
Allicin protects spinal cord neurons from glutamate-induced oxidative stress through regulating the heat shock protein 70/inducible nitric oxide synthase pathway
Shu-Guang
Liu
ac,
Peng-Yu
Ren
b,
Guo-Yu
Wang
a,
Shu-Xin
Yao
c and
Xi-Jing
He
*a
aDepartment of Orthopaedics, The Second Affiliated Hospital, School of Medicine, Xi'an Jiaotong University, Xi'an, Shaanxi 710004, China. E-mail: hexijing_xjtu@163.com; Fax: +86-029-87679002; Tel: +86-029-87679001
bDepartment of Neurosurgery, The Third Affiliated Hospital, School of Medicine, Xi'an Jiaotong University, Xi'an, Shaanxi 710068, China
cDepartment of Orthopaedics, Hong Hui Hospital, the Affiliated Hospital, School of Medicine, Xi'an Jiaotong University, Xi'an, Shaanxi 710004, China
First published on 23rd October 2014
Abstract
Allicin, the main biologically active compound derived from garlic, exerts a broad spectrum of pharmacological activities and is considered to have therapeutic potential in many neurological disorders. Using an in vitro spinal cord injury model induced by glutamate treatment, we sought to investigate the neuroprotective effects of allicin in primary cultured spinal cord neurons. We found that allicin treatment significantly attenuated glutamate-induced lactate dehydrogenase (LDH) release, loss of cell viability and apoptotic neuronal death. This protection was associated with reduced oxidative stress, as evidenced by decreased reactive oxygen species (ROS) generation, reduced lipid peroxidation and preservation of antioxidant enzyme activities. The results of western blot analysis showed that allicin decreased the expression of inducible nitric oxide synthase (iNOS), but had no effects on the expression of neuronal NOS (nNOS) following glutamate exposure. Moreover, allicin treatment significantly increased the expression of heat shock protein 70 (HSP70) at both mRNA and protein levels. Knockdown of HSP70 by specific targeted small interfere RNA (siRNA) not only mitigated allicin-induced protective activity, but also partially nullified its effects on the regulation of iNOS. Collectively, these data demonstrate that allicin treatment may be an effective therapeutic strategy for spinal cord injury, and that the potential underlying mechanism involves HSP70/iNOS pathway-mediated inhibition of oxidative stress.
Introduction
Spinal cord injury is defined as an injury to the spinal cord that is caused by trauma, vertebral stenosis, tumor or ischemia.1 Ischemic spinal cord injury is one of the most serious complications after open surgery and thoracic endovascular aortic repair, and can cause paraplegia or paralysis in up to 40% of patients.2,3 Although various adjunctive procedures for spinal cord protection have been introduced, including improving surgical technique, progress in monitoring and anaesthesia, or use of steroids, barbiturates, or free-radical scavengers, postoperative paraplegia remains a devastating complication.4–6 The exact molecular mechanism underlying the neuronal injury in the spinal cord and delayed neurological dysfunction is not fully understood.Allium sativum, usually known as garlic, is a species in the onion genus that has been used for thousands of years for medicinal purposes. Ancient Chinese and Indian medicine used garlic to treat many conditions, including hypertension, infections and snakebites.7 Previous studies have isolated hundreds of chemical substances from fresh or dried garlic, among which allicin is the main component responsible for the biological activity of garlic.8,9 Allicin is formed during the chopping, crushing or chewing of garlic cloves through a chemical interaction between alliin, a sulfur-containing amino acid, and the enzyme alliinase.10 Previous in vitro and in vivo experiments have shown that allicin possesses a variety of biological/medical effects, such as anti-inflammatory, antimicrobial, antifungal, antiparasitic, antihypertensive and anticancer activities.7,11,12 Allicin protects against oxidative stress via inducing the generation of antioxidant products, thereby diminishing the concentrations of cytotoxic substances and scavenging free radicals. The cardiovascular benefits and antioxidant properties of allicin have been studied extensively with very promising results.13 A previous study indicated that allicin decreases ROS generation and increases the level of glutathione through its antioxidative activity in endothelial cells.14 More recently, allicin was shown to exert neuroprotective effects through regulating the nitric oxide synthase (NOS) signaling pathway in traumatic neuronal injury and brain trauma models.15,16
Considering these properties of allicin, we speculated that allicin may exert beneficial effects on spinal cord injury through suppressing oxidative stress. In the present study, a well-characterized neuronal injury model, induced by glutamate treatment in spinal cord neurons, was used to mimic neuronal damage in spinal cord injury in in vitro conditions. We examined the protective effects of allicin and the potential underlying mechanisms with a focus on the heat shock protein 70 (HSP70)/iNOS pathway.
Materials and methods
Spinal cord neuron culture
Spinal cord neurons were obtained from embryonic day 14 rat spinal cords by gentle trituration, according to previously described protocols.17,18 Briefly, rat spinal cords were removed from embryos at 16–18 days, stripped of meninges and blood vessels and minced. Tissues were cut into small pieces and dissociated by 0.25% trypsin digestion for 15 min at 37 °C. After adhering at 37 °C for 30 min to eliminate glial cells and fibroblasts, neurons were resuspended in Neurobasal medium containing 2% B27 supplement (Invitrogen, CA, USA) and 0.5 mM L-glutamine and plated at a density of 3 × 105 cells cm−2. Before seeding, 96-well plates and 6 cm dishes were coated with poly-L-lysine (PLL) at room temperature overnight. Neurons were maintained at 37 °C in a humidified 5% CO2 incubator and half of the culture medium was changed every other day. Under these culture conditions, a purity of >85% spinal cord neuronal population was obtained at the seventh day in vitro. Allicin (purity >98%) was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Spinal cord neuronal cultures were treated with 50 μM allicin for 30 min and 5 mM glutamate for an additional 24 h.All experimental protocols were approved by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University and performed in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications no. 80-23, revised 1996). All efforts were made to minimize animal number and their suffering.
H&E staining
After allicin and glutamate treatment, spinal cord neurons were washed with phosphate buffered saline (PBS) and then fixed with 4% paraformaldehyde (PFA) in PBS for at least 0.5 h. Neurons were deparaffinized with xylene, stained by H&E, and then assessed by a pathologist for neuronal loss under an Olympus U-DO3 light microscope (Tokyo, Japan).Neuronal viability
A neuronal viability assay was performed by using the Cell Proliferation Reagent WST-1 following the manufacture's protocol (Roche, Basel, Switzerland). Briefly, spinal cord neurons were cultured at a concentration of 5 × 104 in microplates in a final volume of 100 μL per well culture medium. After various treatments, 10 μL cell proliferation reagent, WST-1, was added into each well and these were incubated for 4 h at 37 °C. Then, 100 μL culture medium and 10 μL WST-1 was added into one well in the absence of neurons, and its absorbance was used as a blank position for the ELISA reader. Cells were shaken thoroughly for 1 min on a shaker and the absorbance of the samples was measured using a microplate reader.Lactate dehydrogenase (LDH) assay
Cytotoxicity was determined by measuring the release of LDH, a cytoplasmic enzyme released from cells. LDH release into the culture medium was detected using a diagnostic kit according to the manufacturer's instructions (Jiancheng Bioengineering Institute, Nanjing, China). Briefly, 50 μL of supernatant from each well was collected to assay LDH release. The samples were incubated with a reduced form of nicotinamide adenine dinucleotide (NADH) and pyruvate for 15 min at 37 °C and the reaction was stopped by adding 0.4 M NaOH. The activity of LDH was calculated from the absorbance at 440 nm and background absorbance from culture medium that was not used for any cell cultures was subtracted from all absorbance measurements. The results are presented as multiples of the control value (fold of control).TUNEL staining
