 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxide diffusion in innovative SOFC cathode materials
Y.
Hu
a,
V.
Thoréton
b,
C.
Pirovano
b,
E.
Capoen
b,
C.
Bogicevic
a,
N.
Nuns
b,
A.-S.
Mamede
b,
G.
Dezanneau
a and
R. N.
Vannier
*b
aLaboratoire Structures, Propriétés et Modélisation des Solides (SPMS), Ecole Centrale Paris, Grande Voie des Vignes, 92295 Châtenay-Malabry, France
bUniv Lille Nord de France, CNRS UMR 8181, Unité de Catalyse et de Chimie du Solide – UCCS, ENSCL, Université Lille 1, CS 90108, 59652 Villeneuve d'Ascq Cedex, France. E-mail: rose-noelle.vannier@ensc-lille.fr; Fax: +33(0)320436814; Tel: +33(0)320436583
First published on 28th July 2014
Abstract
Oxide diffusion was studied in two innovative SOFC cathode materials, Ba2Co9O14 and Ca3Co4O9+δ derivatives. Although oxygen diffusion was confirmed in the promising material Ba2Co9O14, it was not possible to derive accurate transport parameters because of an oxidation process at the sample surface which has still to be clarified. In contrast, oxygen diffusion in the well-known Ca3Co4O9+δ thermoelectric material was improved when calcium was partly substituted with strontium, likely due to an increase of the volume of the rock salt layers in which the conduction process takes place. Although the diffusion coefficient remains low, interestingly, fast kinetics towards the oxygen molecule dissociation reaction were shown with surface exchange coefficients higher than those reported for the best cathode materials in the field. They increased with the strontium content; the Sr atoms potentially play a key role in the mechanism of oxygen molecule dissociation at the solid surface.
Introduction
With the possibility of cogeneration of electricity and heat, solid oxide fuel cells are promising. The first units were commercialised in Japan in 2009 and demonstrators are running in Europe. Their principles are based on a dense ceramic electrolyte which separates two compartments, one containing hydrogen, and the other containing air. Due to a difference in oxygen chemical potential between the two compartments, oxygen molecules are reduced at the cathode into oxide ions, which migrate through the electrolyte to the anode to react with hydrogen molecules and produce water. Hydrogen is usually produced by methane reforming, however, internal reforming of methane is also envisaged. The typical temperatures used range between 700 and 1000 °C. The main electrolyte materials which are currently studied are Yttria Stabilised Zirconia (YSZ), Gadolinia Doped Ceria (GDC) and lanthanum gallates (LSGM). At the anode, a Ni-YSZ cermet containing a 40% volume of nickel is usually used, but it is prone to carbon deposition when used with hydrocarbon fuels. It is also sensitive to sulphur poisoning, and suffers from nickel agglomeration and redox stability with prolonged usage. Alternative materials, such as chromo-manganites (La0.75Sr0.25)Cr0.5Mn0.5O3 or titanates such as lanthanum-doped SrTiO3 have been proposed. At the cathode, strontium-substituted lanthanum manganite (La1−xSrxMnO3−δ, LSM) is conventionally used. However, it displays low oxide ion conductivity, and composites with YSZ have been developed in order to allow use at temperatures lower than 800 °C. To operate at lower temperatures, Mixed Ionic Electronic Conductors (MIEC) are usually preferred in order to allow the oxygen reduction reaction to be sprayed on the whole electrode volume, and not only at the electrode/electrolyte interface. As an alternative to LSM, the La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) perovskite has been widely studied.1 When used with a GDC electrolyte and after optimisation of its microstructure, an area specific resistance (ASR) of only 0.13 Ω cm2 was obtained at 603 °C for this compound,2 the target for the cathode being 0.15 Ω cm2 at 700 °C. This high performance is likely due to the presence of Co(III) in the structure, which is known to be particularly active for the reduction of oxygen molecules into oxide ions.3 Indeed, cobaltites are known to display high oxide ion transport parameters. Among these, cobaltite with a composition of La0.6Sr0.4CoO3−δ is one of the best catalysts for the oxygen molecule reduction reaction with a surface exchange coefficient (k*, cm s−1) of 1.1 × 10−6 cm s−1 associated with an oxygen self-diffusion tracer (D*, cm2 s−1) coefficient of 2.0 × 10−8 cm2 s−1 at 680 °C under 230 mbar.4 One drawback of these materials is their high thermal expansion coefficient (TEC). The aforementioned composition has a value of 23 × 10−6 K−1, which is twice the thermal expansion of YSZ (10 × 10−6 K−1)5 and GDC (12 × 10−6 K−1).6 However, partial substitution of cobalt with iron in LSCF led to a good compromise with a TEC in the range of 14–15 × 10−6 K−1 from 500 to 700 °C.7 More recently, promising performances as SOFC cathodes have been shown for two innovative cobaltites, Ba2Co9O14 and Ca3Co4O9+δ. An ASR of 0.5 Ω cm2 was first measured on a symmetric cell made of a 70 wt% Ba2Co9O14–30 wt% GDC composite electrode deposited on a GDC electrolyte.8 The good electrochemical performance of this compound was later confirmed by Li et al., who obtained ASRs of only 0.133 Ω cm2 and 0.068 Ω cm2 at 750 and 850 °C, and powder densities of 450 mW cm−2 and 770 mW cm−2 on a cell made of a LSGM electrolyte with a Ba2Co9O14–samarium-doped ceria composite cathode at 800 and 850 °C, respectively.3 This compound exhibits a three dimensional structure based on [Co8O8] blocks sandwiched in between [Ba2CoO6] layers (Fig. 1a).9,10 With cobalt atoms at different valences, it displays high electronic conductivity with a value of 240 S cm−1 at 650 °C, but as with other cobaltites, its thermal expansion coefficient is high (22 × 10−6 K−1), which necessitates the use of a composite, at least for mechanical compatibility, between the electrode and the electrolyte. After optimisation of its composition and thickness, an ASR of only 0.08 Ω cm2 at 700 °C was recently measured on a symmetric cell made of a 50 wt% Ba2Co9O14–50 wt% GDC composite electrode with a GDC electrolyte.11 | ||
