 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional graphene materials: preparation, structures and application in supercapacitors
Xiehong
Cao
,
Zongyou
Yin
and
Hua
Zhang
*
School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: HZhang@ntu.edu.sg; Web: http://www.ntu.edu.sg/home/hzhang/
First published on 24th February 2014
Abstract
Three-dimensional (3D) graphene materials (3DGMs) are of great importance due to their unique properties and practical applications. A number of 3DGMs with novel structures have been developed in recent years. This review presents the current progress of 3DGMs. After introducing the preparation strategies of 3DGMs, we summarize the reported 3DGMs based on their different structures, and then focus on the description of their preparation methods, properties and applications. Lastly, the applications of 3D graphene-based materials in supercapacitors are described.
 Xiehong Cao | Xiehong Cao received his B.E. in the Department of Polymer Materials and Engineering in Zhejiang University, China (2008). He completed his PhD under the supervision of Prof. Hua Zhang in the School of Materials Science and Engineering of Nanyang Technological University, Singapore (2012). He is currently working as a Research Fellow in Prof. Hua Zhang's group. His research interests include the synthesis, characterization and applications of two-dimensional nanomaterials. |
 Zongyou Yin | Zongyou Yin studied at Jilin University in China for his B.E. and M.S., and completed his PhD at Nanyang Technological University in Singapore (2008). He joined Prof. Hua Zhang's group in 2008 as a Postdoctoral Fellow, and then worked as a Scientist II in Institute of Materials Research and Engineering at A*STAR of Singapore in 2013. He is currently a Postdoctoral Associate in Massachusetts Institute of Technology in USA. His research interests include the synthesis of low-dimensional nanomaterials and their applications in energy, environment and optoelectronics. |
 Hua Zhang | Prof. Hua Zhang obtained his B.S. and M.S. from Nanjing University in 1992 and 1995, respectively, and completed his PhD with Prof. Zhongfan Liu at Peking University in 1998. As a Postdoctoral Fellow, he joined Prof. Frans C. De Schryver's group at Katholieke Universiteit Leuven (Belgium) in 1999, and then Prof. Chad A. Mirkin's group at Northwestern University in 2001. After working at NanoInk Inc. (USA) and Institute of Bioengineering and Nanotechnology (Singapore), he joined Nanyang Technological University in July 2006. His current research interests focus on two-dimensional nanomaterials including graphene and metal dichalcogenides, and their applications in nano- and bio-sensors, clean energy, water, environment, etc. |
Broader contextIn recent years, the assembly of graphene into macroscopic three-dimensional (3D) structures has been attracting intensive interest, because the utilization of 3D graphene materials (3DGMs) is one of the most effective ways to apply the unique properties of two-dimensional (2D) graphene nanosheets in practical applications. For example, the 3D porous graphene film not only facilitates the access of electrolyte to its surface, but also provides electrically conductive channels for the active materials anchored on it, leading to the enhanced performance of both electric double-layer capacitors and pesudocapacitors. Graphene fibers are highly flexible and easy to be functionalized, which makes them promising for flexible supercapacitors. Until now, a lot of effort has been devoted in order to control the morphologies and properties of 3DGMs, which are mainly aimed at obtaining 3DGMs with a porous structure, large surface area, good electrical conductivity, and high mechanical strength. 3DGMs can also be functionalized and integrated into devices easily, and are compatible with conventional material processing. This review paper summarizes various structures of 3DGMs together with their synthetic methods, properties and applications. In addition, the current challenge of the 3DGMs is also proposed. |
1. Introduction
Graphene, a two-dimensional (2D) carbon sheet, has been used as one kind of promising material.1 Due to its special structure, graphene possesses a lot of unique properties in chemistry, physics and mechanics.2–4 Graphene has a carrier mobility up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1 and a thermal conductivity of 3000–5000 W m−1 K−1 at room temperature,1,5 a high surface area of ∼2630 m2 g−1,6 good optical transparency of ∼97.3% (ref. 7) and excellent mechanical strength with a Young's modulus of 1.0 TPa.8 Various synthetic methods of graphene including mechanical cleavage,1 epitaxial growth,9 graphitization,10 exfoliation11 and chemical vapor deposition (CVD)12,13 have been developed in the past few years, which have promoted the intensive study of graphene-based materials in numerous research areas, such as electronics,14–22 energy storage applications,23–28 sensors29–36 and so on.
000 cm2 V−1 s−1 and a thermal conductivity of 3000–5000 W m−1 K−1 at room temperature,1,5 a high surface area of ∼2630 m2 g−1,6 good optical transparency of ∼97.3% (ref. 7) and excellent mechanical strength with a Young's modulus of 1.0 TPa.8 Various synthetic methods of graphene including mechanical cleavage,1 epitaxial growth,9 graphitization,10 exfoliation11 and chemical vapor deposition (CVD)12,13 have been developed in the past few years, which have promoted the intensive study of graphene-based materials in numerous research areas, such as electronics,14–22 energy storage applications,23–28 sensors29–36 and so on.
Recently, three-dimensional (3D) graphene materials (3DGMs) have been attracting much attention, since they not only possess the intrinsic properties of 2D graphene sheets, but also provide advanced functions with improved performance in various applications.37–42 In particular, due to their unique mechanical characteristics, excellent electrical conductivity and large surface area, 3DGMs have emerged as promising candidates for supercapacitors.43–45
Massive production of graphene sheets is of great importance for the fabrication of 3DGMs. The method involving preparation of graphene oxide (GO) followed by a reduction process is the most common way to obtain graphene materials.11 Although the resultant reduced graphene oxide (rGO) sheets contain defects and exhibit low electrical conductivity, the advantages of high throughput and low cost of this method make GO and rGO favorable as building blocks for the fabrication of graphene-based materials.3,46–55 Alternatively, the chemical vapor deposition (CVD) method is an effective way to produce graphene of a high quality, similar to that of pristine graphene.12,13 Recently, a 30 inch graphene film has been prepared through the CVD method, indicating the potential of large-scale production of graphene through this method.56
To date, a number of synthetic methods for 3DGMs, based on the strategies of either self-assembly, template-assisted preparation or direct deposition, have been developed in recent years. Numerous 3DGMs with various structures and unique functions have emerged. In this review, we mainly focus on the description of the structures of 3DGMs, and then introduce their preparation methods and properties. Moreover, the supercapacitor application of 3D graphene-based materials is also reviewed.
2. Preparation methods
In the past few years, tremendous efforts have been devoted to the development of synthetic methods for 3DGMs with various morphologies, structures and properties, in order to satisfy the requirements arising from different applications. In this section, the preparation methods are generally classified as self-assembly, template-assisted preparation and direct deposition.2.1. Self-assembly
Self-assembly is one of the most commonly used strategies to obtain 3DGMs. A lot of methods based on this strategy have been developed. As a typical example, 3D graphene structures can be produced through the gelation process of GO dispersion followed by reduction to convert GO to rGO.57 In a stable GO dispersion, there is a force balance between the van der Waals attractions from the basal planes of GO sheets and the electrostatic repulsions from the functional groups of GO sheets, which makes GO sheets well dispersed in an aqueous solvent. Gelation of the GO dispersion occurs once the force balance is broken. During the gelation process, GO sheets partially overlap to form GO hydrogels with 3D architectures. After further reduction of the GO hydrogels, 3D rGO networks are obtained. There are many ways to trigger the gelation of a GO dispersion, such as addition of cross-linkers,58 changing the pH value of the GO dispersion,57 or ultrasonication of the GO dispersion.59 Polyvinyl alcohol (PVA) is a cross-linker first reported by Shi et al., which can enhance the attraction of GO sheets and promote the gelation process of a GO dispersion.58 Moreover, a lot of other materials have also been used as the cross-linkers for the self-assembly of GO sheets, such as DNA,60 metal ions,61–63 polymers,64 organic molecules,65 and so on. Besides the methods based on the gelation process of a GO dispersion, the self-assembly of GO sheets into 3D structures has also been achieved by other methods, such as direct freeze-drying,66 tape casting,67 controlled filtration68 and centrifugation of GO dispersions,69 electrochemical deposition,70 sol–gel reaction,71,72 and so on. Alternatively, 3D rGO architectures can be directly obtained through the hydrothermal73–80 or chemical81 reductions of GO sheets. In these cases, GO sheets are self-assembled to form 3D networks, and at the same time converted to rGO.2.2. Template-assisted preparation
Compared with the self-assembly methods, by using pre-designed 3D templates, 3DGMs with much more controlled morphologies and properties can be obtained. This strategy has been well demonstrated by the direct growth of graphene on 3D templates using CVD methods.37,40,82 For example, by using commercially available Ni foam as both the template and catalyst, 3D graphene networks (3DGNs) were successfully synthesized.37,40 Moreover, anodic aluminum oxide (AAO),83 MgO,84 nickel-coated pyrolyzed photoresist films,85,86 metal nanostructures82,87,88 and even metallic salts38,89 have also been used as templates to produce 3DGMs. Compared to the conventional CVD process, which uses flat metal substrates as templates and normally produces a limited amount of graphene, large-amounts of graphene materials can be achieved by using 3D templates. This greatly benefits the applications that require a large quantity of graphene.Alternatively, 3D graphene architectures can be obtained in another convenient way via the assembly of GO sheets onto 3D templates followed by the reduction of GO to rGO. Many assembly techniques have been developed, such as electrophoretic deposition,90 dip-coating,91 refluxing in an autoclave92,93 and template-assisted freeze-drying.94 The templates used in these methods are not limited to metal substrates; non-metals including silica nanoparticles (NPs),95–97 polystyrene (PS) balls,44,98–100 Nafion scaffolds,101 commercially available sponges,102,103 cellulose104 and textile fibers105 have also been reported.