To investigate the effect of allicin on glutamate-induced neuronal apoptosis, TUNEL staining was performed using an in situ cell death detection kit. Briefly, spinal cord neurons were seeded on PLL-coated glass slides at a density of 3 × 105 cells cm−2, and these were subjected to glutamate exposure and various treatments. Then, the neurons were fixed by immersing the slides in a freshly prepared 4% methanol-free formaldehyde solution in PBS for 20 min at room temperature. The neurons were permeabilized with 0.2% Triton X-100 for 5 min. Cells were labeled with fluorescein TUNEL reagent mixture for 60 min at 37 °C, according to the manufacturer's suggested protocol. Subsequently, the slides were examined by fluorescence microscopy and the number of TUNEL-positive (apoptotic) cells was counted. 4′,6-Diamidino-2-phenylindole (DAPI, 10 μg ml−1) was used to stain the nuclei.Measurement of caspase-3 activity
The activity of caspase-3 was measured using a colorimetric assay kit according to the manufacturer's instructions (Cell Signaling Technology, Beverly, MA, USA). Briefly, after being harvested and lysed, 106 cells were mixed with 32 μL of assay buffer and 2 μL of 10 mM Ac-DEVD-pNA substrate. Absorbance at 405 nm was measured after incubation at 37 °C for 4 h. The absorbance of each sample was determined by subtraction of the mean absorbance of the blank and this value was corrected by the protein concentration of the cell lysate. The results are described as relative activity to that of the control group.Measurement of ROS generation
Briefly, the spinal cord neurons were incubated with 2,7-dichlorofluorescein diacetate (DCF-DA) (Sigma, St. Louis, MO, USA) (10 μM) for 1 h at 37 °C in the dark, and then resuspended in PBS. Intracellular ROS generation was detected using the fluorescence intensity of the oxidant-sensitive probe 2,7-dichlorodihydro-fluorescein diacetate (H2DCF-DA) under a microscope, and the fluorescence was read using an excitation wavelength of 480 nm and an emission wavelength of 530 nm.Measurement of lipid peroxidation
Malonyldialdehyde (MDA) and 4-hydroxynonenal (4-HNE), two indices of lipid peroxidation, were detected by using assay kits from Cell Biolabs and strictly following the manufacturer's instructions. The absorbance of the samples was measured with a microplate (ELISA) reader. The results are presented as multiples of the control value.Measurement of antioxidant enzyme activity
The enzymatic activities of manganese superoxide dismutase (MnSOD) and catalase (CAT) were measured by use of commercially available assay kits following the manufacturer's instructions (Cayman Chemical, Ann Arbor, MI, USA). Protein concentration was determined by using a BCA protein kit (Jiancheng Bioengineering Institute, Nanjing, China). The enzyme activities were then normalized to the corresponding protein concentration for each sample.Measurement of complex I activity
Mitochondria were purified by Percoll density gradient centrifugation in extraction buffer (50 mM Tris HCl, pH 7.5, 500 mM NaCl, 0.03% reduced Triton X-100, 1 mM EDTA, 1 mM PMSF, 0.5 mM benzamidine, and 1 mg ml−1 each of pepstatin-A, leupeptin and aprotinin). All the samples were subjected to three freeze–thaw cycles to disrupt membranes and expose enzymes before analysis. The enzymatic activity of mitochondrial respiratory chain complex I was measured at 37 °C as previously described.19 The data are expressed as percentages of the control value.Short interfering RNA (siRNA) and transfection
The specific siRNA-targeted HSP70 (Si-HSP70, sc-270278, Santa Cruz, CA, USA) and control siRNA (Si-Con, sc-37007, Santa Cruz, CA, USA), which should not knock down any known proteins, were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The above siRNA molecules were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in 6-well plates for 72 h. After transfection, the spinal cord neurons were treated with glutamate with or without allicin and subjected to various measurements.Real-time RT-PCR
Total RNA was isolated from spinal cord neurons using Trizol reagent (Invitrogen, Carlsbad, CA, USA). After equalization of the RNA quantity in each group, the mRNA levels were quantitated using a Bio-Rad iQ5 Gradient Real-Time PCR system (Bio-Rad Laboratories, Richmond, CA, USA), and GAPDH was used as an endogenous control. Primers used for all real-time PCR experiments are as follows: hsp70: forward: 5′-ACCAGGACACTGTTGAGTTC-3′, reverse: 5′-ACTCATCTCCGAGTTCACAC-3′; GAPDH: forward: 5′-AAGGTGAAGGTCGGAGTCAA-3′, reverse: 5′-AATGAAGGGGTCAT TGATGG-3′. Samples were tested in triplicates and data from five independent experiments were used for analysis.Western blot analysis
Cultured spinal cord neurons were washed with ice-cold PBS and harvested in lysis buffer containing protease inhibitors. The protein content was determined using a BCA protein assay kit. 60 μg protein was resolved on 10% SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk and incubated with the following primary antibodies: iNOS (sc-7271, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, Santa Cruz), nNOS (sc-5302, 1:800, Santa Cruz), HSP70 (sc-373867, 1:1000, Santa Cruz) and β-actin (ac-47778, 1:600, Santa Cruz). The membranes were then washed and incubated for 1 h at room temperature with secondary antibodies. ImageJ analysis software was used to quantify the optical density of each band.
:1000, Santa Cruz), nNOS (sc-5302, 1:800, Santa Cruz), HSP70 (sc-373867, 1:1000, Santa Cruz) and β-actin (ac-47778, 1:600, Santa Cruz). The membranes were then washed and incubated for 1 h at room temperature with secondary antibodies. ImageJ analysis software was used to quantify the optical density of each band.
Statistical analysis
Statistical analysis was performed using SPSS 16.0 (SPSS Inc., Chicago, IL, USA), a statistical software package. Statistical evaluation of the data was performed by one-way analysis of variance (ANOVA) followed by Bonferroni's multiple comparisons. A value of p < 0.05 was considered statistically significant.Results
Allicin attenuates glutamate-induced excitotoxicity in spinal cord neurons
To investigate the potential protective effects of allicin against glutamate-induced neuronal injury, primary cultured spinal cord neurons were pretreated with 50 μM allicin 30 min before 5 mM glutamate treatment. The results of H&E staining showed that allicin pretreatment significantly attenuated the neuronal loss induced by glutamate (Fig. 1A). Allicin significantly increased the neuronal viability (Fig. 1B), but decreased the LDH release (Fig. 1C), after glutamate treatment. The neuronal apoptosis was detected by TUNEL staining (Fig. 1D), and the results showed that allicin inhibited neuronal apoptosis induced by glutamate treatment (Fig. 1E). As shown in Fig. 1F, a similar result on caspase-3 activity was also observed.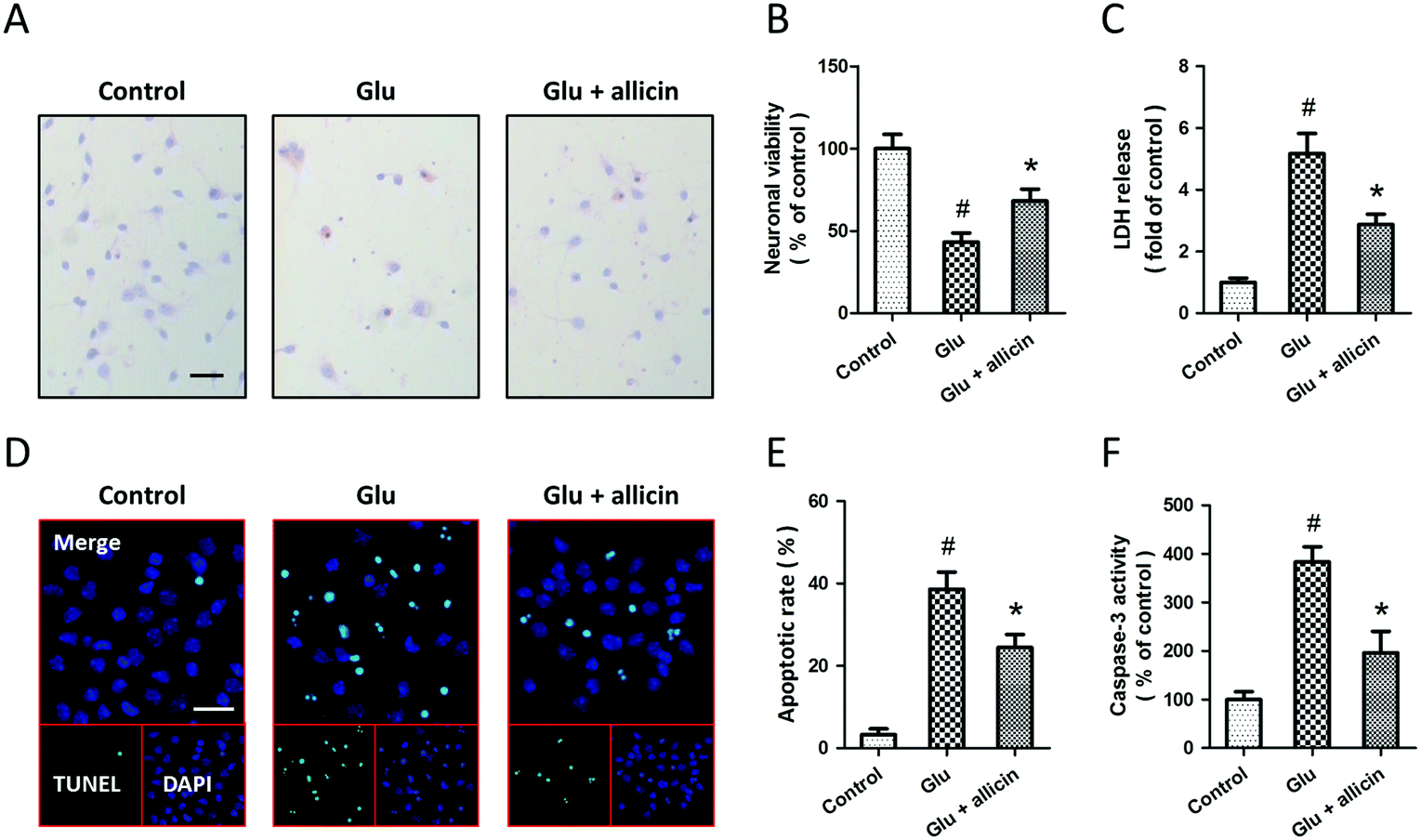 | ||
| Fig. 1 Allicin protects spinal cord neurons against glutamate-induced excitotoxicity. Rat spinal cord neurons were pretreated with 50 μM allicin 30 min before treatment with 5 mM glutamate. The neuronal loss was detected by H&E staining (A). The neuronal viability was measured with the WST assay (B), and the cytotoxicity was measured with the LDH release assay (C). The neuronal apoptosis was detected by TUNEL staining (D), and the apoptotic rate was calculated (E). The activity of caspase-3 was measured (F). Scale bars = 50 μm. The data are either representative of three similar experiments or are shown as mean ± SD of five experiments. #p < 0.05 vs. control. *p < 0.05 vs. Glu. | ||
To determine the protection induced by allicin was mediated by antioxidative activity, we first measured the intracellular ROS generation after glutamate treatment (Fig. 2A). The increased ROS generation after glutamate treatment was reduced by allicin pretreatment (Fig. 2B). Reduction of MDA and 4-HNE formation was observed in allicin pretreated neurons (Fig. 2C and D), indicating that allicin appears to limit lipid peroxidation induced by oxidative stress. We also tested the effects of allicin on the endogenous antioxidant system, and the results showed that the decreased enzymatic activities of MnSOD and CAT were preserved by allicin pretreatment (Fig. 2E). As shown in Fig. 2F, a similar result on mitochondrial complex I activity was also observed.