| Fig. 1 Crystal structures of (a) Ba2Co9O14 and (b) Ca3Co4O9+δ. | ||
In contrast, a thermal expansion coefficient of only 10 × 10−6 K−1 was measured for the well-known Ca3Co4O9+δ thermoelectric compound,12 which has a misfit structure composed of CdI2-type [CoO2] layers sandwiched in between [Ca2CoO3−δ] rock salt slabs (Fig 1b).13,14 With an electronic conductivity of 100 S cm−1 at 700 °C and oxygen vacancies in the rock salt layers, mixed ionic electronic conductivity was expected for this compound. When tested in a symmetrical cell with a GDC electrolyte, ASRs of 4.94 Ω cm2 for the pure compound and 1 Ω cm2 for a 70 wt% Ca3Co4O9+δ–30 wt% GDC composite electrode were first measured.15,16 After optimisation of the composition and thickness, this was decreased to 0.5 Ω cm2 at 700 °C for a 50 wt% Ca3Co4O9+δ–50 wt% GDC composite electrode with a thickness of 21 μm, and further decreased to 0.35 Ω cm2 when calcium was partly substituted with 10% of strontium.17 These good performances were confirmed by Samson et al.,18 who reported a polarization resistance of 0.64 Ω cm2 at 600 °C for a 50 vol% Ca3Co4O9+δ–50 vol% GDC composite electrode. Using a samarium-doped ceria electrolyte, Zou et al.19 measured a maximum power density of 430 mW cm−2 at 700 °C for a 70 wt% Ca2.9Bi0.1Co4O9+δ–30 wt% Ce1.8Sm0.2O1.95|Ce1.8Sm0.2O1.95|Ce1.8Sm0.2O1.95–Ni button cell.
More recently, oxygen diffusion in this material was confirmed by combining 18O/16O isotope exchange, electrical conductivity relaxation and pulse isotopic exchange techniques.20 Despite low diffusion with a coefficient of 2.7 × 10−10 cm2 s−1 at 700 °C, this study showed high kinetics toward oxygen dissociation with a surface exchange coefficient as high as 1.6 × 10−7 cm s−1.
In the present paper, oxygen diffusion in these two innovative cathodes was investigated, with emphasis placed on the impact of the partial substitution of calcium with strontium on the oxygen transport properties of Ca3Co4O9+δ.
Experimental
Ba2Co9O14 and (Ca1−xSrx)3Co4O9+δ (x = 0.1, 0.2) powders were prepared by solid-state reaction from the corresponding carbonates and oxides BaCO3 (Prolabo – 99.5%, dried at 100 °C), CaCO3 (Prolabo – 99.5%, dried at 100 °C), SrCO3 (Aldrich – 99.8%, dried at 100 °C) and Co3O4 (Alfa Aesar, 99.7%). Reactants were carefully ground in an agate mortar and then calcined repeatedly at 900 °C with intermediate grinding. The purity of the samples was checked by X-ray diffraction (D8 Advance Bruker AXS diffractometer, Cu Kα radiation, 1D LynxEye PSD detector). Fullprof software [v2.05, July 2011] was used for refinement of the unit cell parameters.21 An Accupyc II 1340 micromeritic pychnometer was used to measure the powder density. The absolute oxygen stoichiometry of the sample was determined by iodometric titration.22Thermal analyses were conducted under air, using a TG/DTA Setaram Setsys apparatus. 40 mg of the sample powder was introduced into a Pt crucible, and the experiment was carried out at a heating rate of 5 °C min−1 under flowing air (0.4 L h−1) from room temperature to 950 °C. A blank experiment with an empty crucible was performed in order to correct the data for the effects of buoyancy. Thermogravimetry was also carried out for the evaluation of the oxygen stoichiometry of the sample as a function of pO2. In this case, a Setaram 92-1750 apparatus was used. The experiment was carried out using 50 mg of powder in a Pt crucible under the flow of an O2/N2 mixture (5 L h−1), with three successive oxygen partial pressures (100, 200 and 400 mbar), and the temperature ranging from 600 to 750 °C (50 °C step) for each pressure. For this set-up, the buoyancy was negligible and the data were directly used to derive the thermodynamic factor at each temperature.
Dense ceramics, 3 mm thick, were prepared by Spark Plasma Sintering at the IdF Platform in Thiais (France). A 20 mm (diameter) carbon die was used. The samples were sintered within 15 minutes with a 2 minute dwell at 850 °C, under a 50 MPa (8.8 kN) pressure. To remove carbon residues, the pellets were annealed for 12 h at 800 °C. A Hitachi S-3400N instrument was used to characterise the surface of the samples.
For isotopic exchange and electrochemical conductivity relaxation, sample surfaces were roughly polished in the first step, before they were polished down to a 1/4th of a micron with successive SiC papers and diamond pastes (Ra ∼ 10–20 nm) in the second step. In the case of calcium cobaltites, to avoid the evolution of the surface roughness, samples were annealed for 72 h at 900 °C before the second polishing in order to release residual strains in the material.
The 18O/16O Isotope Exchange Depth Profiling technique was applied to all samples in order to derive the oxygen self-diffusion tracer (D*, cm2 s−1) and surface exchange coefficients (k*, cm s−1). The diffusion time was chosen to get diffusion lengths significantly smaller than the sample dimensions to consider a 1D diffusion model perpendicular to the surface of the dense ceramic in a semi-infinite medium. In these conditions, assuming a first order surface exchange reaction and constant 18O concentration during the exchanges, the solution for a semi-infinite solid to the Fick's 2nd law of diffusion is given by the following equation:23
 | (1) |
The exchange conditions are reported in Table 1. Experiments were carried out in between 650 and 750 °C under a pressure close to 210 mbar with pre-annealing under normal oxygen (search grade oxygen 99.995% ALPHAGAZ) to reach thermodynamic equilibrium prior to the exchange under labelled oxygen. For both annealings, under the 18O enriched atmosphere or pre-annealing, the samples were annealed for the required time, and quenched. To determine the 18O content in the oxygen gas used for the exchange, a silicon wafer was oxidised overnight at 1000 °C under 200 mbar.
| Temperature (°C) | Pressure (mbar) | Pre-annealing time (hours) | Annealing time (min) | 18O ratio in the gas phase |
|---|---|---|---|---|
| Ba 2 Co 9 O 14 | ||||
| 648 | 210 | 240 | 2860 | 0.79 |
| 700 | 210 | 96 | 2765 | 0.80 |
| 750 | 218 | 96 | 2790 | 0.80 |
| (Ca 1−x Sr x ) 3 Co 4 O 9+δ x = 0.10, 0.20 | ||||
| 604 | 207 | 160 | 725 | 0.88 |
| 649 | 211 | 100 | 567 | 0.67 |
| 694 | 219 | 61 | 392 | 0.80 |
| 741 | 210 | 20 | 55 | 0.9 |
A ToF-SIMS5 machine (ION-TOF GmbH, 25 keV Bi+ LMIG analysis gun, DSCS sputter gun) was used to measure the 18O diffusion profiles using the same experimental protocol as described in ref. 20. Both depth analysis from the sample surface (depth profile mode)24 and edge analysis of the cut sample (line scan mode)25 were performed and the profiles merged. The depth of the craters, after SIMS depth profiling, were measured with a KLA Tencor Alpha-step IQ profilometer. The same apparatus was used to measure the samples’ roughness.
Electrical conductivity relaxation was also carried out on strontium-doped Ca3Co4O9+δ. For this purpose, plane-parallel samples (17 mm in length and 1 to 2.4 mm in width) were cut from the strontium substituted dense ceramics using a diamond saw. The electrical conductivity was measured using a 4 point configuration while changing the oxygen concentration of the surrounding atmosphere, as shown in ref. 20. A gold paste was used to paint gold contacts which were sintered at 750 °C for 1 h at a rate of 100 °C h−1 before the experiment. The distance between the two internal electrodes was about 14 mm.
Once the sample was at equilibrium at the given temperature, the pO2 was changed step-wise and the conductivity was monitored as a function of time. The variation in pO2 was performed by an abrupt switch between air and mixtures of O2/Ar with oxygen partial pressures of 0.05 atm and 0.01 atm, respectively, at a 5 L h−1 flow rate. In addition, the temperature-dependent electrical conductivity was measured under air with a 5 L h−1 flow rate at a very slow speed of 25 °C h−1.
To characterise the nature of the atoms at the uppermost surface of the sample, Low-Energy Ion Scattering (LEIS) was also applied to a dense ceramic (Ca1−xSrx)3Co4O9+δ with x = 0.1 composition which had been heated at 800 °C in air during the 12 hours prior to the analysis. The analysis was performed using a Qtac100 (ION-TOF GmbH) apparatus. Before analysis, the sample was subjected in situ to an atomic oxygen treatment (2–3 × 10−4 mbar) to remove possible organic surface contamination. The study was carried out with a 4He+ (3 keV) beam on a 1 mm2 analysed area.