2.3. Direct deposition
As a straightforward strategy, the direct deposition of 3D graphene architectures on conductive substrates, e.g. Au and stainless steel, has been demonstrated by plasma-enhanced CVD (PECVD) methods.42,106–108 The graphene sheets were vertically grown on the substrate and connected with each other to form 3D porous graphene, which firmly adhered to the substrate. Importantly, the numerous active sites at the edges of the vertical graphene sheets make this 3D graphene suitable for sensing applications. For example, Chen et al. fabricated a biosensor, composed of graphene sheets vertically grown on a gold electrode and Au NP–antibody conjugates, that could provide a low detection limit of ∼2 ng mL−1 of immunoglobulin G.42 In addition, the placement of 3DGMs can also be easily controlled by patterning the metal substrate with designed features, which might enable construction of various sensor structures for different applications.1073. Structures
Benefiting from the rapid development of preparation methods in previous years, a number of 3DGMs with different structures, morphologies and properties have been prepared. In addition, these 3DGMs have also shown promising applications in many areas. Table 1 summarizes the most typical structures of 3DGMs, along with their preparation methods, properties and applications.| Structures | Synthetic methods | Properties | Applications | Ref. |
|---|---|---|---|---|
| 3D graphene networks | CVD using NiCl2·6H2O as catalyst precursor | Surface area: ∼560 m2 g−1, electrical conductivity: ∼12 S cm−1 | Absorbent | 38 |
| CVD based on Ni foam template | Surface area: ∼850 m2 g−1, electrical conductivity: 10 S cm−1, tensile strain: ∼95% | 37 | ||
| Self-assembly of GO sheets induced by hydrothermal reaction | Electrical conductivity: ∼0.0025 S cm−1, compressive strength: ∼0.042 MPa, compression modulus: ∼0.26 MPa | Catalysis | 74 | |
| Graphene fibers | Wet-spinning | Surface area: ∼884 m2 g−1, electrical conductivity: 2600–4900 S cm−1, specific tensile strength: 188 kN m kg−1, compression modulus: 3.3 MPa | Conductive wire | 166 |
| Wet-spinning | Electrical conductivity: 8–10 S cm−1, tensile strength: 140–150 MPa | Micro-pump | 168 | |
| Wet-spinning | Electrical conductivity: ∼35 S cm−1, tensile strength: ∼182 MPa, Young's modulus: 8.7 GPa | Conductive wire | 164 | |
| Graphene tubes | Hydrothermal using Cu wire as template | Electrical conductivity: 10 S cm−1, tensile strength: ∼180 MPa | Self-powered micromotor | 173 |
| CVD using AAO as template | Electrical conductivity: 950 S m−1, thermal conductivity: 8.28 W m−1 K−1 | Heat transfer and thermal energy storage | 83 | |
| 3D porous graphene films | Leavening strategy | Sheet resistance: <100 Ω sq−1, tensile strength: ∼3.2 MPa | Supercapacitor | 45 |
| Assembly of chemically modified graphene using PS particles as template | Surface area: 194.2 m2 g−1, electrical conductivity: 1024 S cm−1 | Supercapacitor | 100 | |
| Graphene balls | Aerosol-assisted capillary compression process | Surface area: 82 m2 g−1, compression strength: >55 MPa | Microbial fuel cell | 177 |
| CVD using PS ball as template | Surface area: 508 m2 g−1, electrical conductivity: 6.5 S m−1 | Supercapacitor | 44 | |
| Honeycomb-like 3D graphenes | Freeze-casting | Electrical conductivity: ∼0.12 S m−1, compression strength: ∼8 kPa (plateau state), 18 kPa (80% strain), for sample with density of 5.1 mg cm−3 | 184 | |
| Self-assembly | Electrical conductivity: 649 S m−1 | Supercapacitor | 197 |
3.1. 3D graphene networks (3DGNs)
3DGNs, including graphene foams,41,96,109 sponges,102,110 hydrogels111,112 and aerogels,66,78,94 are one of the most reported 3D graphene structures, which have shown promising properties for numerous applications. As an example, Yu et al. utilized commercially available polyurethane sponge (PU) as a template, and fabricated a 3D rGO–PU sponge based on a simple dip-coating method.102 This rGO–PU sponge showed high sensitivity to the pressure applied on it, which was able to detect a minimum pressure of 9 Pa, making it promising as a flexible and sensitive pressure sensor. As shown in Fig. 1a and b, the artificial skin, which was made of a rGO–PU sponge covered by electrode arrays, was able to measure the spatial distribution of pressure applied on it. In addition, 3D rGO networks were also obtained by electrochemical methods, in which GO sheets were electrochemically reduced and then deposited on the electrodes.70,113 | ||
| Fig. 1 (a) Photograph of the artificial skin made from a rGO–PU sponge. Inset: photograph of the rGO–PU sponge. (b) The mapping profile of resistance change of the rGO–PU sponge in (a), which was caused by pressing the bipod (70 g). Reproduced with permission from ref. 102, copyright 2013, WILEY-VCH Verlag GmbH & Co. KGaA. (c and d) Comparison of the 3DGMs obtained by using templates of NiCl2·6H2O (c) and Ni foam (d). Inset in (c): high-magnification SEM image of the 3DGM in (c). Reproduced by permission from Macmillan Publishers Ltd: ref. 38, copyright 2013. | ||
Although many methods have been developed to prepare 3D rGO networks, the resultant materials normally were of low quality and exhibited poor electrical conductivity. Recently, other approaches, including the CVD method, have been reported for preparing high-quality 3DGNs. In a typical CVD process, graphene is deposited on the surface of metal substrates, e.g. commonly used Cu or Ni films or foils,12,13 where the metal substrates serve as both catalysts and templates. Therefore, it is reasonable to think that 3D graphene architectures can be prepared in the CVD process by using pre-fabricated 3D metal substrates as templates. Since a 3D metal substrate has a larger surface area compared to a flat metal substrate, a higher amount of graphene is expected for the same growth time. Recently, Ni foam with a pore size of several hundred micrometers, commonly used as the current collector in energy storage applications, has been successfully used as the template for preparation of 3DGNs through the CVD method.37,40 The D-band, a characteristic peak of graphene related to the density of disordered carbon,114 was negligible in the Raman spectra of 3DGNs, indicating the high quality of the obtained graphene film.37,40 The surface area of 3DGNs is dependent on the layer number of the graphene film, which is as high as ∼850 m2 g−1 for 3DGNs composed of a 3-layer graphene film.37 Importantly, the 3DGNs have also exhibited unique properties in electrical conductivity,37 mechanical strength37,115,116 and thermal conductivity.117 Our work also showed 3DGNs of ∼0.1 g per batch can be achieved in an atmospheric pressure CVD process using ethanol as the carbon source,40 indicating the potential for massive production of CVD-synthesized, high-quality graphene material at low cost. Until now, a number of graphene composites based on 3DGNs have been also demonstrated, which were fabricated through various methods such as hydrothermal,118,119 electrochemical deposition,120 CVD121,122 and so on. These 3DGN-based composites have shown superior properties and performance in various applications including supercapacitors,40,118,119,123 sensors,120,124 batteries,115,121,125–127 hydrogen evolution reaction (HER),128 oxygen reduction reaction (ORR),129 electromagnetic interference shielding,116 light-emitting diodes (LED),130 solar cells,131 and cell cultures.132
Using Ni foam as a template in the CVD process is an effective way to produce 3DGMs with a controlled morphology. However, the obtained products possessed large pore sizes (hundreds of micrometers) with high porosity (∼99.7%).37 To further reduce the pore size and increase the yield of graphene, other templates have been explored. For example, by using commercially available Ni NPs below 30 μm in size as the template, 3DGNs were obtained.82 During the annealing process at high temperature, Ni NPs were melted and agglomerated together to form a 3D Ni template. The yield of the obtained graphene was ∼2.5% of the weight of the Ni NPs used. Lee et al. further simplified the CVD process for synthesis of 3DGNs by annealing a mixture of Ni powder and poly(methyl methacrylate) (PMMA) at low pressure.133 PMMA has already been demonstrated as an effective solid carbon source for the preparation of graphene on flat metal substrates.134 Compared to common gaseous carbon sources, e.g. CH4, the use of PMMA makes the CVD process cheaper and safer. In addition, the instrument setup is also simplified due to removal of the corresponding gas lines and accessories of the CVD system. The yield of graphene in Lee et al.'s work was 0.5 g if 20 g Ni powder was used.
Recently, a nickel salt (NiCl2·6H2O) was also used as both the catalyst precursor and template for 3DGNs.38 During the annealing of NiCl2·6H2O under a mixture of H2–Ar gas at 600 °C, a mixture of gas containing water vapor and hydrogen chloride (product of the reaction between NiCl2 and H2) was rapidly released, resulting in the formation of a 3D Ni skeleton. After the subsequent CVD process operated at 1000 °C followed by etching of Ni, a 3D graphene macroscopic object (3D-GMO) was obtained (Fig. 1c). As shown in Fig. 1c and d, the pore size of the 3D-GMO (several micrometers) was much smaller than that of the 3DGN obtained from Ni foam (several hundred micrometers).38 The smaller pore size also resulted in a relatively higher density of 3D-GMO ranging from ∼22 to ∼100 mg cm−3, compared with that of 3DGN (∼1 mg cm−3).38 Due to a high surface area (∼560 m2 g−1), the 3D-GMO showed excellent performance in the removal of heavy metal ions.
Alternatively, 3DGNs can also be obtained by microwave irradiation of graphite powders,135 or the direct graphitization process of some carbon-containing substances, such as resin136 and sugar.137
3.2. 3D porous graphene films
Due to the π–π stacking interaction and van der Waals attraction among their basal planes, GO or rGO sheets tend to stack in a graphitic structure, leading to significant loss of their surface area. The restacking and aggregation of GO or rGO sheets also hamper the large-scale usage and process of graphene materials in many applications, especially energy storage devices,138 because it is difficult for electrolyte ions to access the interspace among the densely packed graphene sheets.To solve this problem, fabrication of a 3D porous graphene film by the incorporation of spacer materials between the graphene sheets is an effective way to retain the surface area of the graphene. The spacer materials can be carbon nanomaterials,139–141 nanodiamond,142 polymers,21,143 noble metal nanocrystals,144,145 metal oxides,54,146 mesoporous silica sheet,147 metal organic frameworks (MOF)148 and so on. Moreover, water molecules were also reported by Li et al. as a spacer material for enlarged interspacing between stacked chemically converted graphene (CCG) sheets.68 Although the obtained graphene film contained over 92 wt% water, it still exhibited high electrical conductivity with a sheet resistivity of 1860 Ω sq−1. This is because the CCG sheets in the wet film still have a nearly face-to-face-stacked morphology, which ensures the effective electron transport paths presented in the graphene film. Their recent work showed that non-volatile liquids can also act as spacer materials.149 In their work, the wet CCG film prepared by a filtration method was placed in a mixture containing volatile and non-volatile liquids, e.g. H2O–H2SO4. After the water in the CCG film was completely exchanged by the mixture, the film was placed in a vacuum oven to evaporate the volatile solvent, and the remaining non-volatile liquid prevented the restacking of the CCG sheets. The packing density of the CCG film was also increased after evaporation of the volatile liquid. Compared with the other porous graphene films with a packing density of 0.05–0.75 g cm−3,150,151 the graphene film obtained by Li et al. showed a higher value of ∼1.33 g cm−3. Importantly, the used non-volatile liquids, e.g. H2SO4 and 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIMBF4), can serve as electrolytes, which enables the direct integration of the obtained graphene films into supercapacitor devices.
Other methods without the use of spacer materials, such as tap casting,67 light scribing,152,153 leavening,45 and chemical activation,154–156 through treatments of GO/rGO films have also been reported to fabricate 3D porous graphene films. In a recent work, Chen et al. used a so-called “leavening” strategy, similar to the process of baking bread, to fabricate a porous rGO film, in which the compacted GO film acted as the “dough”.45 After the GO film was first prepared by filtration of a GO dispersion through an AAO membrane, it was peeled off from the AAO membrane and then placed in an autoclave with hydrazine at 90 °C for 10 h. During this process, numerous pores were formed in the rGO film, which was attributed to the rapid release of gaseous species, due to reduction of GO, from the compact film. Because of its porous structure and the hydrophobic nature of rGO, the resultant rGO film exhibited an improved absorption ability to organic solvents, such as motor oil and petroleum, compared with the compact rGO film and graphite.45
Recently, 3D porous graphene films have also been fabricated using PS nanospheres,100,157 silica particles,95 or PMMA spheres158 as templates. For example, the porous chemically modified graphene (CMG) film with a uniform pore size of ∼2 μm was obtained by filtration of an aqueous mixture of PS nanospheres and CMG sheets followed by the removal of PS (Fig. 2).100 The resultant porous film possessed a high electrical conductivity of 1204 S m−1 and a surface area of 194.2 m2 g−1, which was subsequently functionalized with MnO2 and then used as a supercapacitor electrode.
 | ||
| Fig. 2 (a) Schematic illustration of the synthesis process of 3D porous MnO2/CMG composite film. (b) Low- and (c) high-magnification cross-section SEM images of the obtained 3D porous CMG film. Reproduced with permission from ref. 100. Copyright 2012, American Chemical Society. | ||
3.3. Graphene fibers
In previous years, carbon fibers with high mechanical strength and electrical conductivity have been successfully fabricated by assembling carbon nanotubes (CNTs), which have shown great importance in industry and our daily life.159–161 The mechanical, electrical and thermal properties of graphene are superior to that of other carbon materials,1,5,8 suggesting that the fabrication of graphene-based carbon fibers would provide more advantages in the practical applications of carbon fiber materials. In comparison with conventional carbon fibers, graphene fibers (GFs) not only exhibit the ordinary features of carbon fibers such as flexibility and electrical conductivity, but also possess unique properties such as lightweight, facile functionalization and low cost. GFs also exhibited a much lower density (∼0.23 g cm−3) compared with conventional carbon fibers (>1.7 g cm−3) and metal wires (∼20 g cm−3).43 The diameter of GFs is normally in the micrometer range, while the length can be up to several tens of centimeters, which gives GFs a high aspect ratio (>10000).