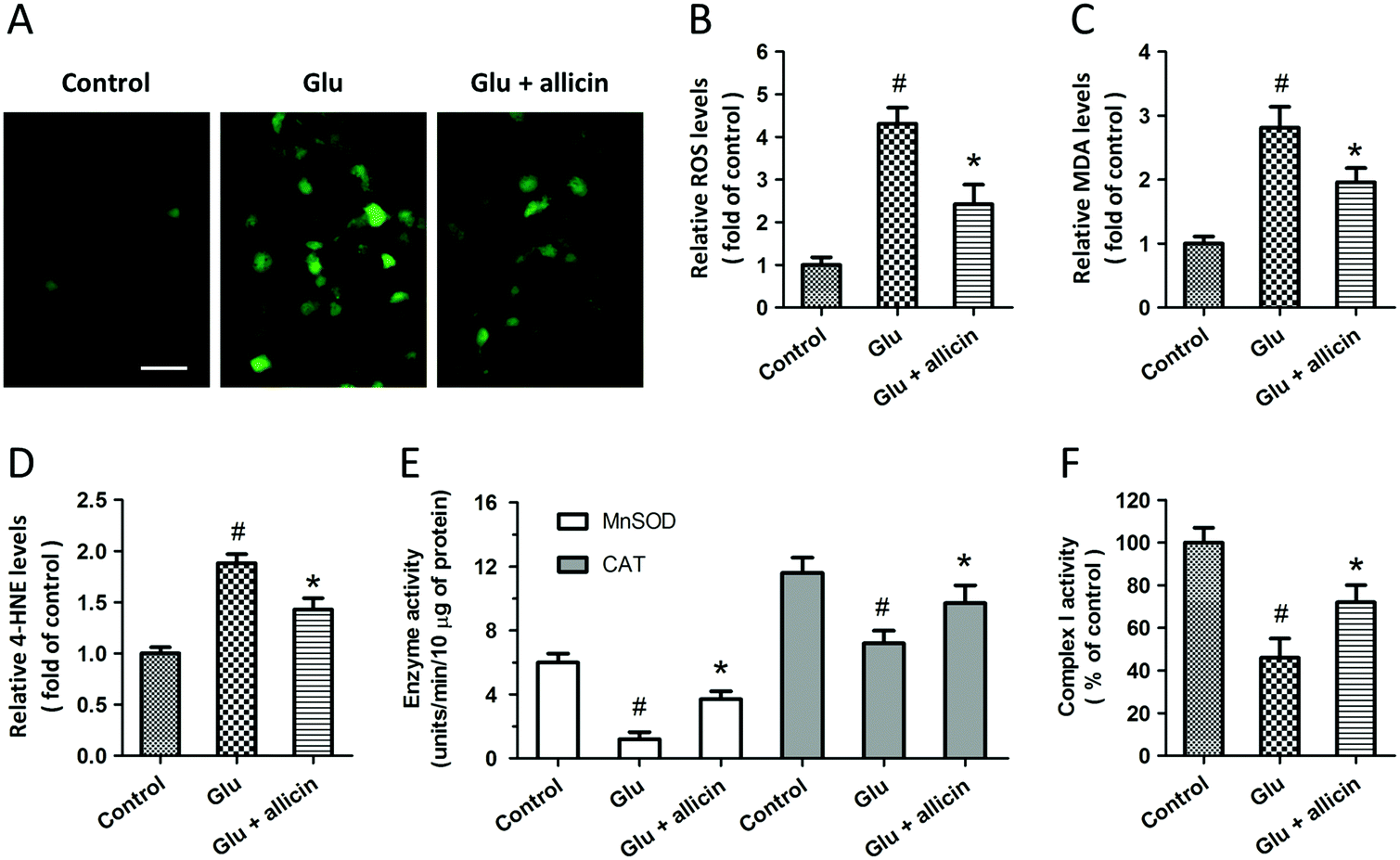 | ||
| Fig. 2 Allicin inhibits glutamate-induced oxidative stress in spinal cord neurons. Rat spinal cord neurons were pretreated with 50 μM allicin 30 min before treatment with 5 mM glutamate. Intracellular ROS generation was detected by DCFDA staining (A) and calculated (B). The lipid peroxidation was determined by measuring the levels of MDA (C) and 4-HNE (D). The enzymatic activity of MnSOD and CAT (E) and the activity of mitochondrial complex I (F) were assayed. Scale bars = 50 μm. The data are either representative of three similar experiments or are shown as mean ± SD of five experiments. #p < 0.05 vs. control. *p < 0.05 vs. Glu. | ||
Expression of iNOS and nNOS and their roles in allicin-induced neuroprotection
Western blot analysis was used to examine the expression of iNOS and nNOS after glutamate treatment (Fig. 3A). The results showed that glutamate significantly increased the expression of iNOS in a time-dependent manner (Fig. 3B), but the expression of nNOS was not altered (Fig. 3C). To investigate the roles of iNOS and nNOS in glutamate-induced neuronal injury, spinal cord neurons were pretreated with the iNOS inhibitor 1400W (10 μM) or the nNOS inhibitor NPLA (5 μM) 30 min before glutamate treatment. Both these two inhibitors overtly attenuated the neuronal viability loss (D), LDH release (E) and apoptotic death (F) induced by glutamate treatment.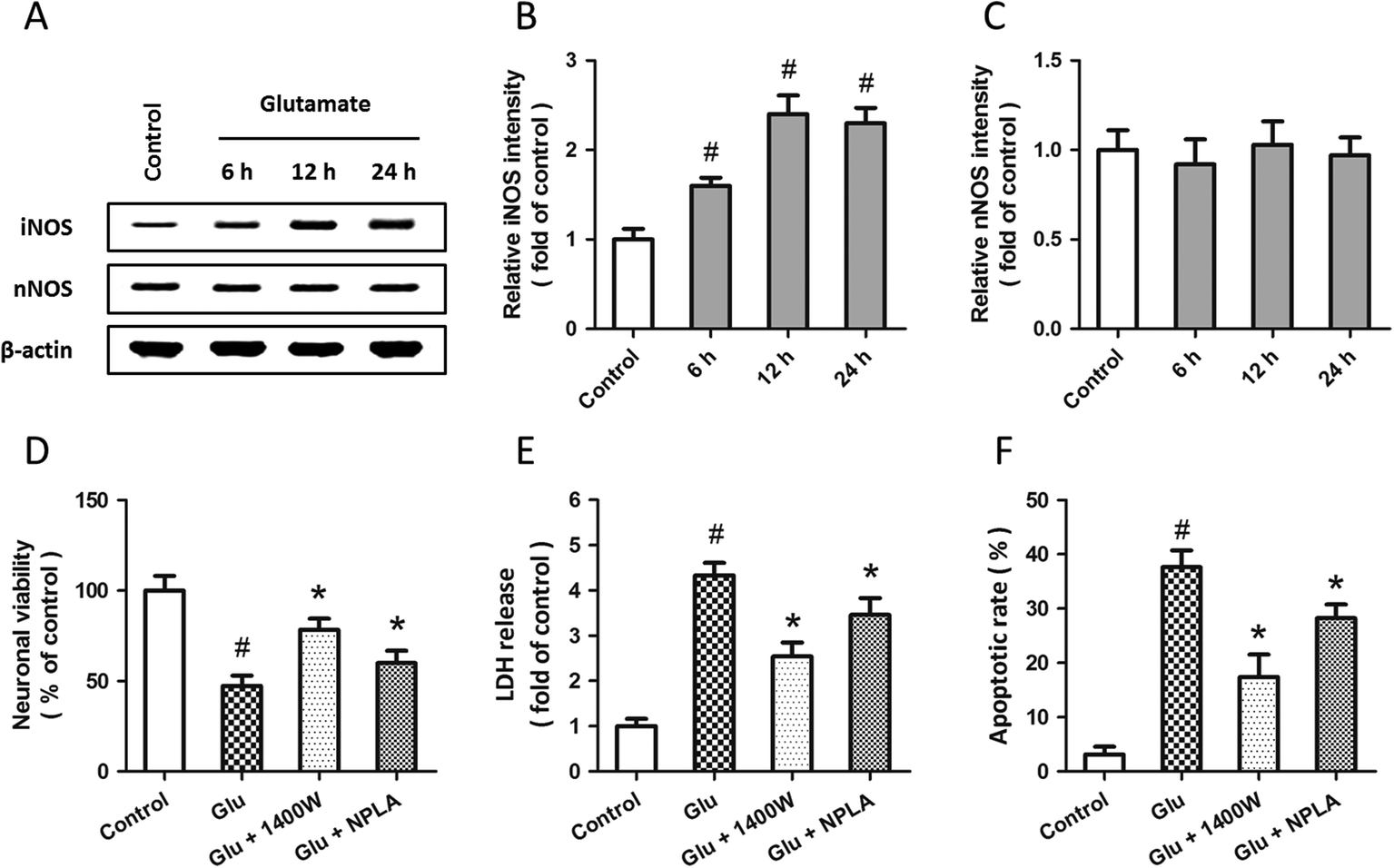 | ||
| Fig. 3 Involvement of iNOS and nNOS in glutamate-induced excitotoxicity in spinal cord neurons. Rat spinal cord neurons were treated with 5 mM glutamate, and the expression of iNOS and nNOS were detected by western blot analysis (A) and calculated (B and C). Rat spinal cord neurons were pretreated with the iNOS antagonist 1400W (10 μM) or the nNOS antagonist NPLA (5 μM) 30 min before treatment with 5 mM glutamate. The neuronal viability (D), the LDH release (E) and the neuronal apoptosis (F) were assayed 24 h after injury. The data are shown as mean ± SD of five experiments. #p < 0.05 vs. control. *p < 0.05 vs. Glu. | ||
To further examine the possible involvement of iNOS and nNOS in allicin-induced neuroprotection, the expressions of these two proteins were detected by western blot after allicin and glutamate treatment (Fig. 4A). As shown in Fig. 4B, the increased expression of iNOS induced by glutamate was mitigated by allicin. Neither glutamate nor allicin significantly affected the expression of nNOS in spinal cord neurons (Fig. 4C). In addition, by using the inhibitors of iNOS and nNOS before glutamate and allicin treatment, we determined the roles of these two proteins in allicin-induced neuroprotection. We found that the nNOS inhibitor NPLA, but not the iNOS inhibitor 1400W, markedly enhanced the protection induced by allicin, as evidenced by further increased neuronal viability (Fig. 4D), decreased LDH release (Fig. 4E) and apoptotic rate (Fig. 4F).
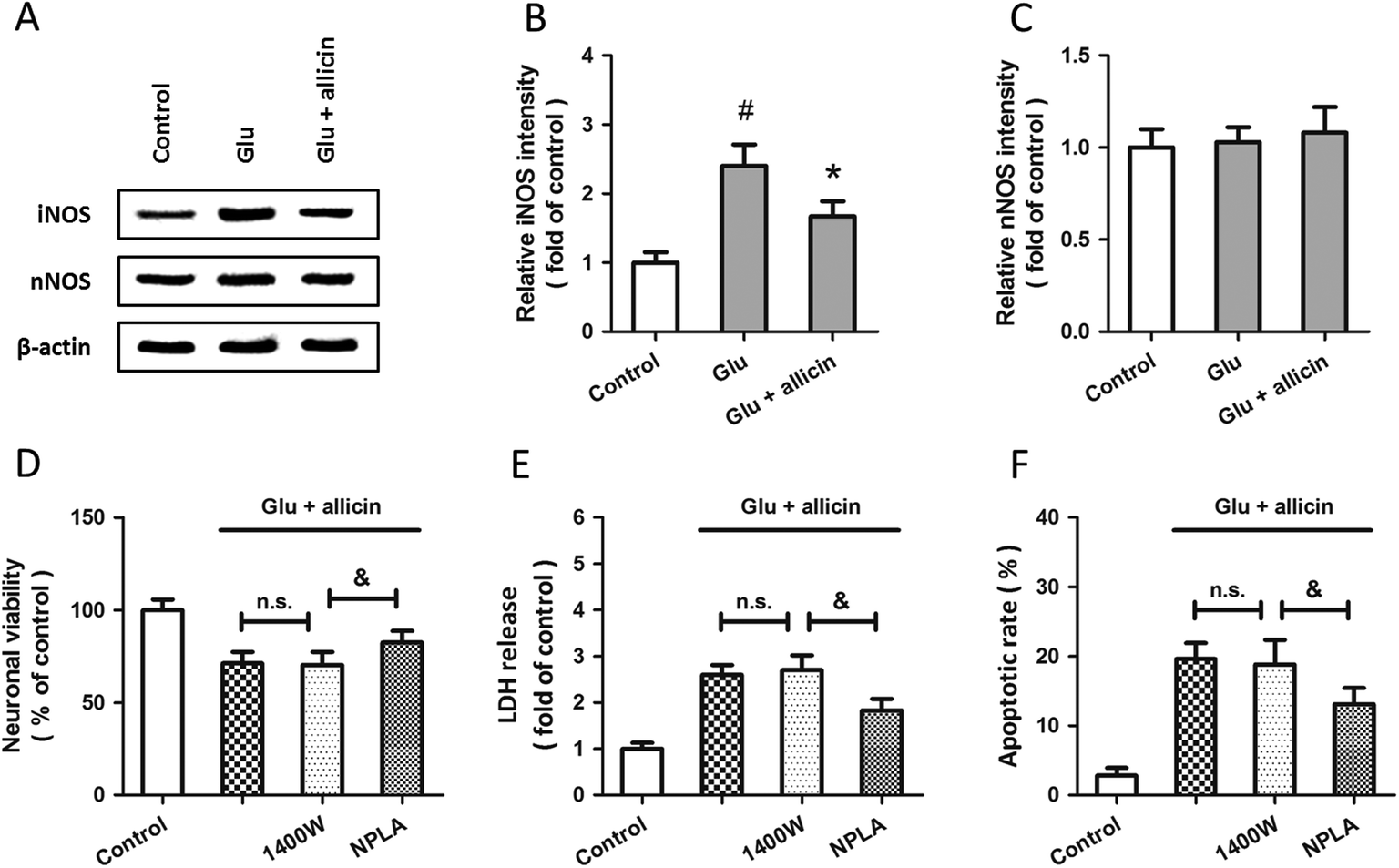 | ||
| Fig. 4 Involvement of iNOS and nNOS in allicin-induced neuroprotection against glutamate-induced excitotoxicity in spinal cord neurons. Rat spinal cord neurons were pretreated with 50 μM allicin 30 min before treatment with 5 mM glutamate. The expression of iNOS and nNOS were detected by western blot analysis (A) and calculated (B and C). Rat spinal cord neurons were pretreated with 50 μM allicin in the presence or absence of 1400W (10 μM) or NPLA (5 μM) 30 min before treatment with 5 mM glutamate. The neuronal viability (D), the LDH release (E) and the neuronal apoptosis (F) were assayed 24 h after injury. The data are shown as mean ± SD of five experiments. #p < 0.05 vs. control. *p < 0.05 vs. Glu. &p < 0.05. n.s. = not statistically significant. | ||
Expression of HSP70 and its role in allicin-induced neuroprotection
Western blot and RT-PCR were used to examine the expression of HSP70 after glutamate treatment in spinal cord neurons. The results showed that HSP70 expression was significantly increased after glutamate treatment at both mRNA (Fig. 5A) and protein (Fig. 5B) levels. The specific targeted siRNA (Si-HSP70) was used to downregulate the expression of HSP70, and the expression of HSP70 in Si-HSP70 transfected neurons was obviously decreased compared to control siRNA (Si-Con) transfected neurons (Fig. 5C). Our data also showed that glutamate-induced neuronal injury in spinal cord neurons was significantly aggravated by HSP70 knockdown, as evidenced by decreased neuronal viability (Fig. 5D), and increased LDH release (Fig. 5E) and apoptotic rate (Fig. 5F).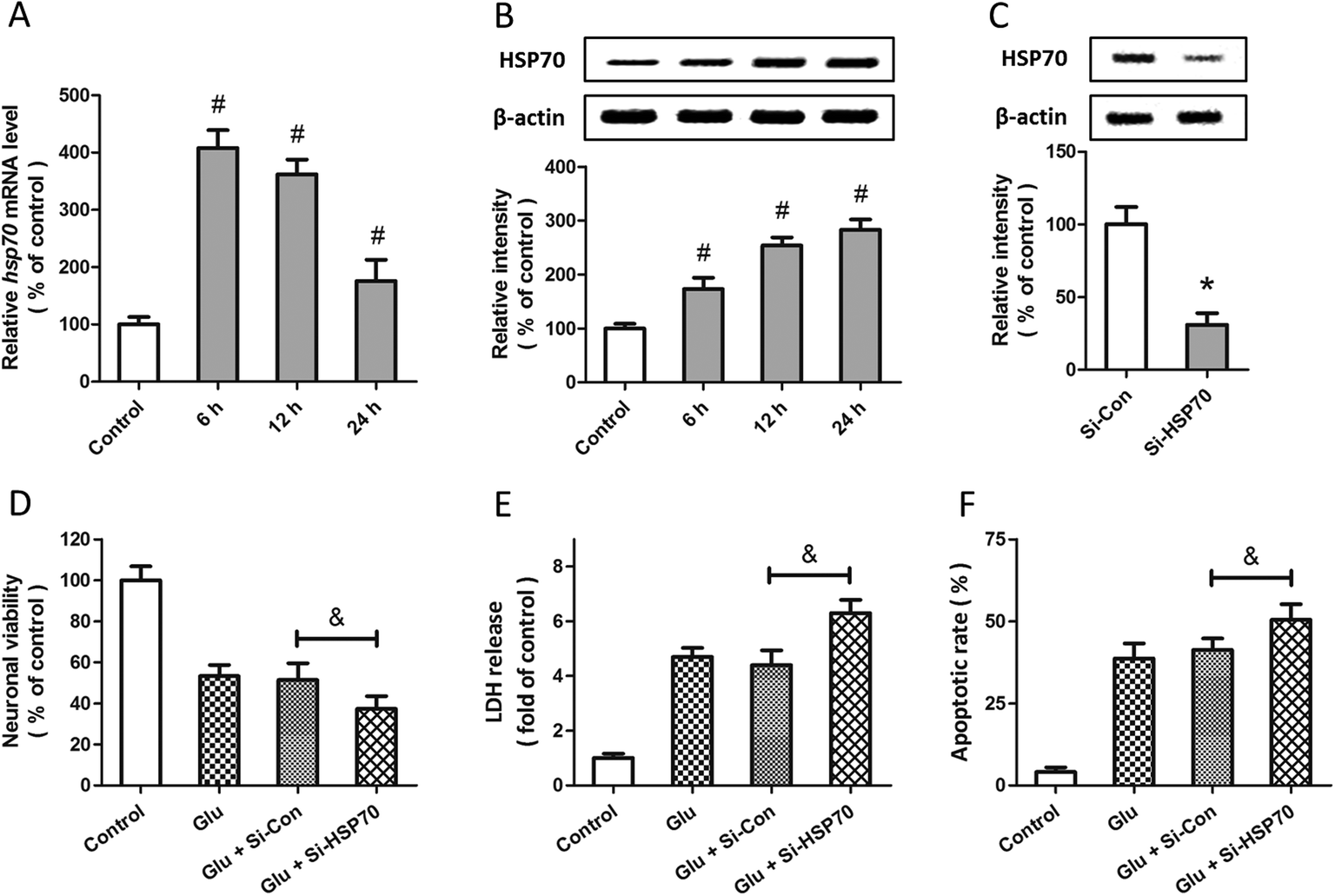 | ||
| Fig. 5 Involvement of HSP70 in glutamate-induced excitotoxicity in spinal cord neurons. Rat spinal cord neurons were treated with 5 mM glutamate, and the expression of HSP70 was detected by RT-PCR (A) and western blot analysis (B). Rat spinal cord neurons were transfected with HSP70 targeted siRNA (Si-HSP70) or control siRNA (Si-Con) for 72 h, and the expression of HSP70 was detected by western blot (C). After transfection and glutamate treatment, the neuronal viability (D), the LDH release (E) and the neuronal apoptosis (F) were assayed 24 h after injury. The data are shown as mean ± SD of five experiments. #p < 0.05 vs. control. *p < 0.05 vs. Si-Con. &p < 0.05. | ||
To determine whether HSP70 is involved in allicin-induced protection in spinal cord neurons, the expression of HSP70 was detected by western blot after allicin and glutamate treatment. We found that the increased expression of HSP70 induced by glutamate treatment was further enhanced by allicin pretreatment (Fig. 6A). We also determined the neuronal viability (Fig. 6B), LDH release (Fig. 6C) and apoptotic rate (Fig. 6D) after HSP70 knockdown and glutamate treatment, and the results showed that the neuroprotective effects of allicin against glutamate-induced neuronal injury were partially nullified by Si-HSP70 transfection.
