon is explained by the results of XRD. With a low content of PVP, the portion of HPW that does not or only weakly interact with PVP is leached into the solution more easily. Therefore, the conversion of cellobiose declines noticeably during recycling.
By replacing PVP with 2-methylimidazole (2-MIm), N-methylimidazole (N-MIm), and N-methylpyrrolidone (Pyro), HILs were synthesized in a manner similar to the synthesis of PVP–HPW. Based on the performance of these HILs in the catalyzed butanolysis of cellobiose, the conversion of cellobiose is observed to decrease with increasing pH value of the aqueous solution. This result indicates that the acidic intensity of HIL decreases when HPW interacts with a cation precursor that has a high basicity. Therefore, the high activity of PVP–HPW (1/5:1) benefits from the low basicity of PVP (Fig. 4(d)). Selectivity is affected by the basicity of the cation precursor, which may be caused by the change in the Brønsted and Lewis acidic sites.26 Interestingly, PVP and Pyro share many common features in terms of molecular structure, but Pyro-based HILs are dissolved in alcohol because of the bounding effect from the carbon chain of PVP. In essence, the IL-like structure has a key role in catalyzing the performance of PVP–HPAs.
4. Conclusions
Through multiple characteristic methods, the existence of an IL-like structure in PVP–HPAs is proven. The content of PVP has a significant influence on the micro/nanostructure and acidic intensity of PVP–HPAs. In the alcoholysis of cellulosic saccharides, PVP–HPAs associate the high activity of inorganic liquid acids with the easy recyclability of solid acids. HSiW is a better alternative for the synthesis of PVP–HPAs. By optimizing the content of PVP and the species of HPAs, PVP–HSiW (1/5:3/4) exhibits outstanding catalyzing performance for 19 cycles in the butanolysis of cellobiose and for 5 cycles in the butanolysis of cellulose. The gradual leaching during recycling is proven to be responsible for the deactivation of PVP–HPAs. Based on the control experiments, the low basicity of the PVP retains the acidity of the HPAs, and the bounding effect from the carbon chain of the PVP results in the PVP–HPAs remaining in the gel state. The IL-like structure has a key role in catalyzing the performance of PVP–HPAs. Given the customizable properties of PVP–HPAs, their application is anticipated to extend to other reactions where the acidic intensity of the catalyst needs to be tuned, for instance, in the selective synthesis of polyether from aldehyde.
Acknowledgements
This work was supported by the National Basic Research Program of China (2011CBA00508), the Natural Science Foundation of China (20923004 and 21173175), the Research Fund for the Doctoral Program of Higher Education (20110121130002) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1036).Notes and references
- C. B. Field, J. E. Campbell and D. B. Lobell, Trends Ecol. Evol., 2008, 23, 65–72 CrossRef PubMed.
- K. Garves, J. Wood Chem. Technol., 1988, 8, 121–134 CrossRef CAS.
- N. Villandier and A. Corma, ChemSusChem, 2011, 4, 508–513 CrossRef CAS PubMed.
- R. E. Reeves, W. M. Schwartz and J. E. Giddens, J. Am. Chem. Soc., 1946, 68, 1383–1385 CrossRef CAS.
- W. P. Deng, M. Liu, Q. H. Zhang, X. S. Tan and Y. Wang, Chem. Commun., 2010, 46, 2668–2670 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- I. A. Ignatyev, P. G. N. Mertens, C. Van Doorslaer, K. Binnemans and D. E. de Vos, Green Chem., 2010, 12, 1790–1795 RSC.
- N. Villandier and A. Corma, Chem. Commun., 2010, 46, 4408–4410 RSC.
- J. Tian, J. H. Wang, S. Zhao, C. Y. Jiang, X. Zhang and X. H. Wang, Cellulose, 2010, 17, 587–594 CrossRef CAS PubMed.
- Y. Leng, J. Wang, D. R. Zhu, X. Q. Ren, H. Q. Ge and L. Shen, Angew. Chem., Int. Ed., 2009, 48, 168–171 CrossRef CAS PubMed.
- A. Romero, A. Santos, J. Tojo and A. Rodriguez, J. Hazard. Mater., 2008, 151, 268–273 CrossRef CAS PubMed.
- Y. B. Xiong, J. J. Liu, Y. J. Wang, H. Wang and R. M. Wang, Angew. Chem., Int. Ed., 2012, 51, 9114–9118 CrossRef CAS PubMed.
- Y. Leng, X. Q. Leng, P. P. Jiang and J. Wang, Chin. J. Catal., 2012, 33, 1224–1228 CAS.
- S. Uchida, K. Inumaru and M. Misono, J. Phys. Chem. B, 2000, 104, 8108–8115 CrossRef CAS.
- A. Travert, A. Vimont, A. Sahibed-Dine, M. Daturi and J.-C. Lavalley, Appl. Catal., A, 2006, 307, 98–107 CrossRef CAS PubMed.
- A. Sidorenko, S. Minko, K. Schenk-Meuser, H. Duschner and M. Stamm, Langmuir, 1999, 15, 8349–8355 CrossRef CAS.
- P. A. Jalil, M. A. Al-Daous, A. R. A. Al-Arfaj, A. M. Al-Amer, J. Beltramini and S. A. I. Barri, Appl. Catal., A, 2001, 207, 159–171 CrossRef CAS.
- S. Ganapathy, M. Fournier, J. F. Paul, L. Delevoye, M. Guelton and J. P. Amoureux, J. Am. Chem. Soc., 2002, 124, 7821–7828 CrossRef CAS PubMed.
- M. Hino, S. Kobayashi and K. Arata, J. Am. Chem. Soc., 1979, 101, 6439–6441 CrossRef CAS.
- C. Thomazeau, H. Olivier-Bourbigou, L. Magna, S. Luts and B. Gilbert, J. Am. Chem. Soc., 2003, 125, 5264–5265 CrossRef CAS PubMed.
- M. Pudipeddi, E. A. Zannou, M. Vasanthavada, A. Dontabhaktuni, A. E. Royce, Y. M. Joshi and A. T. M. Serajuddin, J. Pharm. Sci., 2008, 97, 1831–1842 CrossRef CAS PubMed.
- W. P. Deng, M. Liu, Q. H. Zhang and Y. Wang, Catal. Today, 2011, 164, 461–466 CrossRef CAS PubMed.
- K. Almdal, J. Dyre, S. Hvidt and O. Kramer, Polym. Gels Networks, 1993, 1, 5–17 CrossRef CAS.
- K. Nishinari, in Gels: Structures, Properties, and Functions, Springer, Berlin Heidelberg, 2009, vol. 136, ch. 12, pp. 87–94 Search PubMed.
- S. Dianat, S. Tangestaninejad, B. Yadollahi, A. K. Bordbar, M. Moghadam, V. Mirkhani and I. Mohammadpoor-Baltork, J. Mol. Liq., 2012, 174, 76–79 CrossRef CAS PubMed.
- K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya and A. Satsuma, Green Chem., 2009, 11, 1627–1632 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The results of TG, FTIR and other characterisations. See DOI: 10.1039/c3gc40994e |
| This journal is © The Royal Society of Chemistry 2014 |