The current methods for the preparation of GFs are mainly based on the strategy of controlled assembly of GO sheets,43,162–169 such as hydrothermal treatment of a GO dispersion in a confined container165 and wet-spinning of a concentrated GO liquid crystal.163 Recently, the Yu group developed a method via injection of a GO dispersion into cetyltrimethylammonium bromide (CTAB) solution, leading to the self-assembly of GO sheets into a fiber-like structure.164 The formation of GFs followed the “curliness-fold” formation mechanism, in which the positively-charged CTAB molecules played an essential role (Fig. 3a). After the adsorption of CTAB molecules on the negatively-charged surface of GO sheet, the electrostatic repulsion among the GO sheets was reduced, leading to the curling and folding of the GO sheets. The GO fibers were obtained after continuous adsorption of CTAB molecules on the GO sheets and repeating the process of curling and folding of GO sheets. The resultant GFs had a long length of ∼1.6 m, and also possessed good mechanical strength (∼182 MPa), high electronic conductivity (∼35 S cm−1), and flexibility. Functionalizations of the GFs by polymers or CNTs were also achieved, which further enhanced the mechanical strength of the GFs and also extended the applications of GF-based materials. For example, the GF composited with poly(N-isopropylacrylamide) (PNIPAM) was used as a thermosensitive device.164
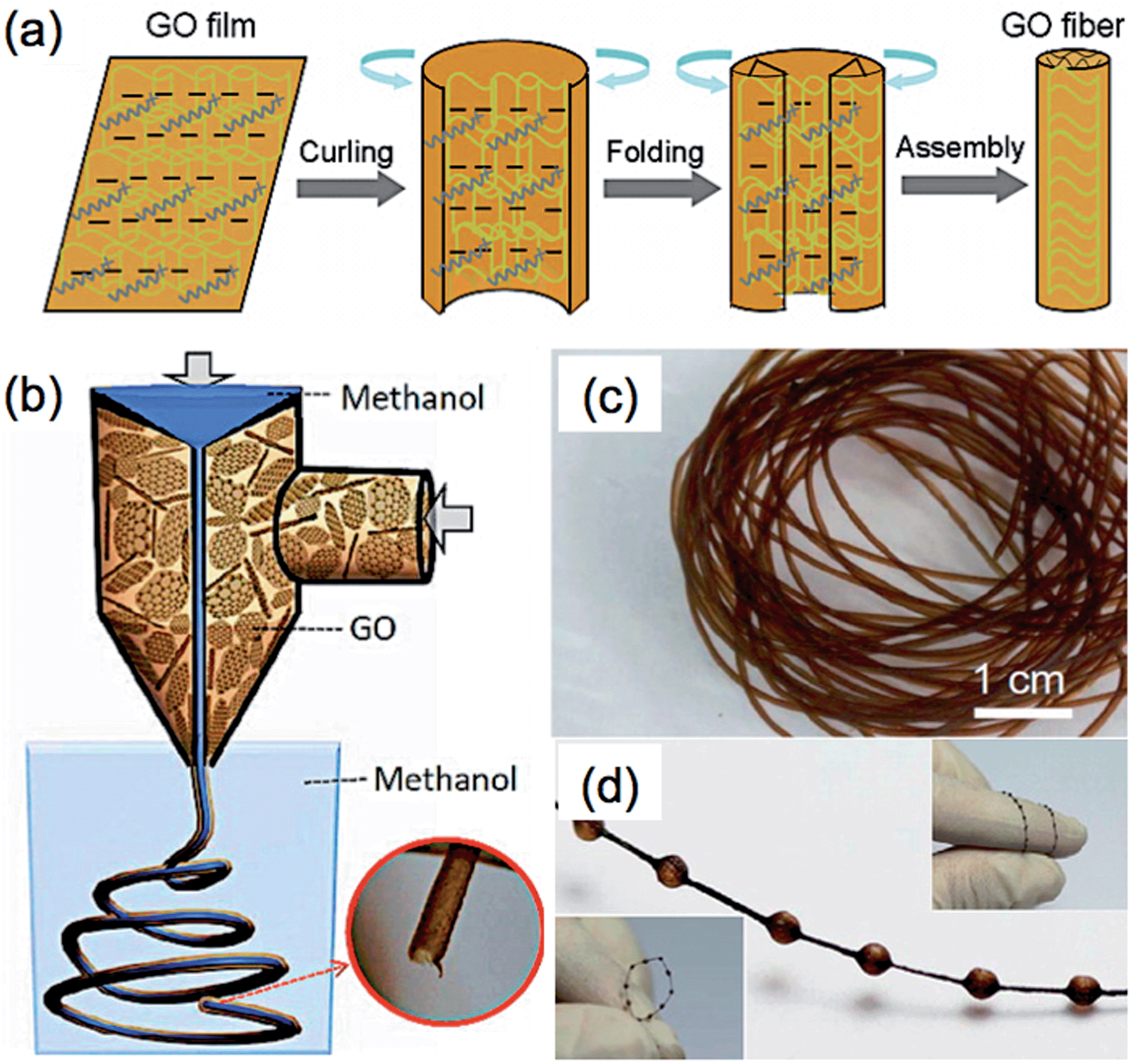 | ||
| Fig. 3 (a) Schematic illustration of the formation mechanism of GFs. Reproduced by permission from Macmillan Publishers Ltd: ref. 164, copyright 2012. (b) Schematic illustration of the fabrication process of GO hollow fibers (GO-HFs). (c and d) Photographs of the obtained GO-HFs (c) and necklace-like fibers (d). Reproduced with permission from ref. 168. Copyright 2013, American Chemical Society. | ||
Similar to the aforementioned Yu's method,164 hollow GFs have been fabricated by Qu et al., in which a coaxial two-capillary spinneret was used to simultaneously inject the GO suspension and methanol into the coagulation bath.168 The obtained GO hollow fibers (GO-HFs) showed a pipe-like morphology with a large diameter of ∼700 μm (Fig. 3b and c), and exhibited a tensile strength of ∼140 MPa. After reduction of the GO-HFs, the resultant rGO-HFs had an electrical conductivity of 8–10 S cm−1. It is worth mentioning that formation of the GO-HFs was very fast. It only took 30 s to produce 1 meter length GO-HFs. Interestingly, necklace-like fibers were also obtained (Fig. 3d), which may have a potential application in a micro pump.168
A GF with a unique core–shell structure, in which the core was composed of porous graphene film and the shell was made of densely stacked graphene sheets, was achieved by Gao et al.166 To prepare this “porous core@dense shell” GF, GO liquid crystals were firstly flowed through a capillary tube and injected into liquid nitrogen, then subjected to freeze-drying and reduction processes. Due to its unique structure, the GF not only exhibited high porosity with a specific surface area as high as 884 m2 g−1, but also possessed excellent electrical conductivity and mechanical strength. Attributed to its densely packed shell, the GF exhibited an electrical conductivity of 2.6 × 103 S m−1, which can be increased further to 4.9 × 103 S m−1 after an annealing treatment, which is much higher than that of graphene aerogels.72,73 The porous core of this GF also enabled the fabrication of GF-based composites, since the additives can be readily infiltrated into the pores. GFs composited with a polymer, Ag and Pt nanocrystals have been demonstrated, which might be promising in catalysis and energy storage.166
The preparation method of GFs is not limited to the assembly of GO sheets; GF can also be fabricated using CVD-produced graphene through a simple “drawing” method.170 After etching of the metal template, the graphene film was first floated on the surface of a mixture of ethanol and water, and subsequently drawn out by tweezers. After evaporation of the solvents, GF with a porous structure was obtained. Benefiting from the use of a high-quality graphene film produced by the CVD method, this GF possessed a high electrical conductivity of ∼1000 S m−1.
3.4. Graphene tubes
Materials with tube-like structures have exhibited great potential as vessels in applications such as separation, purification and fluidics.171,172 Graphene tubes show a similar morphology to that of CNTs but possess a larger inner diameter. This makes functionalization of the inner wall of a graphene tube much easier. Moreover, the walls of graphene tubes are composed of stacked graphene layers, which endow the graphene tube with high electrical conductivity and excellent mechanical strength.Templates are normally required to prepare graphene tubes.83,87,109,173 For example, Wang et al. used Ni nanowires with a diameter of ∼70 nm as both the catalyst and template for the synthesis of graphene tubes by the CVD method.87 The Ni nanowires were prepared by electrodeposition of Ni into an anodic aluminum oxide (AAO) membrane followed by etching of the AAO. To prepare the graphene tubes, a CVD process was first operated to deposit graphene on the surface of Ni nanowires. Graphene tubes were obtained after etching of Ni by 1 mol L−1 FeCl3, and these tubes exhibited a similar diameter to that of Ni nanowires. Recently, graphene was also successfully grown on the inner walls of the pores of an AAO membrane in a CVD process.83 The obtained graphene exhibited a tube-like structure with good thermal transport properties. In a recent work by Qu et al., graphene microtubings (μGTs) were fabricated based on the template of Cu wires through the hydrothermal method.173 In this method, Cu wires were placed inside a glass pipeline followed by filling of the pipeline with a GO dispersion (Fig. 4a). The GO sheets were aggregated and wrapped around the Cu wires after the hydrothermal reduction. The μGTs were obtained after removal of the Cu wires and pipeline. Interestingly, by choosing the glass pipeline and the Cu wire with the proper size and shape, it was possible to easily control the shape and diameter of the prepared μGTs. For instance, a μGT spring can be obtained by wrapping the wet μGT around a rod before drying (Fig. 4b). Helical and multi-channel μGTs can also be readily fabricated by twisting two Cu wires and inserting several Cu wires together into the glass pipeline, respectively, prior to the hydrothermal treatment (Fig. 4c and d). The μGTs showed a tensile strength of up to 180 MPa, which is comparable to that of the solid GFs.162,163 Importantly, selective functionalization of the outer-wall and inner-wall of the μGTs could also be achieved, which made the μGTs suitable for a wide range of applications. As an example, the μGT decorated with Pt NPs on its inner wall was used as a self-powered micromotor, which might be useful in the application of drug delivery.173 In addition to the aforementioned methods, a recent report showed that graphene tubes can also be obtained by partially unzipping the carbon nanofiber along its longitudinal axis.174
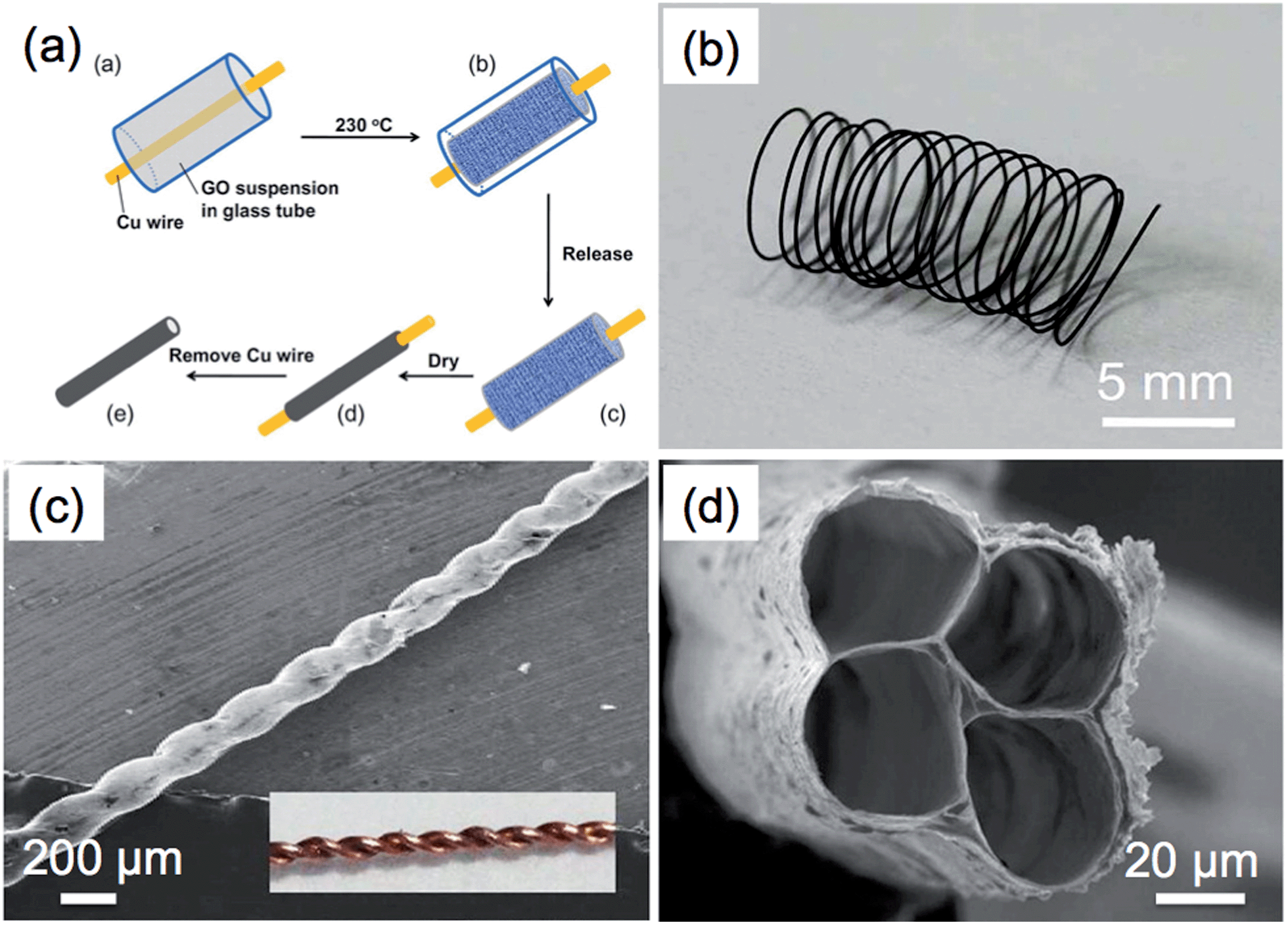 | ||
| Fig. 4 (a) Schematic illustration of the fabrication process of graphene microtubings (μGTs). (b) Photograph of a μGT spring. (c) SEM image of a helical μGT fabricated based on a twist of two Cu wires (inset). (d) SEM image of a 4-channel μGT. Reproduced with permission from ref. 173. Copyright 2012, American Chemical Society. | ||
3.5. Graphene spheres
Graphene spheres (GSs), which normally possess a structure of hollow spheres with a shell of stacked graphene layers, have found promising applications in oil absorption,98 supercapacitors,39,44 and lithium ion batteries.175GSs were mainly prepared based on spherical templates such as metal NPs.88,176 For example, Choi et al. reported the synthesis of hollow GSs via thermal annealing of triethylene glycol (TEG)-coated Ni NPs.88 The synthesis of GSs involve three steps. First, the Ni NPs coated with TEG were annealed at 250 °C to decompose the TEG molecules to carbon atoms. Then the Ni NPs were subjected to another annealing at 500 °C under an argon atmosphere, which led to the transformation of these adsorbed carbon atoms to graphene layers. After removal of Ni by HCl, the GSs with a similar diameter to that of the Ni NP template were obtained without collapse of the structure. Using pre-synthesized Ni NPs and the relatively low-temperature annealing process at 500 °C, it makes this synthetic process simple and scalable.