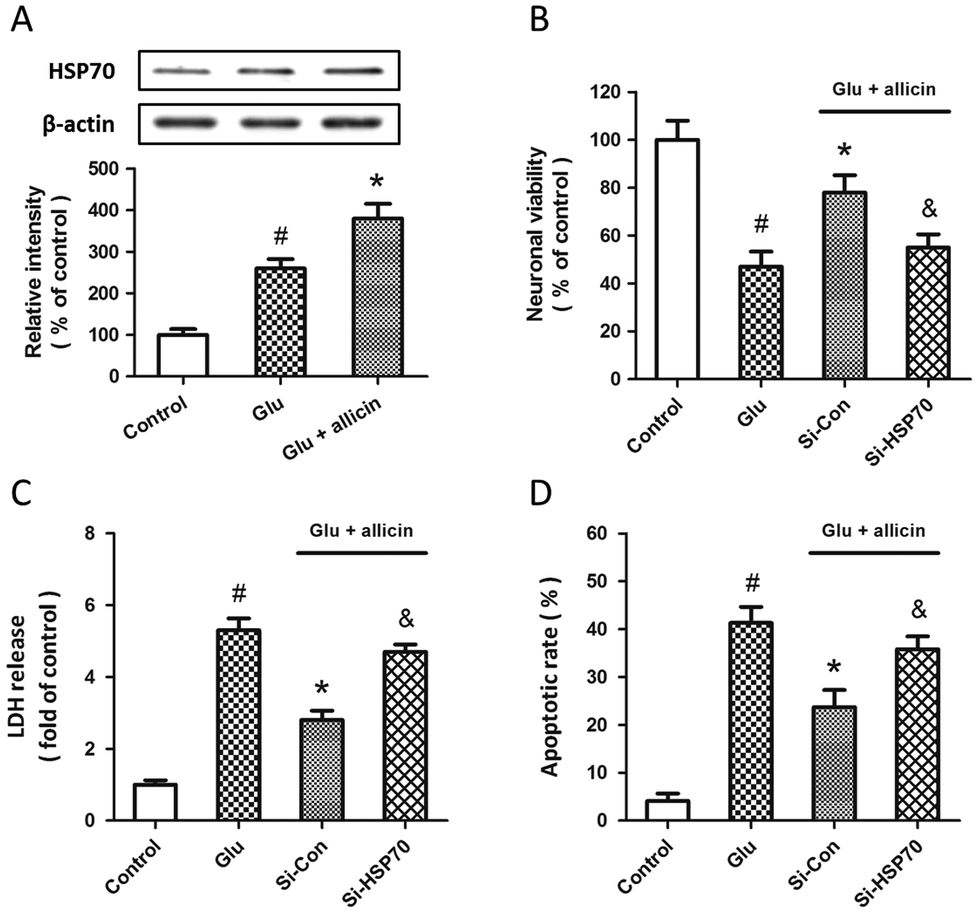 | ||
| Fig. 6 Involvement of HSP70 in allicin-induced neuroprotection against glutamate-induced excitotoxicity in spinal cord neurons. Rat spinal cord neurons were pretreated with 50 μM allicin 30 min before treatment with 5 mM glutamate. The expression of HSP70 was detected by western blot analysis (A). Rat spinal cord neurons were transfected with HSP70 targeted siRNA (Si-HSP70) or control siRNA (Si-Con) for 72 h, and pretreated with 50 μM allicin 30 min before 5 mM glutamate treatment. The neuronal viability (B), the LDH release (C) and the neuronal apoptosis (D) were assayed 24 h after injury. The data are shown as mean ± SD of five experiments. #p < 0.05 vs. control. *p < 0.05 vs. Glu. &p < 0.05 vs. Si-Con. | ||
Effect of HSP70 knockdown on allicin-induced regulation of iNOS
To further elucidate the relationship between allicin-induced regulation of the HSP70 and NOS pathway, the expression of iNOS and nNOS were examined by western blot after siRNA transfection, allicin pretreatment and glutamate treatment (Fig. 7A). The results showed that the allicin-induced inhibition of iNOS activation after glutamate treatment was mitigated by HSP70 knockdown (Fig. 7B), whereas neither allicin nor siRNA transfection altered the expression of nNOS (Fig. 7C). We also determined the changes in HSP70 mRNA and protein after treatment with iNOS and nNOS inhibitors by PCR and western blot. Neither 1400W nor NPLA had effects on the levels of HSP70 mRNA after allicin and glutamate treatment (Fig. 7D). As shown in Fig. 7E, the iNOS inhibitor 1400W, but not the nNOS inhibitor NPLA, further increased the expression of HSP70 protein induced by allicin.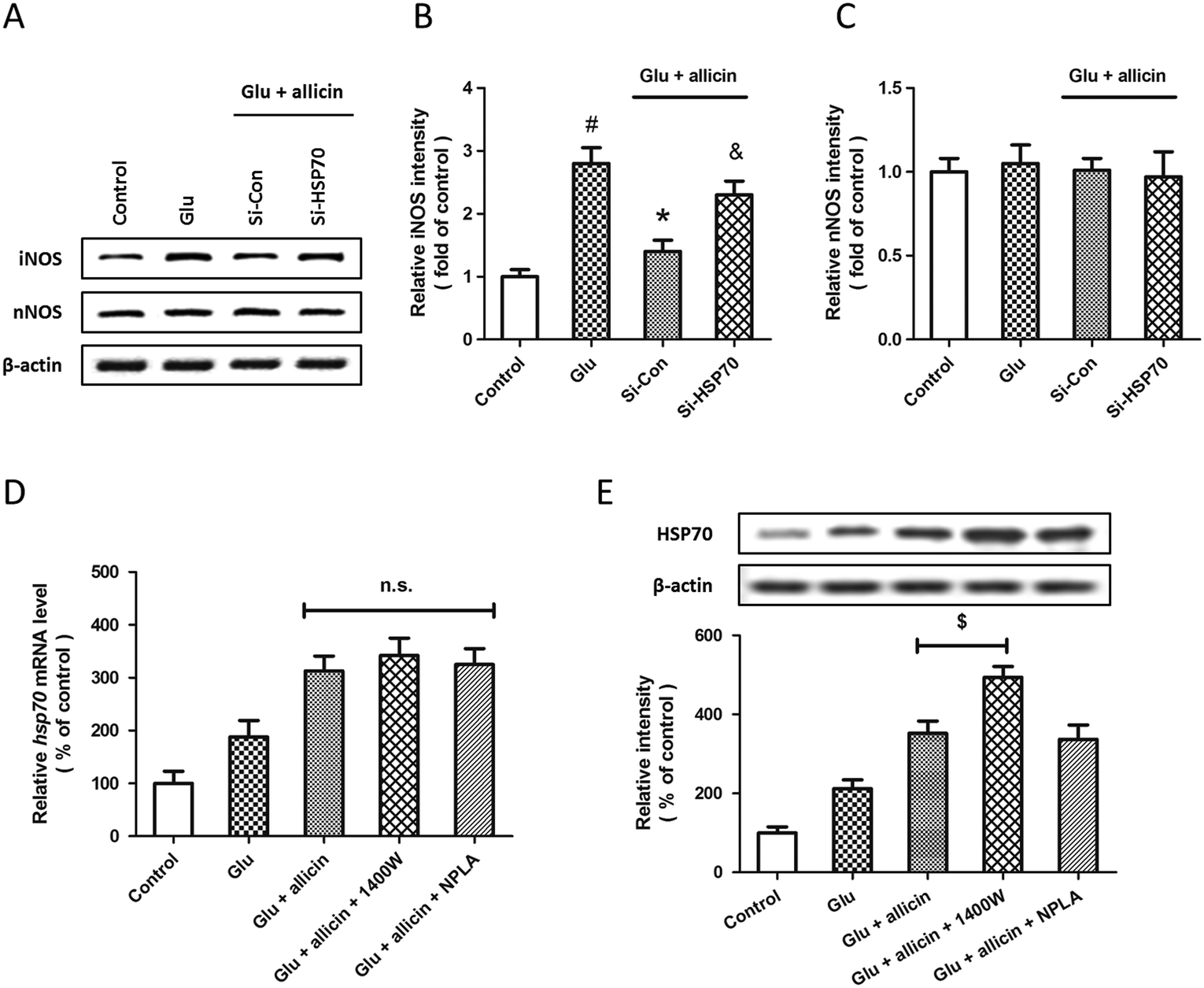 | ||
| Fig. 7 Involvement of HSP70 in allicin-induced regulation of iNOS after glutamate treatment in spinal cord neurons. Rat spinal cord neurons were transfected with HSP70 targeted siRNA (Si-HSP70) or control siRNA (Si-Con) for 72 h, and pretreated with 50 μM allicin 30 min before 5 mM glutamate treatment. The expression of iNOS and nNOS were detected by western blot analysis (A) and calculated (B and C). Rat spinal cord neurons were pretreated with 50 μM allicin with or without 1400W (10 μM) or NPLA (5 μM) 30 min before 5 mM glutamate treatment. The expression of HSP70 was detected by RT-PCR (D) and western blot analysis (E). The data are shown as mean ± SD of five experiments. #p < 0.05 vs. control. *p < 0.05 vs. Glu. &p < 0.05 vs. Si-Con. $p < 0.05. | ||
Discussion
Based on an in vitro experimental model of spinal cord injury, the present study explores the role of allicin in spinal cord neurons. Our results provide novel demonstrations that administration with allicin protects against oxidative stress and apoptotic cell death induced by glutamate treatment in spinal cord neurons. We have further demonstrated that mechanistically, allicin increases the expression of HSP70, and thereby inhibits the activation of iNOS following glutamate treatment.Allicin is a volatile oil found in garlic and other Allium species such as onions and shallots, and it is easily detected with its unique odor.20 Allicin can easily pass through phospholipid bilayers and the blood–brain barrier owing to its hydrophobic nature, and it can interact with various compounds in cellular compartments to form beneficial thiol-containing metabolites, even in an energy-deficient environment.12,21 Thus, allicin can be used directly in cultured spinal neurons to determine its role in our in vitro model. A previous study showed that the biological half-life of allicin is much longer than its chemical half-life, which is approximately 3 h based on chromatographic quantification.22 A freshly prepared allicin solution was used in our experiments because allicin was found to disappear in the cell medium within a short period of time under in vitro conditions.23 The concentration of allicin used in the present study was selected based on previous studies in cortical neurons.15,16 One clove of fresh garlic contained 4–5 mg of allicin, and it was shown that 6–18 μM allicin can be detected in blood when 1–3 cloves of fresh garlic are consumed per day.9,13,14 Therefore, the concentration of allicin used in the present study can easily be obtained from a daily diet for human beings, and this makes our data more relevant to clinical practice.
Although the etiology and pathogenesis of spinal cord injury are not fully understood, it is well known that ROS production and the consequent oxidative stress are important events associated with neuronal injury in the CNS.24–27 Alleviating oxidative stress represents an ideal method for therapeutic intervention, and many compounds, such as methylprednisolone, 21-aminosteroid tirilazad, carotenoids and phenolic compounds have been shown to improve the recovery of spinal cord injury patients in clinical trials.24,28–32 The spinal cord is highly vulnerable to oxidative injury due to its overabundance of polyunsaturated fatty acids that are especially susceptible to peroxidation by ROS.33 Spinal cord trauma or ischemia results in a cascade of secondary events, such as release of excitatory amino acids and disorders of calcium homeostasis, which may cause lipid peroxidation, augmented ROS generation and the breakdown of antioxidant defense systems.34,35 We found in our experiments that glutamate treatment resulted in ROS generation, increased expression levels of MDA and 4-HNE and decreased enzymatic activities of MnSOD and CAT in spinal cord neurons, which are all consistent with observations in in vivo spinal cord injury models.36–38 Allicin has been shown to exert antioxidant properties in the cardiovascular system and brain. An in vitro experiment has shown a dose-dependent effect of allicin on the glutathione levels in endothelial cells, with a maximum increase of up to 8-fold.14 Allicin was also shown to suppress oxidation of lipid molecules and inhibit lipid peroxidation by quenching free radicals.39 In this study, we found that the decrease in oxidative damage and the increase in endogenous antioxidant enzymatic activities might contribute to the protective effect of allicin treatment on spinal cord neurons.
Nitric oxide (NO) is a gaseous biomolecule that plays a role in neurotransmission, and previous studies have shown that the levels of NO increase in rat and rabbit models with spinal cord ischemia.40,41 The role of NO in neuronal injury is complicated depending on its concentration, source, redox state, and concurrence with other molecules.42 The NO released by endothelial NOS (eNOS) plays a protective role, whereas activation of iNOS and neuronal NOS (nNOS) and the following overproduction of NO are detrimental to neuronal cells.43–45 Previous studies showed that the expression of iNOS is significantly increased after spinal cord injury, but the expression of nNOS is not apparently altered.46–48 This was also confirmed in our in vitro model. Inhibiting the activation of iNOS and nNOS through short interfere RNA (siRNA) or selective inhibitors was shown to protect against cerebral ischemia and brain trauma.44,49,50 Similarly, both the iNOS inhibitor 1400W and the nNOS inhibitor NPLA were shown to produce protective effects in spinal cord neurons in the present study. Emerging evidence has illustrated the effects of allicin on the regulation of NOS. A previous study showed that allicin attenuated immune-mediated liver damage through inhibiting the iNOS and NF-κB pathways.51 More recently, allicin was shown to increase the activation of eNOS and decrease the expression of iNOS in traumatic brain injury models.15,16 In the present study, we found that an iNOS-dependent nNOS-independent mechanism might contribute to the protective effects of allicin in spinal cord neurons.
HSP70 is a ubiquitously expressed and highly conserved molecular chaperone in cell survival signaling and protein homeostasis.52 It binds to its protein substrates and stabilizes them against denaturation or aggregation in response to stress. HSP70 acts at multiple steps in a protein's life cycle, including the processes of folding, trafficking, remodeling and degradation.53 Genetic studies (downregulation or overexpression) have routinely linked HSP70 and its cochaperones to the accumulation of misfolded or misprocessed proteins in neurodegenerative diseases.54 Bonini and colleagues demonstrated that α-syn-induced degenerative cell death is prevented if the flies are fed a diet enriched in geldanamycin, a chemical activator of HSP70.55 Moreover, exogenously delivered HSP70 was shown to protect against hippocampal neurodegeneration induced by endogenous glutamate in vivo.56 In the present study, we found that the expression of HSP70 was significantly increased at both mRNA and protein levels after glutamate treatment, and knockdown of HSP70 aggravated glutamate-induced neuronal injury in spinal cord neurons, indicating the protective role of HSP70 in spinal cord injury. Among various downstream signaling factors of HSP70, iNOS is one of the most important ones that mediates oxidative stress in neuronal cells. HSP70 was shown to inhibit iNOS gene expression by transcriptional mechanisms that involve the NF-κB/IκB pathway.57 More recently, HSP70 was shown to protect against seizure-induced neuronal cell death in the hippocampus via inhibition of iNOS expression.58 Our present data show that allicin treatment significantly increases the expression of HSP70 even following glutamate treatment, and both allicin-induced protection and inhibition of iNOS are all partially ablated by downregulation of HSP70, suggesting that allicin-induced neuroprotection against spinal cord neuronal injury is associated with the HSP70/iNOS pathway.
Although we clearly demonstrated the protective effects of allicin against spinal cord neuronal injury, these findings have some limitations. First, the injury model used in this study is established under in vitro conditions, in which the cellular microenvironment is missing and the neuronal damage following spinal cord injury cannot be completely mimicked. Although the in vitro model offers many advantages for studying the molecular mechanisms at subcellular levels,26,59 some in vivo experiments might be helpful to confirm our findings of allicin-induced protective mechanisms. Second, the neuronal viability, LDH release, apoptosis and oxidative stress markers were only detected at 24 h after injury. The neuronal injury caused by spinal cord trauma/ischemia occurs in two phases, an immediate phase and a delayed phase.60–62 The delayed neuronal injury and neurological dysfunction became evident at 24 hours to 7 days.63,64 Further studies at more time points after injury might be helpful to elucidate the effects and mechanisms of allicin on delayed paraplegia after spinal cord injury.
Conclusion
In conclusion, findings from our current study revealed that pretreatment with allicin, an active substance of garlic, protects against glutamate-induced oxidative stress and apoptotic cell death in primary cultured spinal cord neurons. These protective effects were partially dependent on activation of HSP70 and following inhibition of iNOS. Our results imply that the natural product allicin may be useful in the therapy of spinal cord injuries.References
- H. Q. Cao and E. D. Dong, Neurosci. Bull., 2013, 29, 94–102 CrossRef PubMed.
- E. S. Crawford, J. L. Crawford, H. J. Safi, J. S. Coselli, K. R. Hess, B. Brooks, H. J. Norton and D. H. Glaeser, J. Vasc. Surg., 1986, 3, 389–404 CrossRef CAS.
- K. Tabayashi, Surg. Today, 2005, 35, 1–6 CrossRef PubMed.
- M. Isaka, H. Kumagai, Y. Sugawara, K. Okada, K. Orihashi, M. Ohtaki and T. Sueda, J. Vasc. Surg., 2006, 43, 1257–1262 CrossRef PubMed.
- M. Hamaishi, K. Orihashi, M. Isaka, H. Kumagai, S. Takahashi, K. Okada, M. Ohtaki and T. Sueda, Ann. Vasc. Surg., 2009, 23, 128–135 CrossRef PubMed.
- R. Chiesa, G. Melissano, E. Civilini, M. L. de Moura, A. Carozzo and A. Zangrillo, Ann. Vasc. Surg., 2004, 18, 514–520 CrossRef PubMed.
- L. Bayan, P. H. Koulivand and A. Gorji, Avicenna J. Phytomedicine, 2014, 4, 1–14 CrossRef PubMed.
- E. J. Staba, L. Lash and J. E. Staba, J. Nutr., 2001, 131, 1118S–1119S CAS.
- L. D. Lawson and Z. J. Wang, J. Agric. Food Chem., 2005, 53, 1974–1983 CrossRef CAS PubMed.
- L. D. Lawson and C. D. Gardner, J. Agric. Food Chem., 2005, 53, 6254–6261 CrossRef CAS PubMed.
- R. Hunter, M. Caira and N. Stellenboom, Ann. N. Y. Acad. Sci., 2005, 1056, 234–241 CrossRef CAS PubMed.
- J. W. Zhu, T. Chen, J. Guan, W. B. Liu and J. Liu, Neurochem. Int., 2012, 61, 640–648 CrossRef CAS PubMed.
- J. Y. Chan, A. C. Yuen, R. Y. Chan and S. W. Chan, Phytother. Res., 2013, 27, 637–646 CrossRef CAS PubMed.
- L. Horev-Azaria, S. Eliav, N. Izigov, S. Pri-Chen, D. Mirelman, T. Miron, A. Rabinkov, M. Wilchek, J. Jacob-Hirsch, N. Amariglio and N. Savion, Eur. J. Nutr., 2009, 48, 67–74 CrossRef CAS PubMed.
- Y. F. Zhou, W. T. Li, H. C. Han, D. K. Gao, X. S. He, L. Li, J. N. Song and Z. Fei, Brain Res. Bull., 2014, 100, 14–21 CrossRef CAS PubMed.
- W. Chen, J. Qi, F. Feng, M. D. Wang, G. Bao, T. Wang, M. Xiang and W. F. Xie, Neurochem. Int., 2014, 68, 28–37 CrossRef CAS PubMed.
- N. K. Liu, Y. P. Zhang, W. L. Titsworth, X. Jiang, S. Han, P. H. Lu, C. B. Shields and X. M. Xu, Ann. Neurol., 2006, 59, 606–619 CrossRef CAS PubMed.
- T. Chen, W. Liu, X. Chao, Y. Qu, L. Zhang, P. Luo, K. Xie, J. Huo and Z. Fei, Neuroscience, 2011, 183, 203–211 CrossRef CAS PubMed.
- E. Estornell, R. Fato, F. Pallotti and G. Lenaz, FEBS Lett., 1993, 332, 127–131 CrossRef CAS.
- N. Izigov, N. Farzam and N. Savion, Free Radical Biol. Med., 2011, 50, 1131–1139 CrossRef CAS PubMed.
- T. Miron, A. Rabinkov, D. Mirelman, M. Wilchek and L. Weiner, Biochim. Biophys. Acta, Biomembr., 2000, 1463, 20–30 CrossRef CAS.
- H. Fujisawa, K. Suma, K. Origuchi, H. Kumagai, T. Seki and T. Ariga, J. Agric. Food Chem., 2008, 56, 4229–4235 CrossRef CAS PubMed.
- U. Sela, A. Brill, V. Kalchenko, O. Dashevsky and R. Hershkoviz, Nutr. Cancer, 2008, 60, 412–420 CrossRef CAS PubMed.
- Z. Jia, H. Zhu, J. Li, X. Wang, H. Misra and Y. Li, Spinal Cord, 2012, 50, 264–274 CrossRef CAS PubMed.
- T. Lam, Z. Chen, M. M. Sayed-Ahmed, A. Krassioukov and A. A. Al-Yahya, Spinal Cord, 2013, 51, 656–662 CrossRef CAS PubMed.
- T. Chen, F. Fei, X. F. Jiang, L. Zhang, Y. Qu, K. Huo and Z. Fei, Free Radical Biol. Med., 2012, 52, 208–217 CrossRef CAS PubMed.
- T. Chen, Y. F. Yang, P. Luo, W. Liu, S. H. Dai, X. R. Zheng, Z. Fei and X. F. Jiang, Cell. Signalling, 2013, 25, 2863–2870 CrossRef CAS PubMed.
- E. D. Hall and J. M. Braughler, J. Neurosurg., 1982, 57, 247–253 CrossRef CAS PubMed.
- D. K. Anderson, R. D. Saunders, P. Demediuk, L. L. Dugan, J. M. Braughler, E. D. Hall, E. D. Means and L. A. Horrocks, Cent. Nerv. Syst. trauma: J. Am. Paralysis Assoc., 1985, 2, 257–267 CAS.
- D. K. Anderson, J. M. Braughler, E. D. Hall, T. R. Waters, J. M. McCall and E. D. Means, J. Neurosurg., 1988, 69, 562–567 CrossRef CAS PubMed.
- T. Y. Yune, J. Y. Lee, C. M. Cui, H. C. Kim and T. H. Oh, J. Neurochem., 2009, 110, 1276–1287 CrossRef CAS PubMed.
- T. Chen, W. Liu, X. Chao, L. Zhang, Y. Qu, J. Huo and Z. Fei, Brain Res. Bull., 2011, 84, 163–168 CrossRef CAS PubMed.
- K. Hamann, A. Durkes, H. Ouyang, K. Uchida, A. Pond and R. Shi, J. Neurochem., 2008, 107, 712–721 CrossRef CAS PubMed.
- K. Hamann and R. Shi, J. Neurochem., 2009, 111, 1348–1356 CrossRef CAS PubMed.
- T. Y. Yune, S. M. Lee, S. J. Kim, H. K. Park, Y. J. Oh, Y. C. Kim, G. J. Markelonis and T. H. Oh, J. Neurotrauma, 2004, 21, 1778–1794 Search PubMed.
- C. Temiz, I. Solmaz, O. Tehli, S. Kaya, O. Onguru, E. Arslan and Y. Izci, Turk. Neurosurgery, 2013, 23, 67–74 Search PubMed.
- N. van Duijnhoven, E. Hesse, T. Janssen, W. Wodzig, P. Scheffer and M. Hopman, Eur. J. Appl. Physiol., 2010, 109, 1059–1066 CrossRef PubMed.
- K. M. Carrico, R. Vaishnav and E. D. Hall, J. Neurotrauma, 2009, 26, 1369–1378 CrossRef PubMed.
- Y. Okada, K. Tanaka, I. Fujita, E. Sato and H. Okajima, Redox Rep., 2005, 10, 96–102 CrossRef CAS PubMed.
- J. H. In, E. J. Lee, B. H. Lee, Y. G. Lim and M. H. Chun, Mol. Cells, 2003, 15, 406–411 CAS.
- Y. Zhou, Y. N. Zhao, E. B. Yang, E. A. Ling, Y. Wang, M. M. Hassouna and P. Mack, J. Surg. Res., 1999, 87, 185–193 CrossRef CAS PubMed.
- C. Santoscoy, C. Rios, R. E. Franco-Bourland, E. Hong, G. Bravo, G. Rojas and G. Guizar-Sahagun, Neurosci. Lett., 2002, 330, 94–98 CrossRef CAS.
- T. M. Dawson and V. L. Dawson, Annu. Rev. Med., 1996, 47, 219–227 CrossRef CAS PubMed.
- A. F. Samdani, T. M. Dawson and V. L. Dawson, Stroke, 1997, 28, 1283–1288 CrossRef CAS.
- M. E. Ross and C. Iadecola, Methods Enzymol., 1996, 269, 408–426 CAS.
- J. Die, K. Wang, L. Fan, Y. Jiang and Z. Shi, Brain Res., 2010, 1346, 251–261 CrossRef CAS PubMed.
- J. Y. Yang, H. S. Kim and J. K. Lee, Spinal Cord, 2007, 45, 731–738 CrossRef PubMed.
- C. Liu, A. Jin, C. Zhou and B. Chen, Chin. Med. J., 2002, 115, 740–742 CAS.
- M. T. Heneka and D. L. Feinstein, J. Neuroimmunol., 2001, 114, 8–18 CrossRef CAS.
- G. J. Southan and C. Szabo, Biochem. Pharmacol., 1996, 51, 383–394 CrossRef CAS.
- R. Bruck, H. Aeed, E. Brazovsky, T. Noor and R. Hershkoviz, Liver Int., 2005, 25, 613–621 CrossRef CAS PubMed.
- C. G. Evans, L. Chang and J. E. Gestwicki, J. Med. Chem., 2010, 53, 4585–4602 CrossRef CAS PubMed.
- S. Patury, Y. Miyata and J. E. Gestwicki, Curr. Top. Med. Chem., 2009, 9, 1337–1351 CrossRef CAS.
- S. N. Witt, Biopolymers, 2010, 93, 218–228 CrossRef CAS PubMed.
- P. K. Auluck and N. M. Bonini, Nat. Med., 2002, 8, 1185–1186 CrossRef CAS PubMed.
- G. X. Ayala and R. Tapia, Neuropharmacology, 2008, 55, 1383–1390 CrossRef CAS PubMed.
- J. Y. Chan, C. C. Ou, L. L. Wang and S. H. Chan, Circulation, 2004, 110, 3560–3566 CrossRef CAS PubMed.
- C. C. Chang, S. D. Chen, T. K. Lin, W. N. Chang, C. W. Liou, A. Y. Chang, S. H. Chan and Y. C. Chuang, Neurobiol. Dis., 2014, 62, 241–249 CrossRef CAS PubMed.
- Y. Garcia and J. Diaz-Castro, Animal, 2013, 7, 1651–1658 CrossRef CAS PubMed.
- T. Kiyoshima, S. Fukuda, M. Matsumoto, Y. Iida, S. Oka, K. Nakakimura and T. Sakabe, Anesthesia and Analgesia, 2003, 96, 839–846 Search PubMed , table of contents.
- M. Kakinohana, K. Kida, S. Minamishima, D. N. Atochin, P. L. Huang, M. Kaneki and F. Ichinose, Stroke, 2011, 42, 2302–2307 CrossRef CAS PubMed.
- M. C. Kao, S. K. Tsai, M. Y. Tsou, H. K. Lee, W. Y. Guo and J. S. Hu, Anesth. Analg. (N.Y.), 2004, 99, 580–583 CrossRef PubMed , table of contents.
- P. K. Liu, Curr. Top. Med. Chem., 2001, 1, 483–495 CrossRef CAS.
- L. Zhou, X. Wang, W. Xue, K. Xie, Y. Huang, H. Chen, G. Gong and Y. Zeng, Brain Res., 2013, 1517, 150–160 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |