 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of DGT as a long-term water quality monitoring tool in natural waters; uranium as a case study†
Geraldine S. C.
Turner
a,
Graham A.
Mills
b,
Michael J.
Bowes
c,
Jonathan L.
Burnett
d,
Sean
Amos
d and
Gary R.
Fones
*a
aSchool of Earth and Environmental Sciences, University of Portsmouth, Burnaby Building, Burnaby Road, Portsmouth, Hampshire, UK. E-mail: gary.fones@port.ac.uk; Fax: +44 2392 842244; Tel: +44 2392 842252
bSchool of Pharmacy and Biomedical Sciences, University of Portsmouth, St Michael's Building, White Swan Road, Portsmouth, Hampshire PO1 2DT, UK
cSchool Centre for Ecology and Hydrology, Maclean Building, Benson Lane, Crowmarsh Gifford, Wallingford, Oxfordshire OX10 8BB, UK
dAWE Aldermaston, Reading, Berkshire RG7 4PR, UK
First published on 13th January 2014
Abstract
The performance of the diffusive gradient in thin film technique (DGT) was evaluated as a tool for the long-term monitoring of water quality, using uranium as a case study. DGTs with a Metsorb™ (TiO2) sorbent were deployed consecutively at two alkaline freshwater sites, the River Enborne and the River Lambourn, UK for seven-day intervals over a five-month deployment period to obtain time weighted average concentrations. Weekly spot samples were taken to determine physical and chemical properties of the river water. Uranium was measured in these spot samples and after extraction from the DGT devices. The accuracy of the DGT device time weighted average concentrations to averaged spot water samples in both rivers was 86% (27 to 205%). The DGT diffusive boundary layer (DBL) (0.037–0.141 cm – River Enborne and 0.062–0.086 cm – River Lambourn) was affected by both water flow and biofouling of the diffusion surface. DBL thicknesses found at both sites were correlated with flow conditions with an R2 value of 0.614. Correlations were also observed between the DBL thickness and dissolved organic carbon (R2 = 0.637) in the River Lambourn, indicating the potential presence of a complex zone of chemical interactions at the surface of the DGT. The range of DBL thicknesses found at the River Lambourn site were also attributed to of the development of macro-flora on the active sampling surface, indicating that the DBL thickness cannot be assumed to be water flow dependant only. Up to a 57% under-estimate of uranium DGT concentration was observed compared to spot sample concentrations if the DBL was neglected. This study has shown that the use of DGT can provide valuable information in environmental monitoring schemes as part of a ‘tool-box’ approach when used alongside conventional spot sampling methods.
Environmental impactPassive samplers provide time weighted average (TWA) concentrations of pollutants in water and are becoming important tools in regulatory compliance monitoring and environmental risk assessments. The diffusive gradient in thin film technique (DGT) is frequently used to measure TWA concentrations of trace metals in surface waters. We investigated the impact of the thickness of the diffusive boundary layer (DBL) on the uptake of uranium into the DGT over a five month period in two rivers with different flow regimes and water chemistry. If the device is to be used as a long-term monitoring tool then it is recommended that the thickness of the DBL is determined with each deployment in order to improve the confidence of the measurements. |
Introduction
Currently, monitoring of water quality relies on the collection of low volume spot (grab or bottle) water samples, usually on monthly, or at most weekly, time intervals. This approach has a number of limitations, being both expensive and time consuming, the possibility for introducing contamination in sample handling or during storage1 and the potential to miss fluctuations in contaminant concentrations. For analytes having low aqueous concentrations, such as radionuclides, often large volumes (5–20 L) of water need to be collected and pre-concentrated to ensure good instrumental limits of detection.2 To overcome some of these drawbacks, continuous in field auto-samplers3 (active samplers) that are programmed to collect samples at set time intervals or during particular flow or meteorological conditions can be used.4 This approach is costly and can also be associated with errors in terms of sample stability for monitoring both metals and nutrients.5,6The use of in situ pre-concentration techniques, such as passive sampling devices, can overcome many of these errors associated with spot sampling7 and can be beneficial in investigations where concentrations of a pollutant fluctuate widely, for instance from increased surface water flow as a result of a storm event, or with large tidal fluctuations.7,8 Passive samplers have the advantage of being relatively low-cost, non-mechanical, require no power and little maintenance and can be deployed in a range of field sites.
Designs of passive sampler are varied and have been developed to measure a wide range of organics and metals. Examples include the Gaiasafe,9 Chemcatcher® for both metals,10 organics11 and organometallics,12 permeable liquid membrane devices13,14 and diffusive gradients in thin films (DGT).15 DGT measures the labile, dissolved fraction of analytes in situ and is the most widely used technique for measuring time weighted average (TWA) concentrations of a number of metals and inorganic substances in a variety of aquatic environments. The device consists of three layers: (i) a binding agent containing a resin with functional groups selective to the target ions, being held in a thin layer of hydrogel (binding gel); (ii) a layer of hydrogel of known thickness, which serves as the diffusive layer; and (iii) a protective outer membrane with a known pore size. A diffusive boundary layer (DBL) that forms on the exposed face of the device must also be accounted for and added to the overall diffusive layer. After deployment, metal ions accumulated in the resin layer are eluted (e.g. with nitric acid) and the resultant extract analysed by a sensitive instrumental technique e.g. inductively coupled plasma-mass spectrometry (ICP-MS).
DGTs have been used for monitoring metals in the aquatic environment in a number of single short-term deployment studies (e.g. 4 days,16 14 days17 and 31 days18). DGTs have also been deployed in the same location during two seasons with longer-term deployment periods (ranging from 13 to 36 days19) to show inter-seasonal variations of pollutants in the Sava River, Croatia. DGTs were also used for one-month deployments over five consecutive months5 in Lake Llyn, Trawsfynydd, UK. The concentration of metals in highly fluctuating, transitional environments, such as estuaries, have been monitored using DGT in short-term studies.7,20,21 Dunn et al.22 showed that in highly fluctuating environments concentrations of metals can change significantly over 24 h and that these variations would therefore be missed by the use of infrequent spot sampling. There is little published data for freshwater systems, however, on the effects of long-term environmental changes (for instance seasonal changes in biological activity and water chemistry and flow rate) on the operational effectiveness of DGT devices. If DGT is to be used by regulatory agencies and to be a fit for purpose monitoring tool, further long-term field testing is required in conjunction with recognised standards such as ISO 5667.1 In an attempt to investigate this, we used DGTs to monitor the concentrations of uranium continuously over a six-month period at two freshwater sites (River Enborne and the River Lambourn, Berkshire, UK) and compared the results against those from weekly spot water sampling. The purpose of this study was to therefore evaluate the usefulness of the DGT technique and to assess any issues (such as the measurement of the DBL, changing river chemistry and seasonal changes in biological activity) that could arise as part of its use as a regulatory environmental monitoring tool. The two river sites were chosen, as they were included in a routine environmental monitoring programme undertaken by Centre for Ecology and Hydrology (CEH). This provided weekly data to aid the interpretation of the DGT results. Both sites were also secure and located on private property, which ensured no interference to the devices over the deployment period. Uranium has a complex aqueous chemistry and was therefore selected to demonstrate that the DGT technique can accumulate a highly reactive analyte in a system with fluctuating water quality.
Uranium is not a priority substance in the European Union's Water Framework Directive23 due to the high concentrations that occur naturally. Environmental monitoring of anthropogenic and naturally occurring radionuclides in natural waters is a requirement of the environmental permits issued by the various environment agencies in the UK, and by the Industrial Pollution and Radiochemical Inspectorate for all users and holders of radioactive materials, under the Environmental Permitting Regulations (England and Wales) 2010 and Radioactive Substances Act 1993.24 These permits require the nuclear industry to continually undertake risk assessments of their discharges to ensure environmental impacts are as low as is reasonably practicable.25 This includes considering the use of new monitoring technologies such as DGT. Uranium has been measured by DGT in artificial and natural waters in eight reported studies.16,17,26–31 There are a number of candidate binding phases effective for uranium. The TiO2-based resin, Metsorb™ used in this study showed a high capacity for uranium.17 Isotopic ratios (235/238U) of uranium were also measured over the field trials to ascertain if the technique could be used as a tool to identify sources of radioactive pollution.