In addition, it is cheaper and more convenient to use metal salts instead of metals as the templates for the synthesis of GSs.44,89 As an interesting example, a nano-frame graphene structure (3D-NFG) was obtained using NiCl2 as a catalyst and polyvinyl alcohol (PVA) as a solid carbon source through a CVD process.89 The prepared GSs were interconnected with few-layer graphene sheets, which exhibited a low sheet resistance of ∼700 Ω sq−1. The resultant 3D-NFG could be used as an alternative counter electrode of Pt for dye sensitized solar cells (DSSC). Recently, graphene nanoballs with meso-pores (MGBs) were achieved in a precursor-assisted CVD process using FeCl3 and PS balls as the catalyst precursor and carbon source, respectively (Fig. 5a–d).44 The obtained MGBs possessed a high specific surface area of 508 m2 g−1 and average pore size of ∼4.27 nm. To prepare the MGBs, the PS balls were first functionalized with –COOH and –SO3H groups to achieve a good dispersion of PS balls in FeCl3 aqueous solution. Then the PS balls were annealed at 1000 °C under a H2–Ar atmosphere. During this process, 3D iron nanoframes were formed, arising from the aggregation of these iron ions adsorbed on the surface of PS balls. Meanwhile, PS balls were decomposed and served as a solid carbon source for the growth of graphene on 3D iron nanoframes. After removal of the iron by HCl, MGBs were obtained.
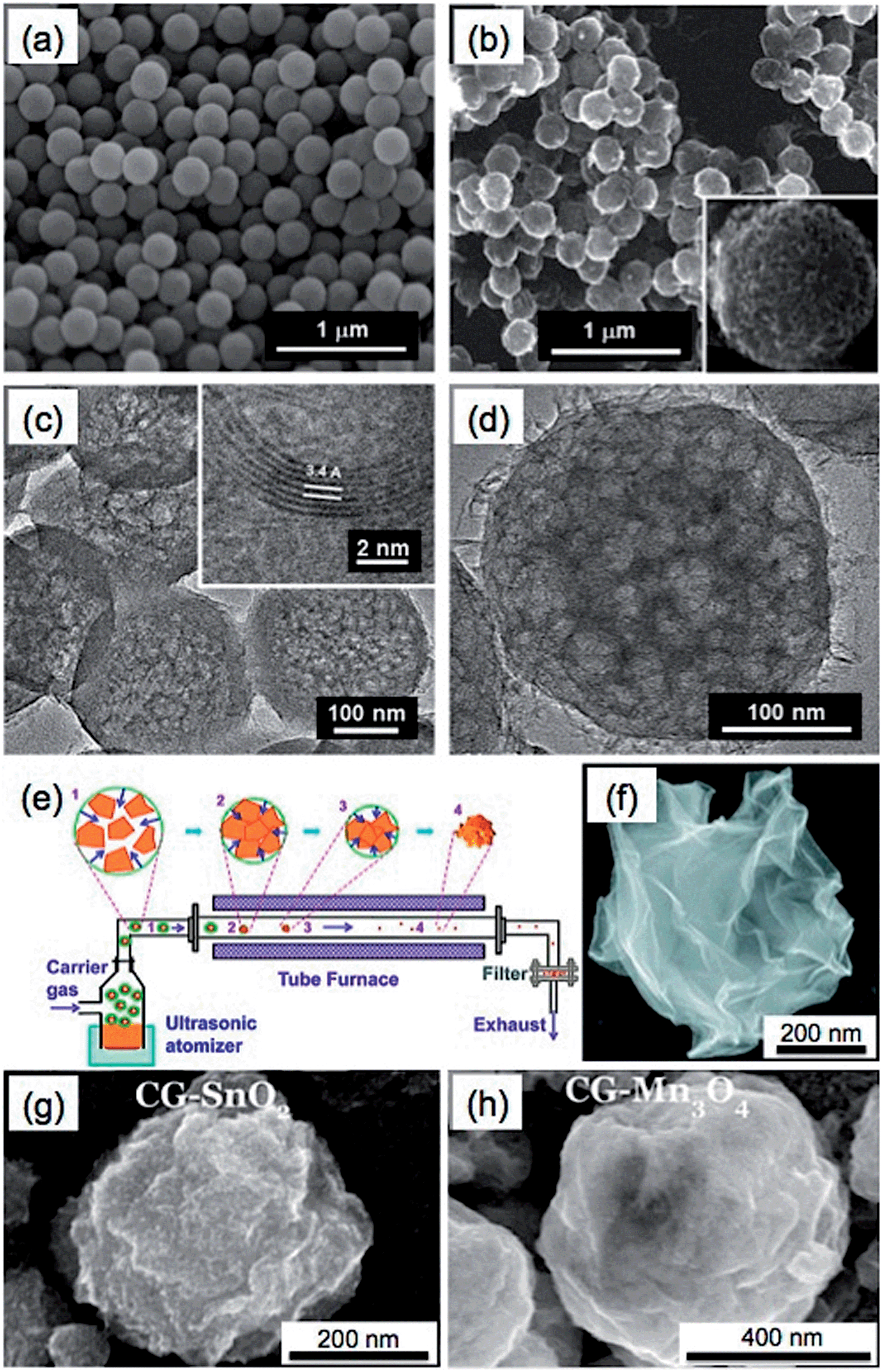 | ||
| Fig. 5 SEM images of (a) the PS balls functionalized with –COOH and –SO3H groups and (b) the graphene nanoballs with meso-pores (MGBs). Inset in (b): high-magnification SEM image of a MGB. (c and d) TEM images of MGBs. Inset in (c): high-resolution TEM image of a MGB shows an interlayer spacing of 0.34 nm. Reproduced with permission from ref. 44. Copyright 2013, American Chemical Society. (e) Schematic illustration of the synthetic process of paper-ball-like GS. (f) SEM image of a GS. Reproduced with permission from ref. 177. Copyright 2011, American Chemical Society. (g and h) SEM images of the crumpled graphene (CG) balls composited with SnO2 (g) and Mn3O4 (h). Reproduced with permission from ref. 39. Copyright 2012, American Chemical Society. | ||
In addition to the CVD methods, GSs have been also prepared by directly assembling GO sheets into spherical structures.39,98,177–181 Through use of an aerosol-assisted capillary compression method, Huang's group fabricated a paper-ball-like 3D graphene structure that was composed of crumpled rGO sheets.177 To prepare it, a GO aqueous solution was first sprayed into a tube furnace heated at 800 °C with the carrying gas of N2 (Fig. 5e). During the rapid evaporation of the solvent of GO droplets, GO sheets were compressed and aggregated to form the 3D graphene structure with a morphology similar to that of crumpled paper balls (Fig. 5f). In this process, the GO was simultaneously thermally reduced to rGO. Importantly, the GSs showed high compressive strength, which gave them an interesting aggregation-resistant property. As we know, flat graphene sheets are easily stacked to form a graphitic structure, which makes dispersion of dried graphene materials extremely difficult. However, even after the GSs were compressed at a high pressure of 2 GPa, they could still be redispersed in a solvent. Moreover, various GS-based composites, e.g. the crumpled graphene (CG) spheres composited with SnO2 or Mn3O4 (Fig. 5g and h), have also been obtained by spraying the metals, metal oxides or their precursors together with the GO dispersion into a furnace, which extends the applications of GSs in supercapacitors and lithium-ion batteries.39,175
3.6. Other types of 3D graphene structures
In addition to the aforementioned 3D structures, other types of graphene structures, such as onion rings,182 honeycombs,183,184 scrolls,185–191 nanosacks,192,193 and erythrocyte-like microspheres,194 have also been reported, which exhibited their unique properties and applications.For example, Tour et al. synthesized hexagonal graphene onion rings by a CVD method.182 In this unique structure, a number of graphene nanoribbons were grown beneath a monolayer graphene sheet, and formed a nearly concentric onion ring-like structure (Fig. 6a and b). Importantly, after removing the top monolayer graphene sheet by argon plasma, graphene nanoribbons, a promising graphene material with a width-dependent band gap for electronics,195 can be obtained. The resultant graphene nanoribbons exhibited high binding energy for lithium ions, and therefore have potential usage in lithium ion storage.
 | ||
| Fig. 6 (a) Optical image and (b) structural model of hexagonal graphene onion rings (HGoRs). Reproduced with permission from ref. 182. Copyright 2013, American Chemical Society. (c) Photograph and (d) SEM image of a freestanding 3D honeycomb-like graphene film. Reproduced with permission from ref. 196. Copyright 2013, WILEY-VCH Verlag GmbH & Co. KGaA. | ||
Honeycomb-like 3D graphene structures are mostly prepared by self-assembly of GO sheets,184,196,197 which have been used in the applications of supercapacitors197,198 and dye-sensitized solar cells (DSSC).183 As an interesting example, Li et al. used a freeze-casting method to fabricate a 3D graphene with a honeycomb-like structure, where ice crystals played the role of template.184 The obtained honeycomb-like 3D graphene exhibited excellent mechanical strength and elasticity, which could sustain a compression strain of 80% and fully recover its original shape and size. This 3D graphene material also showed a high porosity of ∼99.98% with a density as low as ∼0.5 mg cm−3. In addition, Chen et al. fabricated a freestanding 3D honeycomb-like graphene film using a complex of dimethyldioctadecylammonium (DODA) and GO sheets (Fig. 6c and d).196 Recently, a honeycomb-like 3D graphene structure was also obtained from the reaction between Li2O powder and CO gas at 550 °C under low pressure for 12–24 h.183 The counter electrode made of this 3D graphene exhibited an energy conversion efficiency of up to 7.8% in a DSSC, which is comparable to that using a Pt electrode (8%).
Carbon nanoscrolls, a novel 3D graphene structure prepared by wrapping graphene sheets into a tubular shape, has been revealed as promising in energy storage186,188 and electronic devices.185,191 Recently, our group developed a scalable method to produce well-aligned GO scrolls on various substrates.185 After the reduction process, the resultant rGO scrolls were used as a gas sensor, which exhibited a detection limit of 0.4 ppm towards NO2 gas.
4. Supercapacitor application
Supercapacitors, promising electrochemical energy storage devices with the advantages of high power density and long cycle life, have been attracting researchers' increasing interests due to their potential applications ranging from portable electronics to electric vehicles.199–202 Based on the different charge storage mechanisms, supercapacitors can be divided into electric double-layer capacitors (EDLCs) and pesudocapacitors. The charge storage mechanism of EDLCs relies on the separation of charges at the interface of the electrolyte and electrode, while pesudocapacitors are operated following the mechanism of reversible redox reactions occurring at the surface or near-surface areas of active materials.199 Carbonaceous materials, such as activated carbons, carbon nanotubes and graphene, have been widely employed for the construction of supercapacitor electrodes, due to their low cost, electrical conductivity and high surface area.203–211 Recently, 3D graphene-based materials have been proven as promising candidates for supercapacitors.40,155 The unique properties and porous structures of 3DGMs not only improve the accessibility of electrolyte to the surface of the electrode, but also provide electrically conductive channels for the active materials decorated on them, which enhance the performances of both EDLCs and pesudocapacitors. In this section, the supercapacitor applications of different types of 3D graphene structures and their composites are summarized. The flexible supercapacitor, especially the fiber-based supercapacitor, is also reviewed.3DGMs with various structures, such as hydrogels,111,112 aerogels,94,212 sponges110 and porous films,149,154,155 have been extensively explored in the application of supercapacitors. As a typical example, a 3D porous graphene with extremely high surface area of 3100 m2 g−1, produced by chemical activation of microwave exfoliated graphene oxides (MEGO), was used for constructing a symmetrical supercapacitor with ionic liquid and organic electrolytes.155 The activated MEGO (a-MEGO) contained numerous small pores with sizes ranging from ∼1 to 10 nm, and also possessed a high electrical conductivity of ∼500 S m−1. The supercapacitor delivered a specific capacitance of 150 F g−1 at a current density of 0.8 A g−1 with a small equivalent series resistance (ESR) of 4.6 ohms. Moreover, their subsequent work further demonstrated that the a-MEGO-based supercapacitor was capable of operation below room temperature (down to −50 °C), giving a specific capacitance over 100 F g−1.213 Although more effective surface area of graphene can be obtained by construction of 3D graphene structures, the specific capacitances in previously reported graphene electrodes are still far from the theoretical capacitance of graphene, e.g. 550 F g−1 calculated based on the intrinsic capacitance and theoretical specific surface area of graphene.214,215
3D porous graphene films composited with pseudo-capacitive materials that have high theoretical capacitances, such as metal oxides and conducting redox polymers, have been widely explored for improving the supercapacitor performance of 3DGMs.216–218 Higher specific capacitance and energy density, better rate capability and longer cycling life have been observed in the composite electrodes.112,212 For example, the Ni(OH)2/graphene composite prepared by Dai et al. exhibited a specific capacitance of ∼877 F g−1 at 40 mV s−1, while only 339 F g−1 for the physical mixture of these two components was obtained.219 Even at a high current density of 45.7 A g−1, a specific capacitance of ∼953 F g−1 was still retained for the Ni(OH)2/graphene composite. After a cycling test at 28.6 A g−1 for 2000 cycles, the electrode of Ni(OH)2/graphene composite showed a negligible change of capacitance. The enhanced supercapacitor performance of composites is generally attributed to the synergetic effect of graphene and the other components.216 First, the pseudo-capacitive materials not only contribute pseudo-capacitance to the whole composite electrode, but also act as spacer materials to enlarge the interspace between graphene sheets, resulting in the improved accessibility of electrolyte to the electrode. Second, the interconnected graphene sheets provide electrically conductive channels to the composite, which enables fast charge transport in the composite and preserves the good performance of electrode at a high charge–discharge current density.
Graphene spheres (GS) have a relatively stable structure with high surface area in aqueous solvent.88,177 Since GS can be prepared by scalable methods, it makes GS suitable for the construction of supercapacitor electrodes.39,44,180 For example, mesoporous graphene nanoballs (MGBs) are able to be obtained on the gram scale by a CVD method, which showed a high surface area of 508 m2 g−1 and conductivity of 1.7 S cm−1.44 After a subsequent doping process to increase the conductivity of MGB to 6.5 S cm−1, the p-doped MGB exhibited a specific capacitance of 206 F g−1 at 5 mV s−1 and over 96% retention of the initial capacitance after 10000 cycles at a relatively high current density of 20 A g−1 in an electrolyte of 1 mol L−1 H2SO4. The excellent performance of p-doped MGB is attributed to the high conductivity of the CVD-produced graphene, and its mesoporous structure with a mean pore size of 4.27 nm, which enables the electrolyte to easily access the inner surface of the MGB. The performance of GS-based supercapacitors has been improved further by the combination of GS with pseudo-capacitive materials.39 Chen et al. reported the direct deposition of graphene–Mn3O4 composite spheres on indium tin oxide (ITO) substrate by an aerosolization method. The obtained electrode exhibited a specific capacitance of 1027 F g−1 at a current density of 5 A g−1 and 404 F g−1 at a high current density of 40 A g−1. After testing for 1000 cycles at a current density of 20 A g−1, the GS-based composite electrode still retained 78% capacitance.39
Direct coating of graphene on a 3D substrate followed by the functionalization of graphene with other pseudo-capacitive materials is a convenient and effective way to apply 3D graphene in supercapacitors.91,94,105 For instance, Bao et al. developed a MnO2/graphene/textile composite electrode by using the template of 3D porous textile fibers.105 This composite electrode was fabricated through coating textile fibers with solution-exfoliated graphene sheets, followed by electrodeposition of pseudocapacitive MnO2 NPs onto the surface of the graphene/textile fibers. The coated graphene layer not only increased the electrical conductivity of the fabricated electrodes, but also improved the adhesion of the textile fibers to the active materials. The composite electrode showed a specific capacitance of ∼315 F g−1 at a scan rate of 2 mV s−1. An asymmetrical supercapacitor operated in 0.5 mol L−1 Na2SO4 aqueous electrolyte was also fabricated by using the MnO2/graphene/textile and CNT/textile as the positive and negative electrode, respectively. This asymmetrical supercapacitor delivered a power density of 110 kW kg−1 and an energy density of 12.5 W h kg−1, and also exhibited excellent cycling performance with ∼95% retention of the initial specific capacitance after 5000 cycles.
Recently, 3DGNs,37 prepared by a CVD method based on the template of Ni foam, have been demonstrated as a promising material for the fabrication of supercapacitor electrodes.40 To date, a number of materials have been composited with 3DGNs, and some of the 3DGN-based composites have shown excellent supercapacitor performances.118,119,123,220,221 For example, our group used a simple electrochemical deposition method to fabricate a NiO/3DGN composite electrode for supercapacitor, which produced a specific capacitance of 745 F g−1 at a current density of 1.4 A g−1.40 The excellent electrical contacts among the active material of NiO, graphene sheets and the current collector of Ni foam allowed rapid charge transfer within this composite. Meanwhile, the porous structure of 3DGNs facilitated the easy accessibility of electrolyte ions to the active materials. An asymmetric supercapacitor was also fabricated by using Ni(OH)2/3DGN and a-MEGO as the positive and negative electrodes, respectively.221 The obtained supercapacitor showed a high power density of 44.0 kW kg−1 in the aqueous electrolyte of 6 mol L−1 KOH, which is much higher than that of commercially available supercapacitors. Moreover, a hierarchical nanostructure, which was composed of 3DGNs coated with two different components, has been successfully prepared by our group and used as a supercapacitor electrode. In the aforementioned work, the Ni3S2@Ni(OH)2/3DGN was obtained through a one-step hydrothermal reaction, in which the crystalline Ni3S2 nanorods coated with Ni(OH)2 nanosheets were grown on the surface of 3DGNs (Fig. 7a).119 The highly crystalline Ni3S2 nanorods that grew perpendicularly to the surface of 3DGNs played an essential role in the supercapacitor performance of the Ni3S2@Ni(OH)2/3DGN composite, which not only served as electrically conductive channels to improve the electron transport performance of coated Ni(OH)2 sheets, but also increased the contact area of Ni(OH)2 with electrolyte. Due to the unique hierarchical nanostructure, the supercapacitor performance of Ni3S2@Ni(OH)2/3DGN was much better than those of both Ni3S2/3DGN and Ni(OH)2/3DGN, which exhibited high specific capacitances of 1277 F g−1 at 2 mV s−1 and 1037.5 F g−1 at 5.1 A g−1, and also high areal capacitances of 4.7 F cm−2 at 2 mV s−1 and 3.85 F cm−2 at 19.1 mA cm−2. The obtained Ni3S2@Ni(OH)2/3DGN also showed an excellent cycling performance, as it was able to retain 99.1% of the initial specific capacitance even after 2000-cycle operation.
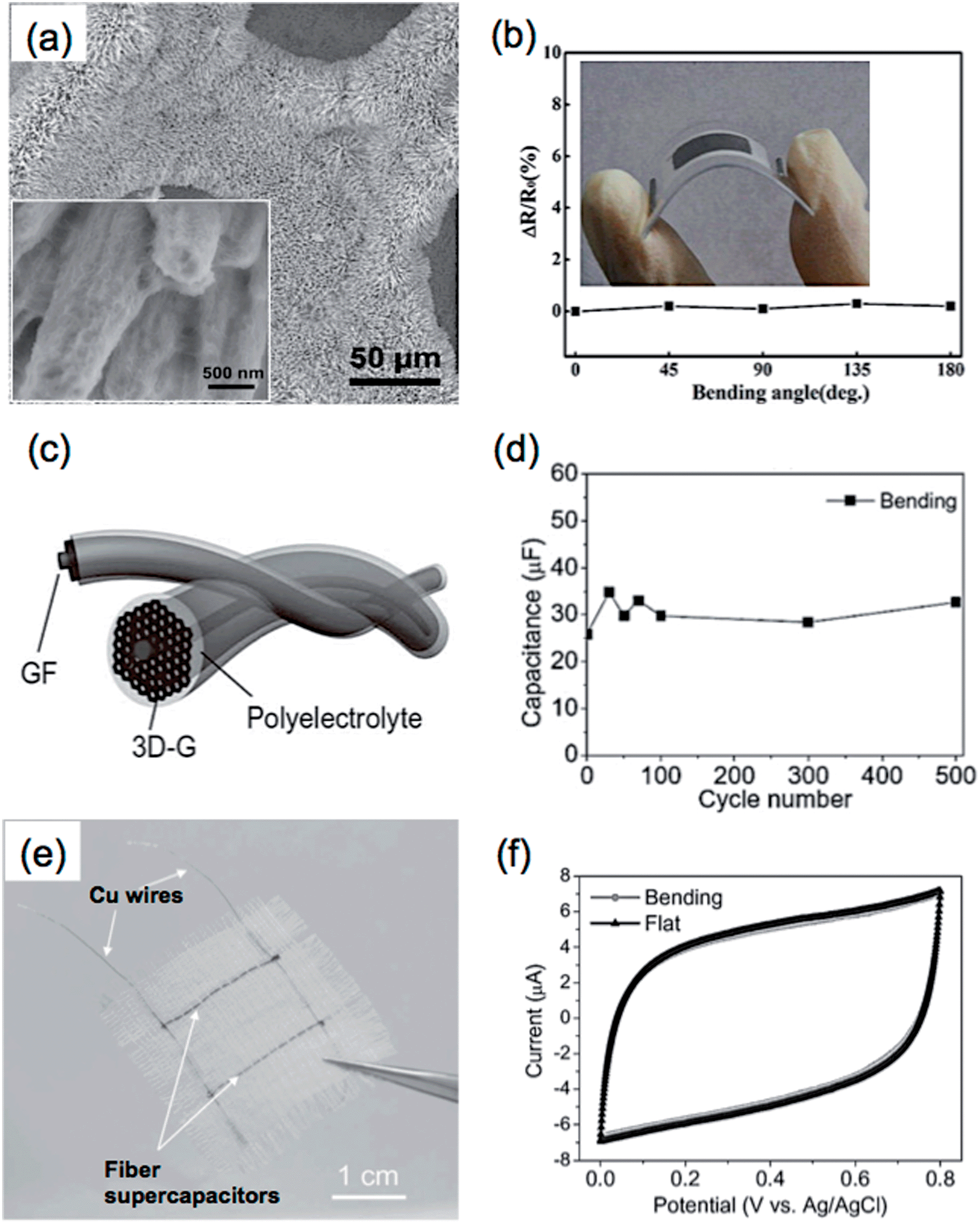 | ||
| Fig. 7 (a) SEM image of Ni3S2@Ni(OH)2/3DGN composite. Inset: high-magnification SEM image of the Ni3S2@Ni(OH)2 coated on 3DGN. Reproduced from ref. 119 with permission from The Royal Society of Chemistry. (b) Resistance changes (ΔR/R) of 3DGN electrodes at different bending angles. Inset: photograph of a flexible MnO2/3DGN composite supercapacitor. Reproduced with permission from ref. 220. Copyright 2013, American Chemical Society. (c) Schematic illustration of the configuration of an all-solid-state GF supercapacitor. (d) Cyclability of a GF supercapacitor after bending for 500 cycles. (e) Photograph of a flexible supercapacitor device fabricated by integrating two GF supercapacitors into a textile. (f) Cyclic voltammetry (CV) curves of the flexible supercapacitor device in (e) the flat and bending states at a scan rate of 50 mV s−1. Reproduced with permission from ref. 43. Copyright 2013, WILEY-VCH Verlag GmbH & Co. KGaA. | ||
Flexible supercapacitors are of great importance, because they are the potential power supplies for next-generation wearable devices, e.g. electronic textiles.222 Due to their excellent mechanical and electrical properties, 3D graphene architectures, especially graphene fibers,43,143,170,223 porous films,45,152 and networks,220,224 have played essential roles in the fabrication of flexible supercapacitor devices. For example, Xie et al. fabricated a flexible and lightweight symmetrical supercapacitor based on the MnO2/3DGN composite.220 Before the growth of 3DGNs by CVD, the Ni foam template was first pressed to form a more compact 3D structure, which reduced the pore size and increased the volume density of the synthesized 3DGNs. Importantly, this process also improved the flexibility of the 3DGNs. The resultant 3DGN electrodes showed negligible resistance variations under bending with the angle up to 180° (Fig. 7b). After electrodeposition of MnO2 nanospheres on the 3DGNs, the obtained MnO2/3DGN composite showed a large specific surface area (392 m2 g−1) and an extremely high mass loading of 92.9% MnO2. The symmetrical supercapacitor based on the MnO2/3DGN composite delivered an areal capacitance of 1.42 F cm−2 and a specific capacitance of 130 F g−1 at a scan rate of 2 mV s−1.
Fiber-based supercapacitors (FSs) are promising candidates for flexible energy storage devices.225,226 Many fiber materials including ZnO227 and CNTs228,229 have been developed and investigated in this area. FSs were prepared by coating graphene on fiber materials to enhance the supercapacitor performance.230–233 However, the flexibility and performance of those reported FSs still require further improvement.
Graphene fibers (GF) have the advantages of flexibility, conductivity, large surface area and easy functionalization, giving them great potential for the construction of FS.234 As a typical example, Qu et al. fabricated an all-solid-state GF supercapacitor, in which two GFs were intertwisted together with H2SO4–polyvinyl alcohol (PVA) gel as electrolyte (Fig. 7c).43 The fabricated GF-based supercapacitors exhibited an areal capacitance of 1.2–1.7 mF cm−2, and a stable electrochemical performance without an obvious change in capacitance after bending for 500 cycles (Fig. 7d). Interestingly, a flexible supercapacitor device was fabricated by weaving two GF-based supercapacitors into a textile, which showed negligible variation in the cyclic voltammetry (CV) curves before and after bending (Fig. 7e and f). The functionalization of GFs with active materials is able to improve the energy density of a GF-based supercapacitor.170,235 In a recent work, Zhu et al. prepared a MnO2-coated GF based on the high electrically conductive CVD-grown graphene film, which exhibited an areal capacitance of 42 mF cm−2 at 10 mV s−1 with an energy density of 1.46 × 10−3 mW h cm−2.235
5. Conclusions
3D graphene materials (3DGMs) have been proven as promising in a wide area of research interests. Up to now, a number of 3DGMs with different structures and varied functions have been reported. These novel materials not only preserve the intrinsic properties of the individual 2D graphene sheet, but also provide a tremendous opportunity to explore graphene in various practical applications. For example, graphene with a porous 3D macroscopic structure, having high electrical conductivity, large surface area and high mechanical strength, can be used directly or after decoration with electroactive materials as a supercapacitor electrode.Although lots of strategies on the construction of 3DGMs with specific morphologies have been demonstrated, there are still some challenges. The properties of 3DGMs are related to their structure, but the precise control of pore size and porosity of 3DGMs has not been achieved. Most of the reported 3DGMs showed a wide pore size distribution ranging from a few hundred nanometers to several hundred micrometers. The fabrication of 3D graphene architectures with uniform meso- or micro-pores is still a challenge. There are also few reports on the preparation of freestanding and macroscopic 3DGMs that are composed of a single-layer graphene film, mostly due to its insufficient mechanical strength. To address these issues, it could be feasible to choose nanoporous graphene sheets including graphene nanomeshes84,236,237 and mesoporous graphene sheets155,238,239 with uniform pore sizes in the graphene, as building blocks, which are then assembled into 3D macroscopic structures. Of course, more efforts and experiments are required to obtain a more controllable and efficient method for the fabrication of 3DGMs for practical applications.
Recently, graphene-like 2D nanosheets, such as transition metal dichalcogenides,240–248 have been attracting increasing attention. The future direction will be to assemble these novel 2D materials into 3D macroscopic structures, and investigate their unique properties and various applications.
Acknowledgements
This work was supported by MOE under AcRF Tier 2 (ARC 26/13, No. MOE2013-T2-1-034), AcRF Tier 1 (RG 61/12, RGT18/13), and Start-Up Grant (M4080865.070.706022) in Singapore. This research is also conducted by NTU-HUJ-BGU Nanomaterials for Energy and Water Management Programme under the Campus for Research Excellence and Technological Enterprise (CREATE), that is supported by the National Research Foundation, Prime Minister's Office, Singapore.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- P. W. Sutter, J.-I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406–411 CrossRef CAS PubMed.
- K. V. Emtsev, A. Bostwick, K. Horn, J. Jobst, G. L. Kellogg, L. Ley, J. L. McChesney, T. Ohta, S. A. Reshanov, J. Rohrl, E. Rotenberg, A. K. Schmid, D. Waldmann, H. B. Weber and T. Seyller, Nat. Mater., 2009, 8, 203–207 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- Q. He, S. Wu, S. Gao, X. Cao, Z. Yin, H. Li, P. Chen and H. Zhang, ACS Nano, 2011, 5, 5038–5044 CrossRef CAS PubMed.
- X. Huang, Z. Zeng, Z. Fan, J. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979–6004 CrossRef CAS PubMed.
- B. Li, X. Cao, H. G. Ong, J. W. Cheah, X. Zhou, Z. Yin, H. Li, J. Wang, F. Boey, W. Huang and H. Zhang, Adv. Mater., 2010, 22, 3058–3061 CrossRef CAS PubMed.
- J. Liu, Z. Lin, T. Liu, Z. Yin, X. Zhou, S. Chen, L. Xie, F. Boey, H. Zhang and W. Huang, Small, 2010, 6, 1536–1542 CrossRef CAS PubMed.
- J. Liu, Z. Yin, X. Cao, F. Zhao, A. Lin, L. Xie, Q. Fan, F. Boey, H. Zhang and W. Huang, ACS Nano, 2010, 4, 3987–3992 CrossRef CAS PubMed.
- J. Liu, Z. Yin, X. Cao, F. Zhao, L. Wang, W. Huang and H. Zhang, Adv. Mater., 2013, 25, 233–238 CrossRef CAS PubMed.
- J. Liu, Z. Zeng, X. Cao, G. Lu, L.-H. Wang, Q.-L. Fan, W. Huang and H. Zhang, Small, 2012, 8, 3517–3522 CrossRef CAS PubMed.
- X. Qi, C. Tan, J. Wei and H. Zhang, Nanoscale, 2013, 5, 1440–1451 RSC.
- Z. Yin, Z. Zeng, J. Liu, Q. He, P. Chen and H. Zhang, Small, 2013, 9, 727–731 CrossRef CAS PubMed.
- Z. Yin, J. Zhu, Q. He, X. Cao, C. Tan, H. Chen, Q. Yan and H. Zhang, Adv. Energy Mater., 2014, 4, 1300574 Search PubMed.
- J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014 DOI:10.1002/smll.201303202.
- M. He, J. Jung, F. Qiu and Z. Lin, J. Mater. Chem., 2012, 22, 24254–24264 RSC.
- J. Song, Z. Yin, Z. Yang, P. Amaladass, S. Wu, J. Ye, Y. Zhao, W.-Q. Deng, H. Zhang and X.-W. Liu, Chem.–Eur. J., 2011, 17, 10832–10837 CrossRef CAS PubMed.
- Z. Yin, S. Sun, T. Salim, S. Wu, X. Huang, Q. He, Y. M. Lam and H. Zhang, ACS Nano, 2010, 4, 5263–5268 CrossRef CAS PubMed.
- Z. Yin, S. Wu, X. Zhou, X. Huang, Q. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307–312 CrossRef CAS PubMed.
- Q. He, S. Wu, Z. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764–1772 RSC.
- S. Wu, Q. He, C. Tan, Y. Wang and H. Zhang, Small, 2013, 9, 1160–1172 CrossRef CAS PubMed.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- Q. He, H. G. Sudibya, Z. Yin, S. Wu, H. Li, F. Boey, W. Huang, P. Chen and H. Zhang, ACS Nano, 2010, 4, 3201–3208 CrossRef CAS PubMed.
- G. Lu, H. Li, C. Liusman, Z. Yin, S. Wu and H. Zhang, Chem. Sci., 2011, 2, 1817–1821 RSC.
- H. G. Sudibya, Q. He, H. Zhang and P. Chen, ACS Nano, 2011, 5, 1990–1994 CrossRef CAS PubMed.
- Z. Wang, J. Zhang, P. Chen, X. Zhou, Y. Yang, S. Wu, L. Niu, Y. Han, L. Wang, P. Chen, F. Boey, Q. Zhang, B. Liedberg and H. Zhang, Biosens. Bioelectron., 2011, 26, 3881–3886 CrossRef CAS PubMed.
- Z. Yin, Q. He, X. Huang, J. Zhang, S. Wu, P. Chen, G. Lu, P. Chen, Q. Zhang, Q. Yan and H. Zhang, Nanoscale, 2012, 4, 293–297 RSC.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- W. Li, S. Gao, L. Wu, S. Qiu, Y. Guo, X. Geng, M. Chen, S. Liao, C. Zhu, Y. Gong, M. Long, J. Xu, X. Wei, M. Sun and L. Liu, Sci. Rep., 2013, 3, 2125 Search PubMed.
- S. Mao, Z. Wen, H. Kim, G. Lu, P. Hurley and J. Chen, ACS Nano, 2012, 6, 7505–7513 CrossRef CAS PubMed.
- X. Cao, Y. Shi, W. Shi, G. Lu, X. Huang, Q. Yan, Q. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- H. S. Ahn, J. M. Kim, C. Park, J.-W. Jang, J. S. Lee, H. Kim, M. Kaviany and M. H. Kim, Sci. Rep., 2013, 3, 1960 Search PubMed.
- S. Mao, K. Yu, J. Chang, D. A. Steeber, L. E. Ocola and J. Chen, Sci. Rep., 2013, 3, 1696 CAS.
- Y. N. Meng, Y. Zhao, C. G. Hu, H. H. Cheng, Y. Hu, Z. P. Zhang, G. Q. Shi and L. T. Qu, Adv. Mater., 2013, 25, 2326–2331 CrossRef CAS PubMed.
- J.-S. Lee, S.-I. Kim, J.-C. Yoon and J.-H. Jang, ACS Nano, 2013, 7, 6047–6055 CrossRef CAS PubMed.
- Z. Niu, J. Chen, H. H. Hng, J. Ma and X. Chen, Adv. Mater., 2012, 24, 4144–4150 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- X. Huang, S. Li, S. Wu, Y. Huang, F. Boey, C. L. Gan and H. Zhang, Adv. Mater., 2012, 24, 979–983 CrossRef CAS PubMed.
- X. Huang, X. Zhou, S. Wu, Y. Wei, X. Qi, J. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 513–516 CrossRef CAS PubMed.
- H. Li, X. Cao, B. Li, X. Zhou, G. Lu, C. Liusman, Q. He, F. Boey, S. S. Venkatraman and H. Zhang, Chem. Commun., 2011, 47, 10070–10072 RSC.
- X. Qi, H. Li, J. W. Y. Lam, X. Yuan, J. Wei, B. Z. Tang and H. Zhang, Adv. Mater., 2012, 24, 4191–4195 CrossRef CAS PubMed.
- X. Qi, K.-Y. Pu, H. Li, X. Zhou, S. Wu, Q.-L. Fan, B. Liu, F. Boey, W. Huang and H. Zhang, Angew. Chem., Int. Ed., 2010, 49, 9426–9429 CrossRef CAS PubMed.
- X. Qi, K.-Y. Pu, X. Zhou, H. Li, B. Liu, F. Boey, W. Huang and H. Zhang, Small, 2010, 6, 663–669 CrossRef CAS PubMed.
- Z. Wang, J. Zhang, Z. Yin, S. Wu, D. Mandler and H. Zhang, Nanoscale, 2012, 4, 2728–2733 RSC.
- S. Wu, Q. He, C. Zhou, X. Qi, X. Huang, Z. Yin, Y. Yang and H. Zhang, Nanoscale, 2012, 4, 2478–2483 RSC.
- S. Wu, Z. Yin, Q. He, G. Lu, Q. Yan and H. Zhang, J. Phys. Chem. C, 2011, 115, 15973–15979 CAS.
- S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. Ri Kim, Y. I. Song, Y.-J. Kim, K. S. Kim, B. Ozyilmaz, J.-H. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed.
- H. Bai, C. Li, X. Wang and G. Shi, J. Phys. Chem. C, 2011, 115, 5545–5551 CAS.
- H. Bai, C. Li, X. Wang and G. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- O. C. Compton, Z. An, K. W. Putz, B. J. Hong, B. G. Hauser, L. Catherine Brinson and S. T. Nguyen, Carbon, 2012, 50, 3399–3406 CrossRef CAS PubMed.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef CAS PubMed.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- Y. Chang, J. Li, B. Wang, H. Luo, H. He, Q. Song and L. Zhi, J. Mater. Chem. A, 2013, 1, 14658–14665 CAS.
- S. Sun and P. Wu, J. Mater. Chem., 2011, 21, 4095–4097 RSC.
- P. M. Sudeep, T. N. Narayanan, A. Ganesan, M. M. Shaijumon, H. Yang, S. Ozden, P. K. Patra, M. Pasquali, R. Vajtai, S. Ganguli, A. K. Roy, M. R. Anantharaman and P. M. Ajayan, ACS Nano, 2013, 7, 7034–7040 CrossRef CAS PubMed.
- H. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554–2560 CrossRef CAS PubMed.
- S. Korkut, J. D. Roy-Mayhew, D. M. Dabbs, D. L. Milius and I. A. Aksay, ACS Nano, 2011, 5, 5214–5222 CrossRef CAS PubMed.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS PubMed.
- F. Liu and T. S. Seo, Adv. Funct. Mater., 2010, 20, 1930–1936 CrossRef CAS.
- K. Sheng, Y. Sun, C. Li, W. Yuan and G. Shi, Sci. Rep., 2012, 2, 247 Search PubMed.
- M. A. Worsley, T. Y. Olson, J. R. I. Lee, T. M. Willey, M. H. Nielsen, S. K. Roberts, P. J. Pauzauskie, J. Biener, J. H. Satcher and T. F. Baumann, J. Phys. Chem. Lett., 2011, 2, 921–925 CrossRef CAS.
- M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher and T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067–14069 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- Z. Tang, S. Shen, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 4603–4607 CrossRef CAS PubMed.
- W. Wei, S. Yang, H. Zhou, I. Lieberwirth, X. Feng and K. Müllen, Adv. Mater., 2013, 25, 2909–2914 CrossRef CAS PubMed.
- X. Xie, Y. Zhou, H. Bi, K. Yin, S. Wan and L. Sun, Sci. Rep., 2013, 3, 2117 Search PubMed.
- Y. Su, Y. Zhang, X. Zhuang, S. Li, D. Wu, F. Zhang and X. Feng, Carbon, 2013, 62, 296–301 CrossRef CAS PubMed.
- Z. Han, Z. Tang, P. Li, G. Yang, Q. Zheng and J. Yang, Nanoscale, 2013, 5, 5462–5467 RSC.
- H. Bi, X. Xie, K. Yin, Y. Zhou, S. Wan, L. He, F. Xu, F. Banhart, L. Sun and R. S. Ruoff, Adv. Funct. Mater., 2012, 22, 4421–4425 CrossRef CAS.
- Y. Tao, X. Xie, W. Lv, D.-M. Tang, D. Kong, Z. Huang, H. Nishihara, T. Ishii, B. Li, D. Golberg, F. Kang, T. Kyotani and Q.-H. Yang, Sci. Rep., 2013, 3, 2975 Search PubMed.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- Z. Chen, W. Ren, B. Liu, L. Gao, S. Pei, Z.-S. Wu, J. Zhao and H.-M. Cheng, Carbon, 2010, 48, 3543–3550 CrossRef CAS PubMed.
- M. Zhou, T. Lin, F. Huang, Y. Zhong, Z. Wang, Y. Tang, H. Bi, D. Wan and J. Lin, Adv. Funct. Mater., 2013, 23, 2263–2269 CrossRef CAS.
- G. Ning, Z. Fan, G. Wang, J. Gao, W. Qian and F. Wei, Chem. Commun., 2011, 47, 5976–5978 RSC.
- X. Xiao, T. E. Beechem, M. T. Brumbach, T. N. Lambert, D. J. Davis, J. R. Michael, C. M. Washburn, J. Wang, S. M. Brozik, D. R. Wheeler, D. B. Burckel and R. Polsky, ACS Nano, 2012, 6, 3573–3579 CrossRef CAS PubMed.
- X. Xiao, J. R. Michael, T. Beechem, A. McDonald, M. Rodriguez, M. T. Brumbach, T. N. Lambert, C. M. Washburn, J. Wang, S. M. Brozik, D. R. Wheeler, D. B. Burckel and R. Polsky, J. Mater. Chem., 2012, 22, 23749–23754 RSC.
- R. Wang, Y. Hao, Z. Wang, H. Gong and J. T. L. Thong, Nano Lett., 2010, 10, 4844–4850 CrossRef CAS PubMed.
- S.-M. Yoon, W. M. Choi, H. Baik, H.-J. Shin, I. Song, M.-S. Kwon, J. J. Bae, H. Kim, Y. H. Lee and J.-Y. Choi, ACS Nano, 2012, 6, 6803–6811 CrossRef CAS PubMed.
- J.-S. Lee, H.-J. Ahn, J.-C. Yoon and J.-H. Jang, Phys. Chem. Chem. Phys., 2012, 14, 7938–7943 RSC.
- X. H. Xia, J. P. Tu, Y. J. Mai, R. Chen, X. L. Wang, C. D. Gu and X. B. Zhao, Chem.–Eur. J., 2011, 17, 10898–10905 CrossRef CAS PubMed.
- J. Chen, K. Sheng, P. Luo, C. Li and G. Shi, Adv. Mater., 2012, 24, 4569–4573 CrossRef CAS PubMed.
- H. Wang, G. Wang, Y. Ling, F. Qian, Y. Song, X. Lu, S. Chen, Y. Tong and Y. Li, Nanoscale, 2013, 5, 10283–10290 RSC.
- T. Zhai, F. Wang, M. Yu, S. Xie, C. Liang, C. Li, F. Xiao, R. Tang, Q. Wu, X. Lu and Y. Tong, Nanoscale, 2013, 5, 6790–6796 RSC.
- S. Ye, J. Feng and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 7122–7129 CAS.
- J.-C. Yoon, J.-S. Lee, S.-I. Kim, K.-H. Kim and J.-H. Jang, Sci. Rep., 2013, 3, 1788 Search PubMed.
- X. Huang, K. Qian, J. Yang, J. Zhang, L. Li, C. Yu and D. Zhao, Adv. Mater., 2012, 24, 4419–4423 CrossRef CAS PubMed.
- G.-h. Moon, Y. Shin, D. Choi, B. W. Arey, G. J. Exarhos, C. Wang, W. Choi and J. Liu, Nanoscale, 2013, 5, 6291–6296 RSC.
- K. Sohn, Y. Joo Na, H. Chang, K.-M. Roh, H. Dong Jang and J. Huang, Chem. Commun., 2012, 48, 5968–5970 RSC.
- H. Wang, D. Zhang, T. Yan, X. Wen, J. Zhang, L. Shi and Q. Zhong, J. Mater. Chem. A, 2013, 1, 11778–11789 CAS.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- L. Estevez, A. Kelarakis, Q. Gong, E. H. Da'as and E. P. Giannelis, J. Am. Chem. Soc., 2011, 133, 6122–6125 CrossRef CAS PubMed.
- H.-B. Yao, J. Ge, C.-F. Wang, X. Wang, W. Hu, Z.-J. Zheng, Y. Ni and S.-H. Yu, Adv. Mater., 2013, 25, 6692–6698 CrossRef CAS PubMed.
- D. D. Nguyen, N.-H. Tai, S.-B. Lee and W.-S. Kuo, Energy Environ. Sci., 2012, 5, 7908–7912 CAS.
- W. Ouyang, J. Sun, J. Memon, C. Wang, J. Geng and Y. Huang, Carbon, 2013, 62, 501–509 CrossRef CAS PubMed.
- G. Yu, L. Hu, M. Vosgueritchian, H. Wang, X. Xie, J. R. McDonough, X. Cui, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 2905–2911 CrossRef CAS PubMed.
- Z. Bo, K. Yu, G. Lu, P. Wang, S. Mao and J. Chen, Carbon, 2011, 49, 1849–1858 CrossRef CAS PubMed.
- K. Yu, P. Wang, G. Lu, K.-H. Chen, Z. Bo and J. Chen, J. Phys. Chem. Lett., 2011, 2, 537–542 CrossRef CAS.
- C. S. Rout, A. Kumar, T. S. Fisher, U. K. Gautam, Y. Bando and D. Golberg, RSC Adv., 2012, 2, 8250–8253 RSC.
- C. Hu, X. Zhai, L. Liu, Y. Zhao, L. Jiang and L. Qu, Sci. Rep., 2013, 3, 2065 Search PubMed.
- Z. Xu, Z. Li, C. M. B. Holt, X. Tan, H. Wang, B. S. Amirkhiz, T. Stephenson and D. Mitlin, J. Phys. Chem. Lett., 2012, 3, 2928–2933 CrossRef CAS.
- P. Chen, J.-J. Yang, S.-S. Li, Z. Wang, T.-Y. Xiao, Y.-H. Qian and S.-H. Yu, Nano Energy, 2013, 2, 249–256 CrossRef CAS PubMed.
- H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 2801–2810 CAS.
- K. Chen, L. Chen, Y. Chen, H. Bai and L. Li, J. Mater. Chem., 2012, 22, 20968–20976 RSC.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
- N. Li, Z. Chen, W. Ren, F. Li and H.-M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17360–17365 CrossRef CAS PubMed.
- Z. Chen, C. Xu, C. Ma, W. Ren and H.-M. Cheng, Adv. Mater., 2013, 25, 1296–1300 CrossRef CAS PubMed.
- M. T. Pettes, H. Ji, R. S. Ruoff and L. Shi, Nano Lett., 2012, 12, 2959–2964 CrossRef CAS PubMed.
- X.-C. Dong, H. Xu, X.-W. Wang, Y.-X. Huang, M. B. Chan-Park, H. Zhang, L.-H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS PubMed.
- W. Zhou, X. Cao, Z. Zeng, W. Shi, Y. Zhu, Q. Yan, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2216–2221 CAS.
- X. Cao, Z. Zeng, W. Shi, P. Yep, Q. Yan and H. Zhang, Small, 2012, 9, 1703–1707 CrossRef PubMed.
- X. Cao, Y. Shi, W. Shi, X. Rui, Q. Yan, J. Kong and H. Zhang, Small, 2013, 3433–3438 CrossRef CAS PubMed.
- Z. Yan, L. Ma, Y. Zhu, I. Lahiri, M. G. Hahm, Z. Liu, S. Yang, C. Xiang, W. Lu, Z. Peng, Z. Sun, C. Kittrell, J. Lou, W. Choi, P. M. Ajayan and J. M. Tour, ACS Nano, 2012, 7, 58–64 CrossRef PubMed.
- W. Wang, S. Guo, M. Penchev, I. Ruiz, K. N. Bozhilov, D. Yan, M. Ozkan and C. S. Ozkan, Nano Energy, 2013, 2, 294–303 CrossRef CAS PubMed.
- F. Yavari, Z. Chen, A. V. Thomas, W. Ren, H.-M. Cheng and N. Koratkar, Sci. Rep., 2011, 1, 166 Search PubMed.
- H. Ji, L. Zhang, M. T. Pettes, H. Li, S. Chen, L. Shi, R. Piner and R. S. Ruoff, Nano Lett., 2012, 12, 2446–2451 CrossRef CAS PubMed.
- X. Cao, B. Zheng, X. Rui, W. Shi, Q. Yan and H. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1404–1409 CrossRef CAS PubMed.
- J. Luo, J. Liu, Z. Zeng, C. F. Ng, L. Ma, H. Zhang, J. Lin, Z. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136–6143 CrossRef CAS PubMed.
- Y.-H. Chang, C.-T. Lin, T.-Y. Chen, C.-L. Hsu, Y.-H. Lee, W. Zhang, K.-H. Wei and L.-J. Li, Adv. Mater., 2013, 25, 756–760 CrossRef CAS PubMed.
- Y. Xue, D. Yu, L. Dai, R. Wang, D. Li, A. Roy, F. Lu, H. Chen, Y. Liu and J. Qu, Phys. Chem. Chem. Phys., 2013, 15, 12220–12226 RSC.
- B.-J. Kim, G. Yang, M. Joo Park, J. Seop Kwak, K. Hyeon Baik, D. Kim and J. Kim, Appl. Phys. Lett., 2013, 102, 161902–161904 CrossRef PubMed.
- B. Tang, G. Hu, H. Gao and Z. Shi, J. Power Sources, 2013, 234, 60–68 CrossRef CAS PubMed.
- N. Li, Q. Zhang, S. Gao, Q. Song, R. Huang, L. Wang, L. Liu, J. Dai, M. Tang and G. Cheng, Sci. Rep., 2013, 3, 1604 Search PubMed.
- C. Shan, H. Tang, T. Wong, L. He and S.-T. Lee, Adv. Mater., 2012, 24, 2491–2495 CrossRef CAS PubMed.
- Z. Sun, Z. Yan, J. Yao, E. Beitler, Y. Zhu and J. M. Tour, Nature, 2010, 468, 549–552 CrossRef CAS PubMed.
- V. Sridhar, I. Lee, H.-S. Yoon, H.-H. Chun and H. Park, Carbon, 2013, 61, 633–639 CrossRef CAS PubMed.
- Y. Li, Z. Li and P. K. Shen, Adv. Mater., 2013, 25, 2474–2480 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, C. Zhi, X. Wang, D. Tang, Y. Xu, Q. Weng, X. Jiang, M. Mitome, D. Golberg and Y. Bando, Nat. Commun., 2013, 4, 2905 Search PubMed.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- D. Yu and L. Dai, J. Phys. Chem. Lett., 2009, 1, 467–470 CrossRef.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, S. E. Moulton, J. M. Razal and G. G. Wallace, ACS Nano, 2013, 7, 3981–3990 CrossRef CAS PubMed.
- Z. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 CAS.
- Y. Sun, Q. Wu, Y. Xu, H. Bai, C. Li and G. Shi, J. Mater. Chem., 2011, 21, 7154–7160 RSC.
- Q. Wu, Y. X. Xu, Z. Y. Yao, A. R. Liu and G. Q. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- C. Liu, K. Wang, S. Luo, Y. Tang and L. Chen, Small, 2011, 7, 1203–1206 CrossRef CAS PubMed.
- C. Tan, X. Huang and H. Zhang, Mater. Today, 2013, 16, 29–36 CrossRef CAS PubMed.
- W. Shi, J. Zhu, D. H. Sim, Y. Y. Tay, Z. Lu, X. Zhang, Y. Sharma, M. Srinivasan, H. Zhang, H. H. Hng and Q. Yan, J. Mater. Chem., 2011, 21, 3422–3427 RSC.
- S. Yang, X. Feng, L. Wang, K. Tang, J. Maier and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 4795–4799 CrossRef CAS PubMed.
- M. Jahan, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707–6713 CrossRef CAS PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2012, 46, 1094–1103 CrossRef PubMed.
- S. Murali, N. Quarles, L. L. Zhang, J. R. Potts, Z. Tan, Y. Lu, Y. Zhu and R. S. Ruoff, Nano Energy, 2013, 2, 764–768 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- V. Strong, S. Dubin, M. F. El-Kady, A. Lech, Y. Wang, B. H. Weiller and R. B. Kaner, ACS Nano, 2012, 6, 1395–1403 CrossRef CAS PubMed.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 Search PubMed.
- C. Wu, X. Huang, G. Wang, L. Lv, G. Chen, G. Li and P. Jiang, Adv. Funct. Mater., 2013, 23, 506–513 CrossRef CAS.
- C.-M. Chen, Q. Zhang, C.-H. Huang, X.-C. Zhao, B.-S. Zhang, Q.-Q. Kong, M.-Z. Wang, Y.-G. Yang, R. Cai and D. Sheng Su, Chem. Commun., 2012, 48, 7149–7151 RSC.
- K. Jiang, Q. Li and S. Fan, Nature, 2002, 419, 801 CrossRef CAS PubMed.
- K. Koziol, J. Vilatela, A. Moisala, M. Motta, P. Cunniff, M. Sennett and A. Windle, Science, 2007, 318, 1892–1895 CrossRef CAS PubMed.
- B. Vigolo, A. Pénicaud, C. Coulon, C. Sauder, R. Pailler, C. Journet, P. Bernier and P. Poulin, Science, 2000, 290, 1331–1334 CrossRef CAS.
- Z. Xu, H. Y. Sun, X. L. Zhao and C. Gao, Adv. Mater., 2013, 25, 188–193 CrossRef CAS PubMed.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571 CrossRef PubMed.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, Sci. Rep., 2012, 2, 613 Search PubMed.
- Z. L. Dong, C. C. Jiang, H. H. Cheng, Y. Zhao, G. Q. Shi, L. Jiang and L. T. Qu, Adv. Mater., 2012, 24, 1856–1861 CrossRef CAS PubMed.
- Z. Xu, Y. Zhang, P. G. Li and C. Gao, ACS Nano, 2012, 6, 7103–7113 CrossRef CAS PubMed.
- Z. Xu, Z. Liu, H. Y. Sun and C. Gao, Adv. Mater., 2013, 25, 3249–3253 CrossRef CAS PubMed.
- Y. Zhao, C. Jiang, C. Hu, Z. Dong, J. Xue, Y. Meng, N. Zheng, P. Chen and L. Qu, ACS Nano, 2013, 7, 2406–2412 CrossRef CAS PubMed.
- L. Chen, Y. L. He, S. G. Chai, H. Qiang, F. Chen and Q. Fu, Nanoscale, 2013, 5, 5809–5815 RSC.
- X. M. Li, T. S. Zhao, K. L. Wang, Y. Yang, J. Q. Wei, F. Y. Kang, D. H. Wu and H. W. Zhu, Langmuir, 2011, 27, 12164–12171 CrossRef CAS PubMed.
- D. Li, J. T. McCann and Y. Xia, Small, 2005, 1, 83–86 CrossRef CAS PubMed.
- Y. Zhao, X. Cao and L. Jiang, J. Am. Chem. Soc., 2007, 129, 764–765 CrossRef CAS PubMed.
- C. G. Hu, Y. Zhao, H. H. Cheng, Y. H. Wang, Z. L. Dong, C. C. Jiang, X. Q. Zhai, L. Jiang and L. T. Qu, Nano Lett., 2012, 12, 5879–5884 CrossRef CAS PubMed.
- M.-S. Wu and Y.-H. Fu, Carbon, 2013, 60, 236–245 CrossRef CAS PubMed.
- J. Luo, X. Zhao, J. Wu, H. D. Jang, H. H. Kung and J. Huang, J. Phys. Chem. Lett., 2012, 3, 1824–1829 CrossRef CAS.
- K. H. Kim, M. Yang, K. M. Cho, Y.-S. Jun, S. B. Lee and H.-T. Jung, Sci. Rep., 2013, 3, 3251 Search PubMed.
- J. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J. M. Gibson and J. Huang, ACS Nano, 2011, 5, 8943–8949 CrossRef CAS PubMed.
- P. Guo, H. Song and X. Chen, J. Mater. Chem., 2010, 20, 4867–4874 RSC.
- D. J. Han, J. H. Jung, J. S. Choi, Y. T. Kim and T. S. Seo, Lab Chip, 2013, 13, 4006–4010 RSC.
- J. Luo, H. D. Jang and J. Huang, ACS Nano, 2013, 7, 1464–1471 CrossRef CAS PubMed.
- X. F. Ma, M. R. Zachariah and C. D. Zangmeister, Nano Lett., 2012, 12, 486–489 CrossRef CAS PubMed.
- Z. Yan, Y. Liu, J. Lin, Z. Peng, G. Wang, E. Pembroke, H. Zhou, C. Xiang, A.-R. O. Raji, E. L. G. Samuel, T. Yu, B. I. Yakobson and J. M. Tour, J. Am. Chem. Soc., 2013, 135, 10755–10762 CrossRef CAS PubMed.
- H. Wang, K. Sun, F. Tao, D. J. Stacchiola and Y. H. Hu, Angew. Chem., Int. Ed., 2013, 52, 9210–9214 CrossRef CAS PubMed.
- L. Qiu, J. Z. Liu, S. L. Y. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- H. Li, J. Wu, X. Qi, Q. He, C. Liusman, G. Lu, X. Zhou and H. Zhang, Small, 2013, 9, 382–386 CrossRef CAS PubMed.
- F. Y. Zeng, Y. F. Kuang, Y. Wang, Z. Y. Huang, C. P. Fu and H. H. Zhou, Adv. Mater., 2011, 23, 4929–4932 CrossRef CAS PubMed.
- X. Xie, L. Ju, X. Feng, Y. Sun, R. Zhou, K. Liu, S. Fan, Q. Li and K. Jiang, Nano Lett., 2009, 9, 2565–2570 CrossRef CAS PubMed.
- F. Y. Zeng, Y. F. Kuang, G. Q. Liu, R. Liu, Z. Y. Huang, C. P. Fu and H. H. Zhou, Nanoscale, 2012, 4, 3997–4001 RSC.
- W. Zhou, J. Liu, T. Chen, K. S. Tan, X. Jia, Z. Luo, C. Cong, H. Yang, C. M. Li and T. Yu, Phys. Chem. Chem. Phys., 2011, 13, 14462–14465 RSC.
- L. M. Viculis, J. J. Mack and R. B. Kaner, Science, 2003, 299, 1361 CrossRef CAS PubMed.
- J. Zheng, H. Liu, B. Wu, Y. Guo, T. Wu, G. Yu, Y. Liu and D. Zhu, Adv. Mater., 2011, 23, 2460–2463 CrossRef CAS PubMed.
- Y. Chen, F. Guo, Y. Qiu, H. Hu, I. Kulaots, E. Walsh and R. H. Hurt, ACS Nano, 2013, 7, 3744–3753 CrossRef CAS PubMed.
- Y. Chen, F. Guo, A. Jachak, S.-P. Kim, D. Datta, J. Liu, I. Kulaots, C. Vaslet, H. D. Jang, J. Huang, A. Kane, V. B. Shenoy and R. H. Hurt, Nano Lett., 2012, 12, 1996–2002 CrossRef CAS PubMed.
- Y. Tian, G. Wu, X. Tian, X. Tao and W. Chen, Sci. Rep., 2013, 3, 3327 Search PubMed.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS PubMed.
- S. Yin, Y. Goldovsky, M. Herzberg, L. Liu, H. Sun, Y. Zhang, F. Meng, X. Cao, D. D. Sun, H. Chen, A. Kushmaro and X. Chen, Adv. Funct. Mater., 2013, 23, 2972–2978 CrossRef CAS.
- S. H. Lee, H. W. Kim, J. O. Hwang, W. J. Lee, J. Kwon, C. W. Bielawski, R. S. Ruoff and S. O. Kim, Angew. Chem., Int. Ed., 2010, 49, 10084–10088 CrossRef CAS PubMed.
- C.-M. Chen, Q. Zhang, X.-C. Zhao, B. Zhang, Q.-Q. Kong, M.-G. Yang, Q.-H. Yang, M.-Z. Wang, Y.-G. Yang, R. Schlogl and D. S. Su, J. Mater. Chem., 2012, 22, 14076–14084 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972–1985 CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805–1834 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983–5992 RSC.
- Y. P. Zhai, Y. Q. Dou, D. Y. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- S. Bose, T. Kuila, A. K. Mishra, R. Rajasekar, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 767–784 RSC.
- A. Ghosh and Y. H. Lee, ChemSusChem, 2012, 5, 480–499 CrossRef CAS PubMed.
- M. J. Zhi, C. C. Xiang, J. T. Li, M. Li and N. Q. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- Z.-S. Wu, Y. Sun, Y.-Z. Tan, S. Yang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 19532–19535 CrossRef CAS PubMed.
- W.-Y. Tsai, R. Lin, S. Murali, L. Li Zhang, J. K. McDonough, R. S. Ruoff, P.-L. Taberna, Y. Gogotsi and P. Simon, Nano Energy, 2013, 2, 403–411 CrossRef CAS PubMed.
- Y. Wang, Y. Wu, Y. Huang, F. Zhang, X. Yang, Y. Ma and Y. Chen, J. Phys. Chem. C, 2011, 115, 23192–23197 CAS.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505–509 CrossRef CAS PubMed.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 CAS.
- R. Ramachandran, V. Mani, S. M. Chen, R. Saraswathi and B. S. Lou, Int. J. Electrochem. Sci., 2013, 8, 11680–11694 CAS.
- Y. He, W. Chen, C. Gao, J. Zhou, X. Li and E. Xie, Nanoscale, 2013, 5, 8799–8820 RSC.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2012, 7, 174–182 CrossRef PubMed.
- J. Ji, L. L. Zhang, H. Ji, Y. Li, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, ACS Nano, 2013, 7, 6237–6243 CrossRef CAS PubMed.
- L. Hu, M. Pasta, F. L. Mantia, L. Cui, S. Jeong, H. D. Deshazer, J. W. Choi, S. M. Han and Y. Cui, Nano Lett., 2010, 10, 708–714 CrossRef CAS PubMed.
- H. H. Cheng, Z. L. Dong, C. G. Hu, Y. Zhao, Y. Hu, L. T. Qu, N. Chena and L. M. Dai, Nanoscale, 2013, 5, 3428–3434 RSC.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- Y. Fu, X. Cai, H. Wu, Z. Lv, S. Hou, M. Peng, X. Yu and D. Zou, Adv. Mater., 2012, 24, 5713–5718 CrossRef CAS PubMed.
- J. Bae, Y. J. Park, M. Lee, S. N. Cha, Y. J. Choi, C. S. Lee, J. M. Kim and Z. L. Wang, Adv. Mater., 2011, 23, 3446–3449 CrossRef CAS PubMed.
- J. Bae, M. K. Song, Y. J. Park, J. M. Kim, M. Liu and Z. L. Wang, Angew. Chem., Int. Ed., 2011, 50, 1683–1687 CrossRef CAS PubMed.
- J. Ren, L. Li, C. Chen, X. Chen, Z. Cai, L. Qiu, Y. Wang, X. Zhu and H. Peng, Adv. Mater., 2013, 25, 1155–1159 CrossRef CAS PubMed.
- K. Wang, Q. Meng, Y. Zhang, Z. Wei and M. Miao, Adv. Mater., 2013, 25, 1494–1498 CrossRef CAS PubMed.
- Y. Cao, M. Zhu, P. Li, R. Zhang, X. Li, Q. Gong, K. Wang, M. Zhong, D. Wu, F. Lin and H. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 19550–19556 RSC.
- Y. Li, K. Sheng, W. Yuan and G. Shi, Chem. Commun., 2013, 49, 291–293 RSC.
- B.-H. Kim, K. S. Yang and J. P. Ferraris, Electrochim. Acta, 2012, 75, 325–331 CrossRef CAS PubMed.
- X. Li, X. Zang, Z. Li, X. Li, P. Li, P. Sun, X. Lee, R. Zhang, Z. Huang, K. Wang, D. Wu, F. Kang and H. Zhu, Adv. Funct. Mater., 2013, 23, 4862–4869 CAS.
- T. Huang, B. Zheng, L. Kou, K. Gopalsamy, Z. Xu, C. Gao, Y. Meng and Z. Wei, RSC Adv., 2013, 3, 23957–23962 RSC.
- X. Li, T. Zhao, Q. Chen, P. Li, K. Wang, M. Zhong, J. Wei, D. Wu, B. Wei and H. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 17752–17757 RSC.
- Z. Zeng, X. Huang, Z. Yin, H. Li, Y. Chen, H. Li, Q. Zhang, J. Ma, F. Boey and H. Zhang, Adv. Mater., 2012, 24, 4138–4142 CrossRef CAS PubMed.
- X. Wang, L. Jiao, K. Sheng, C. Li, L. Dai and G. Shi, Sci. Rep., 2013, 3, 1996 Search PubMed.
- Y. Fang, Y. Lv, R. Che, H. Wu, X. Zhang, D. Gu, G. Zheng and D. Zhao, J. Am. Chem. Soc., 2013, 135, 1524–1530 CrossRef CAS PubMed.
- T. H. Han, Y. K. Huang, A. T. L. Tan, V. P. Dravid and J. X. Huang, J. Am. Chem. Soc., 2011, 133, 15264–15267 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- X. Huang, Z. Zeng, S. Bao, M. Wang, X. Qi, Z. Fan and H. Zhang, Nat. Commun., 2013, 4, 1444 CrossRef PubMed.
- X. Huang, Z. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998–6001 CrossRef CAS PubMed.
- Z. Zeng, C. Tan, X. Huang, S. Mao and H. Zhang, Energy Environ. Sci., 2014, 7, 797–803 CAS.
- H. Li, J. Wu, X. Huang, G. Lu, J. Yang, X. Lu, Q. Xiong and H. Zhang, ACS Nano, 2013, 7, 10344–10353 CrossRef CAS PubMed.
- H. Li, G. Lu, Y. Wang, Z. Yin, C. Cong, Q. He, L. Wang, F. Ding, T. Yu and H. Zhang, Small, 2013, 9, 1974–1981 CrossRef CAS PubMed.
- J. Wu, H. Li, Z. Yin, H. Li, J. Liu, X. Cao, Q. Zhang and H. Zhang, Small, 2013, 9, 3314–3319 CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |