 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransport of hop aroma compounds across Caco-2 monolayers†
A.
Heinlein
a,
M.
Metzger
b,
H.
Walles
b and
A.
Buettner
*ac
aDepartment of Chemistry and Pharmacy, Food Chemistry, University Erlangen-Nuremberg, 91052 Erlangen, Germany. E-mail: andrea.buettner@fau.de
bTissue Engineering and Regenerative Medicine, University Wuerzburg, 97070 Wuerzburg, Germany
cFraunhofer Institute for Process Engineering and Packaging (IVV), 85354 Freising, Germany
First published on 27th May 2014
Abstract
Although being reported and used as a sedative remedy for several years, the bioactive principle of hop preparations is still not decisively clarified. Understanding absorption and transformation processes of potential physiologically active constituents is essential to evaluate the likeliness of biological effects on humans. Therefore, single hop aroma compounds as well as digestive transformation products thereof have been investigated in view of their human intestinal absorption, applying Caco-2 transport experiments as well as investigations on potential biotransformation processes. Selective and sensitive identification and quantification were thereby achieved by application of two-dimensional high resolution gas chromatography-mass spectrometry in conjunction with stable isotope dilution analysis, leading to the determination of apparent permeability values by different mathematical approaches considering sink and non-sink conditions. Overall, calculated permeability values ranged from 2.6 × 10−6 to 1.8 × 10−4 cm s−1 with all mathematical approaches, indicating high absorption potential and almost complete bioavailability for all tested compounds with hydroxyl-functionalities. Considering this high permeability together with the high lipophilicity of these substances, a passive transcellular uptake route can be speculated. Investigated sesquiterpenes and β-myrcene showed flat absorption profiles while the investigated esters showed decreasing profiles. In view of the lipophilic and volatile nature of the investigated substances, special attention was paid to recovery and mass balance determination. Furthermore, in the course of the transport experiments of 1-octen-3-ol and 3-methyl-2-buten-1-ol, additional biotransformation products were observed, namely 3-octanone and 3-methyl-2-butenal, respectively. The absence of these additional substances in control experiments strongly indicates an intestinal first-pass metabolism of the α,β-unsaturated alcohols 1-octen-3-ol and 3-methyl-2-buten-1-ol in Caco-2 cells.
Introduction
Besides its use in the brewing industry, the hop plant (Humulus lupulus L.) has been applied since medieval times in folk medicine to beneficially influence a variety of symptoms.1 Nowadays, it is recommended for the treatment of restlessness, anxiety and sleep disturbances.2 Several in vitro and in vivo studies have been conducted in order to verify these physiological effects.3 However, human trials have so far mainly been focusing on preparations using a fixed combination of valerian and hops, not on single hop dosages.4,5 In the course of these studies, it could be shown that valerian in combination with hops is more effective than valerian alone.5 Further, a recent study suggests that the sedative effect of non-alcoholic beer in healthy female nurses is due to hop components.6 Still there is a lack of conclusive evidence to which single hop compounds a physiological activity can be attributed. With regard to potential pathways underlying these observations, the potency of aroma compounds, amongst them several substances present in hops, to elicit effects on the γ-aminobutyric acid receptor (GABAA), has been reported in several studies.7–11 The GABAA receptor is the most important inhibitory neurotransmitter in the central nervous system and mediates sedation and anxiolysis, amongst others. Thus, it is suspected that the volatile fraction of hops contributes to sedative and anxiolytic effects of hop preparations. It has been shown that, although α-bitter acids are the most active constituents of hop extracts regarding sedative effects in mice, hop essential oil clearly contributes to its activity.12In view of these observations and the necessary preceding uptake mechanisms of the respective substances by the human organism, important steps related to bioavailability are the passage through the gastro-intestinal tract after ingestion, and the subsequent absorption mainly in the small intestine, which poses the primary location of substance uptake. The small intestinal mucosa is being covered by a single layer of high-prismatic epithelial cells.13 These cells build a physical and biochemical barrier to regulate the absorption of nutrients and drugs into the circulatory system.14 As a model for this intestinal barrier, Caco-2 cells are commonly used. This cell line is derived from a human colon carcinoma and has first been characterized by Hidalgo et al. for its use in intestinal epithelial permeability studies.15 In culture, the cells spontaneously differentiate into polarized enterocytes with microvilli and tight junctions – characteristics similar to human small intestinal cells – and form continuous monolayers. Concerning hops, Caco-2 transport experiments have already been applied to study the intestinal permeability of bitter acids, present in the non-volatile fraction of hops. Thereby, Cattoor et al. showed that the membrane permeability of hop α-acids is higher than that of β-acids due to efflux processes and conjugation by phase II metabolism.16 In another study, they furthermore showed that iso-α-acids and tetrahydro-iso-α-acids were transported more efficiently than dihydro-iso-α-acids.17
With regard to aroma compounds, Stadler et al. investigated the absorption and metabolic conversion of 3(2H)-furanones by Caco-2 cell monolayers.18 The authors showed 2,5-dimethyl-4-methoxy-3(2H)-furanone (DMMF) to be absorbed with the highest rate and proposed passive diffusion by paracellular transport to be the primary route for uptake.
In the present study, we focused on substances present in the volatile fraction of hops. Previous studies have shown that several hop aroma compounds with possible sedative properties were not stable during in vitro digestion processes but underwent chemical transformations.19 To further investigate the obstacles the hop aroma compounds as well as their transformation products encounter in the human body, we used Caco-2 transport experiments in the apical to basolateral direction to mimic intestinal uptake from the gut into the blood circulation. Samples were analyzed via two-dimensional high resolution gas chromatography-mass spectrometry (2D-HRGC-MS). Quantification of the odorant transitions was primarily based on stable isotope dilution analysis, and chromatograms were additionally screened for potential metabolites. Apparent permeability values, mass balances and recoveries were calculated and interpreted critically. The obtained in vitro data are discussed in relation to their predictability of human intestinal bioavailability.
Experimental
The Caco-2 cell line as well as collagen I from rat tail was kindly provided by the Chair for Tissue Engineering and Regenerative Medicine, Julius Maximilians University of Wuerzburg, Germany.Minimum essential medium (MEM) with Earle's salts and stable glutamine, fetal calf serum (FCS), non-essential amino acid solution (NEAA), L-glutamine, sodium pyruvate, penicillin/streptomycin, trypsin, EDTA and Mg2+/Ca2+ free phosphate buffered saline (PBS−) were purchased from Biochrom AG, Berlin, Germany.
Phenol-red free powdered minimum-essential medium, rhodamine 123, fluorescein and propranolol-hydrochloride were obtained from Sigma-Aldrich, Steinheim, Germany.
Cell culture flasks and 12-well plates were obtained from Techno Plastic Products AG, Trasadingen, Switzerland. Hanging cell culture inserts as well as the Millicell ERS-2 Voltohmmeter were obtained from Millipore GmbH, Schwalbach, Germany.
Linalool, geraniol, nerol, R-(+)-limonene, β-caryophyllene, α-humulene, 2-methyl-3-buten-2-ol, 3-methyl-2-buten-1-ol, 1-octen-3-one, 3-octanone and 3-methyl-2-butanal were obtained from Sigma-Aldrich, Steinheim, Germany. β-Myrcene, β-farnesene and 1-octen-3-ol were obtained from Fluka, Steinheim, Germany. α-Terpineol was obtained from SAFC, Hamburg, Germany. 2-Methylbutyl-isobutyrate, 3-methylbutyl-isobutyrate as well as the stable-isotopically labelled compounds 2H3-1-octen-3-ol, 2H3-1-octen-3-one, 2H3-α-terpineol, 2H2-limonene, 2H5-linalool, 2H2-geraniol, 2H6-β-myrcene, 2H6-2-methyl-3-buten-2-ol, 2H6-β-farnesene, 2H7-2-methylbutyl-isobutyrate and 2H7-3-methylbutyl-isobutyrate were obtained from Aromalab, Freising, Germany.
