Carbon nanotube catalysts for oxidative desulfurization of a model diesel fuel using molecular oxygen
Wei
Zhang
,
Hong
Zhang
,
Jing
Xiao
,
Zhenxia
Zhao
,
Moxin
Yu
and
Zhong
Li
*
School of Chemistry and Chemical Engineering, South China University of Technology, 510640 Guangzhou, China. E-mail: cezhli@scut.edu.cn; Fax: +86-20-87110608
First published on 22nd October 2013
Abstract
We firstly propose the application of CNTs as novel catalysts and molecular oxygen as the oxidant for the oxidative desulfurization (ODS) of a model fuel containing benzothiophene (BT), dibenzothiophene (DBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT) at atmospheric pressure and low temperature. Results showed that when three CNTs including CNT-SZ, CNT-TS, and CNT-CD were separately used as catalysts with molecular oxygen as the oxidant, the conversion of DBT to its corresponding sulfone reached 100% at 150 °C separately within 40, 120 and 180 min. The CNT-SZ exhibited a superior catalytic activity even at a high fuel-to-catalyst ratio of 7.5 kg fuel per g catalyst. The oxidation reactivity of these benzothiophenic compounds followed the order: 4,6-DMDBT > DBT > BT. The deactivated CNT can be effectively regenerated by heat treatment under an argon atmosphere at 900 °C. Raman spectroscopy analysis revealed that the graphitization degree of the CNT played a decisive role in its catalytic activity for DBT oxidation. The CNT with the higher degree of graphitization had higher catalytic activity for DBT oxidation since its higher electric conductivity benefited the transfer of electrons involved in the oxidation–reduction reaction.
Introduction
Deep desulfurization of liquid fuels has become an increasingly important subject worldwide due to environmental concerns, e.g. SOx emissions and particulate matter (PM) formation. By 2009, the sulfur content of transportation fuels became regulated to be no more than 15 ppmw in many developed countries.1 Currently, the major desulfurization technology in the petroleum refinery is the hydrodesulfurization (HDS) process operated at the temperatures of 320–380 °C and the H2 pressures of 30–70 atm. HDS is highly efficient in removing aliphatic and acyclic sulfur compounds, but it is not efficient in removing refractory benzothiophenic compounds, e.g. 4,6-dimethyldibenzothiophene (4,6-DMDBT), due to their steric hindrance effects.2–4 Hence, HDS has become a less effective or cost-intensive process to achieve an ultra-clean fuel with less than 15 ppmw of sulfur. Therefore, developing alternative desulfurization approaches is highly desirable.5–10So far, oxidative desulfurization (ODS) has been considered to be one of the most promising alternative desulfurization processes. By employing the ODS process, the refractory benzothiophenic compounds can be converted to highly polar sulfoxides or sulfones under mild conditions,11 which would be easily removed by extraction or adsorption.11 Up to now, hydrogen peroxide (H2O2) in aqueous solution has been the most commonly used oxidant in ODS systems.12–17 One drawback of using H2O2 as the oxidant for ODS is that the water involved in these systems created the biphasic problem and limited the mass transfer. In addition, it inevitably led to loss of a certain amount of fuel during the liquid–liquid phase separation process after the ODS reaction. To overcome these shortcomings, oil-soluble oxidants, such as tert-BuOCl18 and tert-BuOOH,19 were used to provide a uniform and homogeneous environment for ODS. However, the reaction's safety and the high cost of the oxidant limited its industrial application.
Molecular oxygen has long been considered a desirable oxidant for deep ODS of liquid fuels, due to its advantages of low cost, large abundance, and environment benignity. However, due to its low oxidizing ability under mild conditions, it is very difficult to achieve efficient removal of benzothiophenic compounds if molecular oxygen is simply used for ODS. It was found that effective oxidation of benzothiophenic compounds by molecular oxygen can be achieved by adding aldehydes as co-oxidants.20,21 However, the addition of co-oxidants not only increases the cost of the oxidation process, but also increases the amount of fuel loss when separating it from the organic acids. Up to now, very few studies have reported on effective ODS in the absence of co-oxidants.22,23
Recently, the application of carbon nanotubes (CNTs) as a catalyst has attracted a great deal of attention of the catalysis community due to their unique textural property and chemical features. CNTs demonstrated a superior catalytic performance in oxidative dehydrogenation (ODH) of aromatic hydrocarbons and light hydrocarbons.24–26 To the best of our knowledge, CNTs have not yet been investigated for ODS of diesel using molecular oxygen as the oxidant.
In this work, we propose the application of CNTs as novel catalysts and molecular oxygen as the oxidant without the addition of any sacrificial agent. Then the catalytic activity of the CNTs for the oxidation of benzothiophenic compounds in a model diesel fuel was tested. The fresh CNTs and the deactivated CNT were characterized. The effects of textural properties and chemical groups of the CNTs on their catalytic activity would be discussed and reported here.
Results and discussion
Dibenzothiophene oxidation with various CNT catalysts
Three kinds of multi-walled CNTs were purchased from Shenzhen Nanotech Port Co. (Shenzhen, China), Tsinghua University (Beijing, China) and Chengdu Institute of Organic Chemistry of the Chinese Academy of Sciences (Chengdu, China), which were labeled as CNT-SZ, CNT-TS and CNT-CD, respectively. The CNTs were used as such without any further purification.
Fig. 1 shows the catalytic performance of various CNTs as catalysts for DBT oxidation. It can be seen that all CNTs exhibited excellent catalytic performance for DBT oxidation when using molecular oxygen. When CNT-SZ, CNT-TS, and CNT-CD were separately applied as catalysts, the conversion of DBT reached 100% separately within 40 min, 120 min, and 180 min. It showed that the catalytic activities of these CNTs as catalysts for DBT oxidation followed the order: CNT-SZ > CNT-TS > CNT-CD. Sampanthar23 reported that the application of 30 mg of Co–Mn catalysts to completely catalyze the oxidation of 400 ppm of DBT in 25 mL of a model fuel using molecular oxygen at 150 °C required 24 h. Zhou22 used iron tetranitrophthalocyanine as a catalyst for the oxidation of 500 ppm of DBT, and reported that at a temperature range of 100–140 °C and 0.3 MPa oxygen pressure, 98.7% conversion of DBT could be obtained within 2 h with the fuel-to-catalyst ratio of 0.1 kg g−1. For a comparison of the catalytic activity of the CNT-SZ with those of the Co–Mn catalyst and the iron tetranitrophthalocyanine catalyst separately reported by Sampanthar23 and Zhou,22 the catalytic reaction rate needs to be estimated. The catalytic reaction rate was defined as the mass of sulfur converted per unit mass of catalyst per unit time (mg S g−1 min−1). The catalytic reaction rate vt can be calculated by the following equation:
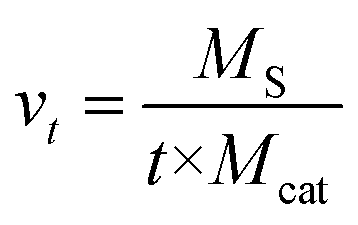
 | ||
| Fig. 1 Catalytic activities of three CNT catalysts for the oxidation reaction of DBT (reaction conditions: 0.01 g of the CNT catalyst, reaction temperature of 150 °C, O2 flow rate of 150 mL min−1). | ||
On the basis of our experimental data, the catalytic reaction rate vt of DBT oxidation in the presence of CNT-SZ at 150 °C was up to 32 mg S g−1 min−1. On the other hand, we also calculated the catalytic reaction rate vt on the basis of the experimental data reported by Sampanthar23 and Zhou.22 Our calculation indicated that their catalytic reaction rates vt of DBT oxidation in the presence of the catalysts Co–Mn and iron tetranitrophthalocyanine are separately around 0.15 and 0.42 mg S g−1 min−1. This indicated that the vt value of DBT oxidation in the presence of CNT-SZ was much higher than those in the presence of the catalysts Co–Mn and iron tetranitrophthalocyanine. The superior ODS activity of the CNTs would be further explained and discussed in the next characterization section.
Effect of reaction conditions on dibenzothiophene oxidation
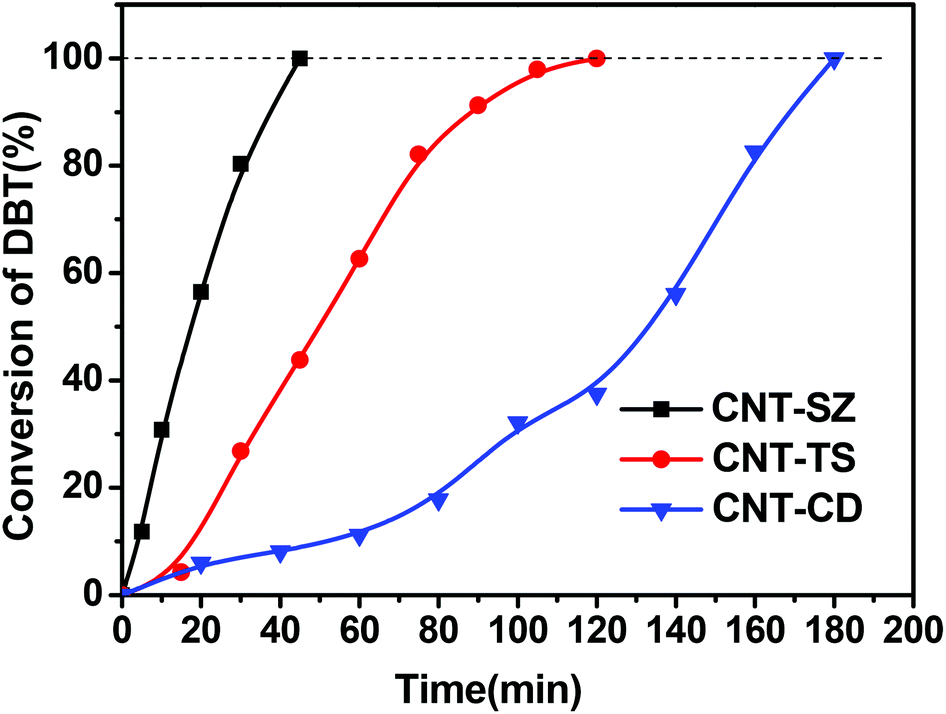
Fig. 2 shows the effect of reaction temperature on DBT oxidation. It indicated that the reaction temperature had a significant influence on DBT oxidation. When the reaction temperature was increased from 120 °C to 150 °C, a remarkable increase in DBT conversion was obtained. At 120 °C, only 77% of DBT was oxidized within 6 h, while at 150 °C almost all of DBT was oxidized within 40 min, suggesting that the reaction temperature has a significant impact on DBT oxidation.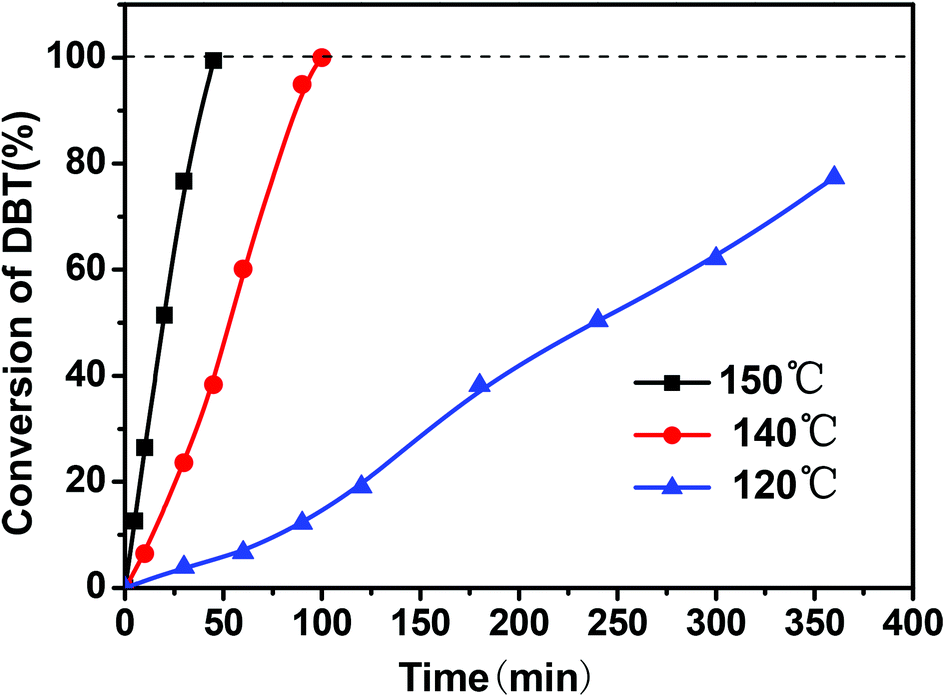 | ||
| Fig. 2 Effect of reaction temperature on the ODS of DBT (reaction conditions: 0.01 g of CNT catalyst, O2 flow rate of 150 mL min−1). | ||
Usually, petroleum diesel is produced from the fractional distillation of crude oil between 200 °C and 350 °C at atmospheric pressure. If CNTs as novel catalysts and molecular oxygen as the oxidant can be applied to the ODS reaction for removing organic sulfur compounds from petroleum diesel at temperatures lower than 200–350 °C in the future, more energy would not be required for the ODS technology proposed in this work because the reaction temperature of 150 °C is lower than that of petroleum diesel from the fractional distillation of crude oil.
Fig. 3 shows the effect of the oxygen flow rate on the conversion of DBT. An increase of the oxygen flow rate from 40 mL min−1 to 150 mL min−1 caused the conversion of DBT to slightly increase due to the introduction of more reactants of molecular oxygen into this reaction system at higher feeding rate.
 | ||
| Fig. 3 Effect of O2 flow rates on the conversion of DBT (reaction conditions: 0.01 g of CNT-SZ catalyst, reaction temperature of 150 °C). | ||
Fig. 4 shows the effect of the amount of the CNT catalyst in the reactor on the conversion of DBT. It was observed that an increase in the amount of the CNT-SZ catalyst from 0.004 g to 0.01 g made the conversion of DBT increase remarkably; however, a further increase in the catalyst amount from 0.01 g to 0.03 g could not further improve the DBT conversion. It implied that there were enough active sites for DBT oxidation in the case of the CNT-SZ catalyst amount of 0.01 g. It was noted that when using 0.004 g of catalyst, the fuel-to-catalyst ratio was as high as 7.5 kg g−1. Under these conditions, a complete conversion of DBT within 60 min was obtained, suggesting that the catalyst CNT-SZ had a superior catalytic performance for the DBT oxidation.
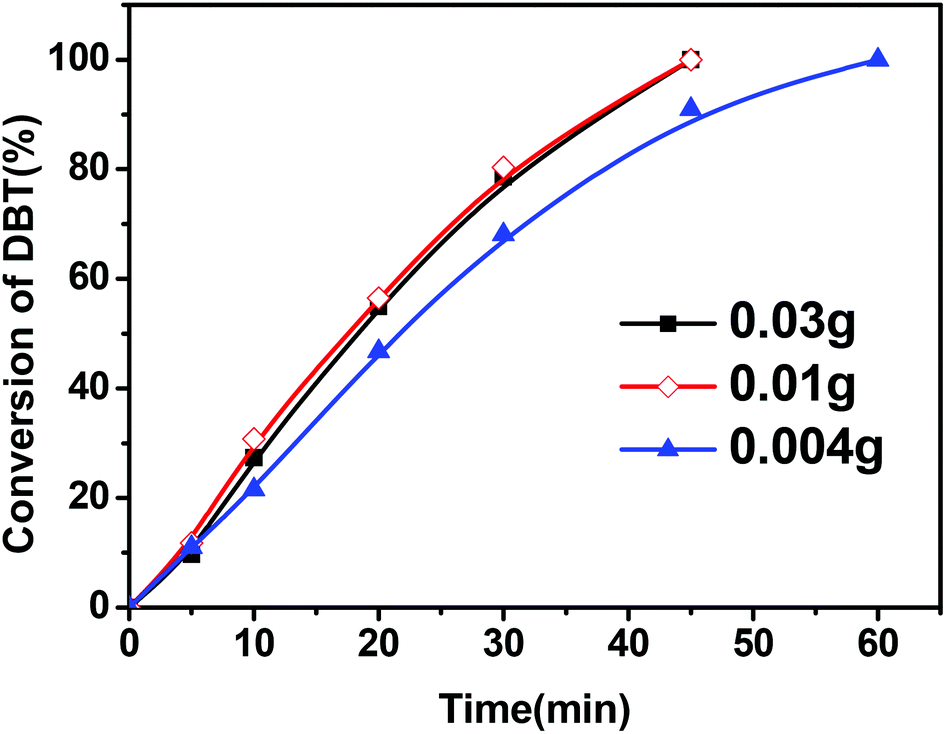 | ||
| Fig. 4 Effect of catalyst amounts of CNT-SZ on the conversion of DBT (reaction conditions: reaction temperature of 150 °C, O2 flow rate of 150 mL min−1). | ||
Oxidation reactivity of various sulfur compounds
Fig. 5 shows the oxidation reactivity of three kinds of sulfur compounds, BT, DBT and 4,6-DMDBT. It indicated that the reactivity of these benzothiophenic compounds followed the order: 4,6-DMDBT > DBT > BT. Generally speaking, the oxidation reactivity of the sulfur compounds increases with an increase in the electron density of their sulfur atoms. Shiraishi et al.27 and Otsuki et al.16 reported that the electron densities on sulfur atoms for BT, DBT, and 4,6-DMDBT are 5.739, 5.758, and 5.760, respectively. That is to say, the electron densities on sulfur atoms for BT, DBT, and 4,6-DMDBT followed the order: 4,6-DMDBT > DBT > BT. Our results about the oxidation reactivity of these benzothiophenic compounds were in good agreement with the order of the electron densities of these sulfur atoms.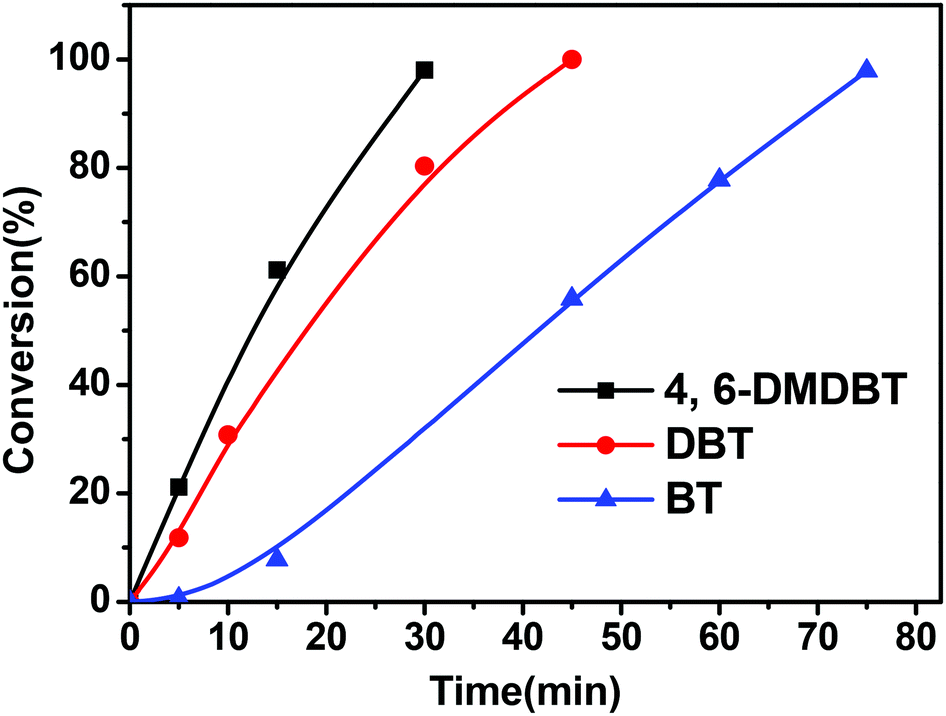 | ||
| Fig. 5 The comparison of the reactivity of three kinds of sulfur compounds (4,6-DMDBT, DBT, BT) for the ODS (reaction conditions: 0.01 g of CNT-SZ catalyst, reaction temperature of 150 °C, O2 flow rate of 150 mL min−1). | ||
Selectivity of the catalyst CNT-SZ for three benzothiophenic compounds
In order to verify the selectivity of the CNT-SZ for BT, DBT and 4,6-DMDBT at the same time, the model fuel containing a mixture of BT, DBT and 4,6-DMDBT was used for the ODS reaction. The model fuel contained the same mole concentration of each substrate with the total sulfur content of 450 ppmw. Then the model fuels were analyzed at different reaction times.Fig. 6 gives the PFPD chromatograms of the model fuels at different reaction times when CNT-SZ was used as a catalyst at 150 °C. It was clearly observed that the peak intensities for BT, DBT and 4,6-DMDBT decreased with reaction time. The disappearance rates of these three sulfur compounds in the model fuel followed the order: 4,6-DMDBT > DBT > BT. It indicated that the oxidation selectivity of the CNT-SZ catalyst for these three sulfur compounds followed the order: 4,6-DMDBT > DBT > BT, which was in accordance with the order of the oxidation reactivity of the three sulfur compounds in the ODS reactions when BT, DBT and 4,6-DMDBT were separately used as a reactant.
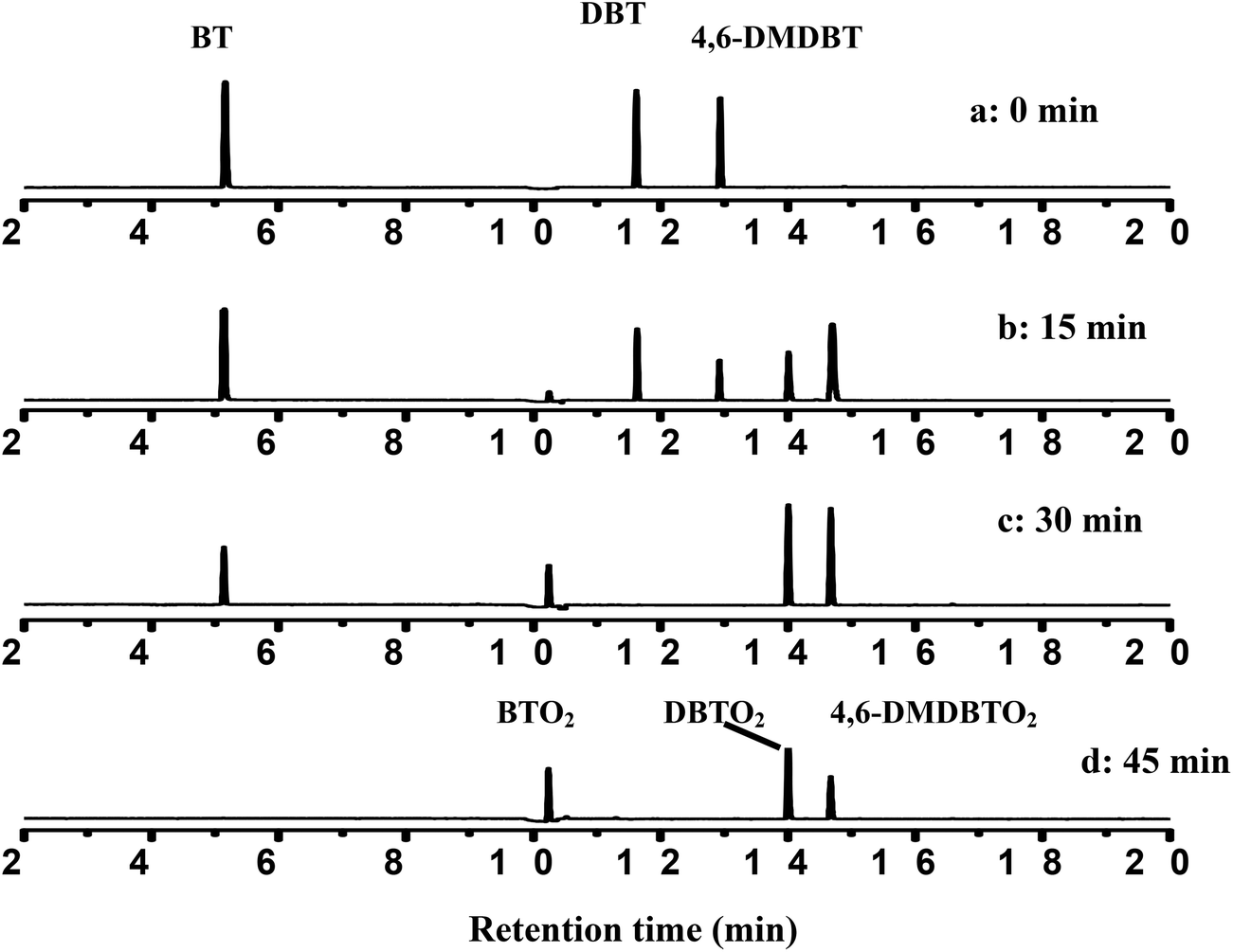 | ||
| Fig. 6 PFPD chromatograms of the model fuel over CNT-SZ at different reaction times (reaction conditions: 0.01 g of CNT-SZ catalyst, reaction temperature of 150 °C, O2 flow rate of 150 mL min−1). | ||
According to the result and discussion above, it can be considered that the oxidation of BT, DBT and 4,6-DMDBT may undergo an electrophilic addition reaction of active oxygen species.28 The sulfur atoms could be preferentially attacked by active oxygen species yielding sulfones or sulfoxides. It was observed from Fig. 6 that when the peak intensity for BT, DBT and 4,6-DMDBT decreased with reaction time, only three new peaks appeared and became gradually stronger with reaction time. The three new peaks were confirmed separately to be BTO2 (retention time: 10.20 min), DBTO2 (retention time: 14.07 min), 4,6-DMDBTO2 (retention time: 14.75 min) by mass spectrum analysis. For example, Scheme 1a and 1b separately give the mass spectrum of the oxidation product of DBT and the standard mass spectrum of DBTO2. The oxidation product of DBT can be confirmed to be DBTO2 by comparing the mass spectrum of the oxidation product with the standard mass spectrum.
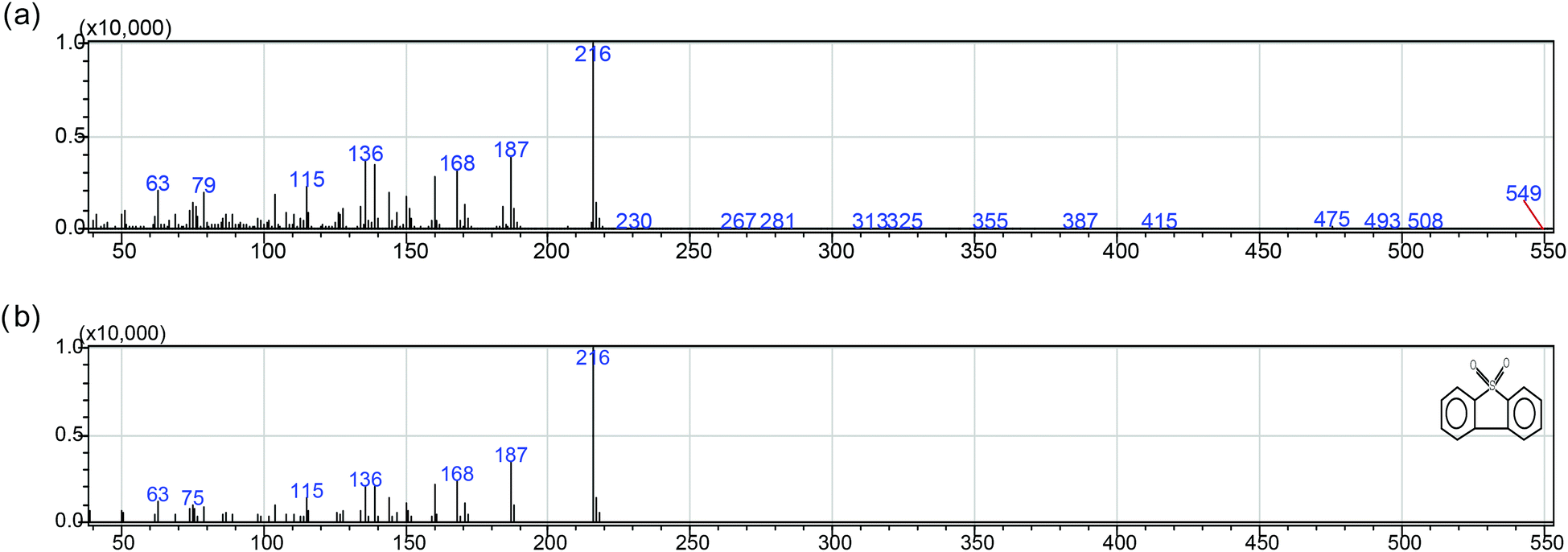 | ||
| Scheme 1 (a) Mass spectrum of the oxidation product of DBT. (b) Standard mass spectrum of DBTO2. | ||
With regard to the selectivity of the CNT-SZ for the formation of the reaction product, it was noticed that during the ODS reaction only three new peaks appeared, which corresponded to BTO2, DBTO2 and 4,6-DMDBTO2, and no other new peak appeared until complete conversion of the sulfur compounds. This suggests that each of the sulfur compounds was oxidized to its corresponding sulfone, respectively, without the generation of any by-product. It meant that the selectivity of the CNT catalyst for the conversion of the sulfur compound into its corresponding sulfone was 100%. Thus, the yield of oxidation reaction products of these sulfur compounds should be equal to the amount converted of these sulfur compounds.
In addition, Fig. 6 also shows that after 45 min of reaction, no peak for BT, DBT and 4,6-DMDBT was observed, suggesting complete conversion of BT, DBT and 4,6-DMDBT. It indicated that the CNT-SZ catalyst exhibited a very high efficiency for oxidation of these three organosulfur substrates.
Regeneration of the deactivated CNT
Fig. 7 shows the catalytic performances of the spent catalyst CNT-SZ-W, the fresh CNT-SZ and the regenerated catalyst CNT-SZ-H. The activity of the spent catalyst CNT-SZ-W was lower than that of the fresh CNT-SZ, indicating that the activity of the CNT-SZ decreased after the ODS reaction. However, the deactivated catalyst CNT-SZ-W can be regenerated by a heat treatment method proposed in this work. It can be seen that the catalytic performance of the regenerated catalyst CNT-SZ-H was very close to that of the fresh catalyst CNT. It implied that the application of heat treatment can effectively regenerate the deactivated catalyst. It can enable the catalytic activity of the catalyst to be almost completely recovered.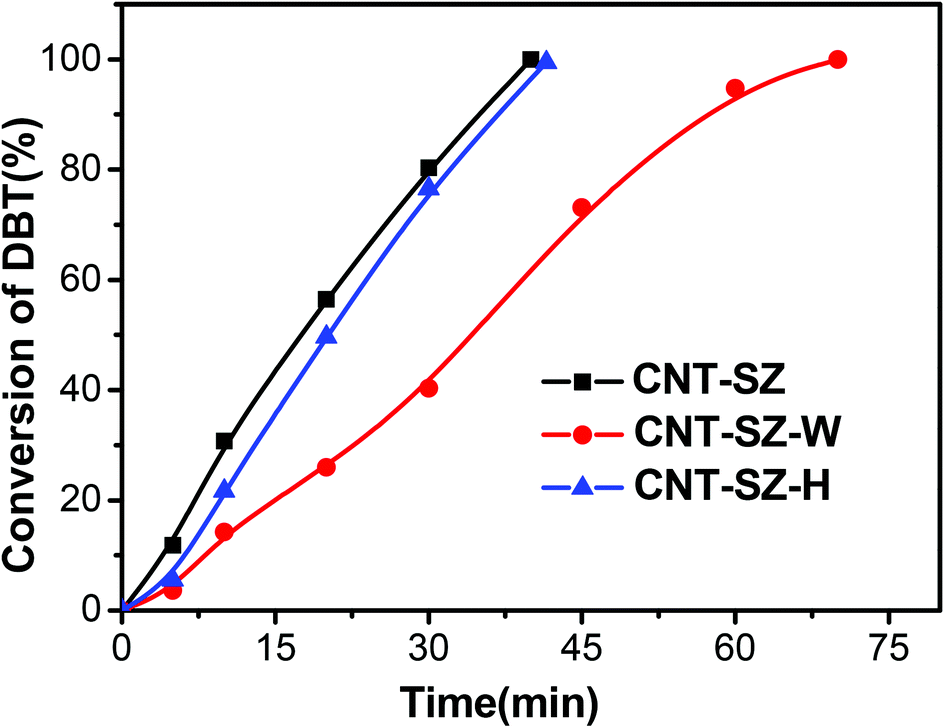 | ||
| Fig. 7 Comparison of catalytic performances of fresh CNT-SZ, spent CNT-SZ-W and regenerated CNT-SZ-H for DBT oxidation (reaction conditions: 0.01 g of each catalyst, reaction temperature of 150 °C, O2 flow rate of 150 mL min−1). | ||
In addition, the recycling experiments of the CNT-SZ for ODS reactions were conducted. It can be seen from Fig. 8 that the activity of the regenerated CNT-SZ-H was basically stable. Although it showed a slight decrease after 5 runs, the activity of the regenerated CNT-SZ-H catalyst was still retained as high as over 95%, suggesting good regenerability of the CNT catalyst.
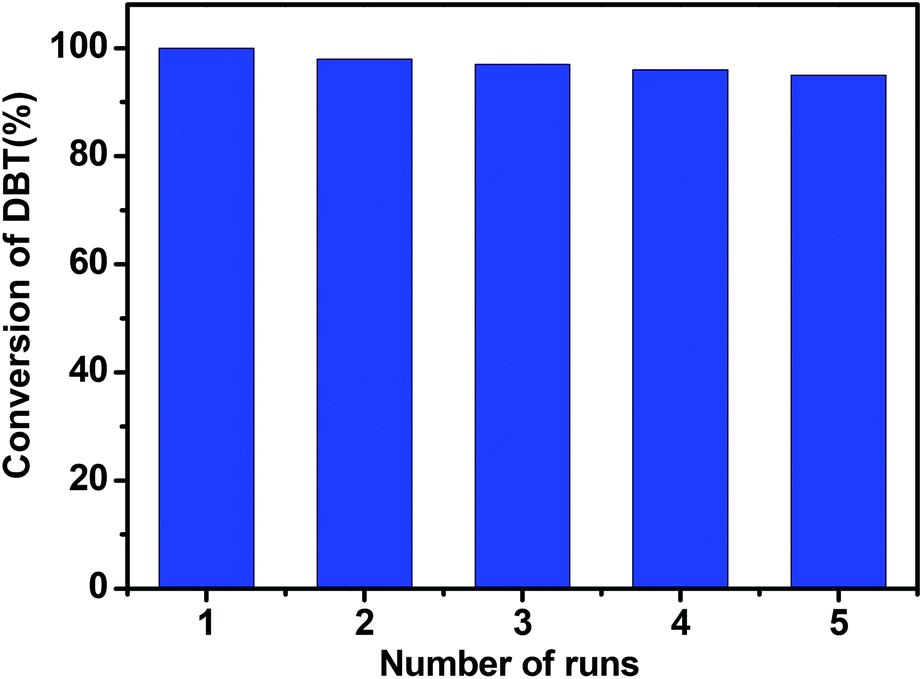 | ||
| Fig. 8 Cycling runs in the oxidation of DBT in the presence of CNT-SZ (reaction conditions: 0.01 g of the catalyst, reaction temperature of 150 °C, O2 flow rate of 150 mL min−1, reaction time: 40 min). | ||
Fig. 9 shows the SEM micrographs of the CNT-SZ before and after reaction. It seems that no obvious change in the morphology of the CNT-SZ before and after the ODS reaction was observed. However, the CNT-SZ catalysts before and after the ODS reaction would be characterized separately by N2 adsorption–desorption isotherms, Raman spectra and XPS characterizations in the next section.
 | ||
| Fig. 9 SEM micrographs of the CNT-SZ: (a) before the ODS reaction and (b) after the ODS reaction. | ||
Textural properties of the CNT catalysts
Table 1 lists the textural properties of three original CNTs, the deactivated catalyst CNT-SZ-W and the regenerated catalyst CNT-SZ-H. The BET surface areas of the three fresh CNTs were very close to those reported in the literature.31 The data in Table 1 indicate that the CNT-SZ and CNT-CD had similar textural properties, i.e. surface area (146.5 m2 g−1 and 140.9 m2 g−1) and average pore sizes (11.2 nm and 15.2 nm). However, their catalytic activities for DBT oxidation were obviously different. The CNT-SZ exhibited a much higher catalytic activity for DBT conversion than the CNT-CD, as shown in Fig. 1. It meant that the textual properties of the catalysts were not crucial factors affecting their catalytic activities. Table 1 indicates that the textural property/parameters of the deactivated catalyst CNT-SZ-W were nearly the same as the regenerated catalyst CNT-SZ-H, but their catalytic activities were obviously different, as shown in Fig. 7. It implied that the partial deactivation of the CNT-SZ during the ODS reaction may not be caused by the pore blockage or collapse of the CNT-SZ catalyst.| Carbon nanotubes | BET surface area (m2 g−1) | Pore size (nm) | Total pore volume (cm3 g−1) |
|---|---|---|---|
| CNT-CD | 146.5 | 15.17 | 0.5558 |
| CNT-TS | 192.9 | 13.42 | 0.6471 |
| CNT-SZ | 140.1 | 11.20 | 0.3947 |
| CNT-SZ-W | 134.7 | 11.46 | 0.3860 |
| CNT-SZ-H | 132.2 | 11.51 | 0.3842 |
DRIFTS
DRIFTS can be used to determine the type of oxygen-containing groups on carbon surfaces.29Fig. 10 shows the DRIFTS spectra of the fresh CNTs. It was observed that there were several characteristic peaks, the broad peak centered around 3446 cm−1 was assigned to the stretching vibration of –OH groups in adsorbed water; two peaks separately centered at 1728 cm−1 and 1302 cm−1 represented the carboxylic acid groups; the peak centered at 1568 cm−1 could be assigned to the vibration of quinone groups; the peak centered at 1438 cm−1 could be assigned to the bending vibrations of OH groups; and two bands separately centered at 1162 cm−1 and 1074 cm−1 represented the C–O stretching vibrations of ether-type groups and alcohols.30 Hence, the DRIFTS analysis suggested the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH and adsorbed water on the surfaces of the CNTs.
O, C–OH and adsorbed water on the surfaces of the CNTs.
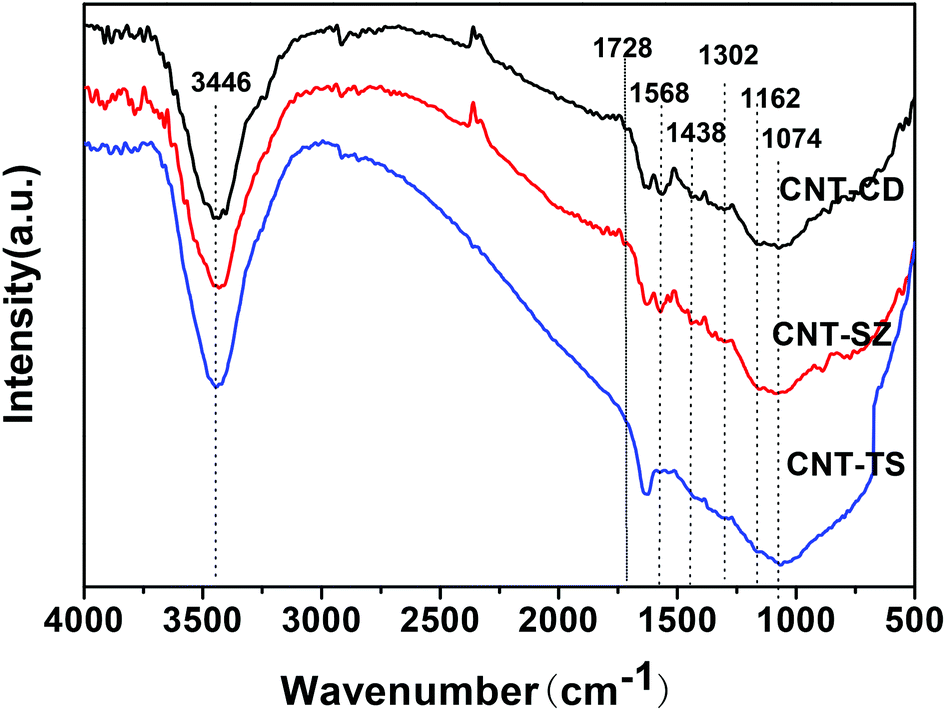 | ||
| Fig. 10 FT-IR spectra of three different CNTs. | ||
XPS
XPS characterization was carried out to quantitatively analyze the types and contents of various oxygen-containing functional groups on the surfaces of CNT catalysts, including CO, –OH and adsorbed water. Fig. 11 shows the O1s spectra of the fresh CNTs and the deactivated CNT-SZ-W samples. The deconvolution of the O1s spectrum of three fresh CNT catalysts revealed three different chemical environments for oxygen, which could be assigned to CO (around 530.5 eV), –OH (around 532.5 eV) and adsorbed water (around 533.6 eV),31,32 which has also been confirmed to be the surface groups of the CNTs by means of the DRIFTS analysis above. It was clearly visible from Fig. 11c and 11d that the CO peak intensity of the CNT-SZ after the ODS reaction decreased obviously, while its C–OH peak intensity increased greatly. Table 2 lists the contents of the CO, –OH and adsorbed H2O on the surfaces of the three fresh CNTs and the deactivated CNT-SZ-W, which were calculated from the peak deconvolution. The data in Table 2 indicated that the contents of the CO, –OH and adsorbed H2O on the surfaces of the CNT-SZ before and after the ODS reaction were also greatly changed. The molar fraction of CO on the surfaces of the CNT-SZ before the ODS reaction was 0.34%, while it was reduced to 0.07% on the surfaces of the CNT-SZ-W after the ODS reaction, having a decrease of 386%. At the same time, the molar concentration of C–OH on the surfaces of the CNT-SZ increased from 0.63% to 2.01%, having an increase of 219%. It implied that most of the oxidizing groups CO were converted to the reducing groups C–OH during the ODS reaction. Normally, the conversion of the oxidizing groups CO to the reducing groups C–OH can only be achieved by reduction rather than oxidation by molecular oxygen. This interesting result strongly suggested that the carbonyl-quinone/hydroxyl functional groups participated in the ODS reaction, and probably acted as the active sites for the ODS reaction, which was consistent with the phenomenon reported in the literature about employing CNT for the oxidative dehydrogenation reaction of hydrocarbons.24,26,31,33–35
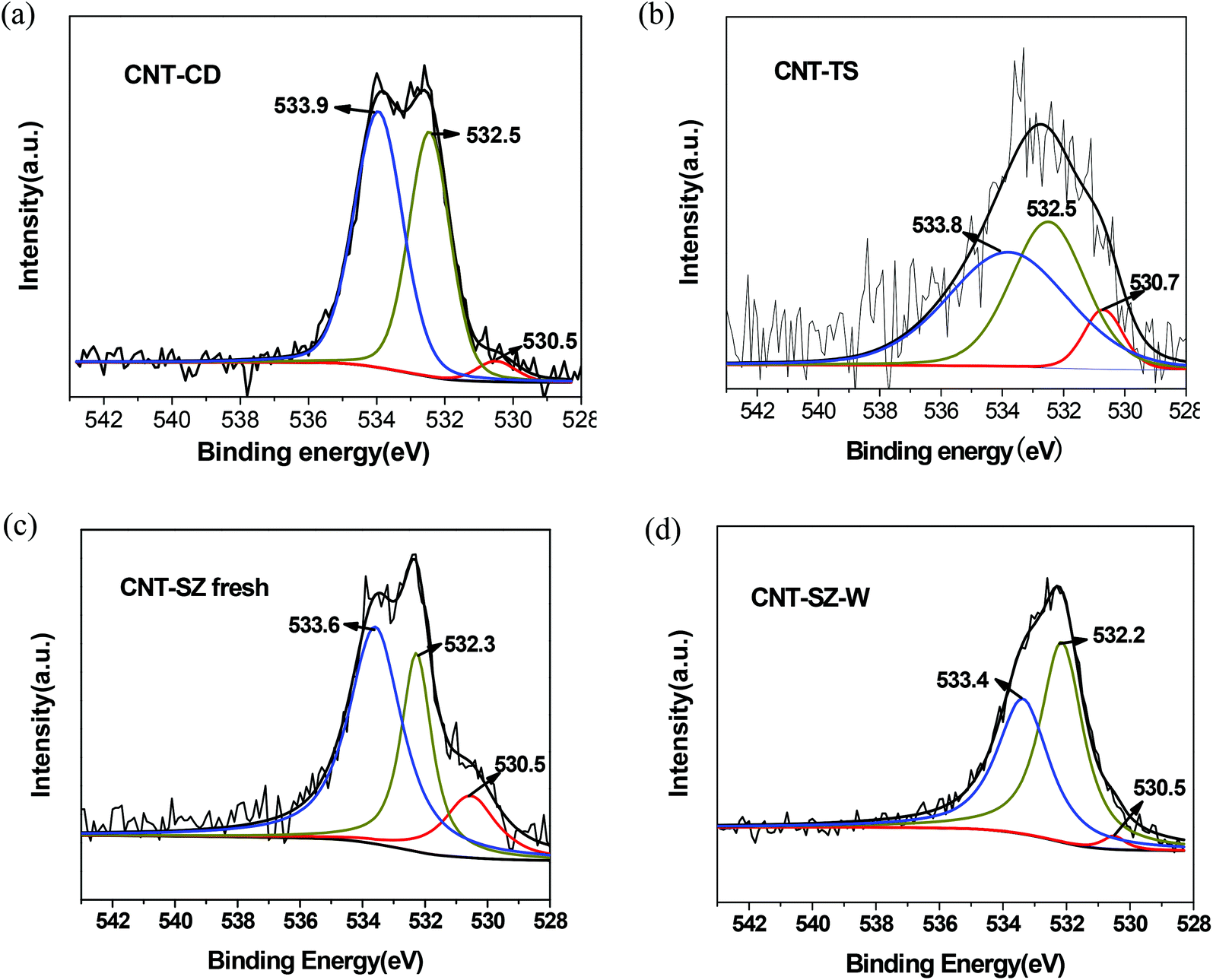 | ||
| Fig. 11 O1s XPS spectra of different CNTs (a, CNT-CD; b, CNT-TS; c, CNT-SZ; d, CNT-SZ-W). | ||
| CNTs |
|
|
H2O (%) |
|
Atom fraction of [O]a (%) |
|---|---|---|---|---|---|
a Atom ratio of the oxygen atoms on the surfaces of the CNTs to all the atoms detected, and this is a sum of the three peaks coming from the oxygen groups, 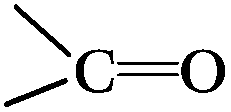 , , 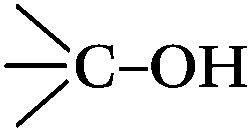 , H2O. , H2O.
|
|||||
| CNT-CD | 0.16 | 1.21 | 1.66 | 1.37 | 3.03 |
| CNT-TS | 0.09 | 0.42 | 0.54 | 0.51 | 1.05 |
| CNT-SZ | 0.34 | 0.63 | 1.20 | 0.97 | 2.17 |
| CNT-SZ-W | 0.07 | 2.01 | 1.66 | 2.08 | 3.75 |
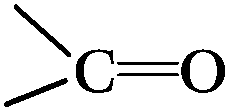
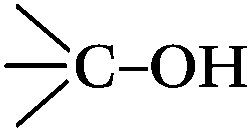
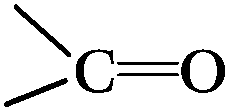
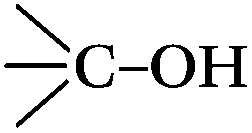
When the catalyst activity and the content of the surface active sites were contrasted on the basis of the data in Table 2 and Fig. 1, it was found that the sum of the two oxygen-containing groups of CO and C–OH for three fresh CNTs followed the order: CNT-CD > CNT-SZ > CNT-TS; the amount of individual CO groups followed the order: CNT-SZ > CNT-CD > CNT-TS; and the amount of individual –OH followed the order: CNT-CD > CNT-SZ > CNT-CD. All the trends were inconsistent with the order of the catalytic activities of these CNTs (CNT-SZ > CNT-TS > CNT-CD). In addition, a similar inconsistency was also found when contrasting the orders of the oxygen-containing groups and the catalytic activities of the original CNT-SZ catalyst and the spent CNT-SZ-W catalyst. Therefore, it was considered that the catalytic activity of the CNTs could not directly or simply be correlated to a pre-existing certain type or total amounts of the oxygen functional groups on the surfaces of the CNTs. It meant that the catalytic activity of the CNTs for DBT oxidation was not mainly governed by the amounts of oxygen-containing groups. It should be mentioned that the polarity of CNTs generally depends on the amount of oxygen-containing groups on the surfaces of CNT.36 It was found that the catalytic activities of the CNTs did not increase or decrease in proportion to the amounts of the total oxygen-containing groups. This meant that the polarity or the amount of oxygen-containing groups was not a dominant factor affecting the catalytic activity of the CNTs.
Raman spectroscopy
Fig. 12 shows the Raman spectra of three kinds of CNTs such as fresh catalyst CNTs, the deactivated catalyst CNT-SZ-W and the regenerated catalyst CNT-SZ-H. It was observed that there were three characteristic bands, namely the D-band at 1324 cm−1, the G-band at 1572 cm−1 and the D′ band at 1608 cm−1.37,38 The D band and D′ band are usually attributed to the presence of amorphous or disordered carbon in these CNT samples, due to the finite graphitic planes and other forms of carbon, such as rings along with defects or oxygen-containing groups on the nanotube walls,39 while the G band can be attributed to ordered sp2-hybridised carbon atoms. Generally speaking, the intensity ratio of the G and D bands, I(G)/I(D), can give a qualitative description of the order or the degree of graphitization of the CNT samples, and the I(G)/I(D) ratio increased with increasing order.33 It can be seen from Fig. 12a that the I(G)/I(D) ratios of the three fresh CNTs followed the order: CNT-SZ(0.99) > CNT-TS(0.64) > CNT-CD(0.55). This trend of the I(G)/I(D) ratios was consistent with the order of the catalytic activity of these three CNTs, CNT-SZ > CNT-TS > CNT-CD, as shown in Fig. 1. It implied that the graphitization degree of the CNTs may be a decisive factor affecting their catalytic activity for DBT oxidation.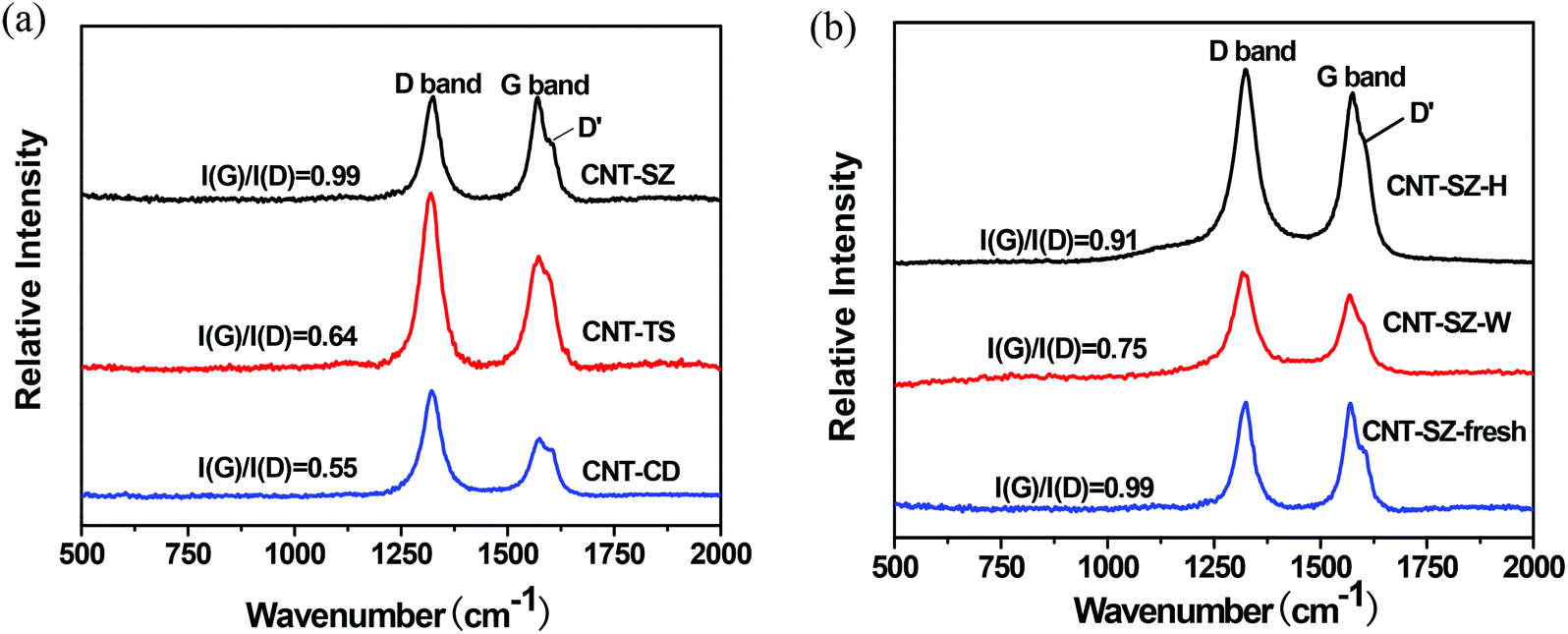 | ||
| Fig. 12 Raman spectra of various CNTs. | ||
Fig. 12b gives a comparison of the Raman spectra of the fresh, deactivated and regenerated CNTs. It was clearly visible that, after the ODS reaction, the I(G)/I(D) ratio decreased from 0.99 for CNT-SZ to 0.75 for the CNT-SZ-W. It indicated that its degree of graphitization decreased, which might be probably due to the formation of defects during the ODS reaction. Correspondingly, the catalytic activity of the CNT-SZ-W also became lower compared to the fresh CNT-SZ as shown in Fig. 7. It also indicated that the catalytic activity of the CNTs was mainly dependent on its graphitization degree. More interestingly, after the deactivated catalyst CNT-SZ-W was regenerated at 900 °C under an argon atmosphere, its graphitization degree was greatly recovered as its I(G)/I(D) ratio increased from 0.75 for CNT-SZ-W to 0.91 for CNT-SZ-H. Correspondingly, its catalytic activity was almost completely recovered, nearly being the same as the fresh CNT-SZ and being much higher than the deactivated catalyst CNT-SZ-W, as exhibited in Fig. 7. This fact further confirmed that the graphitization degree of the CNTs was a decisive factor affecting their catalytic activity for DBT oxidation.
From the foregoing discussion, it can be clearly seen that the graphitization degree of carbon structure of the CNTs employed in this work follows the order: CNT-SZ(0.99) > CNT-SZ-H(0.91) > CNT-SZ-W(0.75) > CNT-TS(0.64) > CNT-CD(0.55), which is in accordance with the order of the catalytic activity of these CNTs, as shown in Fig. 1 and 7. This consistency clearly indicated that the CNTs with higher graphitization degree had higher catalytic activities for DBT oxidation. A similar phenomenon was reported in some other reaction systems by Bégin40 and Su,31 in which the conversion and selectivity of hydrocarbons increased with the graphitization degree when CNT was applied as a catalyst. It can be inferred that the degree of graphitization of the CNTs played a decisive role in the catalytic activity. The reason may be that the higher degree of graphitization would make CNTs have higher electric conductivity, and thus benefit the transfer of electrons involved in the oxidation–reduction reaction.25,41,42 As a result, the catalytic activity of the CNTs is improved.
The possible mechanism
The general reaction equation of DBT oxidation using molecular oxygen in the presence of the CNT is proposed in Scheme 2 on the basis of the experimental results and discussions above.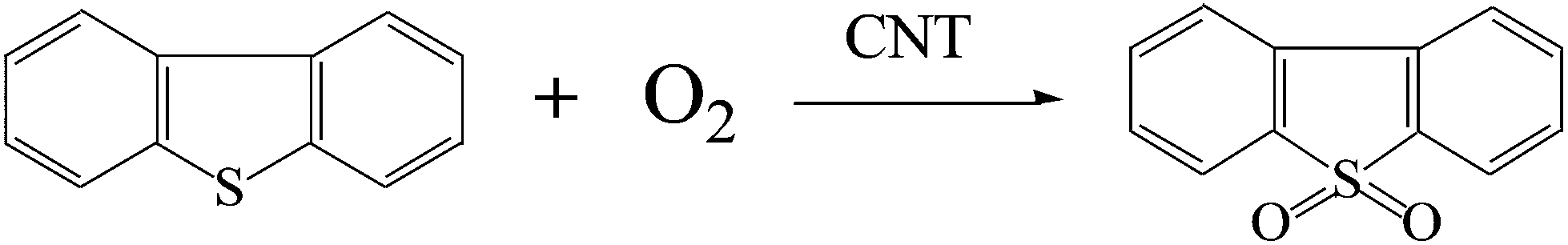 | ||
| Scheme 2 Oxidation reaction pathway of DBT with molecular oxygen as the oxidant and CNT as the catalyst. | ||
This reaction might occur via the following steps: (I) molecular oxygen gains electrons from hydroxyl groups to be activated to active oxygen,26,38 and simultaneously, hydroxyl groups are oxidized to carbonyl groups. (II) DBT is oxidized to DBTO2 by the generated active oxygen while the carbonyl groups gain electrons to be reduced to hydroxyl groups. In this model, the electron-transport rate is mainly governed by the graphitization degree of the CNT. Therefore the graphitization degree is very crucial for the catalytic activity of the CNT (Fig. 13).
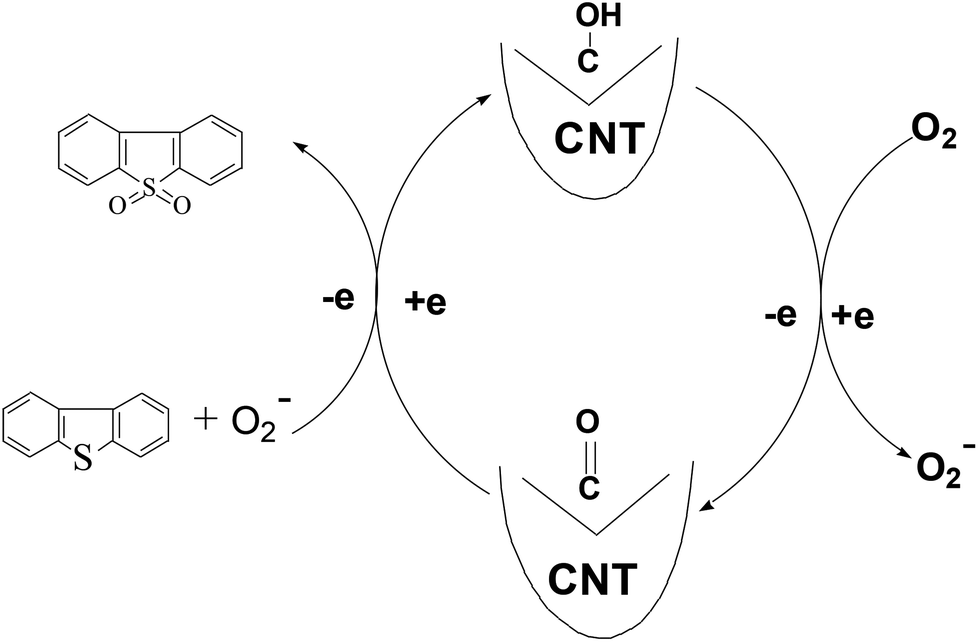 | ||
| Fig. 13 Mechanism of oxidation of DBT by O2 in the presence of CNT. | ||
Conclusion
In this work, CNTs were studied for the first time as ODS catalysts with molecular oxygen as the oxidant. CNTs exhibited a superior catalytic activity for oxidation of benzothiophenic compounds. When the CNT-SZ was used as a catalyst with molecular oxygen as the oxidant, DBT was completely converted to its corresponding sulfone at 150 °C within 40 min. The reactivities of the organic-sulfur compounds followed the order: BT < DBT < 4,6-DMDBT, which was dependent on the electron density on the sulfur atom of the compounds. The organic-sulfur compound with higher electron density of sulfur atoms was preferentially oxidized. The graphitization degree of CNTs played a decisive role in their catalytic activity for DBT oxidation. CNTs with higher graphitization degree had higher catalytic activities for DBT oxidation. The deactivated CNT can be effectively regenerated by heat treatment under an argon atmosphere at 900 °C, and its catalytic activity can be almost completely recovered. Our findings may provide a new perspective on the application of nanostructured carbon materials as a catalyst for the oxidation of organic-sulfur compounds with oxygen molecules as the oxidant.Experimental
Experiments of oxidative desulfurization
The ODS experiments were carried out in a batch reactor. The model fuels were prepared by dissolving BT, DBT, and 4,6-DMDBT into n-dodecane with the sulfur concentration of 427 ppm, separately. In a typical run, 30 g of the model fuel were added in a 100 mL three-necked flask. After the model fuel was heated to 150 °C, 0.01 g of the CNT catalyst was added into the flask and molecular oxygen was bubbled through the reaction solution simultaneously. The mixture was stirred throughout the experiment, and a water-cooled reflux condenser was installed to prevent solvent vaporization. The solution was sampled periodically, and then analyzed using a high-performance liquid chromatograph (HPLC) equipped with an ODS-C18 column and an UV detector (Dalian Elite Analytical Instrument Co., China). Qualitative analysis of the sulfur compounds in a real fuel was conducted using an HP 5890 gas chromatograph (XTI-5 column from Restek, 30 m in length and 0.25 mm in internal diameter) equipped with a pulsed flame photometric detector (PFPD). A gas-chromatograph/mass spectrometer (GCMS-QP 2010, SHIMADZU) equipped with capillary DB-1 (30 m × 0.25 mm × 1 μm) was used to identify the sulfur species in the treated fuel sample. The injector temperature was set at 275 °C and a split ratio of 10 was used. The initial temperature was set at 50 °C for 2 min and was then increased to 250 °C at a heating rate of 10 °C min−1, and held at 250 °C for 10 minutes.Regeneration of the deactivated CNT-SZ catalyst
After the ODS reaction, the used CNT-SZ was washed with methanol for ensuring complete removal of the chemicals from the surface of the catalyst, followed by drying at 110 °C for 8 h. This deactivated catalyst was labeled as CNT-SZ-W. The deactivated CNT-SZ-W was heated under flowing argon (60 cm3 min−1) at 900 °C for 4 h. Then the regenerated catalyst was obtained, which was labeled as CNT-SZ-H. After that, the catalytic activities of the deactivated catalyst CNT-SZ-W and the regenerated catalyst CNT-SZ-H for ODS of the model fuel would be further tested in this work.Characterization of the CNT catalysts
Textural properties of the CNT catalysts were characterized by N2 adsorption–desorption isotherms at 77 K by using a Micromeritics ASAP2010 surface area and porosimetry analyzer. Prior to the textural analysis, the samples were outgassed for 8 h under vacuum at 150 °C. The BET surface was calculated from adsorption isotherms using the standard BET equation. The average pore size was evaluated from the desorption branch of the isotherm using the BJH model.43The diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was carried out by using a FT-IR spectrometer equipped with a diffuse reflectance attachment (Vector 33, Bruker) in order to analyze the oxygen functional groups on CNTs. KBr was used as the background for each sample.29
X-ray photoelectron spectroscopy (XPS) was performed by using a Kratos AXIS Ultra DLD Photoelectron Spectroscope (Britain) equipped with Al Ka anode (hv = 1486.6 eV). All the spectra were calibrated against the major C1s peak at a binding energy (BE) of 284.6 eV. The O1s peak was fitted by combined convoluted Gauss–Lorentz functions.
Raman spectroscopy was carried out using a LabRAM Aramis Raman instrument (HORIBA JobinYvon S.A.S., France) at the excitation wavelength of 632.81 nm.
Acknowledgements
This work was supported by the Science Foundation of Guangdong Province (no. 8251064101000018), the Fundamental Research Funds for the Central Universities (no. 2013ZM0056) and the National Science Foundation of China (no. 20806026).References
- A. Stanislaus, A. Marafi and M. S. Rana, Catal. Today, 2010, 153, 1–68 CAS.
- C. Song and X. L. Ma, Appl. Catal., B, 2003, 41, 207–238 CrossRef CAS.
- I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef CAS.
- D. D. Whitehurst, T. Isoda and I. Mochida, Adv. Catal., 1998, 42, 345–471 CrossRef CAS.
- R. T. Yang, A. J. Hernandez-Maldonado and F. H. Yang, Science, 2003, 301, 79–81 CrossRef CAS PubMed.
- C. S. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- P. S. Kulkarni and C. A. M. Afonso, Green Chem., 2010, 12, 1139–1149 RSC.
- H. S. Gao, C. Guo, J. M. Xing, J. M. Zhao and H. Z. Liu, Green Chem., 2010, 12, 1220–1224 RSC.
- C. Asumana, G. R. Yu, X. Li, J. J. Zhao, G. Liu and X. C. Chen, Green Chem., 2010, 12, 2030–2037 RSC.
- J. Xiao, C. S. Song, X. L. Ma and Z. Li, Ind. Eng. Chem. Res., 2012, 51, 3436–3443 CrossRef CAS.
- P. S. Tam, J. R. Kittrell and J. W. Eldridge, Ind. Eng. Chem. Res., 1990, 29, 321–324 CrossRef CAS.
- Y. Shiraishi, T. Hirai and I. Komasawa, J. Chem. Eng. Jpn., 2002, 35, 1305–1311 CrossRef CAS.
- Y. X. Ding, W. S. Zhu, H. M. Li, W. Jiang, M. Zhang, Y. Q. Duan and Y. H. Chang, Green Chem., 2011, 13, 1210–1216 RSC.
- M. Te, C. Fairbridge and Z. Ring, Appl. Catal., A, 2001, 219, 267–280 CrossRef CAS.
- J. T. Zhang, W. S. Zhu, H. M. Li, W. Jiang, Y. Q. Jiang, W. L. Huang and Y. S. Yan, Green Chem., 2009, 11, 1801–1807 RSC.
- S. Otsuki, T. Nonaka, N. Takashima, W. H. Qian, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232–1239 CrossRef CAS.
- D. S. Zhao, J. L. Wang and E. P. Zhou, Green Chem., 2007, 9, 1219–1222 RSC.
- S. Otsuki, T. Nonaka, W. H. Qian, A. Ishihara and T. Kabe, J. Jpn. Pet. Inst., 2001, 44, 18–24 CrossRef CAS.
- D. H. Wang, E. W. H. Qian, H. Amano, K. Okata, A. Ishihara and T. Kabe, Appl. Catal., A, 2003, 253, 91–99 CrossRef CAS.
- H. Y. Lu, J. B. Gao, Z. X. Jiang, Y. X. Yang, B. Song and C. Li, Chem. Commun., 2007, 150–152 Search PubMed.
- S. Murata, K. Murata, K. Kidena and M. Nomura, Energy Fuels, 2004, 18, 116–121 CrossRef CAS.
- X. R. Zhou, J. Li, X. N. Wang, K. Jin and W. Ma, Fuel Process. Technol., 2009, 90, 317–323 CrossRef CAS PubMed.
- J. T. Sampanthar, H. Xiao, H. Dou, T. Y. Nah, X. Rong and W. P. Kwan, Appl. Catal., B, 2006, 63, 85–93 CrossRef CAS PubMed.
- X. Liu, D. S. Su and R. Schlogl, Carbon, 2008, 46, 547–549 CrossRef CAS PubMed.
- J. Zhang, D. S. Su, A. H. Zhang, D. Wang, R. Schlogl and C. Hebert, Angew. Chem., Int. Ed., 2007, 46, 7319–7323 CrossRef CAS PubMed.
- G. Mestl, N. I. Maksimova, N. Keller, V. V. Roddatis and R. Schlogl, Angew. Chem., Int. Ed., 2001, 40, 2066–2068 CrossRef CAS.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 4362–4375 CrossRef CAS.
- H. Y. Lu, J. B. Gao, Z. X. Jiang, F. Jing, Y. X. Yano, G. Wang and C. Li, J. Catal., 2006, 239, 369–375 CrossRef PubMed.
- M. X. Yu, Z. Li, Q. N. Ji, S. W. Wang, D. G. Su and Y. S. Lin, Chem. Eng. J., 2009, 148, 242–247 CrossRef CAS PubMed.
- D. S. Su, N. I. Maksimova, G. Mestl, V. L. Kuznetsov, V. Keller, R. Schlogl and N. Keller, Carbon, 2007, 45, 2145–2151 CrossRef CAS PubMed.
- D. S. Su, N. Maksimova, J. J. Delgado, N. Keller, G. Mestl, M. J. Ledoux and R. Schlogl, Catal. Today, 2005, 102, 110–114 CrossRef PubMed.
- H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116–8121 CrossRef CAS.
- K. W. R. Gilkes, S. Prawer, K. W. Nugent, J. Robertson, H. S. Sands, Y. Lifshitz and X. Shi, J. Appl. Phys., 2000, 87, 7283–7289 CrossRef CAS.
- J. A. Macia-Agullo, D. Cazorla-Amoros, A. Linares-Solano, U. Wild, D. S. Su and R. Schlogl, Catal. Today, 2005, 102, 248–253 CrossRef PubMed.
- M. F. R. Pereira, J. J. M. Orfao and J. L. Figueiredo, Appl. Catal., A, 2001, 218, 307–318 CrossRef CAS.
- M. H. Li, M. Boggs, T. P. Beebe and C. P. Huang, Carbon, 2008, 46, 466–475 CrossRef CAS PubMed.
- J. Robertson, Mater. Sci. Eng. R Rep., 2002, 37, 129–281 CrossRef.
- N. Keller, N. I. Maksimova, V. V. Roddatis, M. Schur, G. Mestl, Y. V. Butenko, V. L. Kuznetsov and R. Schlogl, Angew. Chem., Int. Ed., 2002, 41, 1885–1888 CrossRef CAS.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS PubMed.
- D. Begin, G. Ulrich, J. Amadou, D. S. Su, C. Pham-Huu and R. Ziessel, J. Mol. Catal. A: Chem., 2009, 302, 119–123 CrossRef CAS PubMed.
- D. Mattia, M. P. Rossi, B. M. Kim, G. Korneva, H. H. Bau and Y. Gogotsi, J. Phys. Chem. B, 2006, 110, 9850–9855 CrossRef CAS PubMed.
- Y. J. Kim, T. S. Shin, H. D. Choi, J. H. Kwon, Y. C. Chung and H. G. Yoon, Carbon, 2005, 43, 23–30 CrossRef CAS PubMed.
- W. Zhang, H. Y. Liu, Q. B. Xia and Z. Li, Chem. Eng. J., 2012, 209, 597–600 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |