 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism†
J.
Durst
*,
A.
Siebel
,
C.
Simon
,
F.
Hasché
,
J.
Herranz
and
H. A.
Gasteiger
Technische Universität München, Lehrstuhl für Technische Elektrochemie, Lichtenbergstr. 4, 85748 Garching, Germany. E-mail: julien.durst@tum.de
First published on 11th April 2014
Abstract
The effect of pH on the hydrogen oxidation and evolution reaction (HOR/HER) rates is addressed for the first time for the three most active monometallic surfaces: Pt, Ir, and Pd carbon-supported catalysts. Kinetic data were obtained for a proton exchange membrane fuel cell (PEMFC; pH ≈ 0) using the H2-pump mode and with a rotating disk electrode (RDE) in 0.1 M NaOH. Our findings point toward: (i) a similar ≈100-fold activity decrease on all these surfaces when going from low to high pH; (ii) a reaction rate controlled by the Volmer step on Pt/C; and (iii) the H-binding energy being the unique and sole descriptor for the HOR/HER in alkaline electrolytes. Based on a detailed discussion of our data, we propose a new mechanism for the HOR/HER on Pt-metals in alkaline electrolytes.
Broader contextThe future of electromobility relies on the development of cost effective and durable energy conversion systems such as fuel cells and electrolyzers. These devices, based on proton-exchange membranes (PEMs), operating at pH 0, offer high power densities, but require large amounts of noble metal for the oxygen reduction reaction (ORR) in fuel cells and the oxygen evolution reaction (OER) in electrolyzers. For the hydrogen oxidation/evolution reaction (HOR/HER), only small amounts of Pt are required to activate this reaction, without contributing to any efficiency loss of the fuel cell or electrolyzer system. In alkaline electrolyte, non-noble metal catalysts catalyze the ORR and OER at similar rates to Pt electrodes. Therefore, a replacement of the noble-metal PEM technology by alkaline membrane technology seems promising, and will offer new challenges in the field of electrocatalysis at high pH. In particular, for as yet unclear reasons, the HOR/HER kinetics on Pt are much slower in alkaline than in acid electrolytes. It is therefore critical to elucidate the reasons for the poor HOR/HER activity of Pt in alkaline electrolytes, and this will certainly come only with a deeper fundamental understanding of this reaction and its mechanism, as we are presenting in this study. |
Fuel cells and electrolyzers are important for renewable energy conversion and storage. They are currently based on proton-exchange membranes (PEMs) operating at low pH (pH ≈ 0), which offer high power densities, but require large amounts of platinum for the oxygen reduction reaction (ORR) in fuel cells1 and of Ir for the oxygen evolution reaction (OER) in electrolyzers.2 For the hydrogen oxidation/evolution reaction (HOR/HER) only very small amounts of Pt are required due to its extremely high activity for the HOR/HER.3 The H2 anode performance in PEMFCs suggested exchange current densities (i0) in the order of 102 mA cmPt−2,4 which was confirmed by mass-transport-free fuel cell measurements3,5 and microelectrode data.6 Until then, 100-fold lower i0-values for Pt in acid were reported erroneously, generally based on rotating disk electrode (RDE) measurements7,8 from which, however, the kinetics of reactions with i0-values much above the diffusion limited RDE current density (≈2–3 mA cmdisk−2) cannot be quantified.9
In an alkaline electrolyte, non-noble metal catalysts are very active for the ORR10,11 and for the OER,12,13 so that in conjunction with alkaline membranes (OH−-exchange membranes14,15) a replacement of the noble-metal intensive PEM technology by alkaline membrane technology seems promising. Unfortunately, for yet unclear reasons, the HOR/HER kinetics on Pt are much slower in alkaline than in acid electrolytes, and large amounts of Pt are needed to catalyze the HOR/HER in an alkaline environment.9 Therefore, it is critical to develop alternative HOR/HER catalysts for alkaline electrolytes and – to guide this search – to elucidate the reasons for the poor HOR/HER activity of Pt in alkaline electrolytes.