Cell maintenance
Cultures were seeded at a density of 1 × 104 cells per cm2 in 75 cm2 cell culture flasks and maintained in growth medium at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Growth medium consisted of minimum essential medium supplemented with 20% FCS, 1% non-essential amino acids and 1 mM sodium pyruvate. The medium was changed every 2–3 days and cells were subcultured at a maximum confluence of 80% using trypsin–EDTA-solutions in PBS−.Preparation of cell monolayers for transport studies
For transport studies, Caco-2 cells were seeded at a density of 8 × 104 cells per cm2 on hanging cell culture inserts (0.1 μm pore size, 1.13 cm2 seeding area). The inserts were coated with 100 μg mL−1 rat-tail collagen for 15 minutes prior to cell seeding.The cell-seeded inserts were maintained in 1.5 mL growth medium on the basolateral (lower) and 0.5 mL on the apical (upper) side of the insert on 12-well plates. Since the plates are more susceptible to infections than cell culture flasks, growth medium was additionally supplemented with 1% penicillin–streptomycin. Media were changed every 2–3 days. Transport experiments were performed 15 days post-seeding. Only cells of passages 39–49 were used in this study since a defined interval of passages is recommended to ensure a consistent cellular phenotype in all experiments.13,20
Transport studies
Stock solutions of test compounds in dimethylsulfoxide (DMSO) were prepared and stored in glass vials at −25 °C. On the day of the experiment, stock solutions were thawed and diluted in sterile plastic tubes with transport medium (i.e. serum- and phenolred-free minimal essential medium supplemented with 1 mM sodium pyruvate) to a final concentration of 100 μM of tested compounds, containing not more than 0.3% DMSO in final test solutions. The concentration of 100 μM of tested compounds was applied in compliance with physiological relevant concentrations of single aroma compounds in pharmaceutical hop preparations, based on following considerations: as elucidated elsewhere, an average amount of a single aroma compound ingested would be 0.5 mg.19 The digestion procedure would further dilute this amount with digestion fluids to about 13 mg L−1 in the small intestine. Given a medium molecular weight of 154 g mol−1 (i.e. linalool), the resulting molar concentration would be 84 μM in the small intestine. For better comparability a concentration of 100 μM was applied for all tested compounds. The overall range of molecular weight for the tested compounds was from 86.13 g mol−1 (2-methyl-3-buten-2-ol, 3-methyl-2-buten-1-ol) to 222.37 g mol−1 (β-caryolanol, humulol) resulting in a concentration range of 8.613 to 22.237 μg mL−1 of tested substances in the test solutions of 100 μM.Each compound was tested individually.
Prior to each experiment, cell growth medium was replaced by transport medium, consisting of serum- and phenol red-free minimum essential medium supplemented with 2 mM L-glutamine and 1 mM sodium pyruvate, and inserts were kept at 37 °C for 30 minutes. After cooling to room temperature, transepithelial electrical resistance (TEER) values were determined (see below).
To start the experiment, medium was replaced with 1500 μL fresh transport medium (initial experimental conditions) or FCS (altered experimental conditions, for further explanation see the Results section) in the acceptor (basolateral) compartment. 600 μL of the respective test solution were applied to the donor (apical) compartment. Thereof, 100 μL were immediately drawn as a donor control sample (DStart), resulting in a donor volume of 500 μL.
Samples of 100 μL were drawn every 20 minutes from the acceptor compartment for up to 120 min. The removed sample volumes were substituted with fresh transport medium or FCS, respectively.
In between samplings, cell plates were placed on an orbital shaker at 150 rpm in the CO2 incubator at 37 °C.
Additionally, samples of 100 μL were drawn from the donor compartment at the end of the experiment (DEnd) to calculate the mass balance.
TEER-values were checked again upon completion of the experiment.
All experiments were done in triplicate on 3 individual cell monolayers. As control, each test solution was additionally applied to an insert without Caco-2 cells and incubated in the same way as described above. Samples were drawn at the beginning of the experiment from the donor compartment as well as at the end of the experiment from donor and acceptor medium.
Calculations
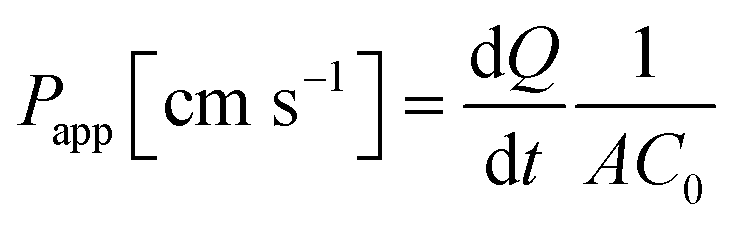
| (1) |
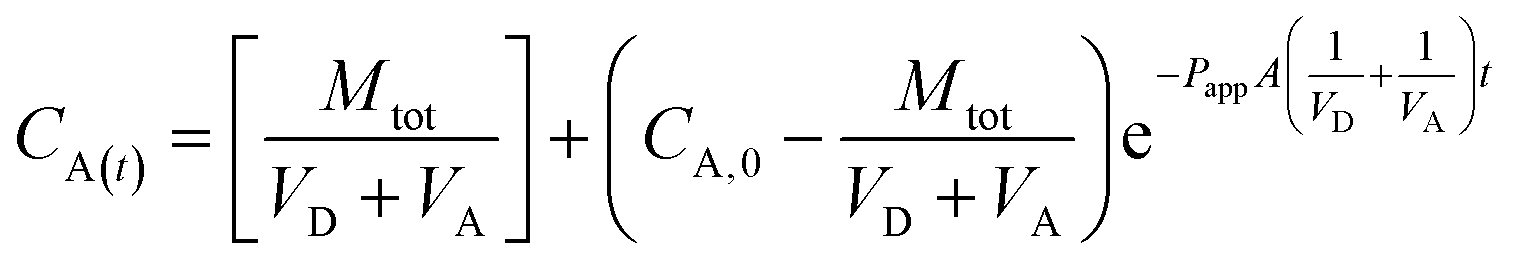 | (2) |
 | (3) |
 | (4) |
Quality control of Caco-2-cells and monolayer integrity
To ensure good performance of the Caco-2-cell assay in our laboratory, Papp values of the well characterized reference compounds fluorescein, rhodamin 123 and propranolol–HCl were determined prior to experiments in the apical to basolateral direction (ab). The Papp-value of rhodamine 123, a P-gp-substrate, was additionally determined in basolateral to apical direction (ba). Cells were considered to be of good quality when they met following requirements: Papp (ab) for fluorescein <1 × 10−6 cm s−1 and for propranolol–HCl >5 × 10−6 cm s−1; Papp (ba) for rhodamine >4 × 10−6 cm s−1; ratio of ba/ab for rhodamine >2.Fluorescein and rhodamine 123 concentrations were determined using a micro-plate reader (Viktor3V, Perkin-Elmer, Rodgau Germany) at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Propranolol–hydrochloride was analyzed via HPLC-UV by Fraunhofer IGB, Stuttgart, Germany as described before.25
Fluorescein Papp-values were additionally determined in parallel to each experiment to ensure continuous good monolayer integrity in all passages.
Furthermore the transepithelial electrical resistance (TEER) of each monolayer was monitored. Only inserts with a TEER-value >200 Ω cm2 were used.
The TEER-value was calculated as:
| TEER [Ω cm2] = (Rsample − Rblank)A | (5) |
Extraction of volatiles
To extract the volatiles into an organic phase for analysis, 200 μL of a dichloromethane-solution containing the respective internal standard was added to the individual samples. Samples were mixed for 10 min at 22 °C at 1400 rpm on a Thermomixer comfort (Eppendorf AG, Hamburg, Germany) to extract the volatiles from the inorganic phase. Subsequently, the organic phase was partitioned off, dried over anhydrous sodium sulfate and transferred directly into vials for GC analysis.Two-dimensional high resolution gas chromatography-mass spectrometry (2D-HRGC-MS)
Samples were analyzed using a 2-dimensional gas chromatography system consisting of two Varian 450 GCs in combination with a Varian Saturn 220 mass spectrometer (Varian Inc., Darmstadt, Germany). The first GC was equipped with a CIS 4C cold on-column injector (Gerstel GmbH & Co.KG, Mühlheim, Germany), an MCS 2 multicolumn switching system (Gerstel) and connected to the second GC via a CTS I CryoTrap (Gerstel).Capillaries used were ZB-FFAP (30 m length, 0.32 mm inner diameter, 0.25 μm film thickness Zebron, Phenomenex, Aschaffenburg, Germany) and Rxi-5HT (30 m length, 0.25 mm inner diameter, 0.25 μm film thickness, Restek GmbH, Bad Homburg, Germany) in the first and the second oven, respectively.
The temperature program of the first oven was: 40 °C held for 2 min, a ramp of 15 °C min−1 to 240 °C and held for 5 min. The program for the second oven was 40 °C with no hold time, 15 °C min−1 to 250 °C and held for 5 min.
As a detector for the first oven, a flame ionization detector (FID, 240 °C) was used, and the mass spectrometer for the second oven. Mass spectra were generated in electron impact (EI) or chemical ionization (CI) mode with methanol as reactant gas.
Quantification of the respective substances in the samples was primarily accomplished by stable isotope dilution analysis (SIDA). The respective stable-isotopically labelled standards used are listed above. For quantification of α-humulene and β-caryolanol, β-caryophyllene was used as an internal standard, for quantification of β-caryophyllene and humulol, α-humulene was used as an internal standard and for quantification of nerol and terpin, geraniol and α-terpineol were used, respectively.
All samples were screened for the presence of metabolites by GC-MS on an Agilent system described previously.19 Identification of metabolites was based on reference compounds by comparison of mass spectra and linear retention indices26 on two different capillary columns, DB-5 and FFAP. Concentrations of metabolites were determined semi-quantitatively via respective peak areas of standard and metabolite.
Results
An overview of chemical structures of all tested substances is displayed in Fig. 6.In the first phase of the investigations, transport experiments were carried out for all substances at a concentration of 100 μM with transport buffer as acceptor medium for an experimental time of 120 min. The amounts of transported substance over time of α-terpineol, geraniol, nerol, linalool, terpin, 2-methyl-3-buten-2-ol, 3-methyl-2-buten-1-ol, 1-octen-3-ol, β-caryolanol and humulol are displayed in Fig. 1. The most rapid increase of the detectable substance on the acceptor side of the system was observed for 2-methyl-3-buten-2-ol whereas its isomer 3-methyl-2-buten-1-ol showed a slower increase and an early onset of a steady-state after about 60 min. 1-Octen-3-ol, another aliphatic compound with hydroxyl-function, showed an even smaller increase and additionally a decrease after about 100 min incubation time.
 | ||
| Fig. 1 Cumulative quantity transported in Caco-2-experiments over time for (A) 2-methyl-3-buten-2-ol, 3-methyl-2-buten-1-ol, and 1-octen-3-ol, (B) α-terpineol, geraniol, nerol, linalool, and terpin and (C) β-caryolanol and humulol. All substances were used in a concentration of 100 μM, the experimental time was 120 min, and the acceptor medium was transport buffer; data are the mean ± SD of 3 individual cell monolayers (n = 3). | ||
Regarding the terpenoid structures, α-terpineol, geraniol and nerol exhibited very similar absorption profiles with a high initial increase and the onset of a steady-state after 80–100 min. In contrast to this, linalool and terpin exhibited a slower increase, an earlier onset of a steady-state after 60 min and an overall lower concentration in the acceptor fluid. The sesquiterpenoid structures humulol and β-caryolanol both displayed very similar profiles with a moderate, steady increase that did not yet reach a steady-state after 120 min incubation.
Based on these findings, permeability values were first calculated using the classical approach described in formula (1) (Table 1). In this formula, the permeability coefficient is dependent on the slope of the function of cumulative quantity absorbed vs. time. As can be seen in Fig. 1, the obtained curves often flatten with increasing incubation time. Therefore only the initial slope with R2 values >0.9 was used to calculate Papp-values. The time intervals used for each individual test compound are indicated in Table 1. For a clearer overview of the data, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Papp values are also given in Table 1 and plotted against their logD7.4 values (see ESI†).
Papp values are also given in Table 1 and plotted against their logD7.4 values (see ESI†).
Papp) calculated via classical approach and via non-sink conditions for fixed and recalculated donor concentrations; in the classical approach, the time interval from which the slope was calculated is indicated; for non-sink conditions all available data points were considereda
| Test substance | logD7.4b |
Classical approach | Non-sink conditions | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| From recalculated donor concentration | From fixed donor concentration | Time (min) | From recalculated donor concentration | From fixed donor concentration | ||||||
| P app ± SD [cm s−1] | logPapp |
P app ± SD [cm s−1] | logPapp |
P app ± SD [cm s−1] | logPapp |
P app ± SD [cm s−1] | logPapp |
|||
|
a n.d. = not determined.
b logD7.4 values were retrieved from SciFinder®;29 and calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994–2013 ACD/Labs).
|
||||||||||
| 1-Octen-3-ol | 0.89 | 1.75 × 10−5 ± 8.67 × 10−7 | −4.76 | 1.07 × 10−5 ± 5.27 × 10−7 | −4.97 | 60 | 1.05 × 10−5 ± 3.96 × 10−6 | −4.98 | 8.48 × 10−6 ± 4.07 × 10−6 | −5.07 |
| 1-Octen-3-one | 2.11 | 2.27 × 10−5 ± 4.32 × 10−6 | −4.64 | 9.50 × 10−7 ± 1.81 × 10−7 | (−6.02) | 40 | 2.60 × 10−6 ± 4.10 × 10−7 | −5.59 | n.d. | |
| 2-Methylbutyl-isobutyrate | 3.02 | n.d. | n.d. | n.d. | n.d. | |||||
| 2-Methyl-3-buten-2-ol | 1.08 | 4.63 × 10−5 ± 5.35 × 10−6 | −4.33 | 4.32 × 10−5 ± 4.99 × 10−6 | −4.36 | 100 | 1.00 × 10−4 ± 3.17 × 10−5 | −4.00 | 1.80 × 10−4 ± 2.60 × 10−5 | −3.74 |
| 3-Methylbutyl-isobutyrate | 3.02 | n.d. | n.d. | n.d. | n.d. | |||||
| 3-Methyl-2-buten-1-ol | 1.05 | 3.73 × 10−5 ± 3.19 × 10−6 | −4.43 | 1.94 × 10−5 ± 1.66 × 10−6 | −4.71 | 80 | 6.19 × 10−5 ± 1.02 × 10−5 | −4.21 | 4.06 × 10−5 ± 1.77 × 10−6 | −4.39 |
| α-Terpineol | 3.33 | 3.46 × 10−5 ± 4.46 × 10−6 | −4.46 | 2.23 × 10−5 ± 2.88 × 10−6 | −4.65 | 100 | 6.05 × 10−5 ± 8.56 × 10−6 | −4.22 | 5.75 × 10−5 ± 8.93 × 10−6 | −4.24 |
| α-Humulene | 6.59 | n.d. | n.d. | n.d. | n.d. | |||||
| β-Caryolanol | 4.48 | 3.12 × 10−5 ± 1.01 × 10−5 | −4.51 | 1.43 × 10−5 ± 4.61 × 10−6 | −4.84 | 120 | 5.15 × 10−5 ± 8.33 × 10−6 | −4.29 | 3.08 × 10−5 ± 3.83 × 10−6 | −4.51 |
| β-Caryophyllene | 6.41 | n.d. | n.d. | n.d. | n.d. | |||||
| β-Farnesene | 6.14 | n.d. | n.d. | n.d. | n.d. | |||||
| β-Myrcene | 4.25 | n.d. | n.d. | n.d. | n.d. | |||||
| Geraniol | 3.47 | 5.24 × 10−5 ± 1.28 × 10−6 | −4.28 | 2.45 × 10−5 ± 5.98 × 10−7 | −4.61 | 100 | 9.48 × 10−5 ± 1.22 × 10−5 | −4.02 | 5.14 × 10−5 ± 3.13 × 10−6 | −4.29 |
| Humulol | 4.98 | 1.67 × 10−5 ± 6.52 × 10−6 | −4.78 | 1.45 × 10−5 ± 4.03 × 10−6 | −4.84 | 120 | 7.77 × 10−5 ± 8.24 × 10−5 | −4.11 | 1.06 × 10−5 ± 3.09 × 10−7 | −4.97 |
| Limonene | 4.55 | n.d. | n.d. | n.d. | n.d. | |||||
| Linalool | 3.38 | 5.17 × 10−5 ± 1.16 × 10−5 | −4.29 | 1.33 × 10−5 ± 2.98 × 10−6 | −4.88 | 80 | 6.95 × 10−5 ± 3.90 × 10−5 | −4.16 | 1.80 × 10−5 ± 6.99 × 10−6 | −4.74 |
| Nerol | 3.47 | 2.85 × 10−5 ± 1.61 × 10−6 | −4.55 | 1.79 × 10−5 ± 1.01 × 10−6 | −4.75 | 100 | 5.15 × 10−5 ± 1.06 × 10−5 | −4.29 | 4.56 × 10−5 ± 3.01 × 10−6 | −4.34 |
| Terpin | 1.06 | 1.85 × 10−5 ± 2.41 × 10−6 | −4.73 | 1.85 × 10−5 ± 2.41 × 10−6 | −4.73 | 100 | 7.77 × 10−5 ± 8.24 × 10−5 | −4.11 | 1.03 × 10−5 ± 4.00 × 10−7 | −4.99 |
Secondly, permeability values were calculated as proposed by Tavelin et al. (formula (2)).23 Since for all substances the criteria for non-sink conditions were fulfilled (e.g. the concentration in the acceptor compartment exceeded 10% of the concentration in the donor compartment), this approach likely leads to a more accurate determination of permeability values.
Another goal of our study was to assess how the use of the originally intended donor concentration (100 μM – fixed) in comparison to the use of the actually determined recovery values (see Table 2) from the donor start control samples (DStart – recalculated) influences the Papp values (Table 1).
| Test substance | Recovery (%) | Mass balance (%) |
|---|---|---|
| 1-Octen-3-ol | 60.8 ± 2.9 | 24.9 ± 3.6 |
| 1-Octen-3-one | 3.5 ± 0.7 | 40.3 ± 9.1 |
| 1-Octen-3-one (1 mM) | 4.4 ± 0.4 | 51.7 ± 0.4 |
| 2-Methylbutyl-isobutyrate (serum) | 17.6 ± 3.6 | 19.4 ± 0.3 |
| 2-Methyl-3-buten-2-ol | 97.2 ± 20.2 | 91.5 ± 19.5 |
| 3-Methylbutyl-isobutyrate (serum) | 24.7 ± 2.5 | 21.3 ± 0.3 |
| 3-Methyl-2-buten-1-ol | 50.8 ± 5.0 | 70.6 ± 6.1 |
| α-Terpineol | 64.5 ± 1.7 | 83.8 ± 3.7 |
| α-Humulene (serum) | 45.4 ± 4.4 | 69.9 ± 12.2 |
| β-Caryolanol | 45.7 ± 2.1 | 76.5 ± 10.6 |
| β-Caryophyllene (serum) | 18.1 ± 0.4 | 59.3 ± 12.4 |
| β-Farnesene (serum) | 30.9 ± 13.1 | 67.4 ± 33.8 |
| β-Myrcene (serum) | 12.3 ± 5.1 | 47.6 ± 19.8 |
| Geraniol | 45.8 ± 4.1 | 95.1 ± 3.1 |
| Humulol | 79.1 ± 4.0 | 67.4 ± 14.5 |
| Limonene (1 mM) | 8.2 ± 2.5 | n.d. |
| Linalool | 25.6 ± 2.2 | 74.1 ± 12.5 |
| Nerol | 58.8 ± 9.9 | 70.6 ± 4.9 |
| Terpin | 32.9 ± 9.2 | 102.1 ± 28.0 |
Under the initial experimental conditions with transport buffer as acceptor fluid and 120 min experimental time, no substance was detectable in the acceptor compartment in the case of the esters 2-methylbutyl-isobutyrate and 3-methylbutyl-isobutyrate as well as the monoterpenes β-myrcene and limonene and the sesquiterpenes α-humulene, β-caryophyllene and β-farnesene. To enhance lipophilicity of the acceptor compartment and, concurrently with this, simulating a more physiologically accurate environment, the acceptor medium was changed from transport buffer to fetal calf serum (FCS). Serum as acceptor fluid has been used in other transport studies before.27,28 Since it was suspected that the high volatility of the aroma substances might lead to evaporation over time accompanied by a loss of substance, the experimental time was reduced from 120 to 80 min. Under these altered conditions the respective compounds, with the sole exception of limonene, were detectable in the acceptor fluids. Results of the experiments under these conditions are shown in Fig. 2. The cumulative quantity found in the acceptor compartment after 20 minutes decreased with incubation time for both esters 2-methylbutyl-isobutyrate and 3-methylbutyl-isobutyrate (Fig. 2A) in a similar manner (potential reasons for these decreases will be addressed in the discussion). Therefore, permeability values could not be determined. In the case of α-humulene, β-myrcene, β-caryophyllene and β-farnesene the quantity found in the acceptor compartment appeared to be almost constant over time (Fig. 2B), indicating a very early steady-state. Accordingly the calculation of permeability values was also not possible.
 | ||
| Fig. 2 Cumulative quantity transported in Caco-2-experiments over time for (A) 2-methylbutyl-isobutyrate and 3-methylbutyl-isobutyrate and (B) α-humulene, β-myrcene, β-caryophyllene, and β-farnesene. All substances were used in a concentration of 100 μM, the experimental time was 80 min, and the acceptor medium was fetal calf serum; data are the mean ± SD of 3 individual monolayers (n = 3). | ||
In the transport experiments of 1-octen-3-one under the initial conditions, an extremely low recovery of about only 3.6% in the donor control sample at the start of the experiment (DStart) was observed. Therefore we raised the concentration from 100 μM to 1 mM and used transport buffer as acceptor fluid and an experimental time of 80 min. Recovery was still extremely low (4.2%) yet the target substance was detectable in the acceptor compartment. The results of this experiment are shown in Fig. 3. Overall, a high increase of substance was detectable in the acceptor compartment during the first 40 min of the experiments, afterwards the concentration decreased rapidly.
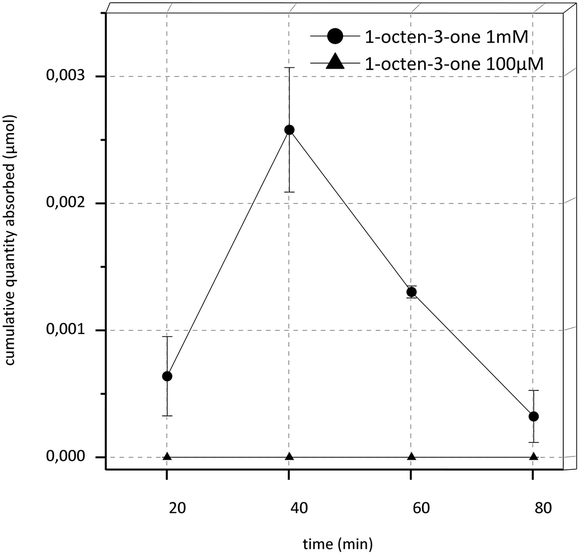 | ||
| Fig. 3 Cumulative quantity transported in Caco-2-experiments over time for 1-octen-3-one in a concentration of 1 mM and 100 μM. The experimental time was 80 min and the acceptor medium was transport buffer; data are the mean ± SD (n = 3). | ||
For limonene, alterations of experimental conditions to a higher concentration of 1 mM as well as the use of serum as acceptor fluid and the shortening of the experimental time to 80 min did not improve results. Furthermore, the substance was neither detectable in donor end nor donor start controls. However, when working with a freshly prepared stock solution and immediate start of the experiment as well as immediate extraction of samples with dichloromethane and analysis via GC, at least a recovery of 8.2% could be achieved in donor start controls. Yet neither in donor nor in acceptor fluid after 10 minutes incubation was any limonene detectable.
It is especially interesting to note that after 40 min incubation time in all samples as well as in acceptor and donor end controls (DEnd) of the transport experiments of 1-octen-3-ol, the component 3-octanone was detectable and showed an increase in concentration over time (Fig. 4A and 5). Similar effects were seen in the transport experiments of 3-methyl-2-buten-1-ol, where 3-methyl-2-butenal appeared over time (Fig. 4B and 5). In donor start controls (DStart) as well as in control experiments, where the substances were incubated under the same conditions yet without Caco-2 cells, no 3-octanone or 3-methyl-2-butenal, respectively, was detectable.
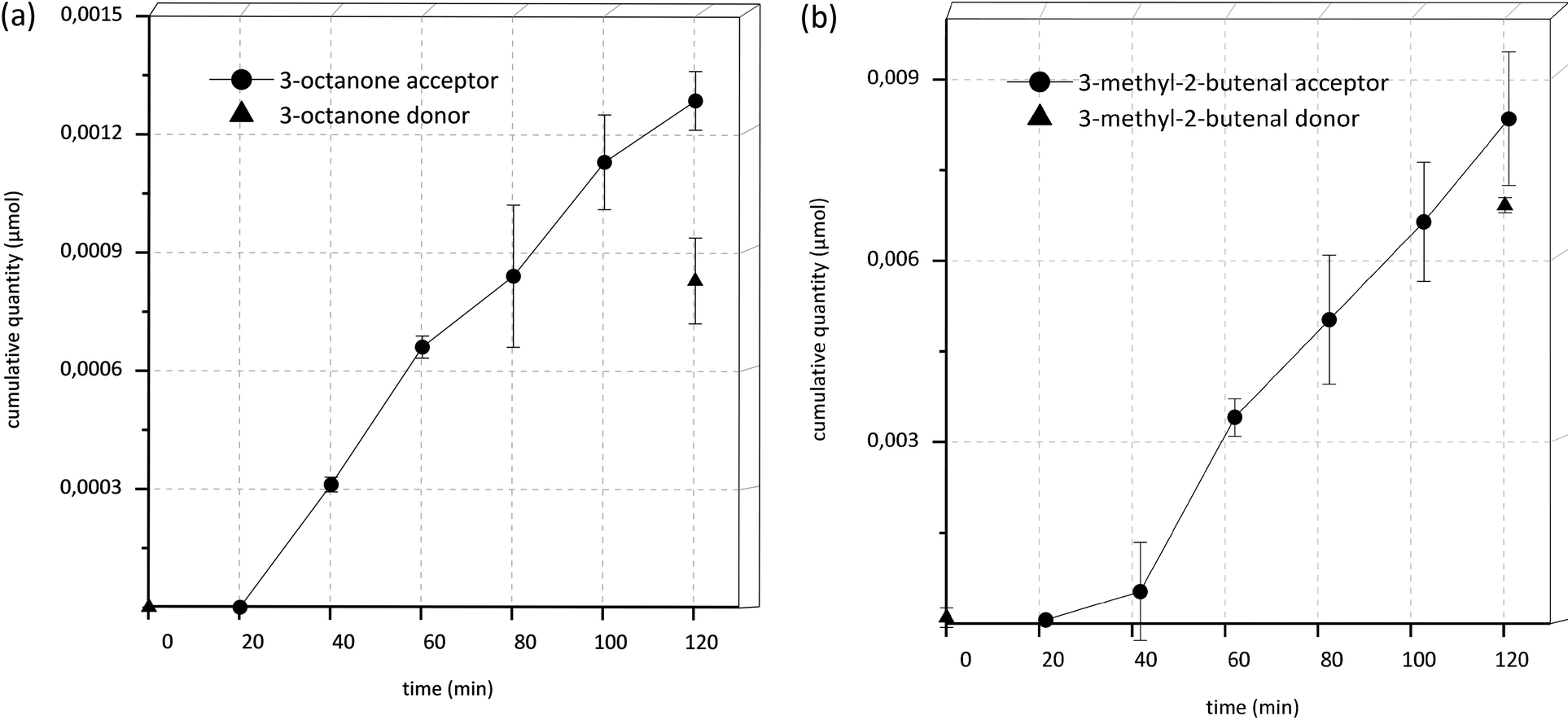 | ||
| Fig. 4 Appearance of metabolites in donor and acceptor compartments. (A) Appearance of 3-octanone in the transport experiments of 1-octen-3-ol and (B) appearance of 3-methyl-2-butenal in the transport experiments of 3-methyl-2-buten-1-ol; data are the mean ± SD (n = 3). | ||
 | ||
| Fig. 5 Intestinal first-pass metabolism observed in Caco-2 cells. | ||
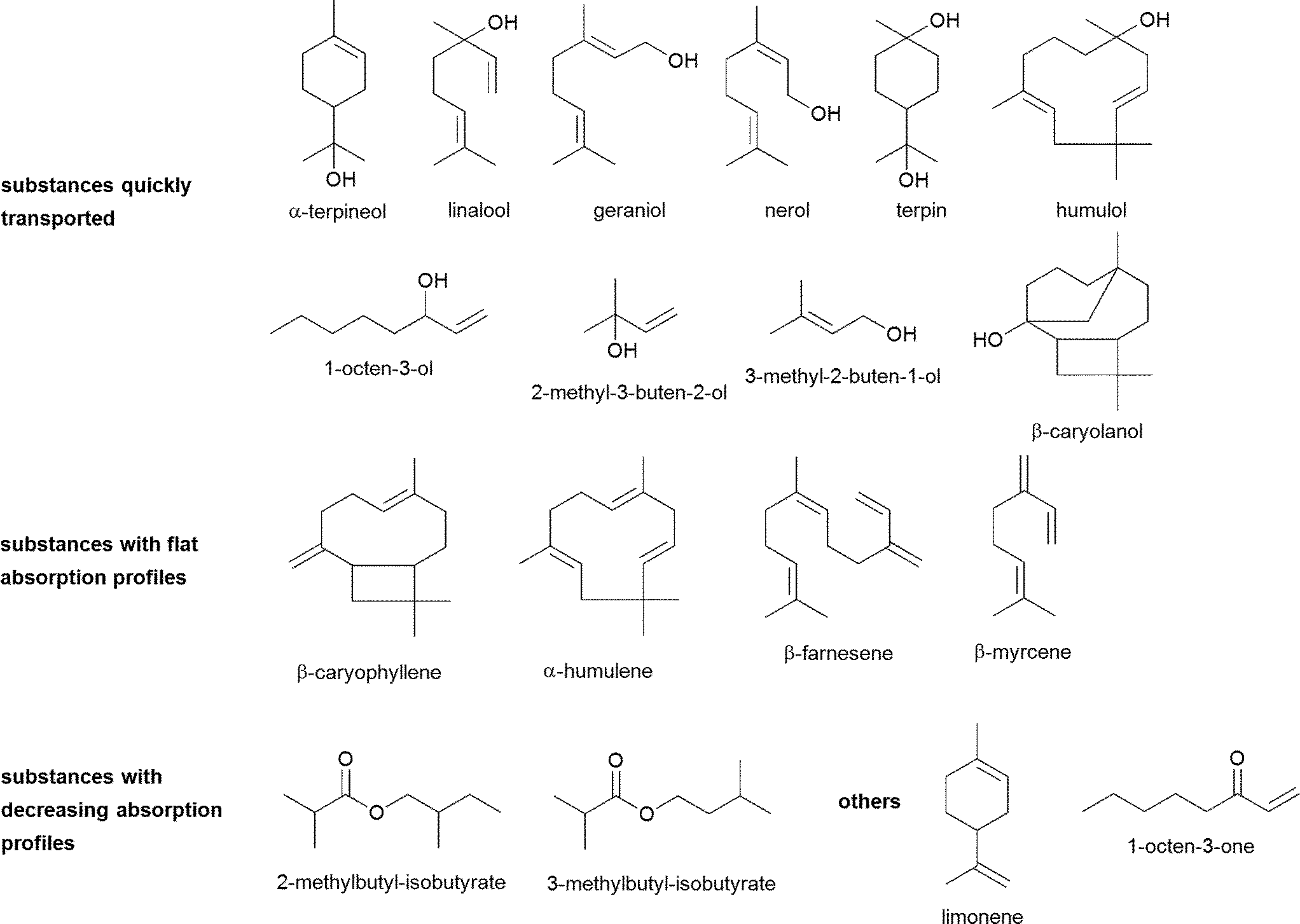 | ||
| Fig. 6 Chemical structures of investigated hop aroma compounds and their digestive transformation products. | ||
Calculation of recovery and mass balance showed huge substance-specific variability (Table 2). The worst recovery values were obtained for limonene and 1-octen-3-one, with values as low as 4% of total recovery. Best results were obtained for 2-methyl-3-buten-2-ol with a recovery of 97%. The mass balance was smallest for the esters 2-methylbutyl-isobutyrate and 3-methylbutyl-isobutyrate with 19% and 21%, respectively. The best mass balance was found for terpin and geraniol with 102% and 95%, respectively.
Discussion
Permeability and bioavailability
Substances with Papp values >1.0 × 10−6 cm s−1 (i.e. logPapp > −6.0) possess high absorption potential according to Ismael et al.,14 and Papp values >2.0 × 10−6 cm s−1 (i.e. logPapp > −5.7) indicate a bioavailability of more than 90%, according to Biganzoli et al.30 All tested substances, where the calculation of Papp-values was possible, namely 1-octen-3-ol, 2-methyl-3-buten-2-ol, 3-methyl-2-buten-1-ol, α-terpineol, β-caryolanol, geraniol, humulol, linalool, nerol and terpin, possess values in the range of 2.6 × 10−6 to 1.8 × 10−4 cm s−1 (i.e. logPapp −5.59 to −3.74). Therefore high absorption potential as well as high bioavailability can be predicted. Low recovery values do not impair these results, since the use of fixed and recalculated values for the initial donor concentration both yield similar results. The same is true for the use of different mathematical approaches considering sink and non-sink conditions. Although single Papp values differ using the different approaches, they are still above the limit of 2.0 × 10−6 cm s−1, and the prediction of the absorption potential and bioavailability is therefore still the same. Only for 1-octen-3-one, where the fixed value of 100 μM resulted in a Papp value of 9.50 × 10−7 cm s−1 (i.e. logPapp −6.02), low absorption potential could be assumed. Low absorption potential is predicted for substances with Papp values <0.5 × 10−6 cm s−1 (logPapp < −6.3)14 and bioavailabilities of <1% for substances with Papp values <0.2 × 10−6 cm s−1 (logPapp < −6.7).30 Yet, when the experimentally determined initial donor concentration of 1-octen-3-one is used for calculations, Papp values >2 × 10−6 cm s−1 result, which leads to the interpretation for the substance as being highly bioavailable. Considering the low recovery of only 4% for 1-octen-3-one, the high permeability value determined with the recalculated donor concentration appears far more reasonable. This result emphasizes the importance of determining the originally applied amount of substance, especially for lipophilic compounds which are prone to be adsorbed to surfaces.
The observed high permeabilities combined with the hydrophobicity of the investigated compounds indicate a passive transcellular uptake route. The passive transcellular route is the most likely pathway for lipophilic compounds since they can easily pass the lipophilic cell membrane which makes up 99% of the surface of the epithelium.14 Yet other transport mechanisms, like a carrier-mediated pathway, cannot be completely ruled out by the presented data and further measurements would be necessary to conclusively clarify this topic.
Remarkably, the correlation between permeability in Caco-2 cells and in vivo drug transport is especially high for passively transported drugs.20 When investigating substances which are quickly absorbed via the passive transcellular pathways, Lennernäs et al. found that permeability values differed only 2–4 fold between the Caco-2 experiments and experiments using a perfusion system on human jejunum in vivo.31 From the obtained in vitro values in the present study, a high in vivo intestinal permeability can therefore be assumed for the aforementioned hop aroma compounds with high permeability values.
In our study, the highly lipophilic compounds α-humulene, β-caryophyllene, β-farnesene and β-myrcene as well as the esters 2-methylbutyl-isobutyrate and 3-methylbutyl-isobutyrate could only be detected in acceptor fluids when enhancing the lipophilicity of the acceptor compartment by applying serum rather than buffer. Serum mimics the human physiological conditions more closely than buffer. Generally, high lipophilicity of the investigated compounds may limit the determination of permeability due to restricted solubility in acceptor and donor fluids, which leads in turn to a higher affinity towards the cellular monolayer resulting in formal disappearance of the target compounds during analysis.32 Alterations to the Caco-2 system like the addition of BSA to basolateral compartments have shown to improve results for highly lipophilic compounds.33,34
The flat absorption profiles of α-humulene, β-caryophyllene, β-farnesene and β-myrcene indicate an early equilibrium state in which the substances are released from the cells and reabsorbed at a constant rate. Since intestinal blood circulation is distinct in the human body, it is likely that the absorbed substances are rapidly carried away under in vivo conditions. Diffusion from the blood stream back into the intestinal lumen occurs therefore probably to a lower extent in vivo than in the static Caco-2 model, hence leading to a higher permeability. This assumption is in good agreement with experimental data obtained previously in in vivo studies: to name one example, for α-humulene, Chaves et al. found a bioavailability of 18% after oral administration in mice.35
Apart from that, a previous study also showed that during in vitro digestion experiments, α-humulene and β-caryophyllene were transformed into humulol and β-caryolanol, respectively.19 Both transformation products possess one hydroxyl group in contrast to their non-substituted hydrocarbon educts and were quickly transported across the Caco-2 monolayer in our study. In these cases, gastric transformation leads to a higher hydrophilicity and hence to an improved permeability in the Caco-2 cells as could be demonstrated by the present study.
In accordance with this, 3-methyl-2-buten-1-ol and terpin, which were also newly built during the in vitro digestion experiments, showed good absorbance through the intestinal wall in the Caco-2 experiments. Furthermore, the compounds 2-methyl-3-buten-2-ol and α-terpineol showed on the one hand a high stability (>75%) during in vitro digestion processes and on the other hand a high absorption rate through the Caco-2-cells. This indicates that these compounds can reach systemic circulation almost unchanged and completely. For other substances with a high stability in the in vitro digestion, namely 1-octen-3-one, α-humulene, β-farnesene and limonene, the uptake through the Caco-2-cells was limited or rather ambiguous. Linalool showed a medium stability in the gastrointestinal tract, yet it was absorbed through the Caco-2-cells quickly. Geraniol and nerol, of which less than 40% of the original substance was available after in vitro digestion, were also transported quickly across the Caco-2-barrier. Both esters 2- and 3-methylbutyl-isobutyrate as well as 1-octen-3-ol showed in the in vitro digestion and Caco-2 experiments a high tendency to adsorption processes, as will be addressed later on.19
Metabolism
In Caco-2 cells several drug-metabolizing enzymes, including carboxylesterase,36,37 cytochrome P monooxygenases, flavine-containing monooxygenases, epoxide hydrolase38 and β-hydroxysteroid dehydrogenase,39 have been found. Yet drug-metabolizing enzymes are often expressed in smaller amounts in Caco-2 cells than in the human intestine.13In the present study, the α,β-unsaturated alcohols 1-octen-3-ol and 3-methyl-2-buten-1-ol have been demonstrated to be oxidized during the transport experiments to 3-octanone and 3-methyl-2-butenal, respectively. Interestingly, the double bond of 1-octen-3-ol was hydrogenated, while the double bond in 3-methyl-2-buten-1-ol was not affected (Fig. 5), since neither 1-octen-3-one was detectable in samples of 1-octen-3-ol nor was 3-methyl-butanal detectable in samples of 3-methyl-2-buten-1-ol.
As no such oxidation products were found in the control samples, it can be excluded that the oxidation was due to oxygen contact of the tested compounds during the transport experiment. The oxidation processes rather appear to be due to enzymatic conversions. Due to the lower expression of drug metabolizing enzymes in Caco-2 cells than in human small intestinal cells, the conversion of 1-octen-3-ol and 3-methyl-2-buten-1-ol might occur to an even larger extent in vivo. Yet, to the best of our knowledge, no studies on the physiological effects of 3-octanone or 3-methyl-2-butenal have been conducted so far. Further studies targeted at the elucidation of the respective enzyme(s) involved in these conversions would be of interest. Alcohol dehydrogenase for example, an enzyme known to convert alcohols to ketones or aldehydes, has been reported not to be expressed in Caco-2 cells.40
Remarkably, no analogous metabolites were detected for geraniol and nerol although these substances also possess primary α,β-unsaturated hydroxyl groups. This lack in biotransformation might be due to steric effects as the respective structural site at the double-bond contains a more voluminous alkyl moiety.
In view of the observed quantitative decrease of the esters, it has been shown that Caco-2 cells express the enzyme carboxylesterase-1 (hCE1) abundantly,37 hence the esters 2-methylbutyl-isobutyrate and 3-methylbutyl-isobutyrate might theoretically be cleaved to isobutyric acid and 2-methyl-1-butanol or 3-methyl-1-butanol, respectively. This might explain the decrease of the esters over time. Unfortunately, none of these cleavage products were detected in the samples. An estimation of the in vivo availability of the esters is therefore difficult.
Recovery and mass balance
Overall, the observed recoveries and mass balances are poor for several of the tested substances, especially for limonene and 1-octen-3-one regarding recoveries and 3-methylbutyl-isobutyrate as well as 2-methylbutyl-isobutyrate regarding mass balance. Reduced mass balances have been reported frequently in transport experiments.41 Generally, in Caco-2 transport experiments, high mass balances are desirable as mass balances of >80% result in an acceptable approximation of the Papp value.13 The lower the mass balance, the likelier the predicted Papp value is error-prone as this error may lead to an underestimation of the Papp values. Hubatsch et al. claimed this to be acceptable for qualitative studies, when aiming at identification of highly permeable compounds.13 In this study, all tested substances for which permeability values could be determined showed a high permeability. An underestimation of the values due to low mass balances would therefore not impair the prediction of a high bioavailability. Furthermore, Krishna et al. have shown that the performance of Caco-2 experiments with highly lipophilic compounds is especially challenging due to high adsorption to plastic and retention in cells.33 To make matters worse, the investigated substances are not only lipophilic but also volatile, potentially leading to even greater losses, especially since incubation at 37 °C for 120 or 80 minutes, respectively, was mandatory. Additionally, the presence of phase I and phase II metabolizing enzymes might contribute to the loss of analytes with the formation of novel biotransformation products. The identification and quantification of the metabolites 3-octanone and 3-methyl-2-butenal improved the mass balance from 25% to 33% and from 71% to 125% for 1-octen-3-ol and 3-methyl-2-buten-1-ol, respectively. Since the metabolites were only semi-quantitatively determined, these elevated mass balance values should be regarded as orientating values. For all other tested substances, no metabolites or degradation products were detectable.Apart from that, the adsorption of lipophilic aroma compounds to plastic surfaces has been subject to several studies.42 The extent of adsorption is on the one hand dependent on the polarity of the odorant and on the other hand on the characteristics of the plastic material, as well as on factors like concentration, storage duration, temperature, pH and the presence of other compounds such as proteins. In the present study, the test compounds were confronted with three different kinds of plastic as well as glass surfaces. The transwell inserts were made of polyethylene terephthalate (PET), the 12-well plates were made of polystyrene (PS) and the pipette tips and tubes used for dilution were made of polypropylene (PP). Glass vials were used to store stock solutions and samples taken were stored in glass vials. Van Willige et al. showed that limonene and β-myrcene are adsorbed to LLDPE (linear low density polyethylene) and OPP (oriented polypropylene) easily and quickly.43 A fast adsorption was also shown for nonanone, a structure closely related to octanone. The ester ethyl-2-methylbutyrate encountered a smaller yet noticeable adsorption. In the present experiments, limonene quickly seemingly disappeared in donor solutions and was only detectable when storage times were as short as possible. Yet after 10 minutes of incubation no remaining substance could be recovered. β-Myrcene showed a recovery of only 12% and 1-octen-3-one, which is only one carbon atom shorter than nonanone and should therefore possess similar properties, showed a recovery rate of only 4% in two different concentrations (100 μM, 1 mM). The esters 2-methylbutyl-isobutyrate and 3-methylbutyl-isobutyrate showed recovery rates of only 18 and 25%, respectively. These substances possess longer side-chains than the ester used in the study by van Willige et al., hence they are likely to show greater adsorption due to higher lipophilicity.42,43 The low recovery values for some substances in this study generally underline the necessity to reinvestigate the applied concentration of lipophilic compounds in in vitro studies as was done in the current study, since there might already occur adsorption processes prior to the start of the experiments, for example in tubes and pipette tips used for dilution. Keeping storage times as short as possible may further help to improve recovery.
In the course of the presented studies, attempts were made to quantify the amounts of aroma substances retained by the cell layer. Yet, most likely for reasons like adsorption and volatility, no remaining substances were detectable after the cell lysis procedure.
Nevertheless, our studies show that important and interesting insights can be obtained on the dynamics of intestinal odorant absorption and potential intestinal biotransformation, even when facing the problematic situation of analyzing volatile and lipophilic compounds, requiring special precautions and careful consideration of potential side-effects such as adsorption, evaporation and limited uptake in donor and acceptor media that are commonly used in established Caco-2 monolayer assays. Overall, aroma compounds with their specific physical–chemical properties require specialized testing, unlike other non-volatile and much more hydrophilic compounds that are commonly investigated in vitro.
Regarding the hop aroma compounds, further studies following the route of these substances through the human body would be of great interest when aiming at understanding their potential physiological impact. This is especially true in view of their obviously high bioavailability, but also their capability of generating further derivatives which might also be physiologically active or even more potent than the original plant educts.
To name one important aspect, the potential passage of the blood–brain barrier is another interesting site to be investigated. Generally, the blood–brain barrier builds its own specific structures and enzymes and is fundamentally different to the intestinal barrier.44 Results obtained from Caco-2 experiments can therefore not be directly transferred to in vivo permeability of the blood–brain barrier. Yet Garberg et al. report a high correlation (r2 = 0.86) between in vitro data from Caco-2 cells and in vivo data for the blood–brain barrier obtained via a mouse brain uptake assay when passively transported compounds in an apical to basolateral direction are regarded.45 The correlation was even higher (r2 = 0.96) when the highly protein bound compound warfarine was excluded from the dataset. It is therefore not unlikely that the supposedly passively transported aroma compounds investigated in the present study are able to easily pass the blood–brain barrier as well – that is assuming they are not utterly metabolized beforehand.
Conclusions
Caco-2 assays were applied to various potentially bioactive aroma compounds of hops and digestive transformation products thereof to elucidate their bioavailability after oral ingestion. Of the tested compounds, all substances possessing a hydroxyl-function, i.e. the terpenoids α-terpineol, geraniol, nerol, linalool and terpin as well as 2-methyl-3-buten-2-ol, 3-methyl-2-buten-1-ol, 1-octen-3-ol, β-caryolanol and humulol were able to pass the Caco-2 cell barrier quickly, indicating good bioavailability in the human body. Taking together the observed high permeability and the high lipophilicity, it is likely that they are transported via the passive transcellular pathway. Thereby, drug metabolizing enzymes present in the cells were able to convert 1-octen-3-ol to 3-octanone and 3-methyl-2-buten-1-ol to 3-methyl-2-butanal. However, the highly lipophilic sesquiterpenes α-humulene, β-caryophyllene, β-farnesene as well as the terpene β-myrcene seemed to quickly establish an equilibirium state of release and reabsorption by the cells under the static in vitro conditions. A shift towards higher permeability due to blood flow in vivo is conceivable. For 1-octen-3-one, limonene and the esters 2-methylbutyl-isobutyrate and 3-methylbutyl-isobuyrate, permeability could not be predicted due to substantial substance losses.Overall, our study showed that for such an analytically problematic but physiologically interesting and important substance group (due to their high lipophilicities and high volatilities) special measures and adjustments need to be undertaken to obtain comprehensive data in the respective physiological assays. Nevertheless, our investigations show that the Caco-2 system still remains a valuable tool to predict processes at the human intestinal wall even for volatile and lipophilic substances that necessitate special consideration with regard to poor mass balances and recoveries. Small adjustments like the use of serum as acceptor fluid and the reduction of experimental time turned out to be helpful with these challenging compounds. Additional modifications, like the use of e.g. teflon-coated cell culture equipment might further help to optimize the system. Future experiments could also include the use of three-dimensional dynamic tissue models that mimic human physiological conditions more closely as well as the use of primary human intestinal epithelial cells.25 Animal testing would be of interest to correlate the presented in vitro data with in vivo results. An approach to enhance the bioavailability of lipophilic compounds could be also the use of surface-active and self-emulsifying amphiphilic carriers.46 Likewise, the effect of simultaneously presented food is worth investigating, since the microstructure of food is also able to influence bioavailability in vivo.47
All in all, the presented results indicate that bioactive hop aroma compounds are able to pass the intestinal barrier, underlining their potential benefits and applicability in orally administered phytopharmaceuticals or functional foods.
Acknowledgements
The authors gratefully acknowledge funding from the German National Academic Foundation.Notes and references
- H. R. Stocker, Schweiz. Brau.-Rundsch., 1967, 60–89 Search PubMed.
- M. Blumenthal, The Complete German Commission E Monographs: Therapeutic Guide to Herbal Medicines, American Botanical Concil, Austin, Texas, 1998 Search PubMed.
- P. Zanoli and M. Zavatti, J. Ethnopharmacol., 2008, 116, 383–396 CrossRef CAS PubMed.
- R. Schellenberg, S. Sauer, E. A. Abourashed, U. Koetter and A. Brattstrom, Planta Med., 2004, 70, 594–597 CrossRef CAS PubMed.
- U. Koetter, E. Schrader, R. Kaufeler and A. Brattstrom, Phytother. Res., 2007, 21, 847–851 CrossRef CAS PubMed.
- L. Franco, C. Sanchez, R. Bravo, A. B. Rodriguez, C. Barriga, E. Romero and J. Cubero, PLoS One, 2012, 7 Search PubMed.
- A. Kessler, C. Villmann, H. Sahin-Nadeem, M. Pischetsrieder and A. Buettner, Flavour Fragrance J., 2012, 27, 297–303 CrossRef CAS.
- H. Aoshima, K. Takeda, Y. Okita, S. J. Hossain, H. Koda and Y. Kiso, J. Agric. Food Chem., 2006, 54, 2514–2519 CrossRef CAS PubMed.
- S. J. Hossain, H. Aoshima, H. Koda and Y. Kiso, J. Agric. Food Chem., 2002, 50, 6828–6834 CrossRef CAS PubMed.
- S. J. Hossain, H. Aoshima, H. Koda and Y. Kiso, J. Agric. Food Chem., 2003, 51, 7568–7575 CrossRef CAS PubMed.
- S. J. Hossain, K. Hamamoto, H. Aoshima and Y. Hara, J. Agric. Food Chem., 2002, 50, 3954–3960 CrossRef CAS PubMed.
- K. H. Schiller, Westfälische Wilhelms-Universität Münster, 2002.
- I. Hubatsch, E. G. E. Ragnarsson and P. Artursson, Nat. Protocols, 2007, 2, 2111–2119 CAS.
- I. Hidalgo and J. Li, in The Process of New Drug Discovery and Development, Informa Healthcare, 2nd edn, 2006, pp. 161–186 Search PubMed.
- I. J. Hidalgo, T. J. Raub and R. T. Borchardt, Gastroenterology, 1989, 96, 736–749 CAS.
- K. Cattoor, M. Bracke, D. Deforce, D. De Keukeleire and A. Heyerick, J. Agric. Food Chem., 2010, 58, 4132–4140 CrossRef CAS PubMed.
- K. Cattoor, J. P. Remon, K. Boussery, J. Van Bocxlaer, M. Bracke, D. De Keukeleire, D. Deforce and A. Heyerick, Food Funct., 2011, 2, 412–422 CAS.
- N. C. Stadler, V. Somoza and W. Schwab, J. Agric. Food Chem., 2009, 57, 3949–3954 CrossRef CAS PubMed.
- A. Heinlein and A. Buettner, Food Funct., 2012, 3, 1059–1067 CAS.
- P. Artursson, K. Palm and K. Luthman, Adv. Drug Delivery Rev., 1996, 22, 67–84 CrossRef CAS.
- P. Artursson, Crit. Rev. Ther. Drug Carrier Syst., 1991, 8, 305–330 CAS.
- R. H. Maffly, R. M. Hays, E. Lamdin and A. Leaf, J. Clin. Invest., 1960, 39, 630–641 CrossRef CAS PubMed.
- S. Tavelin, J. Grasjö, J. Taipalensuu, G. Ocklind and P. Artursson, in Epithelial Cell Culture Protocols, ed. C. Wise, Humana Press, Totowa, New Jersey, 2002, pp. 233–272 Search PubMed.
- P. Artursson, J. Pharm. Sci., 1990, 79, 476–482 CrossRef CAS.
- J. Pusch, M. Votteler, S. Göhler, J. Engl, M. Hampel, H. Walles and K. Schenke-Layland, Biomaterials, 2011, 32, 7469–7478 CrossRef CAS PubMed.
- H. van Den Dool and P. D. Kratz, J. Chromatogr. A, 1963, 11, 463–471 CrossRef CAS.
- I. Novakova, E. A. Subileau, S. Toegel, D. Gruber, B. Lachmann, E. Urban, C. Chesne, C. R. Noe and W. Neuhaus, PLoS One, 2014, 9, e86806 Search PubMed.
- K. Cai, A. E. Hagerman, R. E. Minto and A. Bennick, Biochem. Pharmacol., 2006, 71, 1570–1580 CrossRef CAS PubMed.
- SciFinder; Chemical Abstracts Service: Columbus, OH, 2013; predicted logD values (accessed Aug 05, 2013).
- E. Biganzoli, L. A. Cavenaghi, R. Rossi, M. C. Brunati and M. L. Nolli, Farmaco, 1999, 54, 594–599 CrossRef CAS.
- H. Lennernäs, K. Palm, U. Fagerholm and P. Artursson, Int. J. Pharm., 1996, 127, 103–107 CrossRef.
- P. Wils, A. Warnery, V. Phung-Ba, S. Legrain and D. Scherman, J. Pharmacol. Exp. Ther., 1994, 269, 654–658 CAS.
- G. Krishna, K.-j. Chen, C.-c. Lin and A. A. Nomeir, Int. J. Pharm., 2001, 222, 77–89 CrossRef CAS.
- B. Press, in Permeability Barrier: Methods and Protocols, ed. K. Turksen, Springer Science+Business Media, New York, 2011, vol. 763, pp. 139–154 Search PubMed.
- J. S. Chaves, P. C. Leal, L. Pianowisky and J. B. Calixto, Planta Med., 2008, 74, 1678–1683 CrossRef CAS PubMed.
- T. Prueksaritanont, L. M. Gorham, J. H. Hochman, L. O. Tran and K. P. Vyas, Drug Metab. Dispos., 1996, 24, 634–642 CAS.
- T. Imai, M. Imoto, H. Sakamoto and M. Hashimoto, Drug Metab. Dispos., 2005, 33, 1185–1190 CrossRef CAS PubMed.
- J. Borlak and C. Zwadlo, Xenobiotica, 2003, 33, 927–943 CrossRef CAS PubMed.
- A. Buur and N. Mørk, Pharm. Res., 1992, 9, 1290–1294 CrossRef CAS.
- T. Koivisto and M. Salaspuro, Alcohol.: Clin. Exp. Res., 1997, 21, 1599–1605 CrossRef CAS PubMed.
- J. J. Palmgrén, J. Mönkkönen, T. Korjamo, A. Hassinen and S. Auriola, Eur. J. Pharm. Biopharm., 2006, 64, 369–378 CrossRef PubMed.
- E. A. Tehrany and S. Desobry, Food Addit. Contam., Part A, 2004, 21, 1186–1202 CrossRef CAS.
- R. Van Willige, D. Schoolmeester, A. Van Ooij, J. Linssen and A. Voragen, J. Food Sci., 2002, 67, 2023–2031 CrossRef CAS PubMed.
- S. Lundquist, M. Renftel, J. Brillault, L. Fenart, R. Cecchelli and M.-P. Dehouck, Pharm. Res., 2002, 19, 976–981 CrossRef CAS.
- P. Garberg, M. Ball, N. Borg, R. Cecchelli, L. Fenart, R. D. Hurst, T. Lindmark, A. Mabondzo, J. E. Nilsson, T. J. Raub, D. Stanimirovic, T. Terasaki, J. O. Oberg and T. Osterberg, Toxicol. In Vitro, 2005, 19, 299–334 CrossRef CAS PubMed.
- A. T. M. Serajuddin, J. Pharm. Sci., 1999, 88, 1058–1066 CrossRef CAS PubMed.
- J. Parada and J. M. Aguilera, J. Food Sci., 2007, 72, R21–R32 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3fo60675a |
| This journal is © The Royal Society of Chemistry 2014 |