Experimental
Field locations
Two freshwater field sites were used (Fig. 1): site 1 (51.4469 N, −1.3838 W) was located on the River Lambourn at the village of Boxford, Berkshire, UK and site 2 (51.3792 N, −1.1855 W) on the River Enborne near Brimpton, Berkshire, UK. Both rivers are tributaries of the River Kennet. The River Lambourn has a chalk catchment and is a fast flowing shallow channel with an average pH of 7.9–8.32 The mean flow and base flow indices were 1.71 m3 s−1 and 0.97 respectively.33 The River Enborne drains impermeable tertiary sand, silt and clay deposits34 and has a slow flowing deep channel with a pH ∼7.8. The mean flow and base flow indices were 1.32 m3 s−1 and 0.53. Mean monthly meteorological data was obtained from the Met Office Benson meteorological monitoring station (51.62 N, −1.097 W) (http://www.metoffice.gov.uk/public/weather/climate/benson) (Table S2†) and from daily measurements taken by CEH, Wallingford, UK (51.6032 N, −1.1134 W) using a ground flush type rain gauge (Fig. S1†).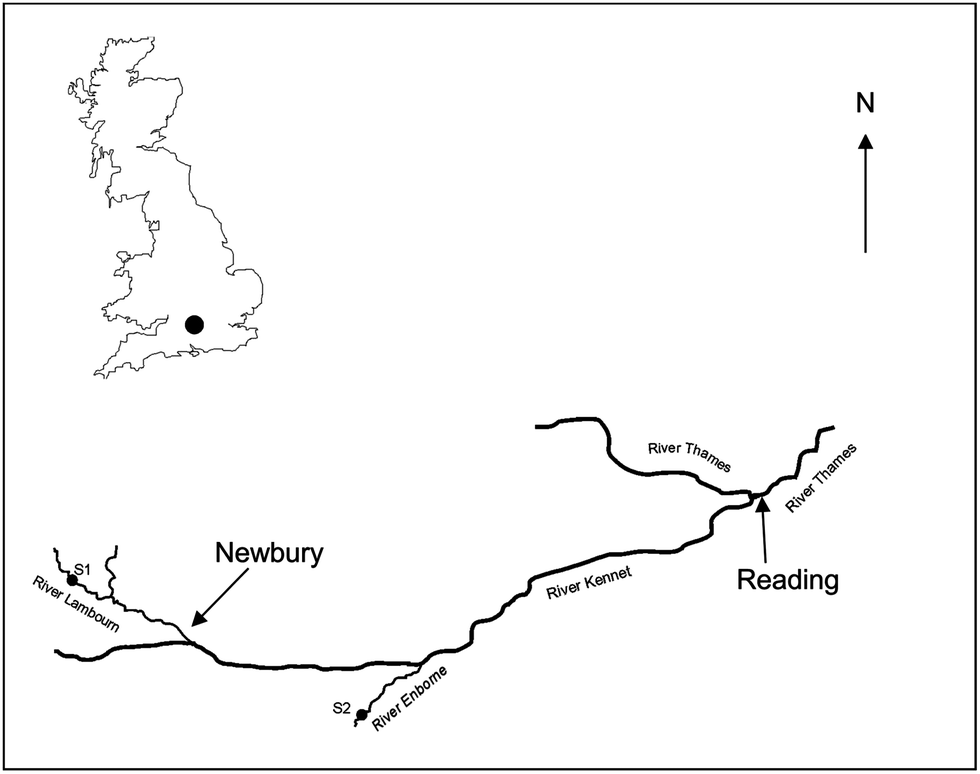 | ||
| Fig. 1 Location of field sites in the UK. Site 1 (S1) is located on the River Lambourn and site 2 (S2) on the River Enborne. Both rivers are tributaries of the River Kennet within the River Thames catchment. | ||
Design of field trial
A continuous monitoring programme was used to assess the performance of DGTs over part of three riverine seasons, from summer through to autumn and winter. During these periods it was expected a wide variation in biological activity, flow regime and water chemistry would occur. DGTs were deployed between Perspex plates (15 × 7 cm, up to 8 devices per plate) (Fig. 2) and attached to a rope and float and weighted to the river bed. The devices were deployed approximately 1.5 m from the river bank, out of the main flow channel to allow for access. Three DGT devices containing Metsorb™ resin gel were removed and replaced every week over a 21 week period from 24/08/2011 to 18/01/2012. Procedural blanks (in triplicate) were exposed to the field environment during deployment and retrieval of each set of samplers. Blanks were analysed in an identical manner to exposed field samplers. | ||
| Fig. 2 Photograph of DGTs held in place by a Perspex plate. The plate held up to eight devices. If more samplers were deployed then two Perspex plates were fixed back to back. The plate was deployed in the rivers a vertical position. | ||
To assess the influence of the DBL on the uptake of uranium, devices containing Metsorb™ were also deployed, with diffusive layer (polyacrylamide (PAM) gel) thicknesses (including 0.015 cm to account for the Supor membrane) of 0.015, 0.055, 0.095 and 0.135 cm, as per Warnken et al.35 The DBLs were measured on 12/10/2011, 07/12/2011, 05/01/2012 and 18/01/2012, corresponding to weeks 7, 15, 19 and 21 of the trial, so as to reflect two autumn and two winter seasonal measurements; with low and average rain fall in the autumn and winter respectively.
Triplicate spot samples of water from the two field sites were collected into acid washed low-density polyethylene (LDPE) bottles (1 L). An aliquot (20 mL) of water was filtered (0.2 μm pore size Supor filter) immediately into a polystyrene (PS) tube (30 mL) and acidified using 6 M HCl (40 μL). The acidified samples were stored in the dark at 4 °C until analysis. Water temperature, depth and flow rate were measured using a temperature YSI Castaway device (Yellow Springs, OH, USA), a rod and hydro-prop type flow meter (with a detectable flow limit of ∼5 cm s−1) respectively. The pH was measured (1 L water sample in the LDPE bottle allowing no headspace for excess CO2 to diffuse into the sample) in the laboratory using a Jenway 3410 Electrochemistry Analyser (Bibby Scientific Ltd., Staffordshire, UK). As part of the CEH Lambourn Observatory Project and the CEH Thames Initiative research platform, the Rivers Lambourn and Enborne were sampled weekly for major anions and cations (Table S1†). Water quality analysis was undertaken at CEH laboratories (see procedures in ESI and Table S1†). Discharge data for each site was obtained from the CEH National River Flow Archive (Fig. S1†), where measurements were taken at the crump weir located 51.3791 N, −1.1855 W, which is approximately 10 m upstream of the River Enborne study site, and at the crump weir monitoring station (51. 24 42 N, 1.1932 W) River Lambourn at Shaw, Berkshire (approximate 13 km downstream of the Boxford deployment site).
Materials and preparation of DGT
Chemicals were of analytical grade or better and supplied by Fisher Scientific Ltd. (Loughborough, UK), unless otherwise specified. Milli-Q (ultra-pure) water (>18.2 MΩ cm, Millipore, Watford, UK) was used as the laboratory water. All uranium ICP-MS standards and were prepared in PS containers from a 1000 mg L−1 in 2% HNO3 (Spex Certiprep, Fisher Scientific Ltd.) stock solution. The ICP-MS internal standard was prepared from a 1000 mg L−1 in 2% HNO3 (Spex Certiprep) bismuth stock solution. All plastic apparatus (including DGT housings) was soaked for 24 h in 10% HNO3 and rinsed three times in Milli-Q water prior to use.PAM diffusive gels (thickness 0.4, 0.8, 1.2 and 1.6 mm) were prepared according to Zhang and Davison.36 The diffusive gels and filter membranes were stored in 0.01 M NaNO3 prior to deployments to ensure ionic equilibrium between the diffusive gel and the deployment environment. The PAM binding gels were prepared with 1 g Metsorb™ HMRP powder (TiO2 with an organic binder, <50 μm; Graver Technologies, Glasgow, USA) according to the method described by Bennett et al.37 A disk of (0.2 μm pore size) Supor polyethylene sulfone (Pall Corporation, Portsmouth, UK) that was first acid washed in 1% HNO3, tripled rinsed in Milli-Q water and stored in 0.01 M NaNO3 was used as the outer membrane. DGT mouldings were obtained from DGT Research Ltd. (Lancaster, UK) and washed for 24 h in 10% HNO3, and then rinsed three times in Milli-Q water prior to use. The devices were assembled according to Davison et al.15 and stored at 4 °C in zip lock plastic bags, containing 1–2 mL of 0.01 M NaNO3 in Milli-Q water (ionic strength matched to freshwater deployment site) to ensure the diffusion properties of the gels were not altered, and to prevent the gels drying out.
Measurement of total uranium
Uranium was determined in all solutions by ICP-MS using an Agilent 7500ce series instrument (Agilent Technologies Inc., Japan). Total uranium was measured under normal plasma conditions in ‘no gas mode’, with the sample introduction system fitted with a micromist nebuliser. The instrument blank for uranium was 6 ng L−1 while the limit of detection (calculated by the Agilent Chemstation software) for uranium was 2 ng L−1, with a measurement relative standard deviation better than 3%. Bismuth (m/z = 209; 25 μg L−1) was used as an internal standard to compensate for any potential instrument drift. The certified fluvial reference material SLRS-5 (National Research Council Canada, Canada) was analysed directly for uranium and found to be within 1% of the stated values. The filtered and acidified spot water samples were analysed directly with no further dilution.Measurement of uranium in DGT
After exposure, the Metsorb™ binding gels were removed from the DGT and eluted (48 h) with 1 M H2O2/HNO3 (2 mL) solution (100 mL made by combining 90 mL 1.1 M HNO3 and 10 mL H2O2). The eluent was then diluted 10 fold with Milli-Q water prior to instrumental analysis. The concentration of uranium (μg L−1) measured by the ICP-MS in the eluent was multiplied by the dilution factor (×10) to give the actual uranium concentration (Ce). The absolute mass (M, ng) of the uranium in the binding gel was calculated using eqn (1), where M is calculated taking into account the gel volume (Vg, cm3), the eluent volume (Ve, mL), the measured concentration of uranium in the eluent (Ce, ng mL−1) and the elution factor (fe).36 For this study the uptake (>90%) and elution factor (83 ± 3%.) for uranium were taken from Turner et al.17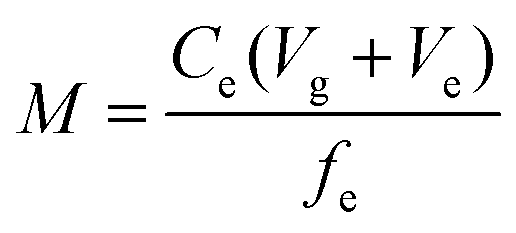 | (1) |
M from eqn (1) is then used to calculated the TWA concentrations (eqn (2)) where the concentration (CDGT, ng mL−1) was calculated using the mass of the analyte in the binding gel (M, ng), the thickness of the diffusive path length (diffusive gel and filter membrane) (Δg, cm), the diffusion coefficient of the analyte (D, cm2 s−1) (as determined for uranium at different pH's by Hutchins et al.16), deployment time (t, s) and the area of the sample exposure window (A, cm2).
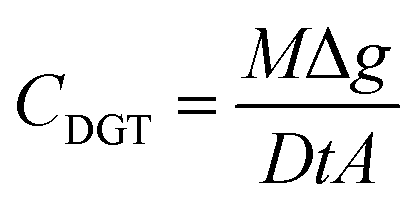 | (2) |
The DBL (δ) thickness was calculated using eqn (3) after Warnken et al.35 A straight line plot of 1/M versus Δg has a slope (m) of 1/(DCDGTAt) and an intercept (b) of δ/(DCDGTAt). The intercept (b) divided by the slope (m) of this plot gives the DBL thickness δ, as per eqn (4). The diffusion coefficients of the uranyl ion in the diffusive gel and the water have a ratio of nearly one,38 and so do not need to be considered for the purposes of this paper.
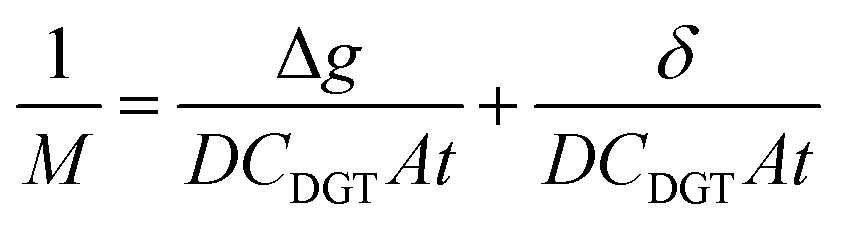 | (3) |
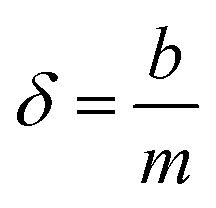 | (4) |
The thickness of the DBL was included in the CDGT calculations for the field trials. The DBL measurements were applied to the weekly DGT field data as follows: DBL data point 1 was applied to weeks 1–7 DGT data; DBL data point 2 was applied to weeks 8–15; DBL data point 3 was applied to weeks 16–19; and DBL point 4 was applied to weeks 20–21 DGT data. An average of all four DBL readings per river was used when an average DBL was applied to the DGT data. The active sampling area (A) was 3.8 cm2 instead of the 3.14 cm2 used in the laboratory trials, as described by Warnken et al.35 to account for lateral spread of the analyte across the surface of the DGT device. The diffusion coefficients from Hutchins et al.16 were used for the TWA calculations and corrected for temperature as per Zhang and Davison.36 Laboratory blanks were measured in triplicate and the average concentration per disk was determined for the Metsorb™ gel disks as 0.03 ± 0.02 ng and 0.30 ± 0.10 ng for 238U and 235U respectively.
Measurement of uranium isotopes
235/238U isotopic ratios were measured with an Agilent microflow (100 μL min−1) PTFE self aspirating nebuliser, to eliminate any signal pulses caused by the peristaltic pump using a micro-mist concentric nebuliser. Isotopic ratios were determined with 3% RSD as low as 0.01 μg L−1 total uranium (0.725 × 10−4 μg L−1 235U). The certified reference material U005a (New Brunswick Laboratories, DoE, Washington, USA) was analysed and was found to be within 99.5% of the isotopic value (0.342 × 10−4 235/238U). The spot water samples were measured directly without any further dilution. For the isotopic signature of uranium found with DGT, the extract was diluted 10 fold prior to analysis.Statistical analysis
The water quality results (including the weekly spot water sample measurements) were averaged over each week (mean of the reading at the beginning and at the end of each deployment week) and then subject to statistical analysis to identify any patterns between the two different techniques used to measure the uranium concentration and fluctuating water quality. All statistical analysis was performed in IBM® SPSS® Statistics Version 20. The non-parametric one sample Shapiro–Wilk test was first used to test the data for normality (normality significance figure ≥0.05). If normality was established a Pearson's product–moment correlation was performed, if the data was not normally distributed then the non-parametric Spearman's ranking correlation coefficient was used (P < 0.05).Results and discussion
Water flow rate was measured at each deployment site (Table S3†) to investigate if this may affect the thickness of the DBL. Flow rates were also back calculated from the discharge data (Fig. S2†). The DBL has been shown previously to be an important factor in the accuracy of the DGT technique in measuring TWA concentrations. Without the inclusion of the DBL in calculations, concentrations can be underestimated by up to 50%.17 We calculated the TWA concentrations of uranium using various DBL values to highlight the importance of including this variable. The TWA concentrations of uranium found with the DGTs were compared to the weekly averaged water quality results to determine if any statistically significant relationships existed. The mean uranium concentrations determined both by DGT and in the spot samples (0.26–0.38 μg L−1) are in line with those reported previously17 and by CEH (Mike Bowes pers com) who reported values of 0.3 μg L−1. The uranium concentrations measured in this study are in line with background uranium concentrations and are not particularly elevated.DBL measurements
Several factors can affect the thickness and measurement accuracy of the DBL. These include fluctuations in water velocity,35 the deposition of particulate matter, bio-fouling by macro-fauna and the growth of bacterial mats39 on the active sampling surface and the dissociation kinetics of organically bound metals at the solute interface of the sampler.40,41Tables 1 and 2 show the thickness of the DBL (calculated from Fig. S5 and S6†) measured in the River Enborne and River Lambourn respectively.| Deployment week | Date | Thickness of DBL (cm) | R 2 of graph | DBL as a ratio of overall diffusive layer thickness (0.095 cm) |
|---|---|---|---|---|
| 7 | 12/10/2011 | 0.141 ± 0.036 | 0.91 | 1.48 |
| 15 | 07/12/2011 | 0.086 ± 0.034 | 0.89 | 0.91 |
| 19 | 05/01/2012 | 0.047 ± 0.008 | 0.99 | 0.49 |
| 21 | 18/01/2012 | 0.037 ± 0.009 | 0.98 | 0.39 |
| Deployment week | Date | Thickness of DBL (cm) | R 2 of graph | DBL as a ratio of overall diffusive layer thickness (0.095 cm) |
|---|---|---|---|---|
| 7 | 12/10/2011 | 0.070 ± 0.022 | 0.93 | 0.74 |
| 15 | 07/12/2011 | 0.070 ± 0.032 | 0.86 | 0.74 |
| 19 | 05/01/2012 | 0.086 ± 0.012 | 0.99 | 0.99 |
| 21 | 18/01/2012 | 0.062 ± 0.018 | 0.99 | 0.65 |
Tables 1 and 2 show that the DBL thickness represents a large component of the overall diffusive layer thickness. The ratio of these values in the River Lambourn throughout the deployment fluctuated between 0.65 to 0.99, and decrease in the River Enborne from 1.48 to 0.39. Fig. 3 and 4 show how the TWA concentrations of uranium calculated over the deployment period vary with different DBL thicknesses; from no DBL accounted for, the average DBL calculated over the entire deployment period, and using the DBL calculated for different times in the trial. The importance of taking the DBL thickness into consideration is clearly demonstrated in Fig. 3b and 4b, as the calculated TWA concentration for uranium is up to 58% less than in the River Enborne (particularly when the calculated DBL was higher) than measurements that account for the periodically measured DBL (Fig. 3c and 4c). For the River Lambourn there was an underestimation of the TWA concentration of uranium by up to 57% when no DBL is accounted for in the calculations, with the TWA calculations using the averaged DBL over the deployment time (Fig. 3a) and the periodically measured DBL (Fig. 3c) within ± 20%.
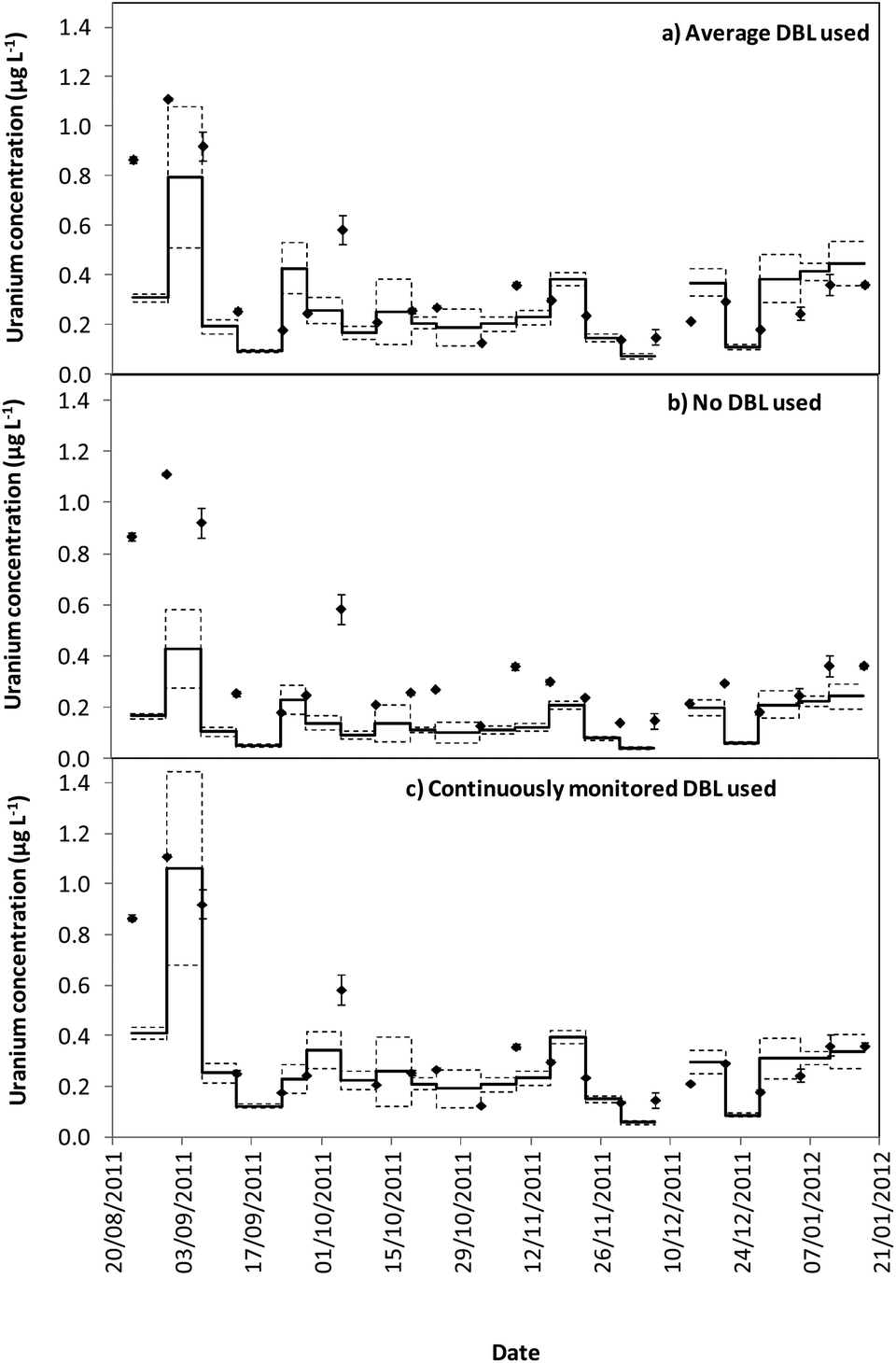 | ||
| Fig. 3 TWA concentrations measured by the DGT (−) and in spot water samples (♦) for uranium (μg L−1) over a 22 week deployment from 24/08/2011 to 18/01/2012 for the River Enborne using different calculated DBL thicknesses. The dashed line represents the standard error of the DGT measurements, as calculated from triplicate samples. (a) Average DBL thickness measured over the entire deployment, plus diffusive layer and filter membrane (0.078 + 0.095 = 0.173 cm). (b) No DBL thickness accounted for, only the diffusive layer and filter membrane (0.095 cm). (c) Different DBL thicknesses calculated over the deployment: 24/8/2011–12/10/2011 (0.141 cm); 12/10/2011–7/12/2011 (0.086 cm); 07/12/2011–05/01/2012 (0.047 cm); 05/01/2012–18/01/2012 (0.037 cm), plus diffusive layer and filter membrane (0.095 cm). | ||
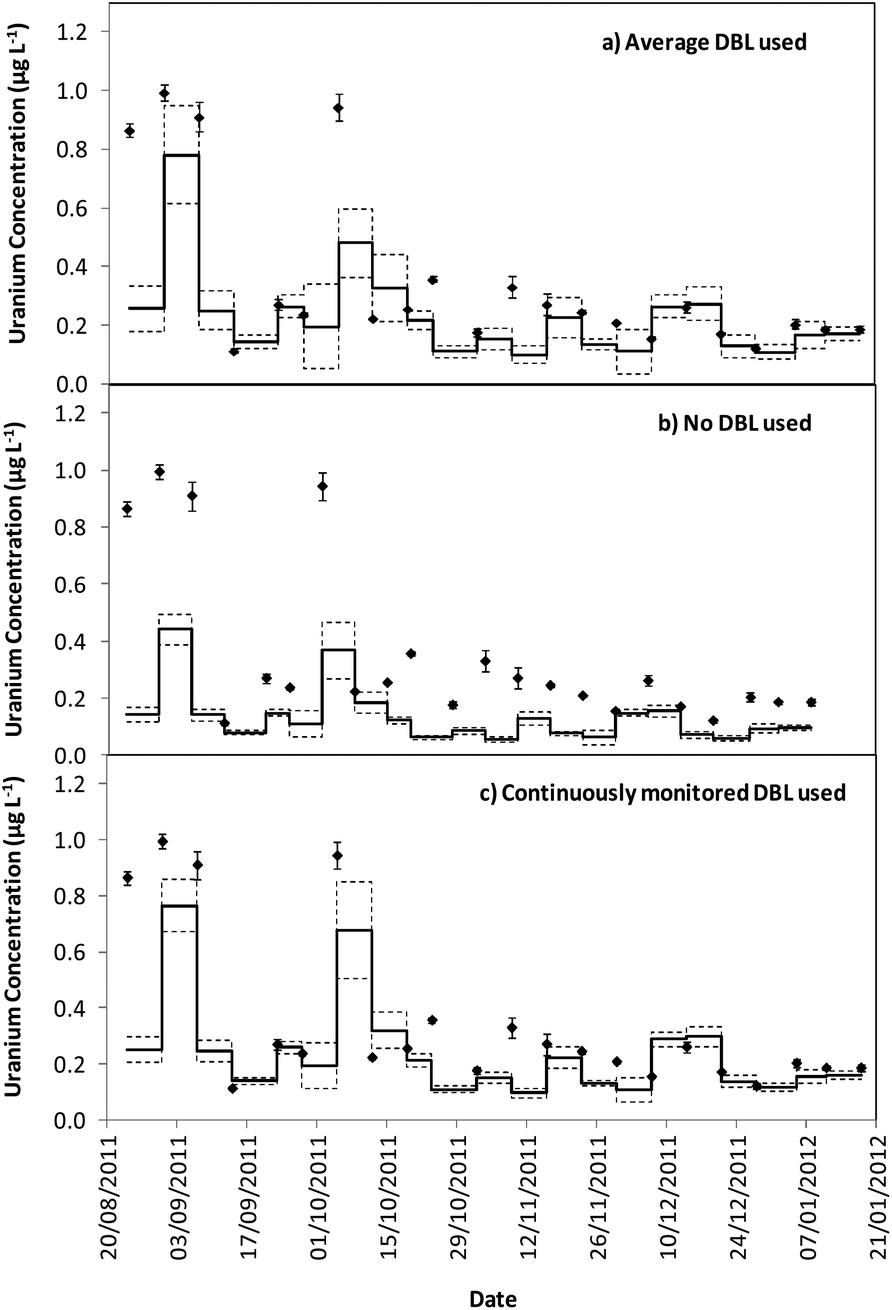 | ||
| Fig. 4 TWA concentrations measured by the DGT (−) and in spot water samples (♦) for uranium (μg L−1) over a 22 week deployment from 24/08/2011 to 18/01/12 for the River Lambourn using different DBL calculations. The dashed line represents the standard error of the DGT measurements, as calculated from triplicate samples (a) average DBL thickness measured over the entire deployment, plus diffusive layer and filter membrane (0.073 + 0.095 = 0.168 cm). (b) No DBL thickness accounted for, only the diffusive layer and filter membrane (0.095 cm). (c) Different DBL thicknesses calculated over the deployment: 24/8/2011–12/10/2011 (0.141 cm); 12/10/2011–7/12/2011 (0.086 cm); 07/12/2011–05/01/2012 (0.047 cm); 05/01/2012–18/01/2012 (0.037 cm), plus diffusive layer and filter membrane (0.095 cm). | ||
Effect of water flow rate on the thickness of DBL
During the first 4 months of the deployment (August to late November 2011) the River Enborne experienced below average precipitation (Table S2†) in conjunction with lower flow rates (Table S3 and Fig. S2†) and discharge (Fig. S1†), and consequentially a larger DBL thickness of 0.141 ± 0.036 cm (Table 1) was measured. The flow rate in September and October 2011 were calculated to be ≤2 cm s−1 (Fig. S1 and S2†), with the river flow where the samplers were sited likely to be even lower, as this was located outside the main channel. The sustained above average precipitation from the second week in December 2011 (Fig. S2†) increased the discharge and reduced the thickness of the DBL to 0.086 ± 0.019 cm in December and then to 0.047 ± 0.008 cm in January 2012 (Table 1). January 2012 experienced average levels of precipitation, and hence a thinner DBL of 0.037 ± 0.006 cm. Fig. S3a & b† show the differences in flow regime at the River Enborne over the deployment period with a potential difference in river height of up to 1.2 m. This demonstrates the need to fully characterise the attributes of a field site prior to deployment, to ensure the devices remain submerged in a reasonably turbulent environment and are always retrievable.It is clear from the field measurements of the DBL at the River Enborne (Table 1) that the changing DBL was closely coupled to the flow rate (the 1/M vs. Δg plots used for each of the DBL measurements can be seen in Fig. S5a–d and the flow rate in Fig. S2†). As only four DBL measurements were taken over the deployment period, it was not possible to perform any statistical tests. A simple correlation could be undertaken and graphed to show that the flow is linked to the size of the DBL, as shown in Fig. S4 in the ESI.† This shows a decrease in DBL thickness with increasing flow rates over the 6 month deployment period in this study. The River Enborne shows a clearer variation in DBL thickness with flow rate than the River Lambourn most likely due to the fact that the River Enborne has a highly fluctuating flow regime. The very large DBL observed in October 2011 (Table 1), when the flow rate of the River Enborne was very low, is concurrent with that found under a laboratory setting by Warnken et al.35 in quiescent conditions, where a large DBL of 0.15 ± 0.013 cm was observed (Table 3). Under laboratory conditions in previous studies, moderate flow rates up to 2 cm s−1 showed a reduction in the associated thickness of the DBL, with Warnken et al.35 reporting a value of 0.044 ± 0.0014 cm, which is similar to the thickness of the DBL found in this study for the January 2012 deployments in the River Enborne.
If the flow rate exceeds 2 cm s−1 (as for well stirred solutions) then it has been shown that the thickness of the DBL is not directly related to the flow rate of water.35,42 Warnken et al.35 found for high flow rates, in a laboratory setting, the thickness of the DBL was 0.023 ± 0.0032 cm, which is in agreement to the DBL thicknesses (0.024 ± 0.002 cm) found by Scally et al.43 The flow rate in this study frequently exceeded 2 cm s−1 in the River Enborne, but the lowest measured DBL was 0.046 cm, which implies other factors than flow rate may contribute to the DBL.
The River Lambourn showed less variability in the thickness of the DBLs (Table 2 and Fig. S6a–d†) most likely as a result of the discharge remaining at a steady state over the course of the deployment period (Fig. S1†). The flow rate for the River Lambourn over the deployment period averages 8 cm s−1 (Table S3 and Fig. S1 and S2†), which is higher for most of the deployment period than in the River Enborne. The consistent and high (despite low precipitation) flow rates experienced by the River Lambourn is due to the chalk catchment and the fact the river catchment is largely ground water fed. The DBL found in October was 0.070 ± 0.022 cm, which is higher than predicted in the laboratory (Table 3) for the flow rate. Over the course of the deployment period, the thickness of the DBL increased to 0.088 ± 0.009 cm in January 2012, and decreased to 0.062 ± 0.018 which is up to two times that measured in the River Enborne and nearly four times that measured under laboratory conditions. Fig. S4† does not indicate that the DBL in the River Lambourn is flow rate controlled. The flow rate therefore does not give a good indication of the thickness of the DBL in the River Lambourn, which means extraneous factors (such as biofouling) must be also taken into consideration.
DBL measurements in the field have been shown to differ significantly from those on the laboratory. Table 4 shows the thickness of DBLs found in the field, although there is a paucity of data. In a well-stirred field environment, Warnken et al.35 found the measured the thickness (0.026 ± 0.0017 cm) of the DBL closely matched their laboratory results. Thicker DBLs in the field have been reported, by Panther et al.44 (0.080 ± 0.013 cm for PO4) and Bennett et al.37 (0.080 ± 0.013 cm for As and Se). Hutchins et al.16 reported a DBL thickness of 0.02 ± 0.001 cm when measuring concentrations of uranium in a freshwater system. Another consideration when comparing the thickness of DBLs found here to other field studies is the length of time the devices were deployed. DGTs are usually deployed for shorter periods (3–5 days) when examining properties of DBL. In this study, the deployment time was 7 days. A longer deployment is favourable when measuring low concentrations (ng L−1) of a pollutant, as this allows more of the analyte to accumulate onto the resin, however, other factors e.g. biofouling may begin to dominate the uptake process. Warnken et al.35 suggested that when flow exceeds the 2 cm s−1 threshold, then the DBL thickness (present at 0.023 cm) could be discounted. Here a sampling area of 3.14 cm2 can used (as opposed to 3.8 cm2 which accounts for lateral diffusion at the DGT face) to offset the error when not accounting for the DBL, and when using a gel thickness of 0.8 mm. However, as is observed here and in other field studies (Table 4), there may be other factors influencing the thickness of the DBL than simply water flow rate. The major contributor to the thickness of the DBL is the flow rate, however, when the flow rate is decreased other influences including the effect of particulates, biological activity and dissolved organic material were found to play an increasing role but their effects are masked by the influence of high flow rate on the DBL.
| Analyte | Water type | Location | Thickness of DBL (cm) | Flow rate | Deployment time (days) | pH | Ref. |
|---|---|---|---|---|---|---|---|
| U | Fresh | River Lambourn, UK | 0.046 ± 0.006 | Fast | 5 | 7.8 | 17 |
| U | Marine | Southampton water, UK | 0.035 ± 0.019 | Fast | 5 | 8.2 | 17 |
| U | Fresh | Coomera river, Australia | 0.020 ± 0.001 | Fast | 4 | 7.5 | 16 |
| Cd, Pb, Zn | Fresh | River Wyre, UK | 0.026 ± 0.002 | Fast | 3 | — | 36 |
| As, Se | Fresh | Gold Coast, Australia | 0.080 ± 0.013 | Fast | 4 | 7.5 | 38 |
| As, Se | Marine | Gold Coast, Australia | 0.067 ± 0.007 | Fast | 4 | 7.9 | 38 |
| PO4 | Fresh | Gold Coast, Australia | 0.080 ± 0.013 | Fast | 4 | 7.5 | 45 |
| PO4 | Marine | Gold Coast, Australia | 0.067 ± 0.007 | Fast | 4 | 7.9 | 45 |
| Cd, Ni | Fresh | Lake Tantare, Canada | 0.031 ± 0.02 | Slow | 13–14 | 5.3–5.6 | 46 |
Effect of particulate matter and bio-fouling on the thickness of DBL
Previous work has shown that biofouling and turbidity35 can have an impact on the effectiveness of passive sampling devices. The River Enborne contained higher and fluctuating concentrations of suspended particulate material (SPM, mg L−1) than the River Lambourn (Fig. S7†). However, when plotted using a scatter graph, no clear trend was apparent. Particulates could potentially act to increase the thickness of the DBL by acting as an additional physical barrier to diffusion across the filter membrane or by supplying a source of dissociating uranium from particulate surfaces. At the diffusive interface (the surface of the filter membrane) where a concentration gradient will be present, there may be a resupply of uranium sorbed to the surface of the suspended particulates. Previous studies showed the presence of organic material in a river increases the sorption of uranium to particle surfaces.46 This is supported by the fact that when the devices were retrieved, there was particulate matter collected on the active sampling surface (Fig. S8†). Supor membranes are designed to inhibit microbial growth. However, if SPM accumulated on the surface of the membranes then this will provide sites for growth, with a microbial matt developing and potentially acting as a sink for the uranium.39 This could account for variability in the measurements on the thickness of the DBL depending on the depth of the microbial mat, but is an area for further work. The lower values of SPM found for the River Lambourn meant that this process may not be a contributing factor to the DBL.DGTs deployed in River Lambourn accumulated algae and macro-flora over the 7 days deployment. Previous work by Turner et al.17 at this site, showed with daily removal of vegetation and for shorter deployment times (5 days) the thickness of the DBL was 0.046 ± 0.006 cm. However, rapid accumulation of macro-flora (Fig. S9a–c†) resulted in high variability of the DBL over the deployment period including large DBL's with associated errors (Table 2). There was little variation in flow rates at this site (Fig. S1 and S2†), due to its high base flow index (0.97); hence any variation occurring in the thickness of the DBL could be attributed to a biological source. Dragun et al.19 also found limitations on the effectiveness of the DGT due to algal biofouling during long-term (13–36 days), single deployments during the spring. Ideally, DGTs should be deployed in a protective cage in areas prone to the build-up of algae and macro-flora, although this would not prevent the accumulation of periphyton on the surface of the devices. This is an area for further research, as care should be taken not to reduce the water flow inside the cage.
Effects of dissolved organic matter and water quality on the thickness of the DBL
DBLs are both a physical layer where advective transport moves to diffusional transport processes, and/or an apparent layer of chemical dissociation of the analyte from a larger molecule such as dissolved organic matter.41,47 Levy et al.47 showed that in the presence of organic ligands, metals demonstrated varying degrees of kinetic limitation dependent on dissociation rates, and therefore exhibited varying apparent diffusive boundary layer (ADBL) thicknesses. The possibility of the presence of a zone of chemical dissociation cannot be ignored in the case of uranium. This is due to its high affinity towards dissolved organic matter;48 particularly when over 90% of the uranium species modelled (using Visual Minteq) were found as humic complexes (fulvic and humic acids) for the River Enborne, and ∼50% of the uranium bound to humates in the River Lambourn. Fig. S9† shows that the River Enborne contains up to ten times more dissolved organic carbon (DOC) than the River Lambourn, thereby affecting uranium speciation. The DOC concentration in the River Enborne increased during periods of increased precipitation due to its susceptibility to the influence of catchment run off. Warnken et al.41 showed that the ADBL increased with metals that formed increasing strong complexes with dissolved organic matter. Uranium at low uranium–humic acid (U–HA) ratios (such as for the Rivers Enborne and Lambourn with U–HA ratios of 4.17 × 10−5 and 1.81 × 10−4 respectively) forms very strong humic acid complexes that have slow dissociation kinetics (kd = 4.9 × 10−5 s−1) compared to higher U–HA ratios (i.e. >0.01) (kd = 10−3 s−1).49 These slow dissociation kinetics may have affected the thickness of the DBL for both rivers, although this would require further studies in both the field and laboratory settings to confirm. This potential zone of dissociation may account for the presence of an extended DBL (Table 1) in the River Enborne even during periods of high flow and discharge, where the thickness of the DBL was 0.037 cm and 0.047 cm, compared to 0.023 cm in a fast moving system under laboratory conditions (Table 3). However, when plotted using a scatter graph (Fig. S11†), no clear trend was apparent DOC and DBL for the River Enborne, potentially because there are other stronger influencing factors such as flow rate, that make the impact of the DOC indistinguishable. Fig. S11† shows a clear trend of increasing DBL thickness with increasing DOC concentrations. This may be because factors that have a greater influence on the DBL thickness such as flow rate and inorganic ligands (e.g. phosphate) are consistent over the deployment period. Further work would be required to establish the relationship between the DBL and DOC when measuring uranium.Another interesting correlation was that of phosphate and the size of the DBL. In both rivers a positive correlation was observed when the DBL was plotted against the phosphate (Fig. S12†) this correlation being highly significant for the River Enborne (R2 = 0.8285), which may be due to the agricultural catchment has fluctuating phosphate concentrations with run off after precipitation events, similar to that found by Evans et al.50 Further work would be required to confirm this, but the presence of phosphate and SPM may act as both source and sink of uranium on the surface of the DGT devices, thereby increasing the thickness of the DBL, acting as zone of association/dissociation.
Calculation of TWA concentrations
The TWA concentrations of uranium were calculated using varying scenarios (Fig. 3 and 4), (a) the average thickness of the DBL measured over the entire deployment period; (b) not accounting for a DBL; and (c) using the changing thicknesses of DBLs measured during the trial. The parameters e.g. water pH and temperature and diffusion coefficient used in these calculations are given in Table S4.†Fig. 3 and 4 shows that TWA concentrations generally fall between weekly spot sampling data points. This was evident when there were rapid, short-lived, increases in the concentration of uranium during weeks 2 and 6 for the River Enborne, and weeks 2, 6 and 7 for the River Lambourn. During periods of relatively stability, the concentration of uranium measured in spot waters samples (weeks 20–22, River Lambourn and River Enborne; and weeks 15–16, River Enborne) corresponded well with the TWA concentrations found with the DGT. This shows the effectiveness of the DGT in measuring accurately, fluctuating concentrations, despite the difficulties of predicting the thickness of the DBL. The only anomaly within the data is Week 1, which shows a much higher spot sample concentration to the TWA DGT concentration at both rivers. This may be attributed to either a high phosphate concentration, very low flow and low precipitation or the use of a DBL that was determined a number of weeks after this deployment. However, these are all unknowns, but again this highlights the need for the DBL to be determined regularly in a water body that has fluctuating flow and water chemistry and also the need for a toolbox approach to environmental monitoring without the reliance on one technique. Murdock et al.5 attempted to validate DGT as an in situ tool for measuring caesium. They found that over the 5 month study, both the concentrations of caesium measured by the DGT and in spot water samples were in close agreement, being within the 1σ margin of error. As there was close agreement between the spot sample and DGT TWA concentration the DBL thickness which was not measured in this study was deemed an unimportant parameter. The study was undertaken in a lake with little variation in flow and there was a constant input of caesium from the Magnox reactor sited there. They found that the longer the deployments, the more measurement errors can be introduced. This includes increased bio-fouling, susceptibility to changing flow rates, and saturation of the binding phase. Mengistu et al.18 used DGT as a risk assessment tool, and undertook a single 31 days and a single 3 days deployment to measure seventeen metals (including uranium) in water polluted by mining tailings. They found 1–2 orders of magnitude reduction in the mass of metals accumulated in the DGT during the long-term deployments compared with the short-term deployments. Turner et al.17 found decreased uptake by DGT after 7 days, due to bio-fouling and saturation of the binding phase. For this reason, 7 days was chosen as the deployment period in this study.DGT has been to measure other analytes in highly fluctuating environments, such as estuaries.20,21,51 Montero et al.20 deployed DGTs for 10 days in 13 estuaries draining into the Bay of Biscay and found a good correlation with previously measured concentrations of cadmium, copper, nickel and zinc using spot water samples. Dunn et al.8 used DGT to examine the effect of tidal cycles on aqueous concentrations of copper, lead, nickel and zinc, finding it to be an accurate and useful tool for short-term deployments (6 h). Neither of these studies measured the presence of a DBL as it was assumed that in a very fast flow environment this would be negligible, however it is recommended that in future studies the DBL is always measured to ensure that its influence is minimal.
In our study there was a reduction in the TWA concentration of uranium by up to 57% when no DBL thickness was taken into consideration (Fig. 3b and 4b). The closest agreement between the concentrations was observed in weeks 19–21 for both deployment sites (Fig. 3a and 4a) when the periodically measured DBL thicknesses over the deployment period were used. When the aqueous concentration of uranium was relatively stable, the TWA estimates (taking into account the measured DBL thickness) were 99–107% and 71–111% of those found with the spot water samples for the Rivers Enborne and Lambourn respectively. When using an averaged DBL thickness over the whole deployment period, this value rose to 124–136% for the Enborne and lowered to 70–103% for the Lambourn. Using an averaged DBL thickness has less impact on the TWA concentrations in the River Lambourn than the River Enborne, most likely due to the fluctuating flow rates at the latter site. The lower flow periods, when the DBL is greater, will increase the averaged DBL thickness and will therefore result in an overestimation of the TWA estimates (Fig. 2a, weeks 17–21, 14/12/2011–8/01/2012).
To give an indication of the reliability of the DGT technique, the ratio of the TWA concentrations of uranium found with the device to the uranium concentrations found in weekly averaged spot water samples was made (Tables S5 and S6†). The closer to one this ratio is the more accurate the technique can be assumed to be, although there is the possibility that the concentrations have fluctuated throughout the week. Results are in agreement with previous work undertaken at these sites,17 approximately 86% of the dissolved uranium could be measured with accuracy. The River Enborne had an average accuracy of ∼94% (38 to 205%) and the River Lambourn ∼78% (27 to 138%). The failure to achieve 100% accuracy can be attributed to factors such as biofouling, variations in concentration of uranium over the 7 days deployment, and an underestimation of the thickness of the DBL as this was not measured every week.
Isotopic ratios of uranium
There are three naturally occurring isotopes of uranium: 238U (99.276%), 235U (0.718%) and 234U (0.0056%).49 Significant quantities of uranium occur naturally in the environment, however, this element needs to be monitored due to its toxicity, mobility and radiological properties.52 Isotopic composition can indicate if the uranium is of natural or anthropogenic origin as the 235:238 ratio is consistent in nature. As shown in Table 5 there is little difference between the isotopic composition of uranium measured in the spot water samples and DGT.| Location & technique | Average isotopic ratio | RSDa (%) | Accuracyb (%) |
|---|---|---|---|
| a Standard deviation calculated as a % of the mean (precision). b Calculated as (actual reading − measured/actual) × 100. | |||
| River Enborne DGT | 0.007302 | 2.8 | −0.72 |
| River Enborne spot | 0.007181 | 1.8 | 0.96 |
| River Lambourn DGT | 0.007314 | 2.9 | −0.88 |
| River Lambourn spot | 0.007260 | 2.6 | −0.15 |
The accuracy of the DGT is within 1%, with a relative standard deviation of 2.85%, which is comparable to Turner et al.,17 where the accuracy and precision were 1% and 10% respectively. The better precision in this study could be as a result of the longer deployment times, thereby allowing greater quantities of uranium to accumulate onto the resin. At present, slight enrichments or depletions in the 235:238 ratio would not be detectable using this technique. Further refinement would be necessary to increase the accuracy. These could include using a different uranium measurement technique (such as multi-collector ICP-MS) or by removing interferences from the eluent by using an additional actinide specific resin extraction technique.
Conclusions
The data presented here shows DGT can be used as a tool in long-term environmental monitoring programmes, even though seasonal variations in water flow and chemistry can have an impact on results. Water bodies with highly fluctuating flows require extensive DBL measurements. The thickness of the DBL is also affected by factors such as amount of SPM and degree of biofouling. Ideally, the DBL needs to be measured for each deployment. For rivers with a high degree of biological activity, samplers should be mounted in a cage, and this particularly is advisable for longer-term deployments (>4 days). In addition, recording other physical parameters such as water temperature and pH are essential in order to obtain a reliable value for the diffusion coefficient over the trial period. These factors aside, DGT can provide valuable information on labile and bio-available concentrations of wide range pollutants over long periods and give information that is complementary to that obtained with spot water sampling. The inclusion of this passive sampler in the ‘tool box’ of techniques for potential use in regulatory water monitoring programmes is justified.Acknowledgements
We acknowledge the University of Portsmouth and AWE for funding the project; Susan Atkins (University of Portsmouth) for laboratory support, Dr Gareth Old (CEH Lambourn Observatory Manager) and the Centre for Ecology and Hydrology, Wallingford, UK for use of their freshwater field site (River Lambourn) and provision of water quality data for this site; and Wasing Estate Ltd. for access to the River Enborne. We thank Graver Technologies Ltd. (http://www.gravertech.com) for the provision of the MetsorbTM resin. We also thank the two anonymous reviewers' for their helpful comments.Notes and references
- British Standards Institution, Guidance on passive sampling in surface waters, British Standards Publication, London, 2011, vol. 1, p. 23 Search PubMed.
- I. W. Croudace, P. E. Warwick and R. C. Greenwood, Anal. Chim. Acta, 2006, 577, 111–118 CrossRef CAS PubMed.
- G. Wilson, N. McAllister-Hewlings and W. G. Beard, in: AWE (Ed.) (Vol. AWE/ASc/L4/PG/EM/11/QA), AWE, 2012, p. 25.
- C. W. Anderson and S. A. Rounds, USGS 2010–5008, 2010, p. 76 Search PubMed.
- C. Murdock, M. Kelly, L.-Y. Chang, W. Davison and H. Zhang, Environ. Sci. Technol., 2001, 35, 4530–4535 CrossRef CAS.
- W. A. House and M. S. Warwick, Sci. Total Environ., 1998, 210, 111–137 CrossRef.
- I. J. Allan, J. Knutsson, N. Guigues, G. A. Mills, A. M. Fouillac and R. Greenwood, J. Environ. Monit., 2007, 9, 672–681 RSC.
- R. J. K. Dunn, P. R. Teasdale, J. Warnken and J. M. Arthur, Environ. Pollut., 2007, 148, 213–220 CrossRef CAS PubMed.
- R. P. Haas and F. Pfeiffer, in: http://www.gaiasafe.de/popsclz.pdf.
- I. J. Allen, J. Knutsson, N. Guigues, G. A. Mills, A. M. Fouillac and R. Greenwood, J. Environ. Monit., 2008, 10, 821–829 RSC.
- R. Aguilar-Martinez, R. Greenwood, G. A. Mills, B. Vrana, M. A. Palacios-Corvillo and M. M. Gomez-Gomez, Int. J. Environ. Anal. Chem., 2008, 88, 75–90 CrossRef CAS.
- R. Greenwood, G. A. Mills, B. Vrana, I. A. R. Greenwood, G. A. Mills, B. Vrana, I. Allan, R. Aguilar-Martinez and G. Morrison, in Comprehensive Analytical Chemistry, ed. R. Greenwood, G. A. Mills and B. Vrana, Elsevier, 2007, vol. 48, p. 486 Search PubMed.
- V. I. Slaveykova, N. Parthasarathy, J. Buffle and K. J. Wilkinson, Sci. Total Environ., 2004, 328, 55–68 CrossRef CAS PubMed.
- B. Vrana, G. A. Mills, I. J. Allan, E. Dominiak, K. Svensson, J. Knutsson, G. Morrison and R. Greenwood, TrAC, Trends Anal. Chem., 2005, 24, 845–868 CrossRef CAS PubMed.
- W. Davison, G. Fones, M. Harper, P. Teasdale and H. Zhang, in In situ monitoring of aquatic systems: chemical analysis and speciation, ed. J. Buffle and G. Horvai, John Wiley & Sons Ltd, 2000, p. 74 Search PubMed.
- C. M. Hutchins, J. G. Panther, P. R. Teasdale, F. Wang, R. R. Stewart, W. W. Bennett and H. Zhao, Talanta, 2012, 97, 550–556 CrossRef CAS PubMed.
- G. S. C. Turner, G. A. Mills, P. R. Teasdale, J. L. Burnett, S. Amos and G. R. Fones, Anal. Chim. Acta, 2012, 739, 37–46 CrossRef CAS PubMed.
- H. Mengistu, O. Roeyset, A. Tessema, T. Abiye and M. Demlie, Water SA, 2012, 38, 15–22 CrossRef CAS.
- Z. Dragun, B. Raspor and V. Roje, Chem. Speciation Bioavailability, 2008, 20, 33–46 CrossRef CAS.
- N. Montero, M. J. Belzunce-Segarra, J. L. Gonzalez, J. Larreta and J. Franco, Mar. Pollut. Bull., 2012, 64, 31–39 CrossRef CAS PubMed.
- M. Wallner-Kersanach, C. F. F. de Andrade, H. Zhang, M. R. Milania and L. F. H. Niencheski, J. Braz. Chem. Soc., 2009, 20, 333–340 CrossRef CAS PubMed.
- R. J. K. Dunn, P. R. Teasdale, J. Warnken and R. R. Schleich, Environ. Sci. Technol., 2003, 37, 2794–2800 CrossRef CAS.
- EC Directive 2000/60/EC, Establishing a framework for Community action in the field of water policy, 23 October 2000 Search PubMed.
- Environmental Permitting (England and Wales) Regulations 2010, Staturoty Instruments 2010 No. 675, Environmental Protection, England and Wales.
- Environmental Permitting Guidance: Radioactive Substance Regulation for the Environmental Permitting (England and Wales) Regulations 2010, DEFRA, September 2011, version 2.
- B. Docekal and M. Gregusova, Analyst, 2012, 137, 502–507 RSC.
- W. J. Li, J. J. Zhao, C. S. Li, S. Kiser and R. J. Cornett, Anal. Chim. Acta, 2006, 575, 274–280 CrossRef CAS PubMed.
- W. J. Li, C. S. Li, J. J. Zhao and R. J. Cornett, Anal. Chim. Acta, 2007, 592, 106–113 CrossRef CAS PubMed.
- H. Vandenhove, K. Antunes, J. Wannijn, L. Duquene and M. V. Hees, Sci. Total Environ., 2007, 373, 542–555 CrossRef CAS PubMed.
- J. Mihalik, P. Henner, S. Frelon, V. Camilleri and L. Fevrier, Environ. Exp. Bot., 2012, 77, 249–258 CrossRef CAS PubMed.
- L. Duquène, H. Vandenhove, F. Tack, M. Van Hees and J. Wannijn, J. Environ. Radioact., 2010, 101, 140–147 CrossRef PubMed.
- H. P. Jarvie, C. Neal, M. D. Jürgens, E. J. Sutton, M. Neal, H. D. Wickham, L. K. Hill, S. A. Harman, J. J. L. Davies, A. Warwick, C. Barrett, J. Griffiths, A. Binley, N. Swannack and N. McIntyre, J. Hydrol., 2006, 330, 101–125 CrossRef CAS PubMed.
- T. J. Marsh and M. L. Lees, Hydrological Data, United Kingdom, Hydrometric Register and Statistics 1996–2000, Centre for Ecology and Hydrology/British Geological Survey, Wallingford, 2003, p. 210 Search PubMed.
- R. M. S. Smith, D. J. Evans and H. S. Wheater, J. Hydrol., 2005, 304, 366–380 CrossRef CAS PubMed.
- K. W. Warnken, H. Zhang and W. Davison, Anal. Chem., 2006, 78, 3780–3787 CrossRef CAS PubMed.
- H. Zhang and W. Davison, Anal. Chem., 2000, 72, 4447–4457 CrossRef CAS.
- W. W. Bennett, P. R. Teasdale, J. G. Panther, D. T. Welsh and D. F. Jolley, Anal. Chem., 2010, 82, 7401–7407 CrossRef CAS PubMed.
- S. Kerisit and C. Liu, Geochim. Cosmochim. Acta, 2010, 74, 4937–4952 CrossRef CAS PubMed.
- M. Kalin, W. N. Wheeler and G. Meinrath, J. Environ. Radioact., 2004, 78, 151–177 CrossRef PubMed.
- K. W. Warnken, W. Davison and H. Zhang, Environ. Sci. Technol., 2008, 42, 6903–6909 CrossRef CAS.
- K. W. Warnken, W. Davison, H. Zhang, J. Galceran and J. Puy, Environ. Sci. Technol., 2007, 41, 3179–3185 CrossRef CAS.
- Ø. A. Garmo, O. Røyset, E. Steinnes and T. P. Flaten, Anal. Chem., 2003, 75, 3573–3580 CrossRef.
- S. Scally, W. Davison and H. Zhang, Environ. Sci. Technol., 2003, 37, 1379–1384 CrossRef CAS.
- J. G. Panther, P. R. Teasdale, W. W. Bennett, D. T. Welsh and H. J. Zhao, Environ. Sci. Technol., 2010, 44, 9419–9424 CrossRef CAS PubMed.
- M. C. A.-D. L. Torre, P.-Y. Beaulieu and A. Tessier, Anal. Chim. Acta, 2000, 418, 53–68 CrossRef.
- E. K. Lesher, B. D. Honeyman and J. F. Ranville, Geochim. Cosmochim. Acta, 2013, 109, 127–142 CrossRef CAS PubMed.
- J. L. Levy, H. Zhang, W. Davison, J. Galceran and J. Puy, Environ. Sci. Technol., 2012, 46, 3335–3342 CrossRef CAS PubMed.
- J. J. Lenhart, S. E. Cabaniss, P. MacCarthy and B. D. Honeyman, Radiochim. Acta, 2000, 88, 345–353 CrossRef CAS.
- J. Zhao, I. I. Fasfous, J. D. Murimboh, T. Yapici, P. Chakraborty, S. Boca and C. L. Chakrabarti, Talanta, 2009, 77, 1015–1020 CrossRef CAS PubMed.
- D. J. Evans and P. J. Johnes, Sci. Total Environ., 2004, 329, 145–163 CrossRef CAS PubMed.
- S. Meylan, N. Odzak, R. Behra and L. Sigg, Anal. Chim. Acta, 2004, 510, 91–100 CrossRef CAS PubMed.
- T. Mathews, K. Beaugelin-Seiller, J. Garnier-Laplace, R. Gilbin, C. Adam and C. Della-Vedova, Environ. Sci. Technol., 2009, 43, 6684–6690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c3em00574g |
| This journal is © The Royal Society of Chemistry 2014 |