Traditionally, the overall reactions have been written either with protons in acid or with hydroxide ions in alkaline media:16
in acid:
| H2 ⇆ 2 H+ + 2 e− | (1) |
in base:
| H2 + 2 OH− ⇆ 2 H2O + 2 e− | (2) |
In acid, reaction (1) is believed to be composed of two out of three microscopic steps (Tafel/Volmer or Heyrovsky/Volmer):16
Tafel step:
| H2 ⇆ 2 Had | (3) |
Heyrovsky step:
| H2 ⇆ Had + e− + H+ | (4a) |
Volmer step:
| Had ⇆ H+ + e− | (4b) |
In base, the Heyrovsky and the Volmer steps have conventionally been written with hydroxides rather than protons as reactants:17
Heyrovsky step:
| H2 + OH− ⇆ Had + e− + H2O | (5a) |
Volmer step:
| Had + OH− ⇆ H2O+ e− | (5b) |
The different proposed reactions in acid and base, where either H+ (reactions (1), (4a), and (4b)) or H2O/OH− (reactions (2), (5a), and (5b)) would be the reacting species, have recently been used to rationalize the ≈100-fold larger i0-value of Pt in acidic vs. alkaline electrolytes.18 Under this assumption, the authors proposed that a combination of Pt (known to be active for H2 dissociation, reaction (3)) with more oxophilic components (e.g., Ru or Ni) or the use of more oxophilic metals (e.g., Ir) would enhance the interaction with H2O/OH− to yield higher HOR/HER activities in base.18
To further elucidate the HOR/HER activity differences in acid vs. base we will compare the i0-values in both electrolytes of carbon supported platinum nanoparticles (Pt/C) with those of two other carbon supported metals: palladium (Pd/C) and the more oxophilic iridium (Ir/C). Particle sizes and specific surface areas of these catalysts are listed in Table 1. A rigorous comparison has not been performed before, since the HOR/HER activity of Pd/C and Ir/C in acid might also be too high for reliable quantification by RDE experiments which has been used in the past.7,19,20 Here, we quantify for the first time the i0-values of Pd/C and Ir/C nanoparticles using mass-transport free kinetic measurements in a H2 pump configuration with proton exchange membrane based membrane electrode assemblies (MEAs).3
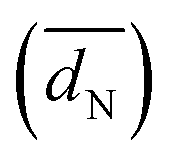 as well as TEM based and H-UPD based electrochemical active surface areas (ECSATEM and ECSAH-UPD, respectively) of Pt/C, Ir/C, and Pd/C catalysts (see the ESI1)
as well as TEM based and H-UPD based electrochemical active surface areas (ECSATEM and ECSAH-UPD, respectively) of Pt/C, Ir/C, and Pd/C catalysts (see the ESI1)
| Sample | Loading on C [wt%] |
|
ECSATEM [m2 g−1] | ECSAH-UPD [m2 g−1] |
|---|---|---|---|---|
| Pt/C | 5 | 2.2 ± 0.7 | 110 | 120 ± 20 |
| Ir/C | 20 | 3.3 ± 0.9 | 68 | 59 ± 10 |
| Pd/C | 10 | 2.8 ± 0.5 | 173 | 105 ± 20 |
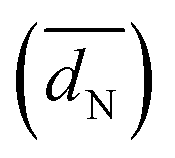
Fig. 1 displays iR-corrected HOR/HER polarization curves in units of mA cmgeo−2 for the Pt/C (black lines), Ir/C (red lines) and Pd/C (blue lines) catalysts recorded in a PEMFC setup (Fig. 1A)3 and in a 0.1 M NaOH electrolyte (Fig. 1B). At both pH values, the high oxophilicity of the Ir/C electrode is demonstrated by the decreasing HOR currents at 0.15–0.30 V vs. RHE (reversible hydrogen electrode) due to the early formation of adsorbed OHads species. This oxophilic behaviour is similar to what was reported for Ru electrodes.21 For a quantitative comparison, the geometric current densities were normalized by the metal areas obtained by H-UPD (see the ESI†) to yield specific current densities (mA cmmetal−2), plotted in Fig. 2A (PEMFC) and B (0.1 M NaOH).
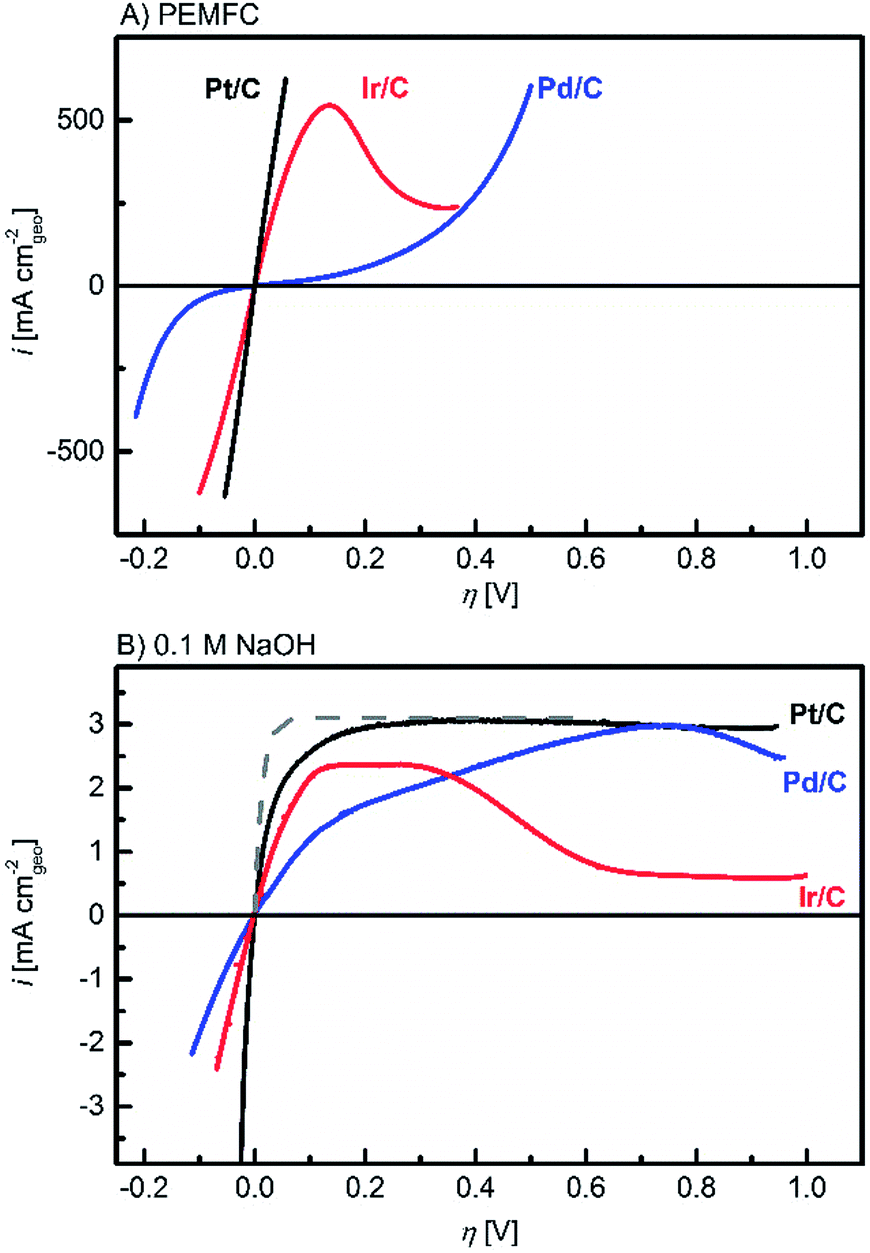 | ||
| Fig. 1 Ohmic loss corrected polarization curves in a H2-saturated atmosphere at ambient pressure and 313 K on Pt/C (black lines), Ir/C (red lines) and Pd/C (blue lines) in: (A) PEMFCs at 2 mV s−1 and (B) 0.1 M NaOH at 10 mV s−1 (the negative going scans are shown) and 1600 rpm. In (B), the diffusion overpotential, calculated according to Sheng et al.9 is also shown (grey dashed line). For metal loadings see the ESI.† | ||
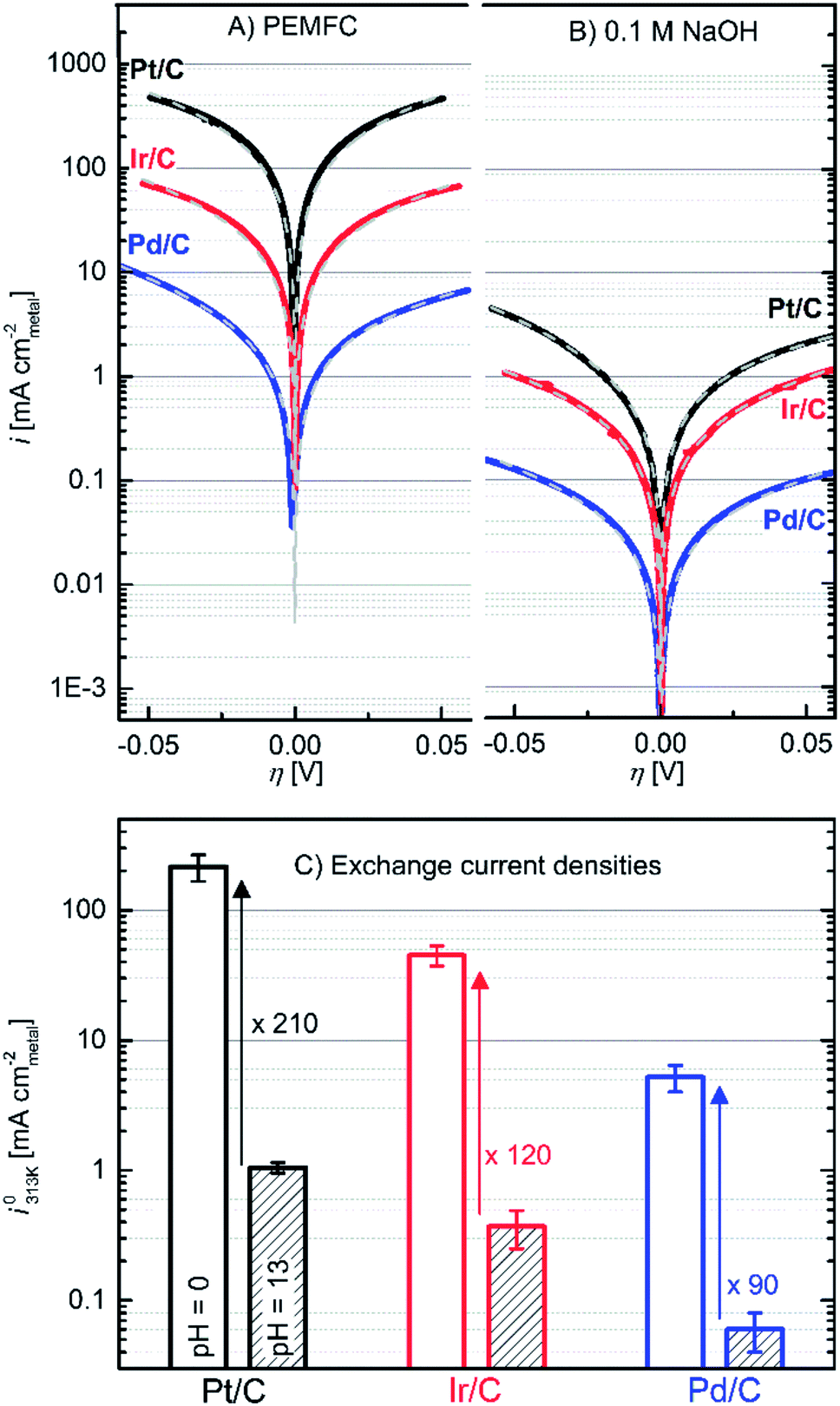 | ||
| Fig. 2 Upper part: HOR/HER Tafel plots of the specific current densities on Pt/C (black lines), Ir/C (red lines) and Pd/C (blue lines) in: (A) PEMFC and (B) 0.1 M NaOH electrolyte. The Butler–Volmer fits are indicated as grey dashed lines. The extracted fitting parameters are reported in the ESI.† Lower part: HOR/HER exchange current densities (i0313K) in PEMFC (empty columns) and 0.1 M NaOH (striped columns) averaged from Butler–Volmer and micropolarization equations. Data recorded in a H2-saturated atmosphere at ambient pressure and 313 K. | ||
The polarization curves were fitted with a simple Butler–Volmer equation to get the i0-values (i0313K, reported in Fig. 2C and Tables S1–S3†).22
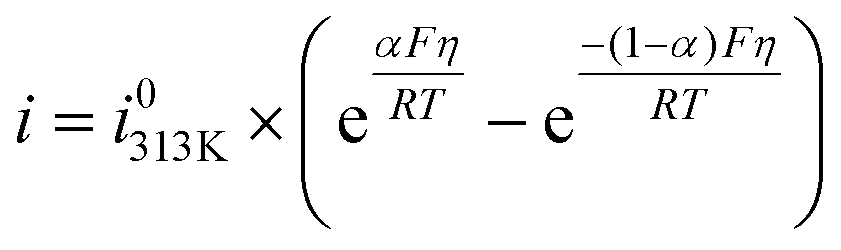 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 A s mol−1). Obviously, all metals are substantially less active in base. The reactivity follows the order Pt > Ir > Pd in each electrolyte. The i0313K values, extracted from fitting eqn (6) to the data, agree well with those obtained from the micropolarization region (−10 to +10 mV vs. RHE) described by the linearized form of the Butler–Volmer equation (see Tables S1–S3†):
485 A s mol−1). Obviously, all metals are substantially less active in base. The reactivity follows the order Pt > Ir > Pd in each electrolyte. The i0313K values, extracted from fitting eqn (6) to the data, agree well with those obtained from the micropolarization region (−10 to +10 mV vs. RHE) described by the linearized form of the Butler–Volmer equation (see Tables S1–S3†):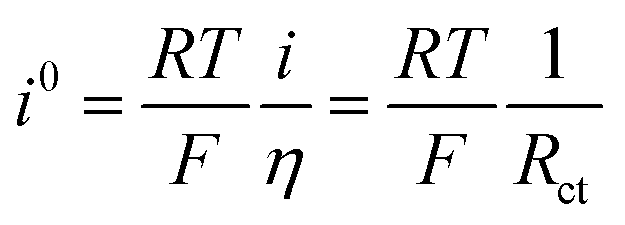 | (7) |
Owing to the large amount of data for platinum, we will first focus our discussion on Pt/C. Historically, the HOR/HER equilibrium has been described by invoking overpotential deposited hydrogen (H-OPD) as the adsorbed reaction intermediate species in the HOR/HER, which forms near and below the reversible H+/H2 potential.23 This was considered to be different in nature from the hydrogen species adsorbed on noble metal surfaces at potentials positive of the reversible H+/H2 potential, the so-called underpotential-deposited hydrogen (H-UPD). Thus, H-OPD was considered a reaction intermediate in the HOR/HER, while H-UPD was regarded as merely a spectator in the reaction. So far, no clear evidence of the difference in the physical nature of H-OPD and H-UPD species has ever been reported. On the other hand, in ab initio computational studies on the HOR/HER reaction, only a single type of adsorbed hydrogen intermediate species has been modeled, considering the three conceivable microscopic reactions, namely the Tafel, Volmer, and/or Heyrovsky reactions (usually in the form of reactions (1), (4a), and (4b)).24 However, no explicit difference has been demonstrated between the adsorbed hydrogen reaction intermediate and the H-UPD. In what follows, we will show for the first time that the HOR/HER and the H-UPD reaction, formally the same as the Volmer reaction (reaction (4b)), have identical rates, so that no H-OPD species have to be invoked to describe the HOR/HER rates.
To support this hypothesis, we will compare the charge transfer resistance for the H-UPD reaction with the exchange current density of the HOR/HER reaction. The reported H-UPD charge transfer resistances from AC impedance studies in aqueous HClO4 are ≈0.03 Ω cm2 for Pt(111)25 and ≈0.05 Ω cm2 for polycrystalline Pt;26 using eqn (7), these equate to i0-values for the Volmer reaction of ≈850 mA cmPt−2 and ≈500 mA cmPt−2, respectively. These are indeed very similar to the HOR/HOR exchange current density of i0313K ≈ 200 mA cmPt−2 on Pt/C obtained from our kinetic measurements (Fig. 2C and Table S1†). Note that an accurate determination of the H-UPD kinetics in an acid electrolyte by impedance spectroscopy is very delicate25 and will also depend on the platinum catalyst morphology (particle size, crystal face), so that a rigorous quantitative comparison can only be made when the HOR/HER exchange current density and the H-UPD charge transfer resistance are measured for the same catalyst (this will be reported in future communications27). To complete the analysis in an acidic electrolyte, Vogel et al. showed by means of gas phase hydrogen–deuterium measurements that the Tafel step (reaction (3)) could be the rate determining step (rds) followed by a fast Volmer step (reaction (4b)).28 If this were true, the Butler–Volmer relationship derived in the case of a rate limiting Tafel step16 would result in HOR and HER Tafel slopes of ≈30 mV per decade (i.e., α = 2) while our mass transport free PEMFC data show 4 times higher Tafel slopes (α = 0.5, see the ESI†), in agreement with a Butler–Volmer relationship derived for a Volmer or a Heyrovsky rate limiting step. Thus, the possibility of a Tafel rate determining step can be clearly discarded.
In 0.05 M NaOH, the charge transfer resistance of 13–54 Ω cm−2 obtained on (stepped) single crystal Pt surfaces equates to 0.2–0.5 mA cmPt−2,17 which is very close to our value of 1.0 mA cmPt−2, obtained for Pt/C in 0.1 M NaOH (Fig. 2C). This again suggests that the HOR/HER kinetics on Pt in an alkaline electrolyte are limited by the Volmer step (reactions (4b)/(5b)).
One possibility for the reduced rate of the Volmer step in base would be a higher H–Pt bond strength, which would slow down both the HOR and the HER rates. A higher H–Pt bond strength is further confirmed by the above discussed charge transfer resistance values for the H-UPD reaction, but also by the reported positive-shift of the H-UPD peaks with increasing pH. These shifts on Pt(553) and Pt(533)29 as well as on polycrystalline Pt30 amount to ≈10 mVRHE pH−1 and ≈11 mVRHE pH−1, respectively. Qualitatively, the same is observed in Fig. 3A for Pt/C, but the H-UPD peaks on Pt/C are too broad to get a sufficiently precise value. Using the values for smooth crystals, the H-UPD peak shift (ΔEH-UPD) would translate into a H-binding energy difference of ΔEbinding ≈12.5–13.5 kJ mol−1 from pH = 0 to pH = 13 (from ΔEH-UPD × F for a 1-electron process). If we assume that the difference in the H-binding energy is proportional to the difference in the activation energy (as described by the Brønsted–Evans–Polany relationship31), then the difference in the HOR/HER rate between pH = 0 and pH = 13 for Pt could be predicted by the Arrhenius equation
| rpH=0/rpH=13 ≈ exp[ΔEbinding/RT] | (8) |
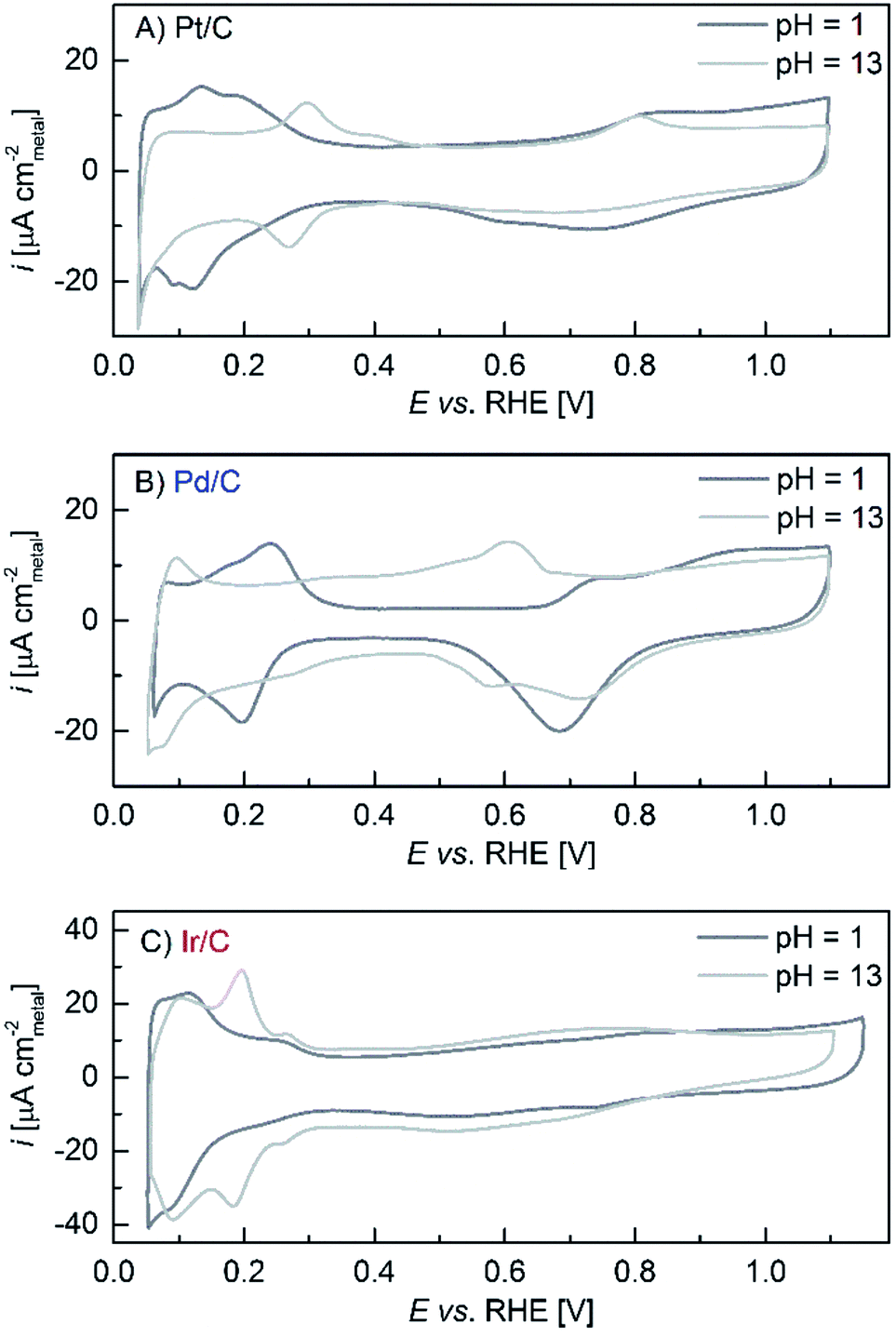 | ||
| Fig. 3 Voltammograms at 293 K recorded in an Ar-saturated atmosphere at 20 mV s−1 in 0.1 M NaOH (light grey line) and 0.1 M HClO4 (dark grey line), except for Ir/C which was recorded in 0.05 M H2SO4 since Ir reacts with ClO4− ions (see Reier et al.2 and ref. 39–41 therein). | ||
Fundamental studies of the HOR/HER on bulk Pd crystals are complicated by the fact that the H-UPD process (adsorption) is difficult to separate from H-absorption.32 Therefore, studies have mostly been focused on Pd monolayers (ML) deposited on Au substrates.33–35 While 5 ML-Pd/Au(111) surfaces also show a large increase in the H-UPD charge transfer resistance between acid36 and base,37 namely from ≈500 Ω cm2 (≡0.05 mA cmPd−2) in 0.1 M HClO4 to ≈8000 Ω cm2 (≡0.003 mA cmPd−2) in 0.1 M NaOH, there is no quantitative agreement with the i0-values shown in Fig. 2C, as was the case for Pt. This may be related to residual H-absorption effects and, as was mentioned by the authors, contamination effects in their NaOH electrolyte.37 Overall, however, these measurements rationalize the here-observed decrease in HOR/HER activity with increasing pH (Fig. 2C) and also point towards the Volmer step as rate limiting.
Iridium being the most oxophilic of the three surfaces (Fig. 1) is an interesting case, as it probes the hypothesis advanced by Strmcnik et al. that a more oxophilic surface might be effective in catalyzing the interaction with H2O/OH− in reaction (2) which they proposed to be the governing reaction in base.18 However, our findings demonstrate explicitly that Ir does not have a higher activity than Pt in base (nor in acid), which is in disagreement with Strmcnik et al.18 and would lead to the conclusion that OH− adsorption or H2O dissociation is not the rate determining step in base. The reason for this discrepancy might be that all polarization curves reported in Fig. 1 were recorded after a stable state of the surface was reached (after a few cyclic voltammograms), while Strmcnik et al. only showed the very first anodic scan.18 These authors also provided HOR/HER first-scan data on Pt0.5Ru0.5 and Pt0.1Ru0.9 bulk crystals, whereby the latter showed the highest activity, supporting their argument that more oxophilic surfaces would promote the HOR/HER kinetics. While Strmcnik et al. considered the strong Pt surface enrichment on their vacuum-annealed Pt–Ru alloys,38 they erroneously quoted from ref. 38 that the surface composition of Pt0.5Ru0.5 and Pt0.1Ru0.9 would be 90 at.% and 50 at.% Pt, respectively, while the correct values are 85 at.% and 90 at.% Pt, respectively (the reasons are discussed in ref. 38). Thus, the most active surface was the one with the lowest Ru surface composition (annealed Pt0.1Ru0.9), i.e., the less oxophilic. Another argument in support of their hypothesis that the HOR/HER in base requires the interaction with H2O/OH− as suggested by reactions (2) and (5b) was based on their kinetic model to describe the pH dependence of the HOR/HER on Pt in unbuffered electrolytes between pH = 2.5 and pH = 10.5. From this they deduced that the reaction mechanism would change when going from acid (reaction (1)) to base (reaction (2)). This conclusion, however, is invalid since recent calculations and experiments by Auinger et al. clearly showed that the observed response in unbuffered solutions is merely due to local pH gradients and does not reflect any changes of the HOR/HER kinetics.39
So far we conclude that there is no substantiated evidence that the HOR/HER reactions in base proceed according to reactions (2) and (5b). In our opinion, reaction (2) merely represents an algebraic exercise, i.e., adding 2 OH− to both sides of reaction (1) (the same is true for the Volmer steps, reactions (4b) and (5b)). Considering that the bulk of aqueous solutions of 0.1 M base and 0.1 M acid contains molecular ratios of H2O/OH− and H2O/H+ of 550/1 (grantedly, this ratio might be somewhat lower at the metal/solution interface) and that H2O is a very fast H+ donor/acceptor, we believe that the microscopic reactions for the HOR/HER are represented by reactions (3) and (4b) (or, alternatively, reactions (4a) and (4b)). While the overall reaction (2) will still hold true, it is, however, more likely composed of coupled surface and bulk reactions:
Σ(surface reactions):
| H2 ⇆ 2 H+ + 2 e− | (9) |
H2O dissociation:
| 2 H+ + 2 OH− ⇆ 2 H2O | (10) |
The sum of reactions (9) and (10), of course, will yield the overall reaction (2). Since H2O is a fast H+-donor/acceptor and since it is the major species in the solution (and at the metal/solution interface), we believe that the microscopic Heyrovsky (reaction (4a)) and Volmer (reaction (4b)) steps do not change with pH. This would be consistent with the observation that the HOR/HER kinetics between acid and base change by a very similar factor for the three studied Pt metals and that the H-binding energy (affected for each metal by the alkalinity of the environment, as can be deduced from the H-UPD shifts shown in Fig. 3) seems to be a good descriptor for the HOR/HER both in acid and base.40 While we cannot definitively prove this hypothesis, it is certainly the simplest mechanism which is consistent with the data.
Conclusions
In conclusion, we have shown that the HOR/HER activity at high pH follows the order Pt > Ir > Pd, with exchange current densities which are much larger than those measured7 and calculated24 previously. We have also shown for the first time that the exchange current densities on carbon-supported Pt, Pd and Ir electrodes are all decreased by about two orders of magnitude when moving from low (pH = 0) to high pH (pH = 13). This result is a clear proof that the oxophilicity of a catalyst (Ir being more oxophilic than Pt and Pd) does not enhance the HOR/HER activities in high pH. From a mechanistic point of view, by comparing our measured HOR/HER kinetics with published H-UPD charge transfer resistance values, we have provided substantial evidence that the Volmer step is the rate determining step on noble metal electrodes and that therefore no H-OPD species have to be invoked to describe the HOR/HER rates. Furthermore, our analysis suggests that the H-UPD reaction is identical to the Volmer reaction, in support of the hypothesis raised by Chen and Kucernak.6 Addressing now the pH effect, our results point toward identical microscopic HOR/HER reaction steps in acid and base and that the H-binding energy is the relevant descriptor in both electrolytes, even though the origin of the difference in H-binding energy is not clear at this point. None of the current DFT models considers any effect of pH on the H-binding energy.24,40,41 In a recent study, Rossmeisl et al. initiated an attempt to address pH in DFT calculation in order to examine its effect on adsorbate coverage and configuration at the Pt(111)/electrolyte interface42 and proposed that the local water configuration might be different in acid vs. base, thus affecting the HOR/HER kinetics; however, its effect on the H-binding energy was not determined even though our data suggest that this might be the origin of the large observed differences.Based on the above analysis, the design of new and advanced electrocatalysts for the hydrogen oxidation and evolution reaction in alkaline fuel cells and electrolyzers would benefit from: (i) improved computational calculations/models which include the pH effect on the H-binding energy, (ii) the development of experimental methods which allow quantification of the H-binding energy, and (iii) the synthesis of electrocatalysts with tuned H-binding energies.
Acknowledgements
The authors would like to thank Dr Marianne Hanzlik for TEM measurements, and Tanaka Kikinzoku Kogyo and Premetek for the supply of the catalysts.Notes and references
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- K. C. Neyerlin, W. Gu, J. Jorne and H. A. Gasteiger, J. Electrochem. Soc., 2007, 154, B631–B635 CrossRef CAS PubMed.
- H. A. Gasteiger, J. E. Panels and S. G. Yan, J. Power Sources, 2004, 127, 162–171 CrossRef CAS PubMed.
- X. Wang, R. Ahluwalia and A. Steinbach, J. Electrochem. Soc., 2013, 160, F251–F261 CrossRef CAS PubMed.
- S. Chen and A. Kucernak, J. Phys. Chem. B, 2004, 108, 13984–13994 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- N. Markovic, B. Grgur and P. Ross, J. Phys. Chem. B, 1997, 101, 5405–5413 CrossRef CAS.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1263–B1268 CrossRef CAS PubMed.
- T. Poux, F. Napolskiy, T. Dintzer, G. Kéranguéven, S. Y. Istomin, G. Tsirlina, E. Antipov and E. Savinova, Catal. Today, 2012, 189, 83–92 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS PubMed.
- C. G. Arges, V. Ramani and P. N. Pintauro, Electrochem. Soc. Interface, 2010, 31 CAS.
- K. Krischer and E. R. Savinova, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Chichhester, 2009, pp. 1873–1905 Search PubMed.
- K. Schouten, M. van der Niet and M. Koper, Phys. Chem. Chem. Phys., 2010, 12, 15217–15224 RSC.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. Van der Vilet, A. P. Paulikas, V. Stamenkovic and N. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef PubMed.
- M. Shao, J. Power Sources, 2011, 196, 2433–2444 CrossRef CAS PubMed.
- S. N. Pronkin, A. Bonnefont, P. S. Ruvinskiy and E. R. Savinova, Electrochim. Acta, 2010, 55, 3312–3323 CrossRef CAS PubMed.
- H. A. Gasteiger, N. M. Markovic and P. N. Ross Jr, J. Phys. Chem., 1995, 99, 16757–16767 CrossRef CAS.
- J. S. Newman, Electrochemical Systems, Prentice Hall, New York, 1991 Search PubMed.
- B. Conway and B. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. Gudmundsdottir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 Search PubMed.
- E. Sibert, R. Faure and R. Durand, J. Electroanal. Chem., 2001, 515, 71–81 CrossRef CAS.
- B. Łosiewicz, R. Jurczakowski and A. Lasia, Electrochim. Acta, 2012, 80, 292–301 CrossRef PubMed.
- P. J. Rheinländer, J. Durst, J. Herranz and H. A. Gasteiger, in preparation.
- W. Vogel, L. Lundquist, P. Ross and P. Stonehart, Electrochim. Acta, 1975, 20, 79–93 CrossRef CAS.
- M. J. van der Niet, N. Garcia-Araez, J. Hernández, J. M. Feliu and M. Koper, Catal. Today, 2013, 202, 105–113 CrossRef CAS PubMed.
- R. Gisbert, G. García and M. Koper, Electrochim. Acta, 2010, 55, 7961–7968 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin and M. Mavrikakis, J. Catal., 2002, 209, 275–278 CrossRef.
- M. Baldauf and D. Kolb, Electrochim. Acta, 1993, 38, 2145–2153 CrossRef CAS.
- L. A. Kibler, A. M. El-Aziz, R. Hoyer and D. M. Kolb, Angew. Chem., Int. Ed., 2005, 44, 2080–2084 CrossRef CAS PubMed.
- C. Gabrielli, P. Grand, A. Lasia and H. Perrot, J. Electrochem. Soc., 2004, 151, A1925–A1936 CrossRef CAS PubMed.
- C. Gabrielli, P. Grand, A. Lasia and H. Perrot, J. Electrochem. Soc., 2004, 151, A1937–A1942 CrossRef CAS PubMed.
- H. Duncan and A. Lasia, Electrochim. Acta, 2007, 52, 6195–6205 CrossRef CAS PubMed.
- M. Martin and A. Lasia, Electrochim. Acta, 2009, 54, 5292–5299 CrossRef CAS PubMed.
- H. Gasteiger, P. N. Ross Jr and E. Cairns, Surf. Sci., 1993, 293, 67–80 CrossRef CAS.
- M. Auinger, I. Katsounaros, J. C. Meier, S. O. Klemm, P. U. Biedermann, A. A. Topalov, M. Rohwerder and K. J. J. Mayrhofer, Phys. Chem. Chem. Phys., 2011, 13, 16384–16394 RSC.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef PubMed.
- J. Rossmeisl, K. Chan, R. Ahmed, V. Tripkovic and M. Bjorketun, Phys. Chem. Chem. Phys., 2013, 15, 10321–10325 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, PEMFC and RDE measurement setups and extraction of exchange current densities from the polarization curves. See DOI: 10.1039/c4ee00440j |
| This journal is © The Royal Society of Chemistry 2014 |