 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An efficient Cu(II)-bis(oxazoline)-based polymer immobilised ionic liquid phase catalyst for asymmetric carbon–carbon bond formation†
Simon
Doherty
*a,
Julian G.
Knight
a,
Jack R.
Ellison
a,
Peter
Goodrich
b,
Leanne
Hall
b,
Christopher
Hardacre
*b,
Mark J.
Muldoon
b,
Soomin
Park
b,
Ana
Ribeiro
c,
Carlos Alberto Nieto
de Castro
c,
Maria José
Lourenço
c and
Paul
Davey
d
aNUCAT, School of Chemistry, Bedson Building, University of Newcastle upon Tyne, Newcastle upon Tyne, NE1 7RU, UK. E-mail: simon.doherty@ncl.ac.uk; Fax: +44 (0)191 222 6929; Tel: +44 (0)191 222 6537
bThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast BT9 5AG, UK. E-mail: c.hardacre@qub.ac.uk; Fax: +44 (0)28 9097 4687; Tel: +44 (0)28 9097 4592
cDepartmento de Química e Bioquímica e Centro de Ciências Moleculares e Materiais Faculdade de Ciências, Universidade de Lisboa, 1749-016, Lisboa, Portugal
dGivaudan, Schweiz AG, Überlandstrasse 138, CH-8600 Dübendorf, Switzerland
First published on 9th December 2013
Abstract
The asymmetric Diels–Alder reaction between N-acryloyloxazolidinone and cyclopentadiene and the Mukaiyama-aldol reaction between methylpyruvate and 1-phenyl-1-trimethylsilyloxyethene have been catalysed by heterogeneous copper(II)-bis(oxazoline)-based polymer immobilised ionic liquid phase (PIILP) systems generated from a range of linear and cross linked ionic polymers. In both reactions selectivity and ee were strongly influenced by the choice of polymer. A comparison of the performance of a range of Cu(II)-bis(oxazoline)-PIILP catalyst systems against analogous supported ionic liquid phase (SILP) heterogeneous catalysts as well as their homogeneous counterparts has been undertaken and their relative merits evaluated.
Introduction
Over the past two decades ionic liquids (ILs) have been shown to be extremely beneficial to a number of transition metal-catalysed reactions including asymmetric catalysis.1 In this respect, a range of chiral metal complexes of Pt, Zn, Cu and Mg have been immobilised in ILs under homogeneous or heterogeneous (Supported Ionic Liquid Phase-SILP) conditions and shown to be excellent catalysts for a host of important asymmetric C–C bond forming reactions such as Diels–Alder and Mukaiyama-aldol reactions, cyclopropanations and oxidations, often with significant and marked enhancements in rate and selectivity compared with the corresponding reaction in conventional solvents.2 In the case of SILP catalysed Diels–Alder reactions (Scheme 1), the presence of an IL film on the silica oxide or carbon support proved crucial in maintaining high conversions, good endo-selectivity and high levels of enantioselectivity (ee).3 However, despite the success of SILP catalysts some issues still limit the potential applications of this concept including their low/poor mechanical stability, leaching of IL into the solvent phase and the high cost of the IL.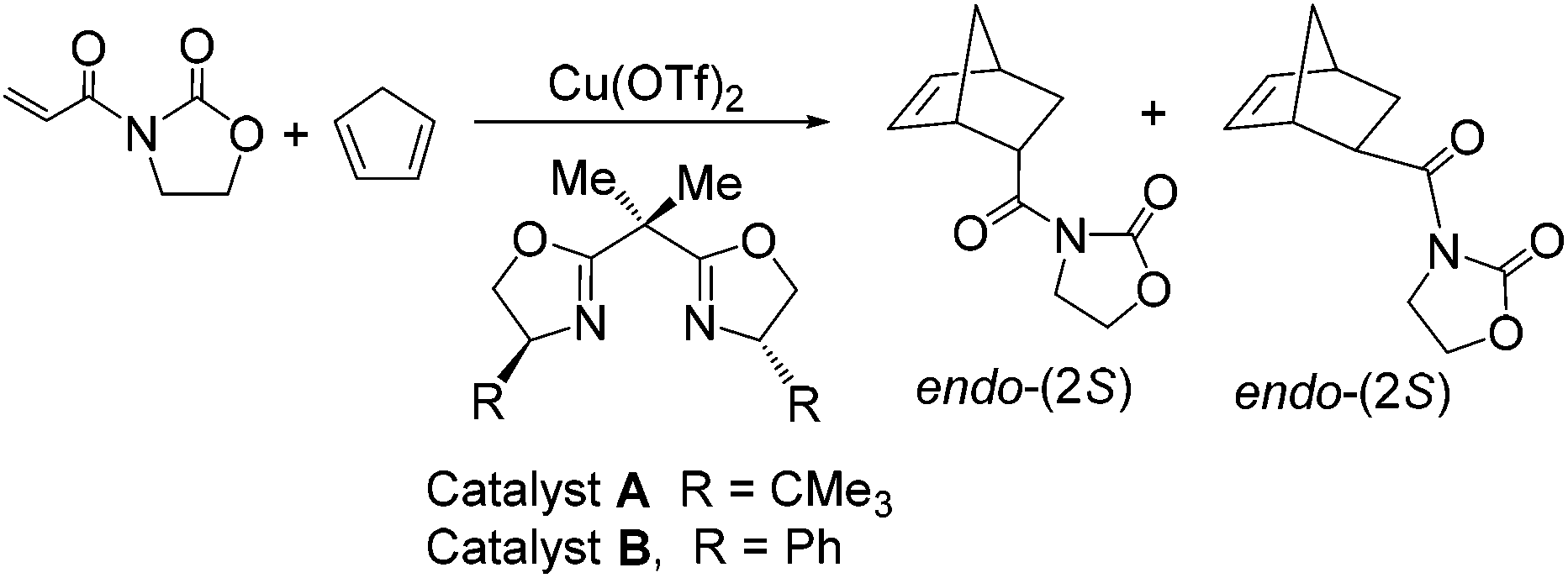 | ||
| Scheme 1 Asymmetric Diels–Alder reaction between N-acryloyloxazolidinone and cyclopentadiene catalysed by 10 mol% Cu(II)-bis(oxazoline) complexes. | ||
Polymers with chirality incorporated into the backbone have been widely studied as catalyst supports for a range of asymmetric C–C bond forming reactions including Diels–Alder cycloadditions, oxidations and hydrogenations.4–8 In contrast, the concept of immobilising an ionic liquid in the form of a cation-decorated polymer, e.g. in the form of a polyelectrolyte, to combine the favourable properties of ionic liquids with the advantages of heterogenisation as well as overcome leaching and improve long term stability in asymmetric catalysis has been far less studied.9,10 Selected examples include the use of polymer-supported imidazolium based ILs in organocatalysed nitroaldol reactions11 and enzyme-catalysed transesterifications.12 A Ru-BINAP complex immobilised on a polymer-supported pyrrolidinium tetrafluoroborate has been applied to the asymmetric hydrogenation of methylacetoacetate.13 Therein, high ee's were obtained but the activity was lower than in methanol under homogeneous conditions. Rhodium and ruthenium complexes of phosphorylated BINAP have been immobilised in polymer electrolytes and used to catalyse the asymmetric hydroformylation of vinyl acetate and styrene14 and the asymmetric hydrogenation of dimethyl itaconate,14 respectively; the latter gave activities and selectivities that matched those obtained under homogeneous conditions.
Having recently developed an efficient and recyclable peroxometalate-based polymer immobilised ionic liquid phase oxidation catalyst using a cation-decorated ring opening metathesis-derived polymer as support,15 we now report that copper(II)-bis(oxazoline)-derived PIILP catalysts A and B either rival or outperform their homogeneous or SILP counterparts for the Diels–Alder (Scheme 1) and the Mukaiyama-aldol reaction (Scheme 2). The ionic polymer structures tested (IP-1–6) as well as polystyrene (P-7) used to support catalysts A and B are shown in Fig. 1.
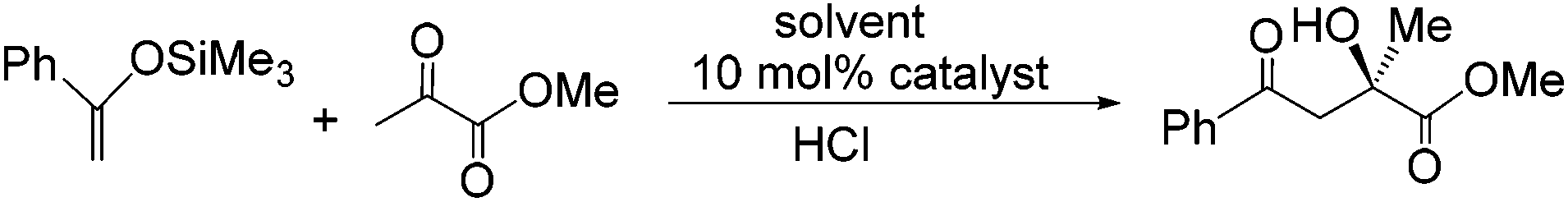 | ||
| Scheme 2 Asymmetric Mukaiyama-aldol reaction between methylpyruvate and 1-phenyl-1-trimethylsilyloxyethene catalysed by 10 mol% Cu(II)-bis(oxazoline) complex A. | ||
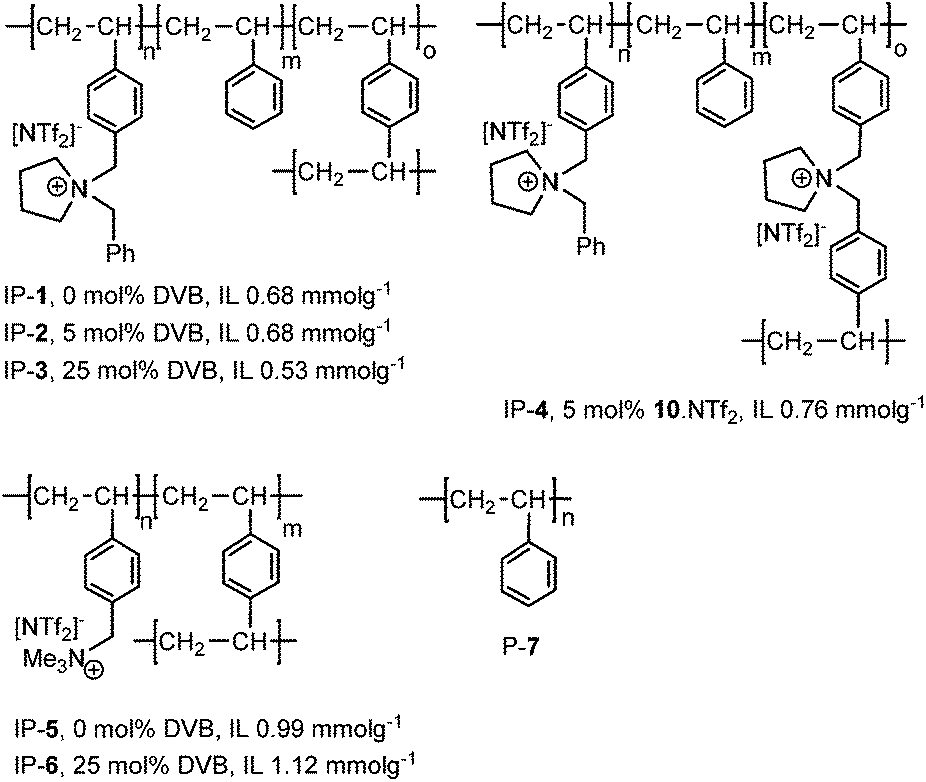 | ||
| Fig. 1 IPs 1–6 and polymer P-7 used in the immobilisation of Cu(II)-bis(oxazoline) catalysts for the asymmetric Diels–Alder reaction. In each case the degree of cross-linking (%DVB) and ionic loading (mmol g−1) is shown. | ||
Results
1-(4-Vinylbenzyl)pyrrolidine 8 was prepared according to a literature procedure and reacted with benzylbromide in acetone to afford 9·Br which was converted into the desired monomer 9·NTf2 by metathesis with lithium bis{(trifluoromethyl)sulfonyl}imide (Fig. 2). The corresponding cross-linker 10·NTf2 (Fig. 2) was prepared in an analogous manner by the reaction of 8 with 4-vinylbenzyl bromide followed by ion exchange of the bromide with lithium bis{(trifluoromethyl)sulfonyl}imide; both monomers were isolated in good yield as air-stable crystalline solids. Ionic polymers IP-1–4 were synthesised by the AIBN-initiated radical co-polymerisation of 1-4-vinylbenzyl-1-benzylpyrrolidinium bis((trifluoromethyl)sulfonyl)imide (9·NTf2) with styrene in methanol at 70 °C for 48 h; the cross-linking was introduced by conducting the polymerisation with the specified amount of divinylbenzene or 1,1-bis(4-vinylbenzyl)pyrrolidinium bis((trifluoromethyl)sulfonyl)imide (10·NTf2) and the degree of cross-linking varied by adjusting the amount of added cross-linker. These in-house synthesised IPs were compared with commercially available microporous gel-type (IP-5) and macroreticular (IP-6) polymers which contain the trimethylammonium cation as well as the polystyrene (P-7) as a non-ionic reference polymer.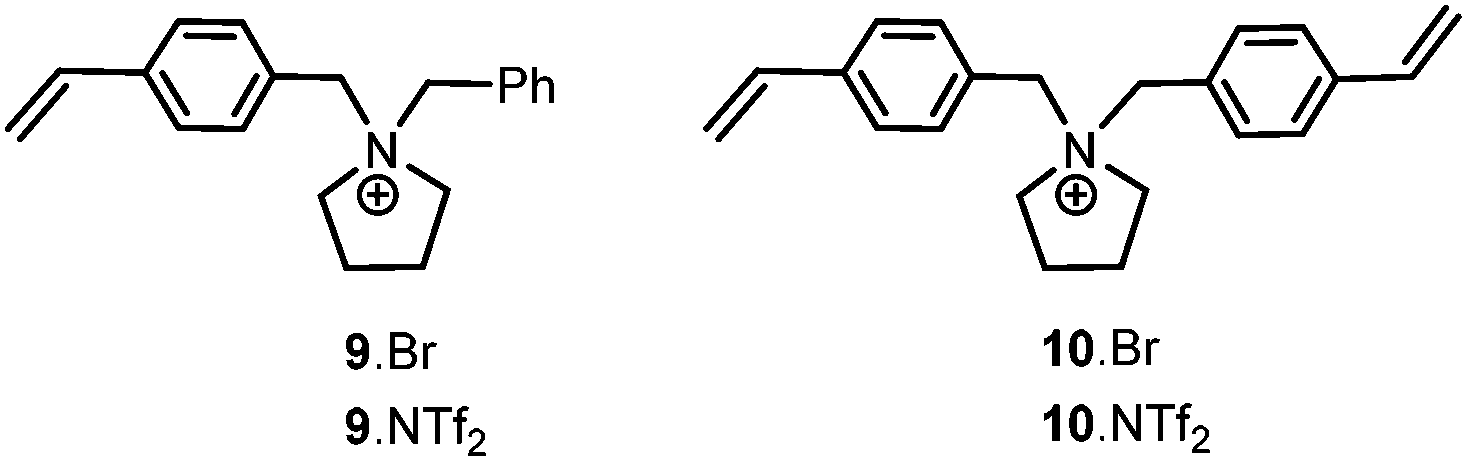 | ||
| Fig. 2 Pyrrolidinium monomers used to synthesise IPs 1–4. | ||
Copper(II)-bis(oxazoline) complexes were selected for immobilisation on the basis that they catalyse a host of asymmetric C–C bond forming reactions and because they have been studied on a variety of support materials which will enable the relative merits of PIILP catalysis to be assessed. The benchmark Diels–Alder reaction between N-acryloyloxazolidinone and cyclopentadiene catalysed by 10 mol% copper(II)-bis(oxazoline) complexes was initially conducted in CH2Cl2, Et2O and 1-ethyl-3-methylimidazolium bis{trifluoromethyl)sulfonyl}imide ([C2mim][NTf2]) under homogeneous, biphasic and SILP conditions, full details of which are summarised in Table 1. With the exception of Et2O, regardless of the choice of solvent employed, catalyst A produced significantly higher ee's and endo selectivities than catalyst B. For both catalysts, under homogeneous conditions, the presence of an IL either as an additive (entries 4 and 5) or as a bulk solvent (entry 3) resulted in higher conversions and ee's compared with similar reactions conducted in molecular solvents (entries 1 and 2).
| Entry | Solvent/support | Time (min) | Catalyst A | Catalyst B | ||||
|---|---|---|---|---|---|---|---|---|
| Conv.a,b (%) | endo eeb (%) | % endob | Conv.a,b (%) | endo eeb (%) | % endob | |||
| a Conversion at 20 °C. b Determined by HPLC. | ||||||||
| 1 | CH2Cl2 | 15 | 78 | 70(S) | 88 | 44 | 16(R) | 82 |
| 2 | Et2O | 15 | 32 | 14(S) | 87 | 38 | 15(R) | 80 |
| 3 | IL | 1 | 100 | 90(S) | 89 | 100 | 19(R) | 88 |
| 4 | IL/Et2O | 5 | 100 | 91(S) | 89 | 100 | 18(R) | 78 |
| 5 | CH2Cl2/IL | 5 | 64 | 82(S) | 96 | 100 | 16(R) | 70 |
| 6 | SiO2 | 5 | 32 | 16(S) | 88 | 76 | 4(R) | 84 |
| 7 | SiO2/IL | 5 | 100 | 87(S) | 85 | 100 | 8(R) | 84 |
| 8 | CNT | 5 | 64 | 66(S) | 87 | 44 | 20(R) | 83 |
| 9 | CNT/IL | 5 | 100 | 92(S) | 87 | 100 | 3(R) | 80 |
With the aim of comparing the efficiency of polymer immobilised ionic liquid supports against conventional SILP-systems, catalysts A and B were also supported on SiO2 or multi-walled carbon nanotubes (CNT) by wet impregnation from dichloromethane using [C2mim][NTf2] as the ionic liquid. Reactions were conducted in diethyl ether as the solvent due to the low rates of reaction for the homogeneous catalysts (entry 2) and the low solubility of [C2mim][NTf2] in diethyl ether compared with dichloromethane, which should reduce ionic liquid/catalyst leaching. A significant increase in both the ee and conversion was observed for reactions conducted using the SILP catalysts (entries 7 and 9) in comparison to the analogous heterogeneous reactions conducted in the absence of a [C2mim][NTf2] film (entries 6 and 8). In the case of the SiO2 support (entry 6) the very low conversion and ee could be due to the role of the surface silanols which can promote cyclopentadiene dimerisation and act as a non-chiral active catalyst.16 Moreover, both SiO2 and CNT supported systems based on catalyst A gave better conversions and markedly higher ee's (entries 7 and 9) than that obtained for homogeneous reactions using molecular solvents (entries 1 and 2). In all cases, the configuration of the Diels–Alder adduct was identical to those obtained under homogenous conditions in both ionic liquid and molecular solvents. These results indicate that heterogenisation of a chiral catalyst in an ionic environment can lead to a marked improvement in performance compared with the corresponding homogeneous and ‘non-ionic’ heterogeneous systems; however, this enhancement appears to be catalyst specific. Although support effects can influence the reaction the enhancement observed in the bulk IL and SILP systems could be, in part, due to the large excess of the less coordinating [NTf2]− anion compared with the [OTf]− anion in the original catalyst.17 Further studies are required to fully understand the role of the anion and nature of the support.
In order to further explore the effect of an ionic environment, catalysts A and B were supported on IPs 1–6 and non-ionic polymer P-7 using wet impregnation from dichloromethane. The efficacy of the resulting PIILP catalysts for the asymmetric Diels–Alder reaction conducted between N-acryloyloxazolidinone and cyclopentadiene in diethyl ether was investigated and compared with the corresponding systems modified with a thin film of IL; the results of which are summarised in Table 2 and will be compared with the systems reported in Table 1. In agreement with previous studies performed using these catalysts in ionic liquids3 the active PIILP catalyst was formed after only 5 min by stirring a dichloromethane solution of Cu(OTf)2 and bis(oxazoline) in the presence of ionic polymer; for comparison much longer aging times (>3 h) are generally required to achieve efficient and reproducible catalysis in molecular solvents.1
| Entry | Polymer | Solvent | Catalyst A | Catalyst B | ||||
|---|---|---|---|---|---|---|---|---|
| Conv.a,b (%) | endo eeb (%) | % endob | Conv.a,b (%) | endo eeb (%) | % endob | |||
| a Conversion at 20 °C after 5 minutes. b Determined by HPLC. | ||||||||
| 1 | IP-1 | Et2O | 86 | 90(S) | 88 | 100 | 35(R) | 78 |
| 2 | IP-1 | IL/Et2O | 100 | 99(S) | 95 | 100 | 27(R) | 76 |
| 3 | IP-2 | Et2O | 80 | 48(S) | 92 | 100 | 32(R) | 79 |
| 4 | IP-2 | IL/Et2O | 65 | 31(S) | 85 | 100 | 26(R) | 74 |
| 5 | IP-3 | Et2O | 69 | 28(S) | 86 | 100 | 38(R) | 73 |
| 6 | IP-3 | IL/Et2O | 77 | 69(S) | 89 | 100 | 15(R) | 76 |
| 7 | IP-4 | Et2O | 100 | 99(S) | 93 | 100 | 41(R) | 76 |
| 8 | IP-4 | IL/Et2O | 60 | 80(S) | 91 | 100 | 31(R) | 73 |
| 9 | IP-5 | Et2O | 27 | 7(S) | 88 | 100 | 16(R) | 92 |
| 10 | IP-5 | IL/Et2O | 98 | 9(S) | 88 | 100 | 18(R) | 79 |
| 11 | IP-6 | Et2O | 47 | 44(S) | 84 | 100 | 38(R) | 80 |
| 12 | IP-6 | IL/Et2O | 91 | 84(S) | 91 | 99 | 18(R) | 91 |
| 13 | P-7 | Et2O | 0 | 0 | 0 | 0 | 0 | 0 |
| 14 | P-7 | IL/Et2O | 74 | 84(S) | 88 | 100 | 30(R) | 73 |
Reference systems generated by adsorbing catalysts A and B onto the non-ionic polymer (P-7) were completely inactive when reactions were conducted in diethyl ether in the absence of additional ionic liquid (entry 13), even after the catalyst was aged for 3 h. In contrast, significant conversions were obtained for reactions performed with PIILP systems based on IP-1–6. In general, higher ee's were obtained with PIILP-systems generated by immobilisation of catalysts A and B onto the in-house synthesised polymers IP-1–4 (entries 1–8) compared with those based on commercially available IP-5–6 (entries 9–12). Moreover, for both catalysts the most efficient PIILP systems were those based on IP-4 with catalyst A giving cycloadduct endo-(2S) in 99% ee and catalyst B giving cycloadduct endo-(2R) in 41% ee at 100% conversion (entry 7). The former is the highest ee reported, to date, for a copper(II)-catalysed reaction of this substrate combination at room temperature under heterogeneous or homogeneous conditions. In addition, the ee of 90% (S) obtained with PIILP catalyst based on IP-1 and catalyst A (entry 1) also matched that obtained in neat ionic liquid and ionic liquid-diethyl ether (Table 1, entries 3–4). The significant difference between the reactions using the ionic and non-ionic polymers is thought to be due to the reduced binding of the complex on the latter support. In this regard, leaching of up to 50% of the copper into the diethyl ether phase for polymer P-7 together with a strong interaction of the cyclopentadiene with the polystyrene support were thought to be responsible for catalyst inactivity.
As supporting catalysts A and B in a thin IL film coated onto CNT and SiO2 supports was shown to have a beneficial effect on reaction activity/selectivity (Table 1), the influence of combining [C2mim][NTf2] with polymers 1–7 was also studied. Encouragingly, in some cases, the introduction of ionic liquid led to a marked increase in catalyst performance. The most significant increase in ee and conversion was obtained for catalyst A immobilised on IP-6 which gave cycloadduct endo-(2S) in 84% ee and 91% conversion in the presence of ionic liquid compared with 44% ee and 47% conversions in the absence of [C2mim][NTf2] (entries 11–12). An enhancement in ee was also achieved when a thin film of [C2mim][NTf2] was added to PIILP systems based on catalyst A immobilised on IP-1 and IP-3 (entries 1–2 and 5–6) with IP-3 giving the most marked enhancement in ee from 28% to 69% (Δee = 41%); however, there was little change in endo/exo-selectivity and conversion. While the addition of ionic liquid to the catalyst immobilised on P-7 also resulted in a marked improvement in conversion (entry 14), it was not possible to quantify the enhancement in ee as both catalysts are completely inactive in the absence of ionic liquid (entry 13). In contrast, the increase in ee from 90% to 99% for the IP-1/catalyst A/[C2mim][NTf2] combination was accompanied by an increase in endo/exo selectivity from 88% to 95%; the performance of this system matched that of IP-4 in diethyl ether. Although IP-5 also showed a marked increase in conversion in the presence of [C2mim][NTf2], there was no significant change in ee which remained very poor, (entry 10). Conversely, for IPs 2 and 4 the presence of a thin layer of ionic liquid led to a significant reduction in ee and endo selectivity (entries 4 and 8). PIILP systems based on catalyst B and IP's 1–6 showed much smaller changes in ee but larger variations in endo/exo selectivity in the presence of an IL film in comparison to the changes observed with catalyst A under comparable conditions. Moreover, in contrast to catalyst A, with the exception of IP-5, all of the IPs showed a decrease in ee upon the addition of an IL thin film. Despite this decrease in ee, all of the PIILP catalyst-IL combinations gave higher or equivalent ee's compared with the same reactions conducted under homogeneous conditions in dichloromethane (Table 1).
While exceptionally high ee's and endo/exo ratios have been achieved using PIILP and PIILP/IL catalysts, the disparate and unpredictable variations in enantioselectivity and endo/exo selectivity as a function of the catalyst and ionic support or support-ionic liquid combination highlights the importance of developing a rational understanding of how catalyst–support interactions influence catalyst efficacy.
Reasoning that a polymer immobilised ionic liquid phase support should effectively retain the catalyst, reusability experiments were conducted on PIILP catalysts derived from A and IP-1 and IP-6 as well as the corresponding catalysts immobilised onto SiO2, with and without a thin film of IL (Table 3). Both PIILP catalysts recycled poorly with a significant decrease in ee and conversion upon successive recycles (entries 1–3 and 6–7).
| Entry | Support | Run | Conv.a,b (%) | endo eeb (%) | % endob |
|---|---|---|---|---|---|
| a Conversion at 20 °C after 5 minutes. b Determined by HPLC. | |||||
| 1 | 1 | 86 | 90(S) | 88 | |
| 2 | IP-1 | 2 | 68 | 73(S) | 86 |
| 3 | 3 | 30 | 7(S) | 85 | |
| 4 | IP-1/IL | 1 | 100 | 99(S) | 95 |
| 5 | 2 | 98 | 74(S) | 77 | |
| 6 | IP-6 | 1 | 47 | 44(S) | 86 |
| 7 | 2 | No reaction | |||
| 8 | IP-6/IL | 1 | 91 | 84(S) | 91 |
| 9 | 2 | 41 | 88(S) | 92 | |
| 10 | SiO2 | 1 | 90 | 54(S) | 85 |
| 11 | 2 | 83 | 43(S) | 84 | |
| 12 | SiO2/IL | 1 | 100 | 87(S) | 86 |
| 13 | 2 | 100 | 84(S) | 85 | |
| 14 | 3 | 93 | 81(S) | 86 | |
ICP analysis of the diethyl ether used to extract the product between recycles revealed that this procedure removed 4.2% and 8.0% of the copper from IP-1 and IP-6 respectively. Ligand leaching of 10.2% from IP-1 and 14.3% from IP-6 was also determined by HPLC. The addition of an IL film to both PIILP catalysts resulted in an increase in conversion and ee (entries 4–5 and 8–9) and a significant reduction in copper leaching to 0.2% for systems based on both IP-1 and IP-6, respectively. Moreover, for the PIILP-IL systems the level of ligand leaching was below the limit of quantification by HPLC analysis. However, these levels of copper/ligand leaching observed with PIILP catalysts in the presence or absence of an IL film do not explain the significant drop in conversion and/or ee observed upon recycle. In addition, no significant change in the polymer structure as determined by infra-red spectroscopy was observed on recycle (see ESI† for data on IP-1 which showed the largest decrease in activity).
The drop in catalyst activity upon recycle for PIILP systems based on catalyst A is thought to be mainly attributed to the build-up of cyclopentadiene dimer18 on the support surface, as has been previously observed in other SILP catalysed Diels–Alder reactions.3 This proposal was supported by a reduction in conversion from 86% to 32% and a drop in ee from 90% to 19% upon addition of cyclopentadiene dimer (10 molar equivalents with respect to substrate) to the ethereal layer of a freshly prepared PIILP system derived from IP-1 and catalyst A. While the PIILP and PIILP-IL catalysts performed poorly upon recycle the SiO2-based SILP system maintained high conversions and good ee's upon recycle.3 This was thought to be associated with more efficient removal of the cyclopentadiene dimer from the hydrophilic silica than from the surface of the lipophilic IP. In this regard, studies are currently underway to explore the effect on recycle efficiency of increasing the hydrophilicity of the polymer by introducing PEG-derived co-monomers or PEG-functionalised pyrrolidinium monomers.
Encouraged by the enhancement in performance obtained with selected PIILP catalysts, testing was extended to include a comparison of the Mukaiyama-aldol reaction (Scheme 2) catalysed by 10 mol% copper(II)-bis(oxazoline) catalyst A under homogeneous conditions in various solvents and under heterogeneous conditions with SILP and PIILP-based catalysts, full details of which are summarised in Table 4. Complete conversion was achieved within 1 min at room temperature with catalyst A in the ionic liquid whereas only moderate conversions were obtained after 15 min in dichloromethane and diethyl ether (entries 1–3). A markedly higher ee in the ionic liquid compared with dichloromethane and diethyl ether was also observed; however, this was offset by the formation of a minor amount (∼10%) of by-product, identified by 1H NMR spectroscopy as 3-hydroxy-1,3-diphenyl-butan-1-one. Interestingly, this by-product was only generated during reactions conducted in ionic liquid and there was no evidence for its formation during reactions catalysed by the PIILP, PIILP/IL or SILP systems. This by-product results from a Mukaiyama-aldol reaction between 1-phenyl-1-trimethylsiloxyethene and acetophenone, the latter of which is generated via hydrolysis of the 1-phenyl-1-trimethylsiloxyethene substrate.2 In addition, both SILP catalysts gave complete conversion in short reactions times with only a slight reduction in ee compared with the homogenous reaction conducted in ionic liquid (entries 5–6). In contrast to the SILP-based catalysts, in general, the ee's and conversions obtained with the PIILP and PIILP/IL system were lower than found under homogeneous conditions. For the PIILP and PIILP/IL catalysts derived from IP-2, ee's of 86% and 90% were obtained, respectively, which are comparable with those obtained under homogeneous IL or SILP conditions, albeit it with slightly lower conversions (entries 9 and 10). For comparison, the reference system generated by supporting catalyst A on the non-ionic polymer P-7 were completely inactive even in the presence of an IL film (entries 15–16). A high ee has previously been achieved for the Mukaiyama-aldol reaction between ketene thioacetal and methyl pyruvate under heterogeneous conditions using an insoluble polymer-bound copper(II)-bis(oxazoline) catalyst; however, while the ee of 91% was comparable to that of the 93% obtained under homogeneous conditions the heterogeneous system was less active and required markedly longer reaction times to reach similar conversions.19
| Entry | System | Time (min) | Conv.a (%) | eeb (%) config |
|---|---|---|---|---|
| a Conversion at 20 °C determined by IH-NMR. b Determined by HPLC. | ||||
| 1 | CH2Cl2 | 15 | 43 | 82(S) |
| 2 | Et2O | 15 | 32 | 80(S) |
| 3 | IL | 1 | 100 | 89(S) |
| 4 | IL/Et2O | 5 | 100 | 84(S) |
| 5 | SiO2/IL | 5 | 100 | 86(S) |
| 6 | CNT/IL | 5 | 100 | 85(S) |
| 7 | IP-1 | 5 | 49 | 24(S) |
| 8 | IP-1/IL | 5 | 47 | 11(S) |
| 9 | IP-2 | 5 | 63 | 86(S) |
| 10 | IP-2/IL | 5 | 56 | 90(S) |
| 11 | IP-4 | 5 | 56 | 13(S) |
| 12 | IP-4/IL | 5 | 69 | 48(S) |
| 13 | IP-6 | 5 | 36 | 40(S) |
| 14 | IP-6/IL | 5 | 55 | 63(S) |
| 15 | P-7 | 5 | 0 | — |
| 16 | P-7/IL | 5 | 0 | — |
Discussion
For a microporous gel-type polymer, and to a lesser extent macroporous polymers, a lack of swelling renders non-surface catalytic sites less accessible.20 The negligible swelling for some of these IPs in diethyl ether could result in poor catalyst access and account for the poor conversions and low ee's obtained with catalyst A observed in the Diels–Alder reaction; however, this explanation is not consistent with the complete conversions observed with catalyst B under the same conditions. In addition, it should be noted that the use of dichloromethane and toluene as solvents to deliberately swell the polymers during the Diels–Alder reaction resulted in complete stripping of both catalysts A and B from PIILP systems derived from IP-1, 4, 5 and 6. Therefore, although the effect of solvent on the polymer structure and the reaction cannot be ignored, the disparate variations in activity and ee are more likely to be due to geometric constraints imposed by the support on catalyst A containing the bulky (S,S)-t-Bu-Box, as previously postulated. For example, it has been suggested that the square planar geometry adopted by catalyst A is significantly more sensitive to geometric changes upon interaction with the support compared with the smaller and more flexible (S,S)-Ph-Box in catalyst B.3,21 For catalyst A this leads to an equilibrium between the copper-bis(oxazoline) complex and non-chiral catalytically active ligand free copper.22 By analogy, the high degree of cross-linking in IP-3 and 6 could disrupt the square planar complex and, thereby, decreases the catalyst selectivity. For example, polymer supported copper-bis(oxazoline) complexes derived from catalysts A and B have been prepared by grafting and their performance in the cyclopropanation of styrene with diethyl azoacetate compared.23 The introduction of cross-linking into this system resulted in a deterioration of trans/cis selectivity and a significant drop in ee compared with the corresponding reaction conducted under homogeneous conditions. It was thought that the low catalytic activity was due to the support morphology, which resulted in little interaction between the Cu(OTf)2 and the covalently tethered bis(oxazoline) which was buried in the cross-linked polymer. A significant drop in ee for Nafion-polymer immobilised catalyst A compared with that obtained for the homogenous reaction was also attributed to steric interactions between the chiral ligand and the support which shifted the metal–ligand equilibrium in favour of the active non-chiral ligand free form. The corresponding Nafion-polymer immobilised catalyst B experienced a much smaller reduction in ee compared with the homogeneous reaction, which may reflect a more favourable metal–ligand equilibrium due to the smaller size of the ligand. Therefore, the improvement in performance for the Diels–Alder reaction achieved by addition of an IL thin film to PIILP systems derived from catalyst A and IP-3 or IP-6 could be due to stabilization of the catalyst and lower metal/ligand leaching. In this regard, catalyst stabilisation is thought to occur by migration of the catalyst away from the polymer surface into the IL film which reduces unfavourable catalyst–support interactions. Moreover, ILs are known to swell polymers and this could also help to improve access to catalytic sites that were previously inaccessible.24 Similarly, catalyst access and poor catalyst stability could also be responsible for the decreased conversions and ee's observed for the Mukaiyama-aldol reaction catalysed by PIILP systems based on IP-4 and IP-6 in combination with catalyst A. An increase in cross-linking has previously been associated with a reduction in ee for the asymmetric Mukaiyama-aldol reaction using a polymer supported chiral Lewis acid catalyst.25 However, in the current study, the IP-2 derived PIILP system outperformed that based on IP-1 despite the former being cross-linked while the latter was based on a linear polymer.For catalyst B smaller changes in the Diels–Alder cycloadduct ee were observed over the range of polymers studied. However, for all PIILP and PIILP-IL systems studied significantly higher conversions were obtained than the corresponding homogeneous reactions (Table 1) which highlights a positive/synergistic influence of the catalyst–polymer–IL interaction compared with the IL–catalyst interaction. It has been well-documented that for catalyst B the metal–ligand complex exists as an equilibrium of several different geometries, all of which are active catalysts,26 and thus the presence of such multiple active geometries could be responsible for the higher conversions observed in comparison to catalyst A. Moreover, these active catalysts can give cycloadducts with opposite configurations which would result in a reduction in the product ee. Such a number of active catalyst geometries could also be the origin of the large differences in endo selectivity which has not been previously observed for similar reactions conducted using SILP based silica and carbon supports.3
As previously established, an ionic environment in SILP conditions is required for higher catalyst performance, therefore, it is also possible that the number and distribution of ionic sites in the polymer can influence the selectivity of the reaction.27 In this study, the concentration of ions at the surface of the polymers is significantly higher than the loading of the Cu. For example, catalyst A/B loadings of 0.1 mmol g−1 on the support are significantly lower than surface concentration of [NTf2]− present. For example, IP-5 has an anion concentration of 0.99 mmol g−1. It may be expected that if the anion were the major determining factor with respect to enantioselectivity, increasing anion concentration would lead to high ee's; however, the converse is observed with higher ee's found for IP-1 and IP-4 which have [NTf2]− concentrations of 0.68 mmol g−1 and 0.76 mmol g−1, respectively, compared with IP-5. This provides further evidence that it is the structure of the support and the support–IL–catalyst interactions that determine the catalyst activity/selectivity rather than simply the concentration of ions present.
Conclusions
In summary, a number of Cu(II)-bis(oxazoline)-derived polymer supported ionic liquid phase systems have been prepared and evaluated as catalysts for the asymmetric Diels–Alder and Mukaiyama-aldol reactions. For the Diels–Alder reaction, in the majority of cases catalysts based on in-house synthesised IPs 1–4 were more active/selective than those prepared from commercial IPs 5–6 and, in some cases, gave higher endo selectivities and ee's compared with the corresponding homogeneous reactions conducted in molecular solvents or IL or under heterogeneous SILP conditions. For the Mukaiyama-aldol reaction, only catalyst A supported on the in-house synthesised IP-2 gave an ee that was comparable to those obtained under homogeneous IL and SILP conditions. The contrasting conversions and ee's for both reactions involving non-covalent immobilisation of Cu-catalysts highlights the complex nature of these PIILP systems. The successful employment of IPs to support copper-bis(oxazoline) catalysts suggests that tailoring the ionic environment on a support may be sufficient to enhance catalyst stability and maximise catalyst efficiency. Therefore, ionic solvation of these metal triflate complexes previously encountered in a bulk IL or under SILP conditions is not paramount to maximise catalyst efficiency. To this end it will be challenging but necessary to develop an understanding of how the ionic microenvironment of the polymer influences catalyst efficiency if this concept is to be fully realised and applied to a wider range of substrate combinations and catalytic processes.Although these PIILP systems showed excellent initial activity/selectivity, deleterious results were observed upon recycle which appeared to be due to the polymer interactions with the highly reactive cyclopentadiene and its dimer. In this regard, it should be possible to develop a highly active and recyclable catalytic system by tailoring the ionic polymer to minimise these negative interactions.
Experimental
Unless otherwise stated all reagents (ex Aldrich) were used as received. Diethyl ether and dichloromethane were dried and degassed prior to use. 1-Ethyl-3-methylimidazolium bis{(trifluoromethyl)sulfonyl}imide, [C2mim][NTf2], was prepared in house and after drying for 24 h at 60 °C under a high vacuum whilst stirring, was found to contain <0.10 wt% water, determined by Karl-Fischer analysis, and <10 ppm halide, determined by suppressed ion chromatography.28SI1320 silica support was obtained from GRACE Davison and calcined at 400 °C prior to use. Carbon nanotubes were obtained from Bayer Material Science. Commercial polymers IRA-400 and IRA-900 were purchased from Aldrich as Cl− salts and exchanged into the [NTf2]− form using LiNTf2 forming the corresponding IPs 5 and 6. Polystyrene P-7 was used as received.
All polymers were ground using a mortar and pestle and particles of <250 μm were used for catalyst immobilisation. For the Diels–Alder reaction the endo selectivity and conversions were determined by HPLC and the ee's based on the endo isomer were calculated from the HPLC profile using a Chiralcel OD-H column (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :propan-2-ol 90:10 flow rate 1 cm3 min−1 at 210 nm. The retention times of the endo enantiomers were major (2S)-enantiomer tR ∼ 18 min and minor (2R)-enantiomer tR ∼ 20 min.
:propan-2-ol 90:10 flow rate 1 cm3 min−1 at 210 nm. The retention times of the endo enantiomers were major (2S)-enantiomer tR ∼ 18 min and minor (2R)-enantiomer tR ∼ 20 min.
For the Mukaiyama-aldol reaction, conversions and selectivity were determined by 1H-NMR. Enantioselectivity was calculated from the HPLC profile using a Chiralcel OD-H column (hexane:propan-2-ol 96:4 flow rate 1 cm3 min−1 at 254 nm. The retention times of the enantiomers were major (2S)-enantiomer tR ∼ 23 min and minor (2R)-enantiomer tR ∼ 19 min.
1-(4-Vinylbenzyl)pyrrolidine (8)
An oven-dried Schlenk flask was charged with hexane (25 cm3) and 4-vinylbenzyl chloride (4.6 cm3, 32.8 mmol) and cooled using ice water. Pyrrolidine (5.4 cm3, 65.6 mmol) was added to the cooled mixture over the course of 1 h causing an instant colour change of cloudy white to clear yellow. Following the complete addition of pyrrolidine the reaction vessel was removed from the ice water and allowed to stir for 19 h at room temperature after which time the mixture was filtered and the solvent removed under reduced pressure to give the product as a yellow oil in 98% yield (6.02 g). 1H NMR (399.78 MHz, CDCl3, δ): 7.31 (m, 4H, Ar–H), 6.66 (dd, J = 17.4, 10.6 Hz, 1H, HaC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHbHc), 5.71 (d, J = 17.4 Hz, 1H, HaCCHbHc), 5.16 (d, J = 10.5 Hz, 1H, HaCCHbHc), 3.56 (s, 2H, Ar–CH2–N), 2.46 (m, 4H, N–CH2), 1.73 (m, 4H, pyrrolidine N–CH2–CH2). 13C{1H} NMR (100.52 MHz, CDCl3, δ): 139.03, 136.55, 128.93, 125.97, 113.20, 60.33, 54.05, 23.34.
CHbHc), 5.71 (d, J = 17.4 Hz, 1H, HaCCHbHc), 5.16 (d, J = 10.5 Hz, 1H, HaCCHbHc), 3.56 (s, 2H, Ar–CH2–N), 2.46 (m, 4H, N–CH2), 1.73 (m, 4H, pyrrolidine N–CH2–CH2). 13C{1H} NMR (100.52 MHz, CDCl3, δ): 139.03, 136.55, 128.93, 125.97, 113.20, 60.33, 54.05, 23.34.
1-Benzyl-1-(4-vinylbenzyl)pyrrolidinium bromide (9·Br)
A round bottom flask was charged with 8 (3.38 g, 18.0 mmol), benzyl bromide (4.2 cm3, 27.0 mmol) and acetonitrile (20 cm3). The yellow solution was allowed to stir at room temperature for 19 h after which time the reaction mixture was added drop-wise to vigorously stirred diethyl ether (ca. 500 cm3) to induce precipitation of the product. The resultant yellow-white solid was isolated by filtration, washed with diethyl ether and dried under high vacuum to give 9·Br as a yellow-white powder in 82% yield (5.32 g). 1H NMR (399.78 MHz, CDCl3, δ): 7.57 (m, 4H, Ar–H), 7.39 (m, 5H, Ar–H), 6.66 (dd, J = 17.4, 10.6 Hz, 1H, HaCCHbHc), 5.79 (d, J = 17.4 Hz, 1H, HaCCHbHc), 5.34 (d, J = 10.6 Hz, 1H, HaCCHbHc), 5.04 (s, 4H, Ar–CH2–N), 3.68 (m, 4H, N–CH2), 2.04 (m, 4H, pyrrolidine N–CH2–CH2); 13C{1H} NMR (100.52 MHz, CDCl3, δ): 139.59, 135.48, 133.46, 133.22, 130.49, 129.15, 127.71, 126.92, 126.75, 116.12, 63.31, 57.64, 21.28; Anal. Calcd for C20H24BrN: C, 67.04; H, 6.75; N, 3.91. Found: C, 67.19; H, 7.11; N, 4.23; HRMS (ESI+): exact mass calcd for C20H24N [M+] m/z 278.1909, found m/z 278.1904.
1-Benzyl-1-(4-vinylbenzyl)pyrrolidinium bis{(trifluoromethyl)sulfonyl}imide (9·NTf2)
A solution of lithium bis{(trifluoromethyl)sulfonyl}imide (4.82 g, 16.8 mmol) in distilled water (30 cm3) was added drop-wise to a rapidly stirred solution of 9·Br (3.00 g, 8.4 mmol) in dichloromethane (30 cm3) and allowed to stir under ambient conditions for 30 min. The organic layer was then extracted and washed with distilled water (20 cm3) repeatedly, checking the aqueous washings for bromide content with AgNO3. After the third wash the aqueous extract did not become turbid upon the addition of AgNO3, the organic layer was then dried in vacuo to give the product as a yellow/white crystalline solid in 92% yield (4.32 g). 1H NMR (399.78 MHz, CDCl3, δ): 7.57 (m, 4H, Ar–H), 7.39 (m, 5H, Ar–H), 6.66 (dd, J = 17.4, 10.6 Hz, 1H, HaCCHbHc), 5.79 (d, J = 17.4 Hz, 1H, HaCCHbHc), 5.34 (d, J = 10.6 Hz, 1H, HaCCHbHc), 5.04 (s, 4H, Ar–CH2–N), 3.68 (m, 4H, N–CH2), 2.04 (m, 4H, pyrrolidine N–CH2–CH2); 13C{1H} NMR (100.52 MHz, CDCl3, δ): 139.59, 135.48, 133.46, 133.22, 130.49, 129.15, 127.71, 126.92, 126.75, 116.12, 63.31, 57.64, 21.28. 19F NMR (376.17 MHz, CDCl3, δ): −78.67 (s, NTf2); Anal. Calcd for C22H24F6N2O4S2: C, 47.31; H, 4.33; N, 5.02. Found: C, 47.49; H, 4.67; N, 5.33; HRMS (ESI+): exact mass calcd for C20H24BrN [M+] m/z 278.1909, found m/z 278.1904.
1,1-Bis(4-vinylbenzyl)pyrrolidin-1-ium bromide (10·Br)
A round bottom flask was charged with 8 (0.66 g, 3.5 mmol), 4-vinylbenzyl bromide (0.82 g, 4.2 mmol) and acetonitrile (4 cm3), the mixture was then allowed to stir under ambient conditions for 18 h. The product was precipitated by the drop wise addition of the reaction mixture into rapidly stirred diethyl ether (ca. 100 cm3) and them isolated by filtration and dried under high vacuum to give 10·Br as a yellow/white powder in 74% yield (1.00 g). 1H NMR (399.78 MHz, CDCl3, δ): 7.55 (m, 4H, Ar–H), 7.42 (m, 4H, Ar–H), 6.67 (dd, J = 17.4, 10.6 Hz, 2H, HaCCHbHc), 5.81 (d, J = 17.4 Hz, 2H, HaCCHbHc), 5.34 (d, J = 10.6 Hz, 2H, HaCCHbHc), 5.03 (s, 4H, Ar–CH2–N), 3.47 (m, 4H, CH2–N–CH2), 2.05 (m, 4H N–CH2–CH2); 13C{1H} NMR (100.52 MHz, CDCl3, δ): 139.6, 135.5, 133.5, 126.9, 126.8, 116.2, 63.1, 57.6, 21.3; Anal. Calcd for C22H26BrN: C, 68.75; H, 6.82; N, 3.64. Found: C, 69.08; H, 7.11; N, 3.89; HRMS (ESI+): exact mass calcd for C22H26N [M+] m/z 304.2065, found m/z 304.2054.
1,1-Bis(4-vinylbenzyl)pyrrolidin-1-ium bis((trifluoromethyl)sulfonyl)imide (10·NTf2)
A solution of lithium bis{(trifluoromethyl)sulfonyl}imide (1.12 g, 3.9 mmol) in distilled water (5 cm3) was added drop wise to a rapidly stirred solution of 10·Br (0.5 g, 1.3 mmol) in dichloromethane (5 cm3) and allowed to stir under ambient conditions for 30 min. The organic layer was then extracted and washed with distilled water (10 cm3) repeatedly, checking the aqueous washings for bromide content with AgNO3. After the third wash the aqueous extract did not become turbid upon the addition of AgNO3, the organic layer was then dried in vacuo to give the product as a off-white crystalline solid in 92% yield (0.69 g). 1H NMR (399.78 MHz, CDCl3, δ): 7.51 (m, 4H, Ar–H), 7.43 (m, 4H, Ar–H), 6.65 (dd, J = 17.4, 10.6 Hz, 2H, HaCCHbHc), 5.79 (d, J = 17.4 Hz, 2H, HaCCHbHc), 5.35 (d, J = 10.6 Hz, 2H, HaCCHbHc), 5.04 (s, 4H, Ar–CH2–N), 3.49 (m, 4H, CH2–N–CH2), 2.07 (m, 4H N–CH2–CH2). 13C{1H} NMR (100.52 MHz, CDCl3, δ): 139.5, 135.4, 133.6, 126.8, 126.7, 116.4, 63.2, 57.7, 21.5. 19F NMR (376.17 MHz, CDCl3, δ): −78.73 (s, NTf2); Anal. Calcd for C22H26F6N2O4S2: C, 49.31; H, 4.48; N, 4.79. Found: C, 49.77; H, 4.67; N, 5.09; HRMS (ESI+): exact mass calcd for C22H26N [M+] m/z 304.2065, found m/z 304.2054.
General procedure for radical initiated polymerisations of 1-benzyl-1-(4-vinylbenzyl)pyrrolidinium bis{(trifluoromethyl)sulfonyl}imide with styrene and cross-linker: synthesis of IP-1–4
In typical procedure, an oven-dried Schlenk flask was charged with the 1-benzyl-1-(4-vinylbenzyl)pyrrolidinium monomer 9·NTf2 (5.2 mmol), styrene (10.3 mmol) and the specified amounts of azobisisobutyronitrile and corresponding cross-linker. After dilution with dry methanol (50 mL) the resulting reaction mixture was degassed with 5 freeze/thaw cycles using liquid nitrogen. Upon warming to room temperature the reaction mixture was heated to 75 °C and allowed to stir for 48 h. In the case of insoluble polymers such as IP-2, IP-3 and IP-4 the solvent was decanted and the resulting precipitate washed with hexane and diethyl ether before drying under high vacuum, whereas the reaction mixture containing soluble IP-1 was added drop-wise to rapidly stirred diethyl ether to induce precipitation of the product which was subsequently isolated by filtration, washed with diethyl ether and finally dried under high vacuum. Each of the polymers was isolated as an off white crystalline solid.General procedure for anion exchange for the commercial resins
A solution of lithium bis{(trifluoromethyl)sulfonyl}imide (1.00 g, 3.49 mmol) in distilled water (30 cm3) was added drop-wise to a rapidly stirred slurry of either IRA-400 or IRA-900 (2.00 g) in distilled water (30 cm3) and allowed to stir under ambient conditions for overnight. The aqueous layer was removed and the resin was washed with distilled water (20 cm3) for 1 h. This process was repeated a further two times with fresh water. After the third wash the resin was dried in vacuo at 60 °C for a minimum of 48 h or until no further mass loss was recorded. The resultant ionic polymer was then used as a heterogeneous support for the Diels–Alder reaction.General procedure for the homogeneous Lewis acid catalyzed Diels–Alder and Mukaiyama-aldol reactions in molecular solvents
A flame dried Schlenk flask was charged with ligand (0.011 mmol), Cu(OTf)2 (0.010 mmol, 10 mol%) and dichloromethane (5 cm3) and the resulting solution stirred at room temperature for 3 h to afford a clear green solution. Thereafter, N-acryloyloxazolidinone (0.0143 g, 0.1 mmol) was added followed by freshly distilled cyclopentadiene (50 μL). For the Mukaiyama-aldol reaction, methyl pyruvate (0.0102 g, 0.1 mmol) followed by (0.0243 g, 0.12 mmol) of 1-phenyl-1-trimethylsiloxyethene. The reaction mixture was stirred at the desired temperature for the specified amount of time and then diluted with 5 cm3 of 1:1 ethyl acetate–hexane.
Following concentration of the Diels–Alder reaction mixture, the crude adduct was dissolved in 5 cm3 of diethyl ether and filtered through a small plug of silica gel to afford unpurified product and analysed directly.
Following concentration of the Mukaiyama-aldol reaction mixture, the residual oil was redissolved in THF (5 cm3) and the corresponding silyl-adduct was hydrolysed by stirring for 1 h using 2 M HCl (10 cm3). The THF was removed under vacuum and the keto-alcohol was extracted from the aqueous by washing with Et2O (3 × 5 cm3). The crude product was analysed directly.
For reactions conducted in diethyl ether, the catalyst was activated in dichloromethane which was then removed under vacuum followed by addition of diethyl ether (5 cm3) and stirred for 30 min after which the substrates were added. The reaction was worked up as previously described to afford the unpurified products which were analysed directly.
General procedure for the homogeneous Lewis acid catalyzed Diels–Alder and Mukaiyama-aldol reaction in 1-ethyl-3-methylimidazolium bis{(trifluoromethyl)sulfonyl}imide ([C2mim][NTf2])
A flame dried Schlenk flask was charged with ligand (0.011 mmol) and Cu(OTf)2 (0.010 mmol, 10 mol%). To this was added dichloromethane (5 cm3) and [C2mim][NTf2] (3.00 g, 2 cm3) and the resulting solution stirred at room temperature for 5 min. The dichloromethane was then removed under high vacuum for one hour leaving the active catalyst dissolved in the ionic liquid. Thereafter, the Diels–Alder or Mukaiyama-aldol substrates were added. The resulting mixture was stirred at room temperature for the specified amount of time after which the ionic liquid was extracted with diethyl ether (3 × 5 cm3) in air. Both the Diels–Alder and Mukaiyama-aldol reaction was worked up as previously described to afford the unpurified products which were analysed directly.General procedure for the liquid–liquid biphasic Lewis acid catalyzed Diels–Alder and Mukaiyama-aldol reactions in [C2mim][NTf2]/diethyl ether
A flame dried Schlenk flask was charged with ligand (0.011 mmol) and metal(II) triflate (0.010 mmol, 10 mol%). To this was added dichloromethane (5 cm3) and [C2mim][NTf2] (0.15 g, 0.23 cm3) and the resulting solution stirred at room temperature for 5 min. Dichloromethane was then removed under high vacuum for one hour leaving the active catalyst dissolved in the ionic liquid. Thereafter, diethyl ether (5 cm3) was added followed by the Diels–Alder or Mukaiyama-aldol substrates. The resulting mixture was stirred at the desired temperature for the specified amount of time after which the diethyl ether was removed and the remaining ionic liquid was extracted with diethyl ether (2 × 5 cm3) in air. The Diels–Alder or Mukaiyama-aldol products were worked up as previously described to afford the unpurified products which were analysed directly.General procedure for the heterogeneous Lewis acid catalyzed Diels–Alder and Mukaiyama-aldol reactions in diethylether/support
A flame dried Schlenk flask was charged with ligand (0.011 mmol), metal(II) triflate (0.010 mmol, 10 mol%) and dichloromethane (5 cm3) and the resulting solution stirred at room temperature for 5 min. To the resulting green solution was added silica, carbon or IP support (0.1 g) and for catalysts using a supported ionic liquid film, 0.1 cm3 of ionic liquid was also added. (For the non-ionic polystyrene polymer 7, the resulting metal triflate–ligand solution was stirred for 3 h before the addition of the polymer support.) After stirring for 5 min, the dichloromethane was removed under vacuum for one hour to afford a free flowing powder after which the flask was charged with diethyl ether (5 cm3) followed by the addition of the Diels–Alder or Mukaiyama-aldol substrates. The reaction mixture was stirred at room temperature for the specified amount of time after which the silica was extracted with diethyl ether (3 × 5 cm3) in air. The reaction was worked up as previously described to afford the unpurified products which were analysed directly.Procedure for recycle of the heterogeneously catalysed reactions
The catalyst/ether slurry solution was left to settle in order to separate the phases after which the ethereal layer was removed by pipette and the heterogeneous catalyst was further extracted with diethyl ether (2 × 5 cm3). After extraction fresh diethyl ether (5 cm3) was added to the heterogeneous catalyst and the resultant slurry was charged with further portions of Diels–Alder substrates. ICP analysis of a portion of the supernatant phase between recycles was conducted using a using a Perkin Elmer Optima 4300DV ICP-OES (Inductively Coupled Plasma Optical Emission Spectrometer).Notes and references
- D. Evans, S. Miller and T. Lectka, J. Am. Chem. Soc., 1993, 115, 6460–6461 CrossRef CAS; D. Evans, M. Kozlowski and J. Tedrow, Tetrahedron Lett., 1996, 37, 7481–7484 CrossRef.
- P. Goodrich, C. Hardacre, C. Paun, V. I. Parvulescu and I. Podolean, Adv. Synth. Catal., 2008, 350, 2473–2476 CrossRef CAS; S. Doherty, P. Goodrich, C. Hardacre, C. Paun and V. I. Parvulescu, Adv. Synth. Catal., 2008, 350, 295–302 CrossRef; S. Doherty, P. Goodrich, C. Hardacre, H.-K. Luo, D. W. Rooney, K. R. Seddon and P. Styring, Green Chem., 2004, 6, 63–67 RSC; L. Lou, K. Yu, F. Ding, W. Zhan, X. Peng and S. Liu, Tetrahedron Lett., 2006, 47, 6513–6516 CrossRef PubMed; M. R. Castillo, L. Fousse, J. M. Fraile, J. I. García and J. A. Mayoral, Chem.–Eur. J., 2007, 13, 287–291 CrossRef PubMed.
- P. Goodrich, C. Hardacre, C. Paun, A. Ribeiro, S. Kennedy, M. J. V. Lourenco, H. Manyar, C. A. Nieto de Castro, M. Besnea and V. I. Parvulescu, Adv. Synth. Catal., 2011, 353, 995–1004 CrossRef CAS.
- C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275–3279 CrossRef CAS PubMed; N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3273 CrossRef PubMed.
- A. Puglisi, M. Benaglia and V. Chiroli, Green Chem., 2013, 15, 1790–1813 RSC.
- B. M. L. Dios, D. E. De Vos, I. V. F. J. Vankelecom and P. A. Jacobs, Adv. Synth. Catal., 2006, 348, 1413–1446 CrossRef CAS; S. Itsuno, K. Watanabe, T. Koizumi and K. Ito, React. Polym., 1995, 24, 219–227 CrossRef.
- Polymeric Chiral Catalyst Design and Chiral Polymer Synthesis, ed. S. Itsuno, John Wiley and Sons, 2011 Search PubMed.
- H. Bae, J. Han, S. Jung, M. Cheong, H. Kim and J. Lee, Appl. Catal., A, 2007, 331, 34–38 CrossRef CAS PubMed.
- P. Barbaro and F. Liguori, Chem. Rev., 2009, 109, 515–529 CrossRef CAS PubMed.
- J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS PubMed.
- M. I. Burguete, H. Erythropel, E. García-Verdugo, S. V. Luis and V. Sans, Green Chem., 2008, 10, 401–407 RSC.
- P. Lozano, E. García-Verdugo, R. Piamtongkam, N. Karbass, T. De Diego, M. I. Burguete, S. V. Luis and J. L. Iborra, Adv. Synth. Catal., 2007, 349, 1077–1084 CrossRef CAS; P. Lozano, E. García-Verdugo, N. Karbass, K. Montague, T. De Diego, M. I. Burguete and S. V. Luis, Green Chem., 2010, 12, 1803–1810 RSC.
- A. Wolfson, J. F. J. Vankelecom and P. A. Jacobs, Tetrahedron Lett., 2003, 44, 1195–1198 CrossRef CAS.
- A. Köckritz, S. Bischoff, V. Morawsky, U. Prüße and K.-D. Vorlop, J. Mol. Catal. A: Chem., 2002, 180, 231–243 CrossRef.
- S. Doherty, J. G. Knight, J. R. Ellison, D. Weekes, R. W. Harrington, C. Hardacre and H. Manyar, Green Chem., 2012, 14, 925–929 RSC.
- D. Rechavi, B. Albela, L. Bonneviot and M. Lemaire, Tetrahedron, 2005, 61, 6976–6981 CrossRef CAS PubMed.
- J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral, D. Carrié and M. Vaultier, Tetrahedron: Asymmetry, 2001, 12, 1891–1894 CrossRef CAS.
- Y. Wan, P. McMorn, F. E. Hancock and G. J. Hutchings, Catal. Lett., 2003, 91, 145–148 CrossRef CAS.
- S. Orlandi, A. Mandoli, D. Pini and P. Salvadori, Angew. Chem., Int. Ed., 2001, 40, 2519–2521 CrossRef CAS.
- B. Corain, M. Zecca and K. Jeřábek, J. Mol. Catal. A: Chem., 2001, 177, 3–20 CrossRef CAS.
- H. Wang, J. Liu, P. Liu, Q. Yang, J. Xiao and C. Li, Chin. J. Catal., 2006, 27, 946–948 CrossRef CAS.
- J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral, S. Gmough and M. Vaultier, Green Chem., 2004, 6, 93–98 RSC.
- M. J. Fernandez, J. M. Fraile, J. I. García, J. A. Mayoral, M. I. Burguete, E. García-Verdugo, S. V. Luis and M. A. Harmer, Top. Catal., 2000, 13, 303–309 CrossRef CAS; J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral and M. A. Harmer, J. Catal., 2004, 221, 532–540 CrossRef PubMed.
- P. Izák, Š. Hovorka, T. Bartovsky, L. Bartovská and J. G. Crespo, J. Membr. Sci., 2007, 296, 131–138 CrossRef PubMed.
- S. Itsuno, S. Arima and N. Haraguchi, Tetrahedron, 2005, 61, 12074–12080 CrossRef CAS PubMed.
- G. Desimoni, G. Faita, A. G. Invernizzi and P. Righetti, Tetrahedron, 1997, 53, 7671–7688 CrossRef CAS.
- M. I. Burguete, E. García-Verdugo, I. Garcia-Villar, F. Gelat, P. Licence, S. V. Luis and V. Sans, J. Catal., 2010, 269, 150–160 CrossRef CAS PubMed.
- C. Villigrán, M. Deetlefs, W. R. Pitner and C. Hardacre, Anal. Chem., 2004, 76, 2118–2123 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR and 19F NMR spectra for the polymer precursors synthesised. TGA, SEM and FTIR traces for the polymers used. HPLC traces of exo and endo-cycloadducts and aldol adducts formed. See DOI: 10.1039/c3gc41378k |
| This journal is © The Royal Society of Chemistry 2014 |