Results and discussion
Characterization of powder and ceramics
The purity of the synthesised powders and dense ceramics was confirmed by X-ray diffraction. As shown in Fig. 2, good pattern matching was obtained for both type of ceramics. | ||
| Fig. 2 The XRD calculated profile compared to experimental data collected on dense pellets of Ba2Co9O14 (a), and (Ca1−xSrx)3Co4O9+δ with x = 0.10 (b). | ||
Corresponding unit-cell parameters are given in Table 2. For Ba2Co9O14, they are in good agreement with the parameters reported by Ehora et al. (5.6963(8) Å, 28.924(6) Å)9 and Sun et al. (5.6958(4) Å, 28.909(4) Å).10 The small differences in parameters we observed between the powders and the ceramics, on one hand, and these authors, on the other hand, are likely due to small differences in the oxygen content due to different thermal histories. For the refinement of (Ca1−xSrx)3Co4O9+δ, the phases were considered as a mixture of two phases, one corresponding to the [CoO2] layers and the second one corresponding to the [Ca2CoO3−δ] rock salt layers, with common a, c and β cell parameters constrained to be the same, and different bH and bRS, refined independently. The evolution of the unit cell parameters with the strontium content is in good agreement with the results reported by Wang et al.,26 who showed almost no variation of the bH unit cell characterising the [CoO2] layer periodicity, and an increase of the bRS unit cell with strontium content, in good agreement with the higher radius of Sr2+ (1.18 Å) compared to Ca2+ (1.00 Å).27
| Powder | Dense ceramics | |
|---|---|---|
Ba
2
Co
9
O
14
(R![[3 with combining macron]](https://www.rsc.org/images/entities/b_char_0033_0304.gif) m) m)
|
||
| a (Å) | 5.6945(3) | 5.700(1) |
| b (Å) | 28.905(2) | 28.926(6) |
| (Ca 1−x Sr x ) 3 Co 4 O 9+δ x = 0.10 | ||
| a (Å) | 4.8429(3) | 4.8433(9) |
| b H (Å) | 2.8255(3) | 2.825(4) |
| b RS (Å) | 4.5968(6) | 4.563(2) |
| c (Å) | 10.8730(4) | 10.881(2) |
| β (°) | 98.137(9) | 98.14(3) |
| (Ca 1−x Sr x ) 3 Co 4 O 9+δ x = 0.20 | ||
| A (Å) | 4.8518(3) | 4.8547(6) |
| b H (Å) | 2.8225(5) | 2.821(1) |
| b RS (Å) | 4.6304(8) | 4.6362(8) |
| c (Å) | 10.8995(7) | 10.904(2) |
| β (°) | 98.123(7) | 98.12(2) |
As shown by Ehora et al.,9 Ba2Co9O14 decomposed into CoO and a cubic BaCoO∼2 form at 1030 °C. To check the stability of the calcium cobaltites, TGA and DTA analyses were carried out on the two strontium-doped compositions. Their thermograms are compared to the signals measured for the non-doped compound in Fig. 3. They exhibit similar behavior. The first transition, which is observed at 130 °C for the pure compound, disappears when strontium is introduced into the structure. The second transition at 550 °C, corresponding to the beginning of a mass loss, is maintained at the same temperature, but the intensity of the associated endothermic peak on heating decreases with the increase in strontium content. All compounds decomposed before 950 °C, and the decomposition temperature was slightly lowered when the strontium content was increased. This decomposition process is reversible, but it is worth noting that recombination happens at a lower temperature when the substitution rate increases.
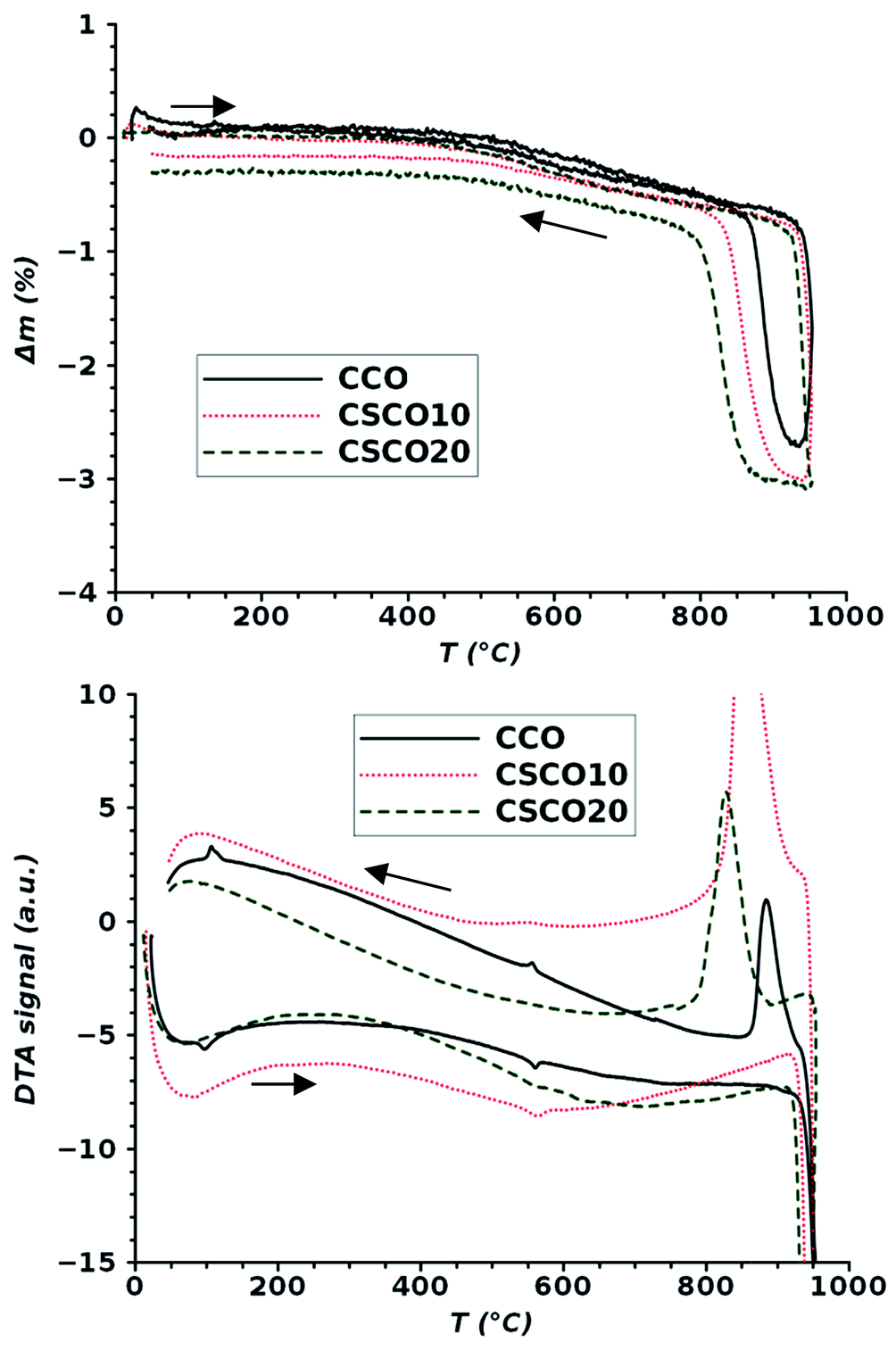 | ||
| Fig. 3 Comparison of TGA and DTA analyses corresponding to the Ca3Co4O9+δ parent compound (CCO, black solid line), (Ca1−xSrx)3Co4O9+δ with x = 0.10 (CSCO10, red line) and (Ca1−xSrx)3Co4O9+δ with x = 0.20 (CSCO20 black dotted line). | ||
Due to the lack of stability at high temperature, it was not possible to sinter these compounds using conventional sintering, but dense ceramics with relative densities higher than 96% were obtained using spark plasma sintering, as confirmed by SEM (see Fig. 4).
 | ||
| Fig. 4 SEM images of Ba2Co9O14 (a), and (Ca1−xSrx)3Co4O9+δ with x = 0.10 (b) and 0.20 (c), showing the dense ceramics. | ||
18O/16O isotope exchange in Ba2Co9O14
The normalised 18O/(16O + 18O) isotope concentrations as a function of the penetration depth are given in Fig. 5 for the three studied temperatures. Both data recorded in the depth-profile and line-scan modes are reported. A good agreement between the two sets of data was observed. However, whatever the temperature of exchange, a high oxygen content was measured at the surface of the samples within the first micron of the depth; the thickness of this high oxygen content layer increased with temperature. It was therefore not possible to fit these data to the diffusion equation (eqn (1)). | ||
| Fig. 5 Normalised 18O diffusion profile measured on Ba2Co9O14 dense ceramics after annealing under labelled oxygen at 650 °C, 700 °C and 750 °C for about 46 hours and corresponding fit (in red) taking into account the data obtained far from the surface. | ||
Taking into account only the data obtained far from the surface, the transport parameters given in Table 3 were derived.
| T (°C) | k* (cm s−1) | D* (cm2 s−1) |
|---|---|---|
| 648 | 1.3 × 10−10 | 2.5 × 10−12 |
| 700 | 4.0 × 10−10 | 5.0 × 10−12 |
| 750 | 2.5 × 10−9 | 6.0 × 10−12 |
Surprisingly, despite very good electrochemical performances, rather low oxygen diffusion and kinetics towards oxygen molecule dissociation were shown. However, one must be careful with these results. The increase in 18O at the sample surface is not clearly understood. It is likely due to an oxidation process which occurs when the sample is cooled, but this hypothesis still has to be confirmed.
Oxygen transport in strontium-doped Ca3Co4O9
The oxygen transport properties in the two strontium-doped Ca3Co4O9+δ materials were characterised by both electrical conductivity relaxation and the 18O/16O isotope depth profile technique. The electrical conductivity at the thermodynamic equilibrium corresponding to the x = 0.10 composition under different oxygen pressures is given in Fig. 6a. An increase in the electrical conductivity was observed when the oxygen partial pressure increased, confirming the p-type behaviour of these materials. As shown in Fig. 6b, the electrical conductivity slightly decreased when strontium was introduced into the structure. The evolution of the electrical conductivity corresponding to the (Ca1−xSrx)3Co4O9+δ (x = 0.1) composition at 750 °C with the changes in surrounding atmosphere is shown Fig. 7. It is worth noting that oxidation and reduction are reversible processes.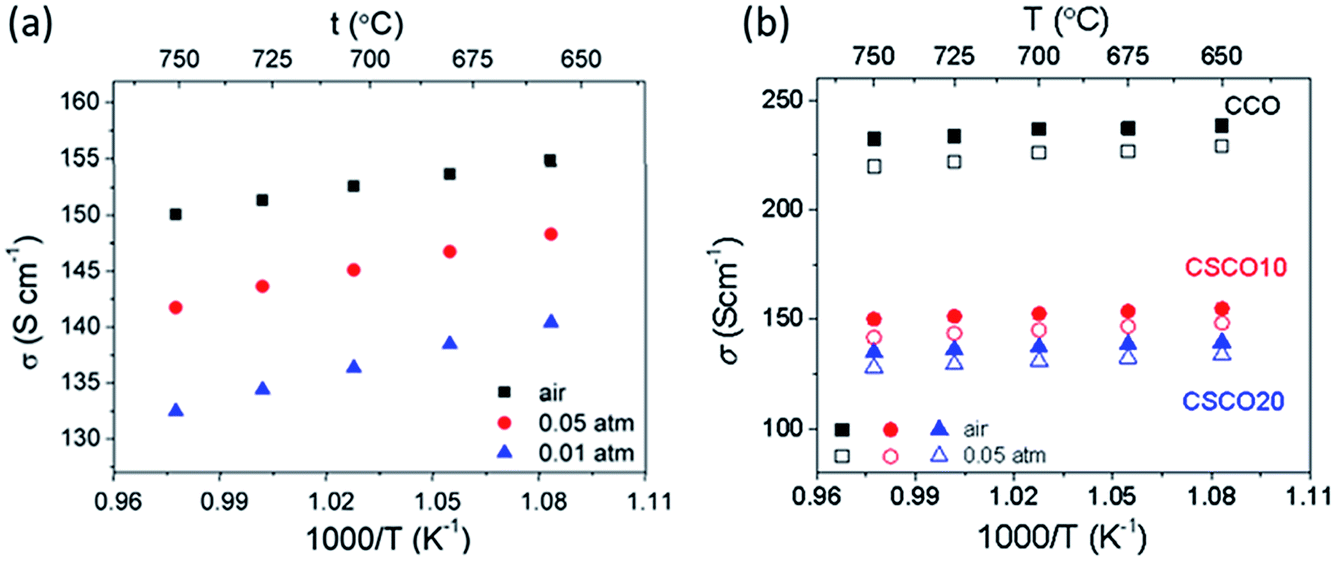 | ||
| Fig. 6 Evolution of the electrical conductivity of (Ca1−xSrx)3Co4O9+δ with an x = 0.10 composition at the thermodynamic equilibrium under different oxygen pressures (a), and comparison of the electrical conductivities of the x = 0 (CCO in black), x = 0.10 (CSCO10 in red) and x = 0.20 (CSCO 20 in blue) compositions as a function of temperature under air and 0.05 atm (b). | ||
 | ||
| Fig. 7 Evolution of the electrical conductivity of (Ca1−xSrx)3Co4O9+δ with an x = 0.10 composition at 750 °C with surrounding atmosphere changes, showing the reversibility of the oxidation and reduction processes. | ||
Data obtained for both compositions in the 650–750 °C range were fitted to the theoretical model of diffusion. In these conditions, only the diffusion coefficient could be derived with precision, reflecting a Dchem/kchem characteristic length that was much smaller than the size of the sample, and suggesting much higher surface exchange kinetics for these materials than for chemical diffusion.28 Corresponding Dchem values are given in Table 4 and shown in Fig. 8. The Dchem values obtained when the air ↔ 0.05 atm and air ↔ 0.01 atm are close, and are similar to what was obtained for the pure material.20 Moreover, the chemical diffusion coefficients are independent of whether the relaxation is a reduction or an oxidation process.
| T (°C) | Air-0.05 atm | 0.05 atm-air | Air-0.01 atm | 0.01 atm-air |
|---|---|---|---|---|
| (Ca 1−x Sr x ) 3 Co 4 O 9+δ x = 0.10 | ||||
| 650 | 1.37 × 10−7 | 1.35 × 10−7 | 1.34 × 10−7 | 1.31 × 10−7 |
| 675 | 1.92 × 10−7 | 1.91 × 10−7 | 1.85 × 10−7 | 1.95 × 10−7 |
| 700 | 2.58 × 10−7 | 2.51 × 10−7 | 2.52 × 10−7 | 2.57 × 10−7 |
| 725 | 3.47 × 10−7 | 3.61 × 10−7 | 3.44 × 10−7 | 3.53 × 10−7 |
| 750 | 4.68 × 10−7 | 4.95 × 10−7 | 4.73 × 10−7 | 4.76 × 10−7 |
| (Ca 1−x Sr x ) 3 Co 4 O 9+δ x = 0.20 | ||||
| 650 | 3.09 × 10−7 | 3.17 × 10−7 | 3.04 × 10−7 | 3.25 × 10−7 |
| 675 | 4.62 × 10−7 | 4.54 × 10−7 | 4.57 × 10−7 | 4.58 × 10−7 |
| 700 | 5.99 × 10−7 | 6.40 × 10−7 | 5.87 × 10−7 | 6.37 × 10−7 |
| 725 | 8.41 × 10−7 | 8.50 × 10−7 | 8.00 × 10−7 | 8.24 × 10−7 |
| 750 | 1.04 × 10−6 | 1.14 × 10−6 | 0.96 × 10−6 | 1.08 × 10−6 |
 | ||
| Fig. 8 Evolution of Dchem values as a function of temperature, measured on the parent compound Ca3Co4O9+δ (CCO, green square), and (Ca1−xSrx)3Co4O9+δ with x = 0.10 (CSCO10, red dot) and x = 0.20 (CSCO20 blue triangle). | ||
As shown in Fig. 8, as expected, the diffusion process is thermally activated, and activation energies of 1.03 ± 0.03 eV and 0.98 ± 0.04 eV were measured for the compositions with x = 0.1 and x = 0.2, respectively, which were slightly lower than the activation energy displayed by the pure compound (1.2 ± 0.02 eV), indicating that diffusion was easier for the strontium-doped compound, which also exhibits higher oxygen diffusion coefficients.
This increase in the oxygen diffusion coefficient with the substitution ratio was confirmed by 18O/16O isotope exchange. The normalized 18O diffusion profile corresponding to the (Ca1−xSrx)3Co4O9+δ with x = 0.1 composition, exchanged at 649 °C over 567 minutes under 211 mbar dry oxygen, is shown in Fig. 9. Again, a good agreement was obtained between data collected in depth profile mode and line scan mode.
 | ||
| Fig. 9 Normalised 18O diffusion profile measured on the (Ca1−xSrx)3Co4O9+δ with x = 0.1 composition, exchanged at 649 °C over 567 minutes under 211 mbar dry oxygen. The calculated profile is given in red. | ||
However, in this case, as shown for the parent compound,20 a small decrease in the 18O content was noticed at the sample surface, which is attributed to a possible carbonation or hydroxylation when back in the ambient atmosphere. In the inset, the same plot has been drawn on a decimal logarithmic scale to compare the experimental and theoretical profiles in depth. As previously noticed for the pure compound, a “tail” can be seen, most likely suggesting anisotropic diffusion.20 Nevertheless, as shown in Fig. 9, a good fit between the calculated and experimental data was obtained. The derived k* and D* parameters are given in Table 5. Their evolution as a function of temperature is shown on an Arrhenius plot in Fig. 10.
| T (°C) | 604 | 649 | 694 | 741 |
|---|---|---|---|---|
| (Ca 1−x Sr x ) 3 Co 4 O 9+δ x = 0.10 | ||||
| k* | 1.4 × 10−8 | 1.9 × 10−7 | 3.0 × 10−6 | 3.0 × 10−6 |
| D* | 1.1 × 10−10 | 2.5 × 10−10 | 4.1 × 10−10 | 1.0 × 10−9 |
| (Ca 1−x Sr x ) 3 Co 4 O 9+δ x = 0.20 | ||||
| k* | 6.7 × 10−8 | 3.0 × 10−7 | 4.0 × 10−6 | 2.0 × 10−6 |
| D* | 3.0 × 10−10 | 2.8 × 10−10 | 6.1 × 10−10 | 6.7 × 10−10 |
 | ||
| Fig. 10 Arrhenius plot of k* and D* values compared to Dchem/γ for the composition (Ca1−xSrx)3Co4O9+δ with x = 0.1 (a) and x = 0.2 (b). | ||
The tracer diffusion can be correlated to the chemical diffusion via the thermodynamic factor γ, according to the following equation:
| Dchem = γD* |
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnpO2 and ΔlnCO are the corresponding variations in the oxygen partial pressure and oxygen content, respectively, and σe and σi are the electronic and ionic conductivities, respectively.
lnpO2 and ΔlnCO are the corresponding variations in the oxygen partial pressure and oxygen content, respectively, and σe and σi are the electronic and ionic conductivities, respectively.
For the sake of comparison, the evolution of D*ECR = Dchem/γ is also reported in Fig. 10. The values are in good agreement. The slightly higher values observed for D*ECR compared to the tracer diffusion coefficient values can be explained by a lack of accuracy in the determination of the thermodynamic coefficient because of small oxygen stoichiometry variations (see the values of the thermodynamic coefficient in Table 6).
| Temp. (°C) | x = 0.1 | x = 0.2 |
|---|---|---|
| 650 | 211 | 313 |
| 675 | 200 | 305 |
| 700 | 189 | 297 |
| 725 | 183 | 299 |
| 750 | 178 | 302 |
An increase in the diffusion coefficient with strontium content is confirmed, and may be explained by the increase in the volume of the rock salt layers, which may facilitate oxygen mobility within these layers. However, with a value in the range of 10−10 cm2 s−1, it is still low, which justifies the addition of gadolinia-doped ceria to improve the electrochemical performance.15–19
With surface exchange coefficients of 3.0 × 10−6 cm s−1 and 4.0 × 10−6 cm s−1 at 700 °C for both the x = 0.1 and x = 0.2 compositions, these materials exhibit very fast kinetics for the oxygen reduction reaction compared to the best cathode materials in the field, since values of only 1.3 × 10−7, 1.1 × 10−6 and 1.3 × 10−7 cm s−1 were reported for La2NiO4+δ at 700 °C,29 La0.6Sr0.4CoO3−δ at 680 °C4 and GdBaCo2O5+δ at 686 °C,30 respectively.
To further understand oxygen transport in these materials, the x = 0.1 composition was characterised by LEIS. The initial spectrum, corresponding to the estimated removal of 0.2 monolayers, is compared to the one measured after the estimated removal of 3 monolayers in Fig. 11. Despite bulk contamination with sodium, which was also observed at the surface of the powders and ceramics of the parent compound,20 mainly strontium and calcium were initially observed at the uppermost sample surface. No cobalt was initially observed, just a sub-surface signal was shown on the low energy side from the surface peak31 in the background, indicating that cobalt atoms are beneath the first measured atomic layer. After ionic sputtering, cobalt was evidenced at the outermost surface, and an increase in calcium was also noticed. However, one must be careful with this technique, since strontium may also segregate at the sample surface. The same behaviour was observed for other compounds. For SmCoO3, Fullarton et al. also showed that samarium was preferentially at the surface with only 5% of cobalt at the outermost surface, and segregation of strontium was evident at the surface for strontium-doped compounds.32 Similarly, Viitanen et al. showed the absence of cobalt and iron in the outermost atomic layer of a La0.6Sr0.4Co0.2Fe0.8O3 membrane.33 We also observed calcium oxide at the uppermost surface of the Ca3Co4O9 parent compound. Here, strontium seems to be mainly at the sample surface, and its presence may partly explain the increase in the kinetics towards the oxygen dissociation reaction when the strontium content increases.
 | ||
| Fig. 11 4He+ (3 keV) LEIS spectra recorded on dense ceramics of (Ca1−xSrx)3Co4O9+δ with x = 0.1 after annealing at 800 °C for 12 hours prior to the analysis. The first spectrum (in black) corresponds to an estimated removal of 0.2 monolayers, and the last spectrum (in red) corresponds to an expected removal of around 3 monolayers. | ||
Conclusions
Although oxygen diffusion was confirmed in the promising material Ba2Co9O14, it was not possible to derive accurate transport parameters because of an oxidation process at the sample surface which still has to be clarified. In contrast, for Ca3Co4O9+δ derivatives, it was shown that oxygen diffusion was improved when calcium was partly substituted with strontium, likely due to an increase in the volume of the rock salt layers of the structure, which may help oxygen mobility. Although the diffusion coefficient remains low, interestingly, fast kinetics towards the oxygen dissociation reaction were shown, with a surface exchange coefficient higher than those reported for the best cathode materials in the field. The surface exchange coefficient is increased when the strontium content increases, which may be correlated to the presence of a higher amount of strontium at the sample surface, which may play a key role in the mechanism of oxygen molecule dissociation at the surface of the solid.Acknowledgements
The “Fonds Européen de Développement Régional (FEDER)”, “CNRS”, “Région Nord Pas-de-Calais” and French “Ministère de l'Education Nationale de l'Enseignement Supérieur et de la Recherche” are acknowledged for funding of X-ray and surface analysis facilities. V.T. is grateful to the University Lille 1 for funding his PhD grant. The authors are also grateful to the Agence Nationale de la Recherche for funding the ANR EVEREST (ANR-07-PANH-005), NanOxyDesign (ANR-10-BLAN-0814) and IDEA-MAT (ANR-12-BS08-0012-01) projects. Maxence Vandewalle, Nora Djelal and Laurence Burylo from UCCS are acknowledged for SEM, TG-DTA analyses and conventional XRD experiments, respectively.References
- Functional materials for sustainable energy applications, Woodhead publishing series in energy, ed. J. A. Kilner, S. J. Skinner, S. J. C. Irvine and P. P. Edwards, Woodhead Publishing Limited, 2012 Search PubMed.
- D. Marinha, J. Hayd, L. Dessemond, E. Evers-Tiffée and E. Djurado, J. Power Sources, 2011, 196, 5084 CrossRef CAS PubMed.
- Y. Li, M. W. Xu and J. B. Goodenough, J. Power Sources, 2012, 209, 40 CrossRef CAS PubMed.
- A. V. Berenov, A. Atkinson, J. A. Kilner, E. Bucher and W. Sitte, Solid State Ionics, 2010, 181, 819 CrossRef CAS PubMed.
- O. Yamamoto, Electrochim. Acta, 2000, 45, 2423 CrossRef CAS.
- K. Kordesch and G. Simader, Fuel Cells and their Applications, VCH Wiley, NY, Weinheim, 1996 Search PubMed.
- B. Fan, J. Yan and X. Yan, Solid State Sci., 2011, 13, 1835 CrossRef CAS PubMed.
- A. Rolle, N. Preux, G. Ehora, O. Mentré and S. Daviero-Minaud, Solid State Ionics, 2011, 184, 31 CrossRef CAS PubMed.
- G. Ehora, S. Daviero-Minaud, M. Colmont, G. André and O. Mentré, Chem. Mater., 2007, 19, 2180 CrossRef CAS.
- J. Sun, M. Yang, G. Li, T. Yang, F. Liao, Y. Wang, M. Xiong and J. Lin, Inorg. Chem., 2006, 45, 9151 CrossRef CAS PubMed.
- I. Kehal, to be published.
- K. Nagasawa, S. Daviero-Minaud, N. Preux, A. Rolle, P. Roussel, H. Nakatsugawa and O. Mentré, Chem. Mater., 2009, 21, 4738 CrossRef CAS.
- H. Muguerra, D. Grebille and F. Bourée, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 676 CAS.
- C. D. Ling, K. Aivazian, S. Schmid and P. Jensen, J. Solid State Chem., 2007, 180, 1446 CrossRef CAS PubMed.
- A. Rolle, S. Boulfrad, K. Nagasawa, H. Nakatsugawa, O. Mentré, J. Irvine and S. Daviero-Minaud, J. Power Sources, 2011, 196, 7328 CrossRef CAS PubMed.
- A. Rolle, V. Thoréton, P. Rozier, E. Capoen, O. Mentré, B. Boukamp and S. Daviero-Minaud, Fuel Cells, 2012, 12(2), 288 CrossRef CAS.
- A. Rolle, to be published.
- A. J. Samson, M. Søgaard, N. Van Nong, N. Pryds and N. J. Bonanos, J. Power Sources, 2011, 196, 10606 CrossRef CAS PubMed.
- J. Zou, J. Park, H. Yoon, T. Kim and J. Chung, Int. J. Hydrogen Energy, 2012, 37, 8592 CrossRef CAS PubMed.
- V. Thoréton, Y. Hu, C. Pirovano, E. Capoen, N. Nuns, A.-S. Mamede, G. Dezanneau, C. Y. Yoo, H. J. M. Bouwmeester and R. N. Vannier, J. Mater. Chem. A, 2014, 2, 19717 Search PubMed.
- J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef CAS.
- L. Karvonen, H. Yamauchi and M. Karppinen, Chem. Mater., 2008, 20, 7143 CrossRef CAS.
- J. Crank, The Mathematics of diffusion, Clarendon Press, Oxford, 2nd edn, 1975 Search PubMed.
- J. A. Kilner, B. C. H. Steele and L. Ilkov, Solid State Ionics, 1984, 12, 89 CrossRef CAS.
- R. J. Chater, S. Carter, J. A. Kilner and B. C. H. Steele, Solid State Ionics, 1992, 53–56, 859 CrossRef CAS.
- L. B. Wang, A. Maignan, D. Pelloquin, S. Hébert and B. Raveau, J. Appl. Phys., 2002, 92, 124 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- M. W. den Otter, H. J. M. Bouwmeester, B. A. Boukamp and H. Verweij, J. Electrochem. Soc., 2001, 148(2), J1 CrossRef CAS PubMed.
- R. Sayers, R. A. De Souza, J. A. Kilner and S. J. Skinner, Solid State Ionics, 2010, 181, 386 CrossRef CAS PubMed.
- A. Tarancón, S. J. Skinner, R. J. Chater, F. Hernández-Ramirez and J. A. Kilner, J. Mater. Chem., 2007, 17, 3175 RSC.
- H. R. J. ter Veen, T. Kim, I. E. Wachs and H. H. Brongersma, Catal. Today, 2009, 140, 197 CrossRef CAS PubMed.
- I. C. Fullarton, J. P. Jacobs, H. E. Benthem, J. A. Kilner, H. H. Brongersma, P. J. Scanlon and B. C. H. Steele, Ionics, 1995, 1, 51 CrossRef CAS.
- M. M. Viitanen, R. G. Welzenis, H. H. Brongersma and F. P. F. van Berkel, Solid State Ionics, 2002, 150, 223 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |