Amylase catalyzed synthesis of glycosyl acrylates and their polymerization†
Wouter M. J.
Kloosterman
,
Danijela
Jovanovic
,
Sander G. M.
Brouwer
and
Katja
Loos
*
Department of Polymer Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: K.U.Loos@rug.nl; Fax: +31-50-3634400; Tel: +31-50-3636867
First published on 26th September 2013
Abstract
The enzymatic synthesis of novel (di)saccharide acrylates from starch and 2-hydroxyethyl acrylate, 2-hydroxyethyl methacrylate and 4-hydroxybutyl acrylate (2-HEA, 2-HEMA and 4-HBA) catalyzed by various commercially available amylase preparations is demonstrated. Both liquefaction and saccharification amylases were tested as biocatalysts. Transglycosidation products were only detected in reaction mixtures containing saccharification amylases. The glycoamylase from Aspergillus niger was found to catalyze the synthesis of 2-(α-glucosyloxy)-ethyl acrylate (Glc-α-EA) from starch. The maltogenic amylase from B. stearothermophilus was found to catalyze the synthesis of 2-(α-maltosyloxy)-ethyl acrylate (Glc-Glc-EA). The transglycosidation product was isolated and purified and its structure was confirmed by 1H-NMR, 13C-NMR and ESI mass spectrometry. The selectivity of the enzyme towards the glycosidation position appeared to be extremely high. Only the anomeric hydroxyl group (C1–OH) of the disaccharide appeared to be glycosidated and the glycosidation products were anomerically pure. Subsequently, the enzymatically synthesized maltosyl acrylate was successfully polymerized by aqueous free radical polymerization to yield a novel acrylic polymer with pendant maltosyl units.
Introduction
Glycopolymers include both natural and artificial carbohydrate containing polymers, as well as synthetically modified natural saccharide-based polymers. Poly(saccharide acrylates) are special types of glycopolymers that combine an apolar acrylic backbone with pending saccharide groups. Since the saccharide side-groups lead to water-soluble, non-toxic, biocompatible and biobased polymers, they have received increased attention over the past decade.1–4 They have been used in diverse applications including as models of biological systems,5,6 macromolecular drugs and drug delivery systems,7 polymeric surfactants8 and many others.9 The synthesis of poly(saccharide acrylates) requires well-defined mono-functionalized saccharide acrylate monomers. The synthesis of such monomers involves a number of challenges, since there are many possibilities for the formation of positional isomers in which different hydroxyl groups of the saccharide become functionalized with the acrylate group. Using conventional synthesis routes, multiple reaction steps are required and all but one hydroxyl group has to be protected to obtain mono-functionalized saccharide acrylate monomers.10 Alternative to these synthesis routes, biocatalytic pathways towards (poly)saccharide synthesis and modification11–16 are beneficial and were explored here for the synthesis of maltosyl acrylates. Since biocatalysts show extremely high enantio-, regio- and chemoselectivity, mono-functionalization of saccharides can usually be achieved easily. Moreover, biocatalysis is advantageous with respect to the ambient reaction conditions used, i.e. atmospheric pressure, low reaction temperature, typically 37 °C, and neutral pH.17 In addition, enzymes are also non-toxic; they are obtained from natural renewable resources and can often be recycled.17 Especially enzymes from the classes of glycosyl transferases and hydrolases (EC 2.4 and 3.2)18 have been used in organic synthesis of (poly)saccharides and glycosides since carbohydrates are their natural substrates. α-Amylases have been applied as catalysts in the organic synthesis of glycosides of various alcohols (methyl-, ethyl-, propyl-, butyl- and benzyl-).19 Glycoamylase has been used as a catalyst for the synthesis of glycosides from hydroxybenzyl alcohol,20 octanol,21 vanillin,22 eugenol and curcumin.23 Nevertheless, the amylase catalyzed synthesis of saccharide acrylates from starch and hydroxyl functional acrylates that we present here has not been described before. Our biocatalytic synthesis routes using glycoamylase from Aspergillus niger or maltogenic amylase from Bacillus stearothermophilus as the catalyst result in anomerically pure, mono-functional (di)saccharide acrylate monomers in one single reaction step from starch. The polymerization of the enzymatically synthesized disaccharide acrylate 2-(α-maltosyloxy)-ethyl acrylate yields a novel acrylic polymer with pendant maltosyl units.Experimental section
Materials
Dinitrosalicylic acid (DNSS), hydroquinone (HQ), monomethyl ether hydroquinone (MEHQ), potassium sodium tartrate (K-Na-tartate), maltose (Glc-2), maltotriose (Glc-3), potato starch, 2-hydroxyethyl acrylate (2-HEA) 96% containing 200 ppm MEHQ, 2-hydroxyethyl methacrylate (2-HEMA) 97% containing 200 ppm MEHQ, lithium bromide (LiBr), sodium azide >99% (NaN3), diphenylamine (DPA) and potassium metabisulfite (K2S2O5) were obtained from Sigma-Aldrich. Maltotetraose (Glc-4), 2-hydroxyethyl methacrylate 97% stabilized with 200 ppm MEHQ (2-HEMA) and acetic acid 99.5% (HAc) were obtained from Acros. 4-Hydroxybutyl acrylate (4-HBA), 95%, containing 200 ppm MEHQ was obtained from ABCR chemicals. D-(+)-Glucose (anhydrous) (Glc), ethyl acetate (EtAc), sodium acetate trihydrate (NaAc), aniline and ammonium persulphate (APS) were obtained from Merck. Soluble starch was supplied by Difco. Silica 230–400 mesh was obtained from Silicycle. Chloroform (CHCl3), methanol (MeOH), propan-2-ol (IPA) and other mentioned organic solvents were obtained from Lab-scan. All chemicals were used as received.Amylases
Various types of amylases were obtained from five different suppliers. α-Amylases from Aspergillus oryzae were obtained from Genencor (Clarase L) and Sigma-Aldrich (A8220). α-Amylases from Bacillus subtilis were obtained from Amano (Neomalz H2O and Kleitase T10S), α-amylases from Bacillus licheniformis were obtained from Genencor (Spezyme FRED, AA, Alpha and Clearflow AA). Thermostable α-amylases were obtained from Novozymes (Liquozyme Supra) and AB enzymes (Gamalpha P120L and Spezial). α-Amylases from Bacillus amyloliquefaciens were obtained from Sigma-Aldrich (A7595) and Amano (Biokleitase L500). β-Amylases from barley were obtained from Sigma-Aldrich (A7130) and Genencor (Optimalt BBA and Spezyme DBA). Glycoamylases were obtained from Sigma-Aldrich (A7095), Novozymes (NS22035 and Spirizyme) and AB Enzymes (Gammadex) and maltogenic amylase from Bacillus stearothermophilus were obtained from Novozymes (Novamyl L) and AB Enzymes (Veron Xtender).Characterization
1H-NMR and 13C-NMR spectra were recorded using a 7.05 T Varian VXR spectrometer (300 MHz and 75 MHz resolution for 1H and 13C-NMR, respectively). Deuterium oxide (99.9 atom% D, Aldrich) was used as the solvent. Thin layer chromatography (TLC) was performed on aluminium sheets Silica gel 60/Kieselguhr F254 (Merck). Three eluent mixtures were used for the separation of the saccharide acrylates: 5/10/4 (v/v/v) n-But–IPA–water, 3/1/1 (v/v/v) EtAc–HAc–water or 2/1 (v/v) CHCl3–MeOH. Spots were visualized as described by Hansen using diphenylamine/aniline reagent.24 Saccharide derivatives appeared as blue/black spots on a white background after spraying the DPA/aniline reagent and heating at 120 °C for 1 hour. Column chromatography was performed on silica gel 60 (400 mesh). The sample was eluted with CHCl3–MeOH 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by CHCl3–MeOH 3:1 and subsequently CHCl3–MeOH 2:1. UV/Vis spectrometry measurements were performed using a PYE Unicam SP-200 UV/Vis spectrophotometer. HPLC was done using a Shimadzu HPLC under reversed phase conditions. An Alltech HP column (Alltima amino, 5 μm, 150 × 4.6 mm) was used for separation. The column temperature was maintained at 40 °C and the injection volume was 15 μL. Compounds were detected using a SPD-M20A photodiode array detector at 205 nm. A gradient of acetonitrile and double distilled water was used as the eluent with a flow rate of 0.5 mL min−1. The water content of the eluent was 15 vol% for 5 minutes followed by a linear increase to 60 vol% in 85 minutes (gradient A) or slowly increased from 5 vol% to 15 vol% in 5 minutes, followed by a linear increase to 60 vol% in 85 minutes (gradient B). The column is brought back to initial conditions by a linear gradient from 60 vol% water to 15 or 5 vol% in 20 minutes and subsequently equilibrated for 10 minutes, before the next injection. Gradient A is used for reaction mixtures containing 2-HEA and gradient B is used for reactions with 2-HEMA. ESI-MS was performed using a Thermo Scientific LTQ Orbitrap mass spectrometer in the positive ion mode. Samples were dissolved in water and diluted with an equal volume of methanol. Gel permeation chromatography (GPC) measurements were performed in N,N-dimethylformamide (DMF) containing 0.01 M LiBr on a Viscotek GPCMAX equipped with TDA detectors (model 302), using PSS-Gram-1000/30 columns. The molar mass was calculated relative to polymethylmethacrylate using narrow disperse standards (Polymer Laboratories). FT-Infrared spectroscopy (FTIR) was done using a Bruker JFS 88 equipped with an ATR-golden gate unit.
:1, followed by CHCl3–MeOH 3:1 and subsequently CHCl3–MeOH 2:1. UV/Vis spectrometry measurements were performed using a PYE Unicam SP-200 UV/Vis spectrophotometer. HPLC was done using a Shimadzu HPLC under reversed phase conditions. An Alltech HP column (Alltima amino, 5 μm, 150 × 4.6 mm) was used for separation. The column temperature was maintained at 40 °C and the injection volume was 15 μL. Compounds were detected using a SPD-M20A photodiode array detector at 205 nm. A gradient of acetonitrile and double distilled water was used as the eluent with a flow rate of 0.5 mL min−1. The water content of the eluent was 15 vol% for 5 minutes followed by a linear increase to 60 vol% in 85 minutes (gradient A) or slowly increased from 5 vol% to 15 vol% in 5 minutes, followed by a linear increase to 60 vol% in 85 minutes (gradient B). The column is brought back to initial conditions by a linear gradient from 60 vol% water to 15 or 5 vol% in 20 minutes and subsequently equilibrated for 10 minutes, before the next injection. Gradient A is used for reaction mixtures containing 2-HEA and gradient B is used for reactions with 2-HEMA. ESI-MS was performed using a Thermo Scientific LTQ Orbitrap mass spectrometer in the positive ion mode. Samples were dissolved in water and diluted with an equal volume of methanol. Gel permeation chromatography (GPC) measurements were performed in N,N-dimethylformamide (DMF) containing 0.01 M LiBr on a Viscotek GPCMAX equipped with TDA detectors (model 302), using PSS-Gram-1000/30 columns. The molar mass was calculated relative to polymethylmethacrylate using narrow disperse standards (Polymer Laboratories). FT-Infrared spectroscopy (FTIR) was done using a Bruker JFS 88 equipped with an ATR-golden gate unit.
Determination of amylase stability in aqueous 2-HEA solution
The stability of the enzymes was determined by measurement of their remaining hydrolytic activity after incubation in aqueous 2-HEA solution. The hydrolytic activity was determined spectrophotometrically using the DNSS method as described in the literature.25 Reaction mixtures for the determination of the amylase stability contained soluble starch (30 wt%/vol%), sodium acetate buffer (7 mL, pH 5.0, 50 mM), 2-HEA (3 mL) and 284 U of the corresponding amylase. Test blank solutions contain soluble starch (30 wt%/vol%), sodium acetate buffer (10 mL, pH 5.0, 50 mM) and 284 U of the corresponding amylase. The reaction mixtures were incubated at 60 °C for 15 minutes before a sample (200 μL) was taken. The sample was diluted with 17.5 mL sodium acetate buffer (pH 6.0, 16 mM) to obtain the right conditions for the determination of the remaining hydrolytic activity. From this solution, 600 μL was used for the determination of the hydrolytic activity as described above. The remaining activity is calculated by the following equation: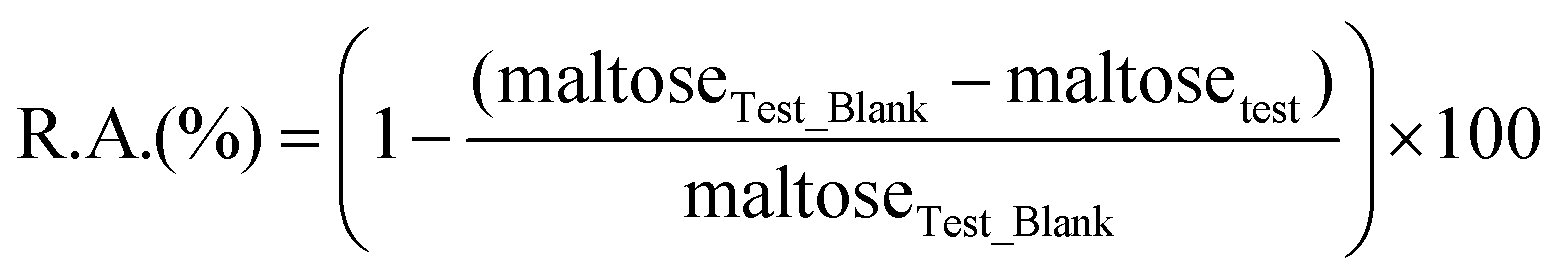
with R.A. = remaining hydrolytic activity of the amylase (%), maltoseTest_Blank = released maltose in test blank reaction (μmol) and maltosetest = released maltose in test reaction (μmol).
Determination of the synthetic activity of α-amylases
Reaction mixtures (total volume 15 mL) contained 3.0 g soluble starch dissolved in 10 mL sodium acetate buffer solution (pH 5.0, 50 mM, 0.02% NaN3). Subsequently 2-HEA (5 mL) was added. The reactions were started by addition of 2000 U of the corresponding amylase and incubated at 55 °C for 3 days. After 3 days the reaction mixtures were analyzed with thin layer chromatography using n-But–iPA–water 5/10/4 as the eluent.Maltogenic amylase catalyzed transglycosidation using hydroxyl functional (meth)acrylates
Reaction mixtures (total volume 15 mL) contained 3.0 g soluble starch dissolved in 10 mL sodium acetate buffer solution (pH 5.0, 50 mM, 0.02% NaN3). Subsequently, 5 mL of the acceptor alcohol (2-HEA, 2-HEMA or 4-HBA) was added. The reactions were started by the addition of 2000 U maltogenic amylase from Bacillus stearothermophilus. The reaction mixtures were incubated at 55 °C for 3 days. After 3 days the reaction mixtures were analyzed with TLC using EtAc–HAc–water 3/1/1 as the eluent. Spots were visualized as described above.Preparative synthesis of 2-(α-maltosyloxy)-ethyl acrylate by maltogenic amylase from Bacillus stearothermophilus
The reaction mixtures (total volume 100 mL) contained 5.0 g soluble starch in sodium acetate buffer solution (pH 5.0, 50 mM, 0.02% NaN3). Subsequently 2-hydroxyethyl acrylate (30 mL) was added. The reaction was started by the addition of maltogenic amylase solution (10 mL 20 kU). The reaction mixture was incubated at 55 °C for 4 days. The reaction mixture was analyzed with TLC. By using the CHCl3–MeOH eluent, three spots were detected with the DPA/aniline reagent: (i) 2-hydroxyethyl acrylate (Rf 1.00, yellow spot); (ii) 2-(α-maltosyloxy)-ethyl acrylate (Rf 0.45, blue spot); (iii) glucose (Rf 0.28, blue spot).Unreacted 2-HEA was removed by extraction with diethyl ether. Hydroquinone was added to the aqueous layer and the solution was concentrated by rotary evaporation at 30 °C. The unreacted starch was subsequently precipitated in isopropanol and removed by filtration. The filtrate was concentrated again by rotary evaporation at 30 °C and a solution containing the product was purified by flash chromatography. Fractions containing the product (Rf 0.45) were pooled, 10 mg hydroquinone was added to prevent thermal polymerization and the eluent was removed by rotary evaporation at 30 °C.
Glc-Glc-EA: transparent syrup, 0.408 g, purity: >85%, yield: 5 mol%; ESI-MS: calculated for C17H28O13 + Na: 463.1422; found: 463.3333. 1H-NMR (D2O) δ in ppm: 6.44 (H11, cis, J = 17.3 Hz), 6.21 (H10, J = 17.3, 10.45 Hz), 5.99 (H11, trans, J = 10.43 Hz), 5.35 (H1′, d, J = 3.76 Hz), 4.96 (H1, d, J = 3.75 Hz), 4.39 (H8), 4.2–3.2 (H2–H6, H2′–H6′, H7, H7′). 13C-NMR (D2O) δ in ppm: 168.6 (C9), 132.8 (C10), 127.7 (C11), 100.1 (C1′α), 98.2 (C1α), 77.5 (C4′), 76.3 (C4), 73.6 (C3), 73.1 (C3′), 72.9 (C2), 72.0 (C2′), 71.2 (C5), 69.5 (C5′), 67.7 (C8), 64.1 (C7), 63.5 (C6′), 60.6 (C6).
Influence of alcohol concentration on the synthesis of 2-(α-maltosyloxy)-ethyl(meth)acrylate catalyzed by maltogenic amylase from Bacillus stearothermophilus
The influence of the alcohol concentration (2-HEA or 2-HEMA) on the concentration of transglycosidation product was measured by HPLC analysis. Reaction mixtures contain soluble starch (30 wt%/vol%), sodium acetate buffer (10 mL, pH 5.0, 50 mM, 0.02% NaN3), maltogenic amylase from B. stearothermophilus (1 mL 2000 U) and 2-HEA or 2-HEMA. The amount of 2-HE(M)A was varied from 0.9 to 70 vol% by the addition of 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, 6.5, 13 or 26 mL. The reaction mixtures were incubated at 50 °C for 3 days. One milliliter of the reaction mixtures was taken, diluted with 24 mL ethanol and measured with HPLC.Polymerization of 2-(α-maltosyloxy)-ethyl acrylate
A novel maltosyl acrylate monomer (Glc-Glc-EA) was synthesized according to the enzymatic synthesis procedure described above. The monomer (0.45 mmol) was dissolved in 10 mL water ([M] = 45 mmol L−1). The reaction mixture was purged with nitrogen for at least one hour. Initiator solutions were prepared: 6 wt% K2S2O5 in 10 mL water and 12 wt% APS in 10 mL water. The reaction mixture was purged with nitrogen and heated to 55 °C. The reaction was initiated by the addition of 0.2 mL APS solution (12 wt%) and 0.2 mL potassium metabisulfite solution (6 wt%). The polymerization reaction was continued for 24 hours under a nitrogen atmosphere. After polymerization, the reaction mixture was precipitated into ten volumes of methanol and subsequently dried under vacuum overnight.Poly(2-α-maltosyloxyethyl acrylate) (P(Glc-Glc-EA)): white powder; 0.15 g, 1H-NMR (D2O) δ in ppm: 5.35 (H1′), 4.96 (H1), 3.44–4.34 (H2–H6, H2′–H6′, H7, H8), 1.70–2.70 (H10b, H11b). 13C-NMR (D2O) δ in ppm: 176.4 (C9), 100.4 (C1), 98.5 (C1′), 77.8 (C4, C4′), 74.8–69.5 (C2–C5, C2′–C5′), 65.7 (C8), 64.5 (C7), 60.7 (C6, C6′), 42.2 (C10b), 34.3 (C11b). FTIR-ATR (cm−1): 3400 (O–H stretch), 2940 (Aliphatic C–H stretch), 1720 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch); 1050 (C–O stretch).
O stretch); 1050 (C–O stretch).
Results and discussion
Activity of various amylases in saccharide acrylate synthesis
We have investigated the catalytic activity of various amylases in the synthesis of saccharide acrylate monomers from 2-hydroxyethyl acrylate and starch according to Scheme 1. α-Amylases that are commonly used for liquefaction of starch were tested as biocatalysts for the transglycosidation reaction. The “liquefaction” α-amylases that we used were endo-type amylases and originated from Aspergillus oryzae, Bacillus subtilis, Bacillus licheniformis and Bacillus amyloliquefaciens. Secondly, amylases that are commonly used for the saccharification of starch were tested as biocatalysts for the transglycosidation reaction between starch and 2-HEA as well. The hydrolytic activities of all amylases used in this study are listed in Fig. S4.† The “saccharification” amylases that we used were exo-type amylases and originated from Aspergillus niger, Bacillus stearothermophilus and barley (β-amylase). The synthetic activity of all amylases was analyzed by TLC. Remarkably, of the ten tested “liquefaction” amylases only the reaction mixture with α-amylase from A. oryzae contained saccharide acrylate glucosides as a result of transglycosidation. Comparable results were obtained by Santamaría in the α-amylase catalyzed transglycosidation of starch with methanol.26 They observed no transglycosidation products when α-amylases from bacterial sources, i.e. B. licheniformis, were used as the biocatalyst. On the other hand, methyl-saccharides could be detected using the α-amylase from A. oryzae as the catalyst for the transglycosidation. We could detect two spots of transglycosidation products with TLC using α-amylase from A. oryzae as the catalyst. The retention factors of the new spots were found to be 0.67 and 0.59, in between the spots of the normally observed hydrolysis products glucose and maltose (Rf 0.53 and Rf 0.43, respectively) and a reference glucoside 2-(β-glucosyloxy)-ethyl acrylate (Rf 0.73) that was synthesized by β-glucosidase from almond catalyzed glycosidation reaction of D-glucose with 2-hydroxyethyl acrylate (2-HEA).27 Although the transglycosidation products can be visually detected by TLC analysis, their concentration in the reaction mixture is rather low, especially compared to the amount of saccharidic products that result from hydrolysis. As a result the transglycosidation products could not be obtained pure from the reaction mixture and could therefore not be analyzed by 1H-NMR and ESI-MS. However, their retention factor in TLC provides information about the physical properties of the two transglycosidation products. First of all, the retention factors lower than the reference glucoside acrylate indicate that the compounds were more polar than 2-(β-glucosyloxy)-ethyl acrylate. Secondly, both compounds (Rf 0.67 and 0.59) were more hydrophobic than glucose, which has a retention factor of only 0.53. Finally, both spots were colored light blue by the TLC reagent, indicating a saccharide derivative. Based on these properties the transglycosidation products with the retention factors 0.67 and 0.59 were most likely 2-(α-maltosyloxy)-ethyl acrylate and 2-(α-maltotriosyloxy)-ethyl acrylate, respectively, but their structure could not be fully characterized as a result of the low product concentrations in the reaction mixture. In general, we might state that the “liquefaction” α-amylases from A. oryzae, B. subtilis, B. licheniformis and B. amyloliquefaciens that were used in this study, were ineffective catalysts for transglycosidation of 2-HEA and starch.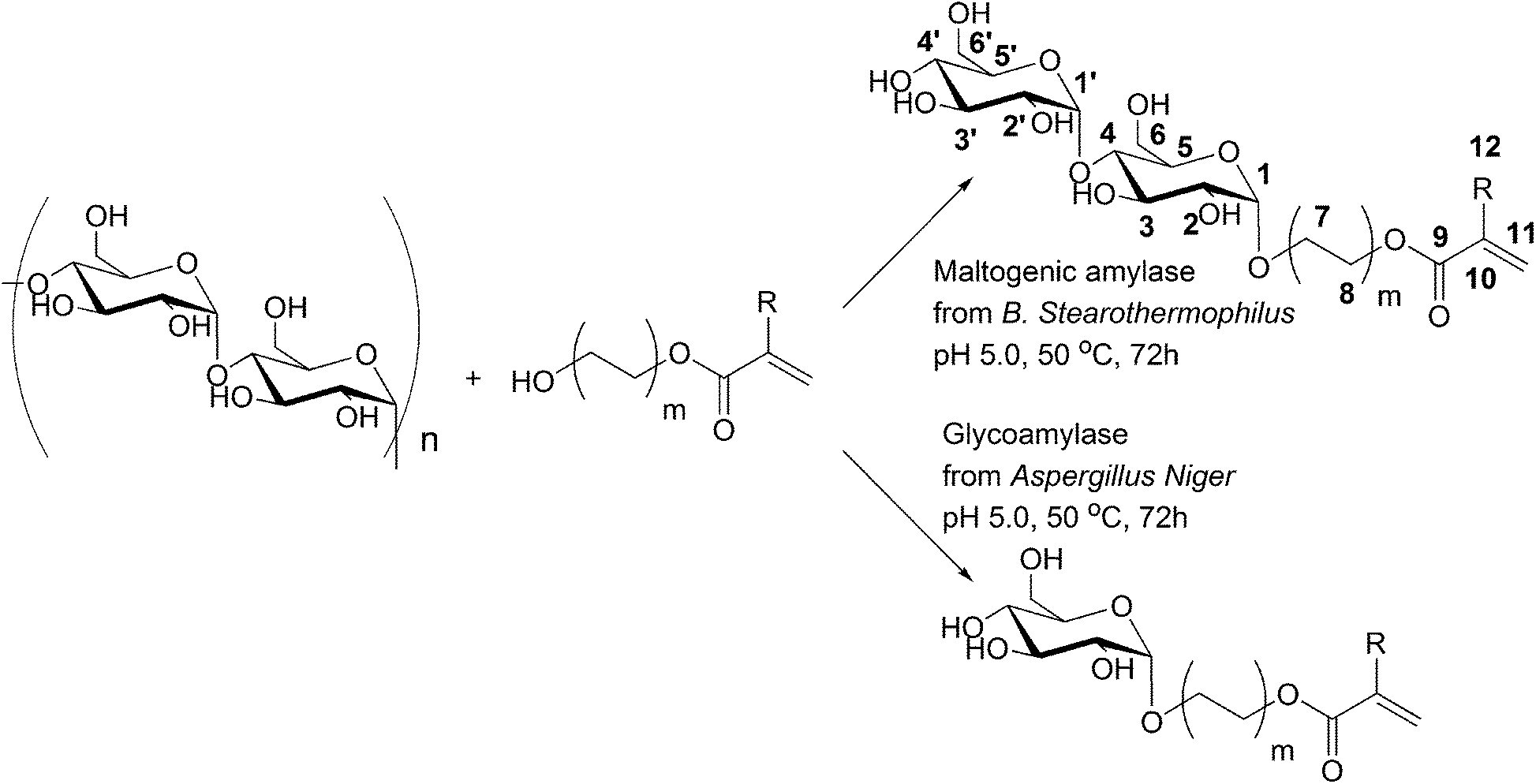 | ||
| Scheme 1 Amylase catalyzed synthesis of novel (di)saccharide acrylates by transglycosidation of starch with 2-HEA (m = 1, R = H), 2-HEMA (m = 1, R = CH3) and 4-HBA (m = 2, R = H). | ||
Much better results were obtained using the “saccharification” amylases from Aspergillus niger, Bacillus stearothermophilus and barley (β-amylase). Almost all commercially available “saccharification” amylase that were used for the transglycosidation reaction between 2-HEA and starch showed new spots in TLC corresponding to saccharide acrylates. Transglycosidation products could be detected in all reaction mixtures with the saccharification α-amylase from B. subtilis as the only exception. When β-amylase was used as the biocatalyst, the retention factor of the product (Rf 0.67) was lower than the retention factor of 2-(β-glucosyloxy)-ethyl acrylate (Rf 0.73) and thus the transglycosidation product must be more polar. Given that the mechanism of β-amylase catalyzed hydrolysis (cleavage of β-maltose from amylose) the transglycosidation product was likely to be 2-(β-maltosyloxy)-ethyl acrylate. However, also for the β-amylase catalyzed transglycosidation, the intensity of the spot of the new compound was very low, indicating an inefficient transfer or enzyme deactivation. On the other hand, both reaction mixtures containing glycoamylases and maltogenic amylases showed intense spots of transglycosidation products. In general, we found that the activity after incubation in aqueous 2-HEA solution of glycoamylases and maltogenic amylases was higher compared to β-amylase from barley (see Fig. S5† for the remaining activity data). The higher enzyme stability in the presence of 2-HEA, might explain the higher concentration of the transglycosidation products.
In the reaction mixtures containing glycoamylase from A. niger a product spot with a retention factor of 0.75 was detected. The retention factor of the new product was identical to the spot of the reference compound; 2-(β-glucosyloxy)-ethyl acrylate. Since the mobility of the product synthesized by glycoamylases was equal to the mobility of the reference compound and the glycoamylase from A. niger is known to be a retaining amylase, the product is very likely to be 2-(α-glucosyloxy)-ethyl acrylate.
The reaction mixtures containing maltogenic amylase showed a new spot with a retention factor of 0.68. The mobility of this glycosidation product indicates a polarity higher than 2-(β-glucosyloxy)-ethyl acrylate and lower than glucose (Rf 0.55). Since maltogenic amylases are known for producing high maltose syrups from liquefied starch,28 it could be expected that the product contained a maltose unit joined to the acceptor alcohol by a glycosidic linkage. Secondly, the maltogenic amylase from B. stearothermophilus is known to be a retaining amylase, resulting in a glycoside in α-configuration. Therefore, the transglycosidation product was expected to be 2-(α-maltosyloxy)-ethyl acrylate. Since the product concentration of the maltogenic amylase catalyzed transglycosidation reaction was relatively high, the product could be isolated, purified and its structure elucidated with NMR and ESI-MS.
Structural analysis of the product synthesized by maltogenic amylase from Bacillus stearothermophilus
The product of the maltogenic amylase catalyzed transglycosidation of starch and 2-HEA was collected by preparative column chromatography. Relative to the starch used, about 8 wt% of transglycosidation product was obtained after the purification steps. The chemical structure of the product was revealed by 1H-NMR (Fig. 1). Two different signals of the anomeric protons were found. The first type of anomeric protons was found at 5.34 ppm. This chemical shift is typically observed for anomeric protons, H-1, of α-(1,4)-linked D-glucopyranose residues in amylose and maltooligosaccharide chains.29 The second type of anomeric protons was found at 4.95 ppm. Anomeric protons with a chemical shift around 4.90 ppm are found for glycosides with α-linked alkyl chains.30 Secondly, the low coupling constant of 3.3 Hz found for this proton is characteristic of a linkage in α-configuration. Thus, we can conclude that the ethyl acrylate moiety was attached to the saccharide by a 1-O-α-linkage. A 1:1 ratio between the α-linked ethyl acrylate moiety (4.95 ppm) and the α-(1,4)-linked D-glucopyranose unit (5.34 ppm) was determined by the integration of the peak areas. This fully agreed with the expected structure of 2-(α-maltosyloxy)-ethyl acrylate. Secondly, the characteristic peaks corresponding to the acrylate double bond were found at 5.97–6.47 ppm. Integration of an acrylic proton peak and a comparison with the two anomeric protons (4.95 and 5.34 ppm) result in a 1.00:0.94:1.08 ratio (see also Fig. 1). This proves that the disaccharide was functionalized with just one acrylate moiety. This is crucial for saccharide acrylate monomers that will be used for the synthesis of linear glycopolymer by aqueous free radical polymerization as described below. Additionally, the structure of 2-(α-maltosyloxy)-ethyl acrylate was confirmed by 13C-NMR and ESI-MS (Fig. S1 and S2,† respectively). Signals corresponding to β-glycosides were not present in the 1H-NMR spectrum or the 13C-NMR spectrum. Thus the biocatalyst was found to catalyze the transglycosidation of starch with retention of the anomeric configuration. Furthermore, using the maltogenic amylase from B. stearothermophilus as the biocatalyst a mono-functional disaccharide acrylate was directly synthesized from starch and 2-HEA in one single reaction step, something which is impossible to achieve using conventional chemistry.
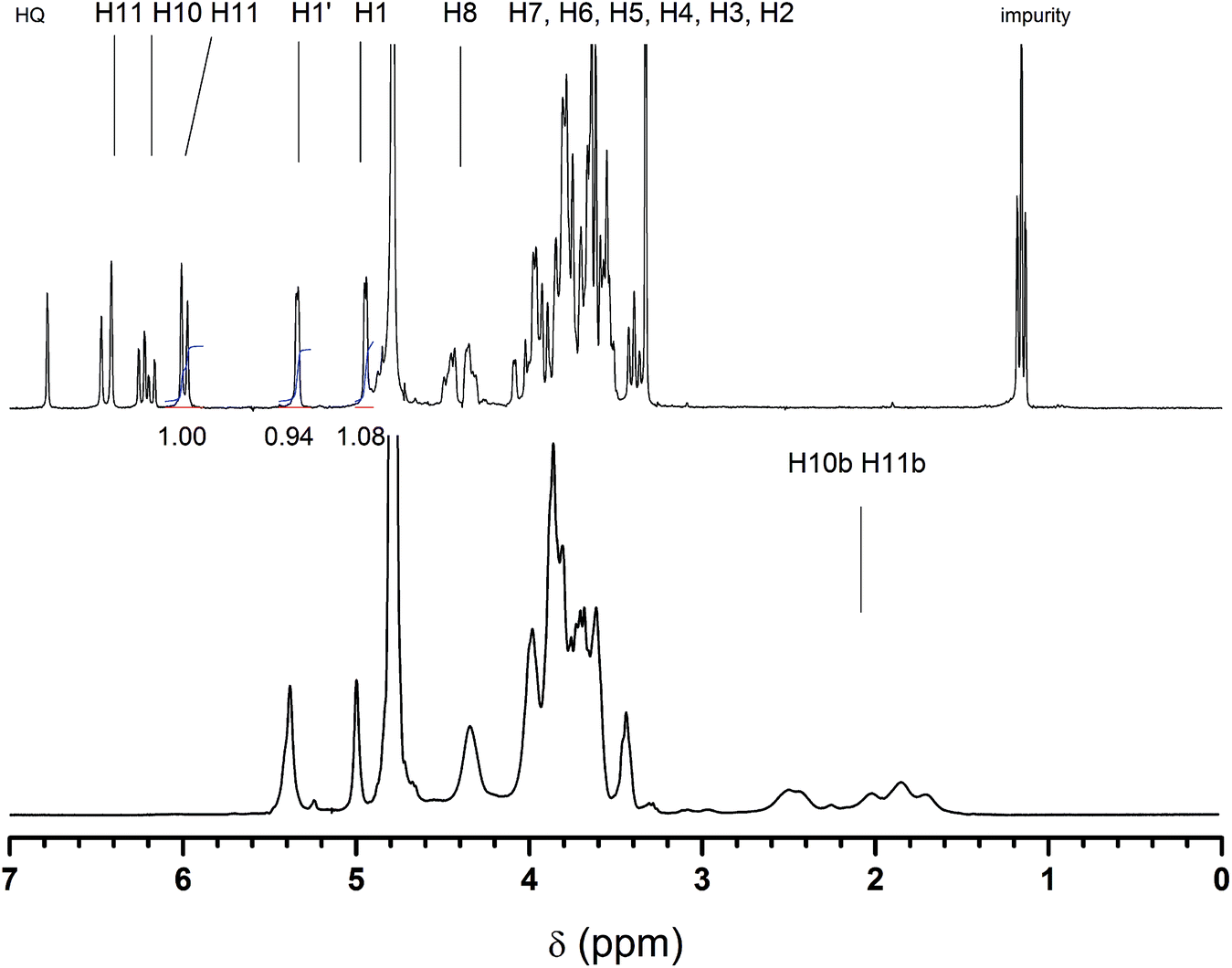 | ||
| Fig. 1 1H-NMR spectra of 2-(α-maltosyloxy)-ethyl acrylate (top) and its corresponding polymer poly(2-(α-maltosyloxy)-ethyl acrylate) (bottom spectrum). | ||
Maltogenic amylase catalyzed transglycosidation with other polymerizable alcohols
The specificity of the maltogenic amylase from B. stearothermophilus was tested with polymerizable alcohols other than 2-HEA as well. Other polymerizable, structurally similar, alcohols used were 2-hydroxyethyl methacrylate (2-HEMA) and 4-hydroxybutyl acrylate (4-HBA). The reaction mixtures containing 30 vol% 2-HEMA and 4-HBA were analyzed by TLC analysis.Transglycosidation products were found as well using 2-HEMA and 4-HBA as the acceptor alcohol (see Fig. 2). The retention factors of the transglycosidation products increased with increasing hydrophobicity of the acceptor alcohol (2-HEA < 2-HEMA < 4-HBA). The novel disaccharide acrylates 2-(α-maltosyloxy)-ethyl methacrylate (Glc-Glc-MA) and 2-(α-maltosyloxy)-butyl acrylate (Glc-Glc-BA) had retention factors of 0.48 and 0.52, respectively. The results showed that the maltogenic amylase from B. stearothermophilus catalyzed synthesis of saccharide acrylates was not limited to 2-hydroxyethyl acrylate, but also other (meth)acrylates with a primary alcohol group can be used as the acceptor alcohol.
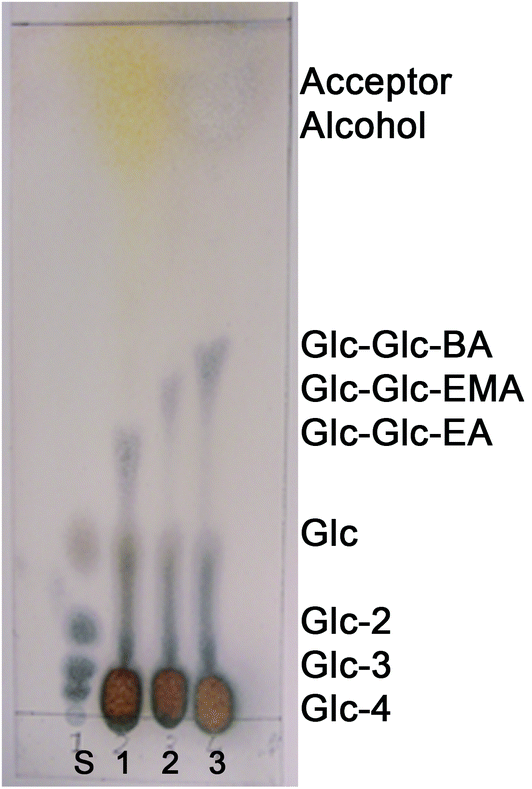 | ||
| Fig. 2 TLC chromatogram of reaction mixtures with 2-HEA (lane 1), 2-HEMA (lane 2) or 4-HBA (lane 3). Lane S contains saccharide standards of glucose (Glc), maltose (Glc-2), maltotriose (Glc-3) and maltotetraose (Glc-4). | ||
The maltogenic amylase is the first enzyme found that catalyzes the synthesis of saccharide acrylates containing more than one glucose unit by direct glycosidation in a single reaction step. Therefore, optimisation of the transglycosidation yield was done by varying the acceptor alcohol concentration.
Influence of alcohol concentration on yield of saccharide acrylate
A quite effective way to improve the glycosidation yield in transglycosidation reactions is to increase the donor or the acceptor concentration.31,32 The influence of the volume fraction of alcohols 2-HEA and 2-HEMA on the concentration of Glc-Glc-EA and Glc-Glc-EMA, was determined. The concentration of the products in the reaction mixtures was determined by HPLC. Using purified products, the peaks corresponding to Glc-Glc-EA and Glc-Glc-EMA were found at 9.25 and 14.20 min, respectively. The amount of transglycosidation product was determined by integration of the corresponding peak areas. Fig. 3 shows the dependence of the volume percentage of the acceptor alcohol on the peak area of the transglycosidation products. The concentration of Glc-Glc-EA was found to increase by increasing the amount of 2-HEA 10-fold from 0.9 vol% to 37 vol%. Further increase of the alcohol concentration in the reaction mixture resulted in a decrease of the amount of product. Most likely, this decrease was caused by the loss of enzyme activity in solutions containing higher amounts of alcohols. The curve of Glc-Glc-EMA shows a similar trend, although the optimal product concentration was found at 20 vol% 2-HEMA rather than 37 vol%. Apparently, the amylase was less active when the hydroxyl functional acceptor was more hydrophobic. A similar dependence on acceptor hydrophobicity was obtained in the amylase catalyzed alkyl glucoside synthesis reported by Larsson.19 Nevertheless, the concentration of Glc-Glc-EMA was increased with a 13-fold increase in the amount of 2-HEMA from 0.9 to 20 vol%.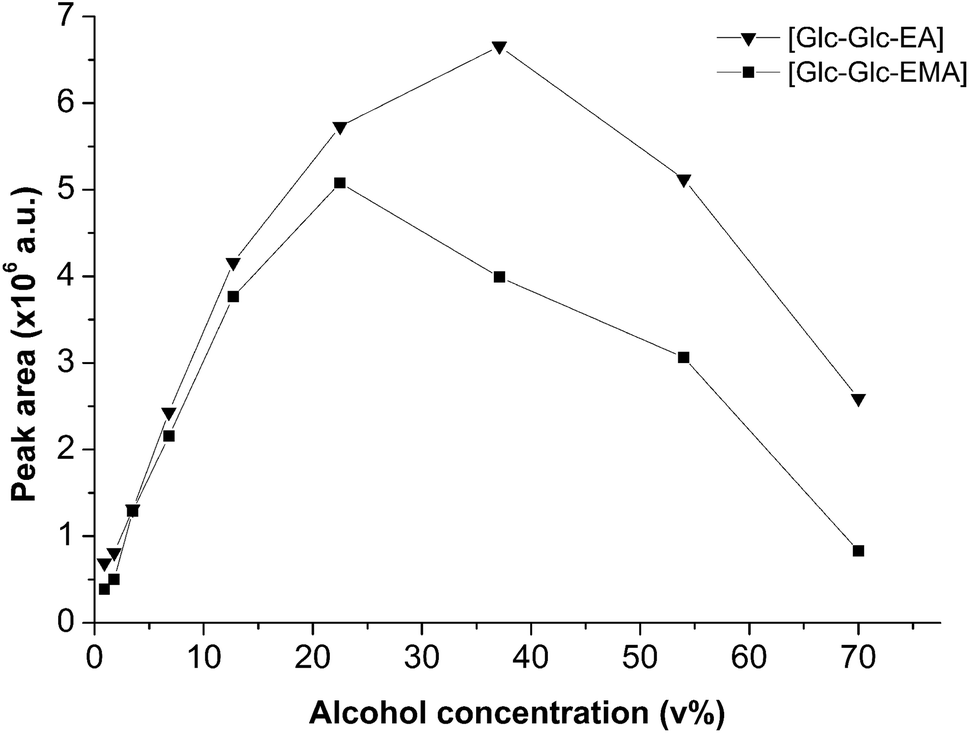 | ||
| Fig. 3 Effect of the volume fraction of 2-HEA and 2-HEMA on the concentration of transglycosidation products 2-(α-maltosyloxy)-ethyl acrylate and 2-(α-maltosyloxy)-ethyl methacrylate, respectively. | ||
Aqueous free radical polymerization of maltosyl acrylate monomer Glc-Glc-EA
The polymerization of the enzymatically synthesized monomer 2-(α-maltosyloxy)-ethyl acrylate was performed in aqueous solutions using an APS/K2S2O8 initiator system according to Scheme 2. Fig. 1 shows the 1H-spectrum of the starting monomer Glc-Glc-EA (top spectrum) and the spectrum of the resulting product poly(Glc-Glc-EA) (bottom spectrum). The three signals at 6 to 7 ppm, which correspond to the protons of the acrylate group, were found to disappear upon polymerization.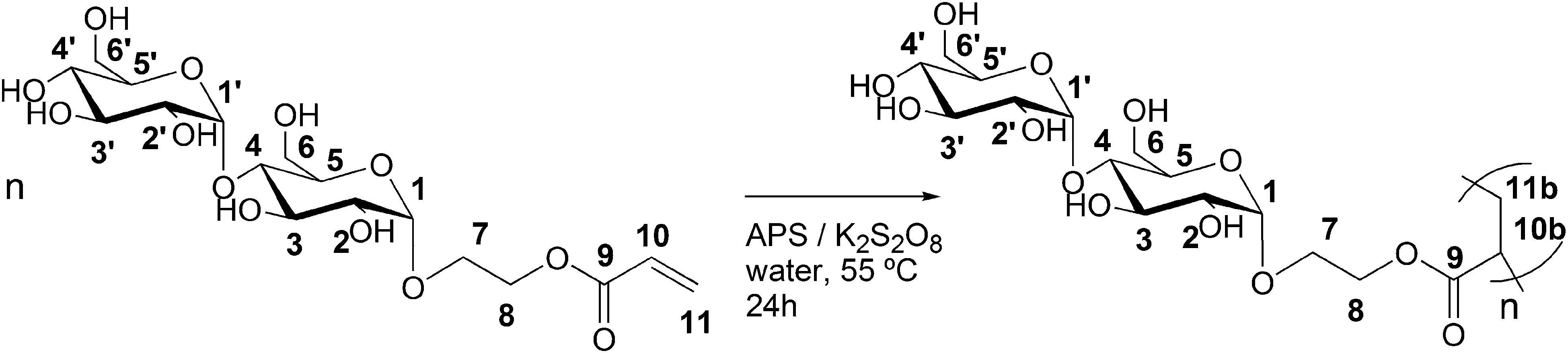 | ||
| Scheme 2 Synthesis of poly(2-(α-maltosyloxy)-ethyl acrylate) by free radical polymerization of 2-(α-maltosyloxy)-ethyl acrylate. | ||
The spectrum of the polymer product shows broad peaks in the region 1.6 to 2.6 ppm that correspond to the aliphatic protons in the backbone of P(Glc-Glc-EA), proving that the maltosyl acrylate monomer was successfully polymerized. In addition, the spectra show that the signals of the anomeric protons 1 and 1′ do not shift, disappear or change in ratio. This proves that the glycosidic part of the monomer Glc-Glc-EA was not changed during the polymerisation and thus, the polymer consists of a carbon–carbon backbone with a 1-O-α-maltosyl group attached by a 2-oxyethyl carboxylate spacer (as depicted in Scheme 2). The polymer structure was further confirmed by 13C-NMR (Fig. S3†). GPC analysis revealed that the molecular weight of the polymer P(Glc-Glc-EA) was in accordance with the relatively high initiator to monomer ratio used; Mn = 4 kg mol−1, Mw = 15 kg mol−1, D = 3.9. In theory, lowering the initiator to monomer ratio or changing the type of initiator should be effective to increase the molecular weight of the polymer. For example, we have already reported that the polymerization of 2-glucosyloxyethyl acrylate monomers using APS or KPS as initiator yields higher molecular weight saccharide acrylate polymers (Mn = 150 kg mol−1).27 Takaragi showed that high molecular weight poly(2-(cellobiosyloxy)-ethyl methacrylate) could be obtained by using the same initiator system in a lower initiator to monomer ratio.33 Therefore, we expect that the molecular weight of P(Glc-Glc-EA) could easily be increased by lowering the initiator to monomer ratio.
Nonetheless 1H-NMR, 13C-NMR and GPC proved the successful polymerization of the maltosyl acrylate monomer we synthesized enzymatically. Successful polymerization of such a maltosyl acrylate was never reported before and the resulting polymers with a high OH functionality might be very interesting as dispersing agents in the field of coatings and biomedicine.34
Conclusions
We have tested the catalytic activity of various amylases in the synthesis of saccharide acrylate monomers from 2-hydroxyethyl acrylate and starch. Liquefaction α-amylases from Aspergillus oryzae, Bacillus subtilis, Bacillus licheniformis and Bacillus amyloliquefaciens were tested as biocatalysts for the transglycosidation reaction. Similarly, saccharification amylases from Aspergillus niger, the maltogenic amylases from Bacillus stearothermophilus and β-amylase from barley were tested as biocatalysts for the transglycosidation reaction. Liquefaction amylases were found to be more stable in aqueous 2-HEA compared to saccharification amylases, but they do not catalyze the transglycosidation of starch and 2-HEA. In contrast, the saccharification amylases do catalyze the transglycosidation reaction between starch and 2-HEA. The glycoamylases from Aspergillus niger and the maltogenic amylases from Bacillus stearothermophilus appear to be the most effective as biocatalysts in the synthesis of saccharide acrylates. The transglycosidation product of the glycoamylase catalyzed reaction was found to have the same retention factor as the reference compound 2-(β-glucosyloxy)-ethyl acrylate. Given the specific properties of glycoamylases, we expect the transglycosidation product to be 2-(α-glucosyloxy)-ethyl acrylate. Similarly, the maltogenic amylase from B. stearothermophilus was found to catalyze the synthesis of a new kind of saccharide acrylate monomer that was identified as 2-(α-maltosyloxy)-ethyl acrylate. Other hydroxyl functional acrylates 2-HEMA and 4-HBA could also be used in the transglycosidation reaction, resulting in 2-(α-maltosyloxy)-ethyl methacrylate and 4-(α-maltosyloxy)-butyl acrylate, respectively. Subsequently the disaccharide acrylate 2-(α-maltosyloxy)-ethyl acrylate was polymerized by aqueous free radical polymerization, yielding a water-soluble glycopolymer. Thus by using maltogenic amylase from Bacillus stearothermophilus as the biocatalyst, different disaccharide monomers containing only one double bond per saccharide molecule can be synthesized in one single reaction step and could subsequently be polymerized to yield a new type of acrylic polymer with pendant maltose groups.Acknowledgements
This research was financed by BASF, Ludwigshafen, Germany. We kindly acknowledge Theodora Tiemersma-Wegman for help with HPLC and ESI-MS analysis.Notes and references
- A. Narumi, H. Kaga, Y. Miura, T. Satoh, N. Kaneko and T. Kakuchi, Polymer, 2006, 47, 2269–2273 CrossRef CAS PubMed.
- R. Narain and S. P. Armes, Biomacromolecules, 2003, 4, 1746–1758 CAS.
- M. L. Cerrada, M. Sánchez-Chaves, C. Ruiz and M. Fernández-García, Biomacromolecules, 2009, 10, 1828–1837 CrossRef CAS PubMed.
- H. F. N. de Oliveira and M. I. Felisberti, Carbohydr. Polym., 2013, 94, 317–322 CrossRef PubMed.
- M. Ambrosi, N. R. Cameron, B. G. Davis and S. Stolnik, Org. Biomol. Chem., 2005, 3, 1476–1480 CAS.
- K. Sasaki, Y. Nishida, T. Tsurumi, H. Uzawa, H. Kondo and K. Kobayashi, Angew. Chem., Int. Ed., 2002, 41, 4463–4467 CrossRef CAS.
- D. Mawad, E. Anne Boughton, P. Boughton and A. Lauto, Curr. Pharm. Des., 2012, 18, 2558–2575 CrossRef CAS.
- J. Raynaud, B. Choquenet, E. Marie, E. Dellacherie, C. Nouvel, J. L. Six and A. Durand, Biomacromolecules, 2008, 9, 1014–1021 CrossRef CAS PubMed.
- S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS.
- M. Ambrosi, A. S. Batsanov, N. R. Cameron, B. G. Davis, J. A. K. Howard and R. Hunter, J. Chem. Soc., Perkin Trans. 1, 2002, 45–52 CAS.
- J. Ciric, J. Oostland, J. W. de Vries, A. J. J. Woortman and K. Loos, Anal. Chem., 2012, 84, 10463–10470 CrossRef CAS PubMed.
- R. Rachmawati, A. J. J. Woortman and K. Loos, Biomacromolecules, 2013, 14, 575–583 CrossRef CAS PubMed.
- J. van der Vlist, M. Faber, L. Loen, T. J. Dijkman, L. Asri and K. Loos, Polymers, 2012, 4, 674–690 Search PubMed.
- J. van der Vlist, M. P. Reixach, M. van der Maarel, L. Dijkhuizen, A. J. Schouten and K. Loos, Macromol. Rapid Commun., 2008, 29, 1293–1297 CrossRef CAS.
- J. van der Vlist, I. Schonen and K. Loos, Biomacromolecules, 2011, 12, 3728–3732 CrossRef CAS PubMed.
- J. Ciric and K. Loos, Carbohydr. Polym., 2013, 93, 31–37 CrossRef CAS PubMed.
- Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley, 2010 Search PubMed.
- J. van der Vlist and K. Loos, in Enzymatic Polymerisation, ed. A. R. A. Palmans and A. Heise, 2010, vol. 237, pp. 21–54 Search PubMed.
- J. Larsson, D. Svensson and P. Adlercreutz, J. Mol. Catal. B: Enzym., 2005, 37, 84–87 CrossRef CAS PubMed.
- H. K. Shin, J. Y. Kong, J. D. Lee and T. H. Lee, Biotechnol. Lett., 2000, 22, 321–325 CrossRef CAS.
- G. Vijayakumar, B. Manohar and S. Divakar, Eur. Food Res. Technol., 2005, 220, 272–277 CrossRef CAS PubMed.
- R. Sivakumar and S. Divakar, Tetrahedron Lett., 2006, 47, 695–699 CrossRef CAS PubMed.
- G. Vijayakumar and S. Divakar, Biotechnol. Lett., 2007, 29, 575–584 CrossRef CAS PubMed.
- S. A. Hansen, J. Chromatogr., A, 1975, 107, 224–226 CrossRef CAS.
- H. U. Bergmeyer, J. Bergmeyer and M. Grassl, Methods of enzymatic analysis, Verlag Chemie, Weinheim, Deerfield Beach, FL, 1983 Search PubMed.
- R. I. Santamaría, G. Del Río, G. Saab, M. E. Rodríguez, X. Soberón and A. López-Munguía, FEBS Lett., 1999, 452, 346–350 CrossRef.
- W. M. J. Kloosterman, S. Roest, S. R. Priatna, E. Stavila and K. Loos, Green Chem., 2013 10.1039/C3GC41115J.
- H. Outtrup and B. E. Norman, Starch - Stärke, 1984, 36, 405–411 CrossRef CAS.
- G. S. Nilsson, L. Gorton, K. E. Bergquist and U. Nilsson, Starch - Stärke, 1996, 48, 352–357 CrossRef CAS.
- S. Koto, M. Hirooka, T. Tashiro, M. Sakashita, M. Hatachi, T. Kono, M. Shimizu, N. Yoshida, S. Kurasawa, N. Sakuma, S. Sawazaki, A. Takeuchi, N. Shoya and E. Nakamura, Carbohydr. Res., 2004, 339, 2415–2424 CrossRef CAS PubMed.
- F. van Rantwijk, M. Woudenberg-van Oosterom and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1999, 6, 511–532 CrossRef CAS.
- C. H. Wong and G. M. Whitesides, Enzymes in synthetic organic chemistry, Pergamon, 1994 Search PubMed.
- A. Takaragi, M. Minoda, T. Miyamoto and J. Watanabe, Macromol. Chem. Phys., 1998, 199, 1119–1126 CrossRef CAS.
- T. Yoshida, T. Akasaka, Y. Choi, K. Hattori, B. Yu, T. Mimura, Y. Kaneko, H. Nakashima, E. Aragaki, M. Premanathan, N. Yamamoto and T. Uryu, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 789–800 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 13C-NMR spectra of Glc-Glc-EA and poly(Glc-Glc-EA); positive ion ESI-MS spectrum of Glc-Glc-EA; initial and remaining enzyme activities. See DOI: 10.1039/c3gc41471j |
| This journal is © The Royal Society of Chemistry 2014 |