A nanoporous molybdenum carbide nanowire as an electrocatalyst for hydrogen evolution reaction†
Received
19th July 2013
, Accepted 16th October 2013
First published on 17th October 2013
Abstract
A highly active and stable electrochemical catalyst of nanoporous molybdenum carbide nanowires (np-Mo2C NWs) has been developed for hydrogen evolution reaction (HER). The np-Mo2C NWs were synthesized simply by pyrolysis of a MoOx/amine hybrid precursor with sub-nanosized periodic structure under an inert atmosphere. The enriched nanoporosity and large reactive surface of these highly dispersed nanowires with uniform Mo2C nanocrystallites provide an efficient electrocatalysis, leading to their superior HER activity with lower onset overpotential and higher current densities than Mo2C microparticles. This study opens a new perspective for the development of highly active non-noble electrocatalysts for hydrogen production from water splitting.
Introduction
Sustainable hydrogen production from water splitting is one of the most attractive methods for energy storage and conversion.1–4 Electrolysis of water has been studied for several decades, but both the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) still remain technical challenges. Although platinum (Pt)-based materials have proved to be the most effective HER electrocatalysts, their high cost and low abundance limit their large-scale applications.5–9 Therefore, some Pt-like materials with cheap and abundant elements have attracted considerable attention as alternative catalysts or catalyst supports for various applications, which sometimes even offer significant advantages over noble metals.10–14 Among these, transition metal carbides display remarkable catalytic activities, owing to their similar electronic and catalytic properties to Pt-group metals (by inducing carbon into the metal lattice), and have been identified as the most promising candidates to replace or reduce Pt employment in catalysis reactions.15–18 Additionally, they have been used as stable supports for precious metals over different ranges of pH.19 Recently, Vrubel and Hu reported that after removal of surface oxides, the commercial molybdenum carbide (Mo2C) was a very active HER catalyst under both acidic and basic conditions.20 However, the use of aggregated Mo2C microparticles may hamper the transport of electrons or protons during the reaction, and thereby result in a loss of activity, given that the performance of HER electrocatalysts greatly depends on their compositions and structures.20 Recently, Sasaki et al. demonstrated that Mo2C nanoparticles on carbon nanotubes were indeed a highly active and durable electrocatalyst for hydrogen production.21 In the present paper, we have developed nanopore-structured Mo2C nanowires to increase the specific surface area for reaction. These nanowires are composed of individual nanocrystallites, each of which is closely interconnected to shorten the electron transfer pathway but free of large aggregation. It is anticipated that such nanowire catalysts with abundant porosity not only possess more exposed active surface but also enable rapid diffusion of ions and electrons favoring a better activity.
The purpose of this work is to demonstrate the prominent HER performance of nanoporous Mo2C nanowires composed of Mo2C nanocrystallites (np-Mo2C NWs). The np-Mo2C NWs were prepared by a strategy developed by our group through simply pyrolyzing precursor MoOx/amine hybrid nanowires under an inert atmosphere.22,23 The sub-nanometer contact between molybdenum oxides and amine molecules in the precursor creates a quasi-homogeneous reaction environment and greatly facilitates the formation of the nanocrystallite-composed nanoporous structure.21 The special features of large active surface areas, nanosized crystallites and enriched nanoporosity of np-Mo2C NWs greatly accelerate their interfacial electrocatalytic reactions while allowing rapid charge transfer kinetics. As a result, superior electrocatalytic activity towards hydrogen production is seen compared with bulk Mo2C particles and other nonprecious metal materials. Using electrochemical impedance spectroscopy, the very high current densities at a np-Mo2C NW-modified electrode are attributed to the considerable increase of electrochemically active surface area as well as the smaller charge-transfer resistances and higher interfacial capacitances.
Results and discussion
Characterization of np-Mo2C NWs
Fig. 1 presents the typical morphology, structure, composition and porosity of the np-Mo2C NW sample, which were prepared by calcination of the organic–inorganic hybrid Mo3O10(C6H8N)2·2H2O nanowire precursor at 725 °C in an inert flow.22 The scanning electron microscopy (SEM) image clearly reveals the uniform 1D morphology of each individual np-Mo2C NW being several micrometers in length and 80–150 nm in width (Fig. 1A). Furthermore, high-resolution transmission electron microscopy (HRTEM) showed that such nanowires were composed of nanocrystallites of about 10–15 nm in size (Fig. 1B and C), significantly differing from the morphology of larger and more irregular bulk commercial Mo2C particles (Fig. S1†).20 Elemental analysis by energy dispersive spectroscopy (EDS) confirmed that the Mo and C contents were consistent with their theoretical values in Mo2C (Fig. 1D), while X-ray diffraction (XRD) confirmed the hexagonal phase structure of the Mo2C nanowires (Fig. 1E). Meanwhile, the results calculated from the N2 sorption isotherm (Fig. 1F) show a BET surface area of 63.9 m2 g−1 and a mesopore size of 3.3 nm resulting from the aggregation of Mo2C nanocrystallites inside the nanowires. Thus, taking into account the evidence from each characterization technique, we can summarize that the np-Mo2C NWs possess uniform one-dimensional morphology, nanosized crystallites, enriched nanoporosity and large surface areas. Such a combination of beneficial attributes would significantly enhance the catalytic potency of np-Mo2C NWs towards HER by not only providing abundant specific catalytic sites but also facilitating the access of electrolytes and allowing rapid charge transfer kinetics. For comparison with both commercial Mo2C and np-Mo2C NWs, we synthesized nanoporous Mo2C flakes composed of nanocrystallites about 20–25 nm in size and bulky Mo2C foams composed of large particles. Their respective SEM and TEM images, as well as XRD patterns, are presented in Fig. S2–S4.† The porosity of the Mo2C flakes and bulky Mo2C foams resulted from the aggregation of crystallites inside the Mo2C. Nitrogen adsorption/desorption isotherms (Fig. S5†) show BET surface areas of 67.6 m2 g−1 and 30.3 m2 g−1, average pore sizes of 4.5 nm and 3.7 nm, and pore volumes of 0.09 cm3 g−1 and 0.04 cm3 g−1 for the Mo2C flakes and bulky foams, respectively.
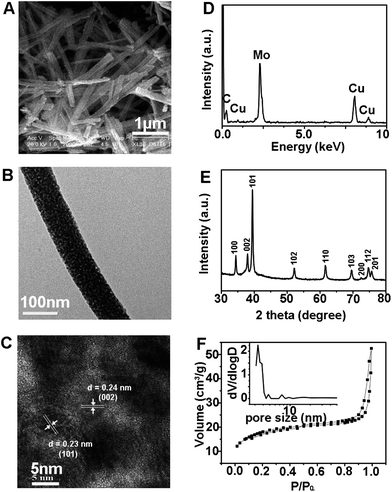 |
| | Fig. 1 (A) SEM image, (B) TEM image, (C) high-resolution TEM image, (D) EDS pattern, (E) XRD pattern and (F) nitrogen adsorption/desorption isotherm and pore size distribution (insert) of np-Mo2C NWs. | |
HER catalytic activities of np-Mo2C NW modified electrodes
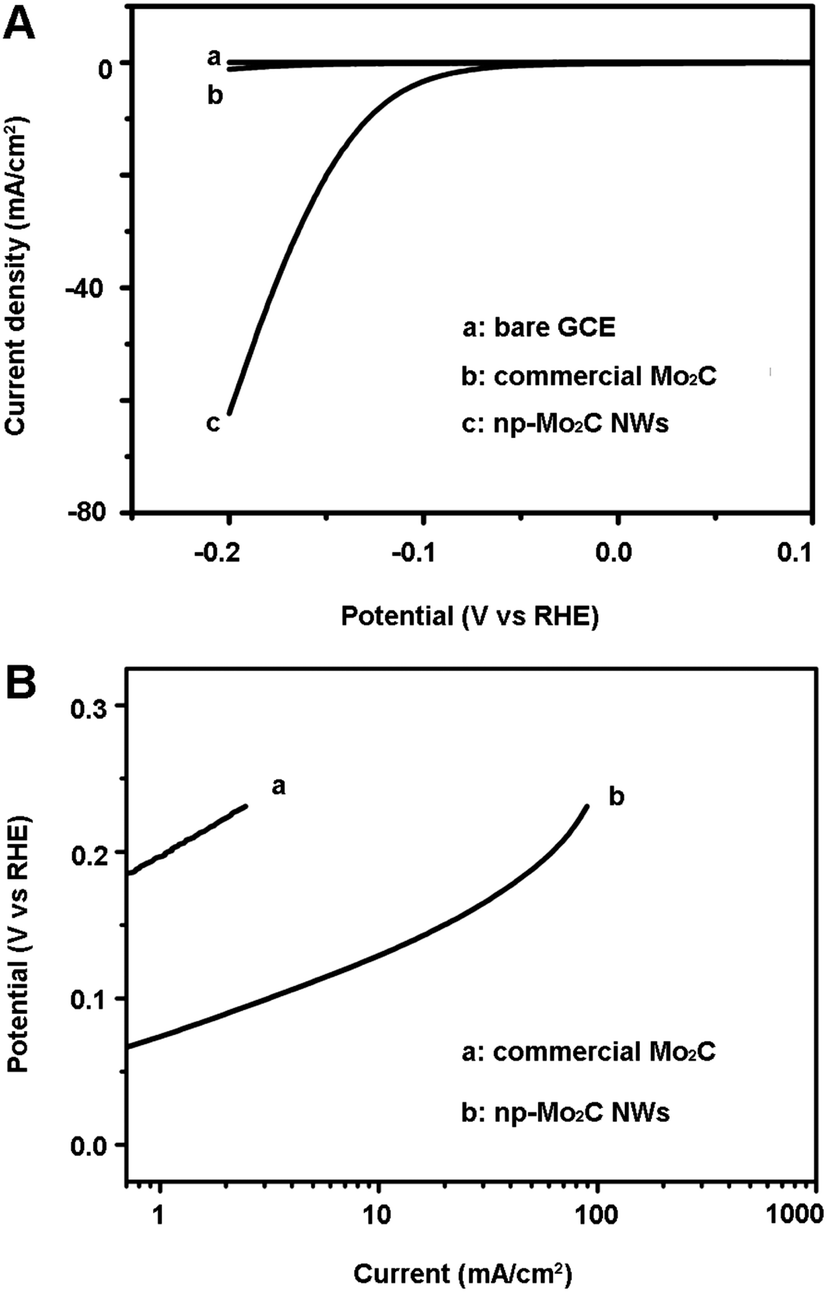
The electrocatalytic HER activity of the np-Mo2C NW sample was investigated by depositing it on a glassy carbon electrode (GCE) with a catalyst loading of 0.21 mg cm−2 (np-Mo2C NW-modified GCE). The experiment was conducted in 0.5 M H2SO4 solution at room temperature using a three-electrode setup. For comparison, GCEs were also modified with commercial Mo2C particles (comm-Mo2C-modified GCE), nanoporous Mo2C flakes (flaky Mo2C-modified GCE), bulky Mo2C foams (bulky Mo2C-modified GCE) as well as a physical mixture of np-Mo2C NWs and Vulcan carbon (np-Mo2C NW/carbon-modified GCE). Fig. 2A shows the polarization curves (i–V plot) recorded for each individual electrode. For a np-Mo2C NW-modified GCE, the catalytic current was observed at a low onset overpotential of about 70 mV (vs. RHE). This current is ascribed to the HER, rather than other processes, due to the similarity of impedance at various HER overpotentials from 70 to 150 mV (Fig. 3). Beyond this, the cathodic current rises rapidly at more negative potentials with substantial current densities of ca. 60 mA cm−2 at an overpotential of 200 mV. Such electrocatalytic activity is one of the best reported for non-noble catalysts in acidic media to date. In contrast, a comm-Mo2C-modified GCE showed much less HER activity, with a current density of only 1.2 mA cm−2 at 200 mV (Fig. 2A), less than 1/50 of those at the np-Mo2C NW-modified GCE. Similarly, a synthesized bulky Mo2C-modified GCE also showed a low current density of 7.3 mA cm−2 at 200 mV (Fig. S6†). Both commercial and bulky Mo2C particles had smaller surface areas and exhibited lower catalytic activities, indicating that the surface area is a crucial structural parameter dominating the HER, as reported widely in the literature. However, the catalytic current density derived from the synthesized nanoporous bulky Mo2C was about 6 times higher than that of the comm-Mo2C-modified GCE. The existence of porosity can prevent the large aggregation between crystallites endowing them with more exposed active surface and also enabling the rapid diffusion of species during the reactions. On the other hand, the flaky Mo2C-modified GCE gave a current density of 21 mA cm−2 at 200 mV (Fig. S6†), higher than that of bulky Mo2C particles while a little lower than that of a np-Mo2C NW-modified GCE. The results showed that the crystallite size was another factor influencing the HER catalysis consistent with the results reported by Sasaki.21 As discussed above, the significantly improved catalytic activity of a np-Mo2C NW-modified GCE derives from a potent combination of specific structural properties, namely large surface areas, high dispersibility, nanosized crystallites and abundant nanoporosity, contributing in turn to the high abundance of available catalytic active sites, less particle aggregation and smaller charge-transfer resistances of the electrode.
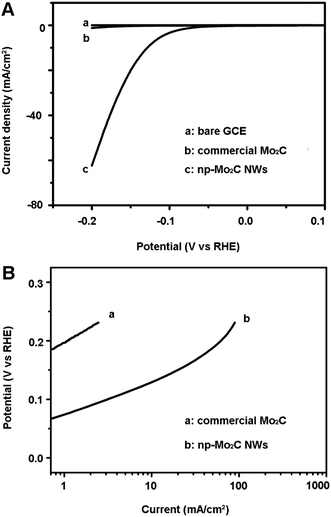 |
| | Fig. 2 (A) Polarization curves obtained from glassy carbon electrodes modified with different Mo2C catalysts, and (B) the corresponding Tafel plots recorded with a catalyst loading of 0.21 mg cm−2; scan rate is 2 mV s−1. | |
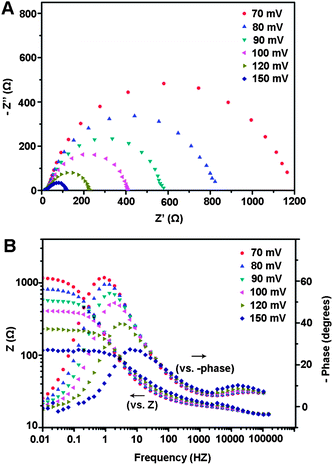 |
| | Fig. 3 Nyquist (A) and Bode (B) plots showing EIS responses of a np-Mo2C NW-modified glassy carbon electrode at various HER overpotentials in 0.5 M H2SO4. | |
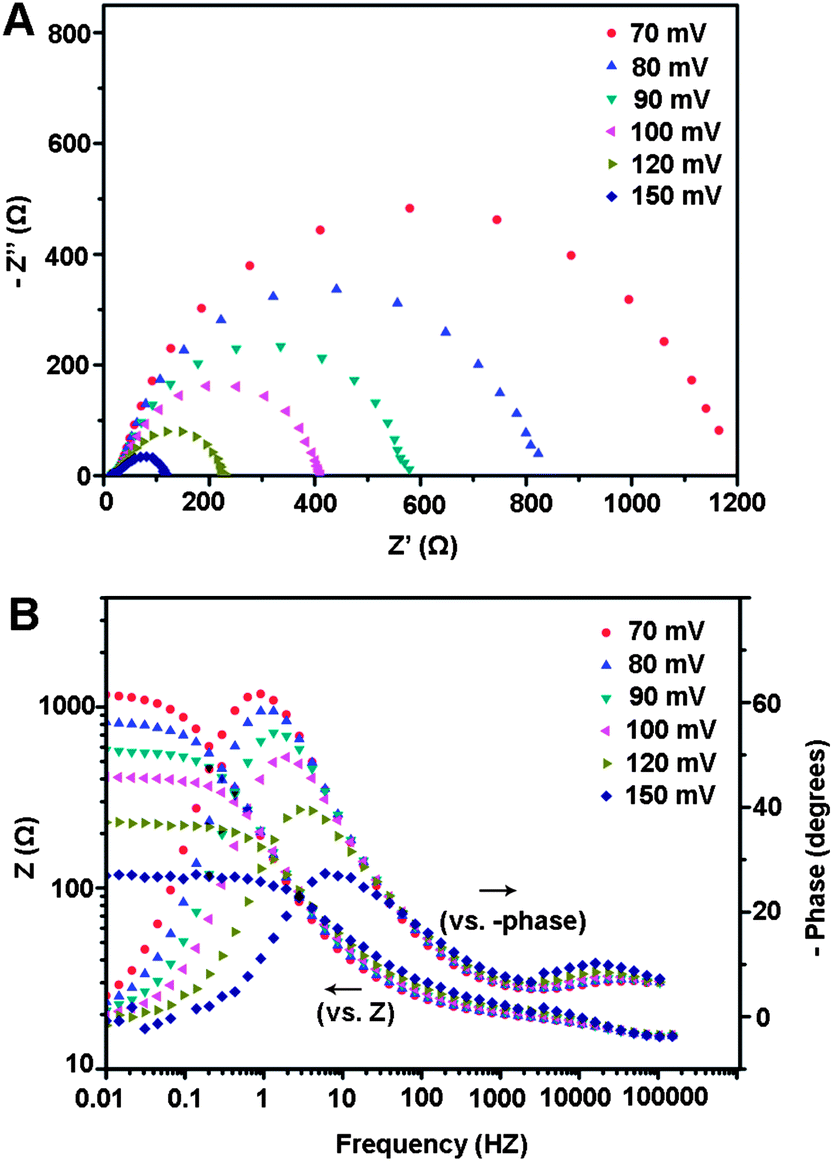
To illustrate the relative enhancement of the catalytic activity for np-Mo2C NWs, electrochemical impedance spectroscopy (EIS) was further applied at various HER overpotentials for this system. The Nyquist and Bode plots of the EIS response are shown in Fig. 3. The np-Mo2C NW-modified electrode exhibited two semicircles at all applied potentials, indicating that the corresponding equivalent circuit for the HER was characterized by two time constants as fitted from the experimental data (Fig. S8†). The first one at high frequencies is related to the porous property of the electrode's surface; whereas another with potential-dependent properties at low frequencies is attributed to the kinetics of the HER process.24–26 Bode plots also display two relaxation times where the high-frequency relaxation time is constant at these overpotentials. The similar impedance properties observed indicated similar electrochemical processes with regard to the HER at all of these overpotentials. Indeed, catalytic currents and hydrogen bubbles were clearly observed at a potential of 70 mV. Such superior electrocatalytic activity of np-Mo2C NWs towards HER was also validated by observation of lower charge-transfer resistance and higher interfacial capacitances derived from the second semicircles (associated with fast reaction kinetics and considerable increases of the electrochemically accessible surface area after nanowire modification). Fig. S9† presents the low-frequency charge transfer resistance (Rct) and constant phase element (CPE) as a function of the HER overpotentials. The recorded values of Rct decrease from 780 Ω at 80 mV to 90 Ω at 150 mV. Application of more negative potentials decreases the double-layer capacitance, from ∼9.5 mF cm−2 (45.2 mF mg−1) at 80 mV to ∼4.3 mF cm−2 (20.5 mF mg−1) at 150 mV, probably due to partial blocking of the electrode surface sites by freshly generated hydrogen species. At more negative potentials, hydrogen bubble evolution is more vigorous at the electrode surface. The high values of double-layer capacitance for the nanoporous np-Mo2C NW-modified electrodes further illustrate their porous character due to being composed of nanosized crystallites with large specific surface areas.27,28 Both lower charge-transfer resistance and higher interfacial capacitance demonstrate the superior electrocatalytic activity of np-Mo2C NWs.
The Tafel slope of the amperometric response for a catalyst-modified electrode is an inherent property indicative of the rate-limiting step for HER. Tafel analysis allows distinction of the possible mechanistic steps occurring during the HER at an electrode surface. In acidic solution, three separate reaction steps are possible when hydrogen is evolved at the catalyst modified electrode surface.
| Discharge reaction: H3O+ + e− + cat → cat-H + H2O (Volmer reaction) |
| Combination reaction: cat-H + cat-H → 2cat + H2 (Tafel reaction) |
| Ion + atom reaction: H3O+ + e− + cat-H → cat + H2 + H2O (Heyrovsky reaction) |
According to previous reports, a Tafel slope of 30 mV per decade suggests that the mechanism proceeds through the Volmer–Tafel mechanism and the recombination step is rate-limiting.29 A Tafel slope of 40 mV per decade suggests hydrogen production via the Volmer–Heyrovsky mechanism and the electrochemical desorption step is rate-limiting.30 Finally, a Tafel slope of 120 mV per decade may arise from various reaction pathways depending on the surface coverage of adsorbed hydrogen.29 The linear portions of the Tafel plots are fitted to the Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj + a, where j is the current density and b is the Tafel slope), yielding Tafel slopes of ∼53 mV per decade (η = 80–100 mV) and ∼78 mV per decade (η = 180–200 mV) for np-Mo2C NWs and commercial Mo2C, respectively (Fig. 2B). Obtaining a Tafel slope, however, is subjective with moderately different values possible by judicious choice of the regions on the current voltage curves to analyse. Additional complications may arise from the different ways in which the iR-drops are corrected. With this in mind, the Tafel slopes of ∼53 and ∼78 mV per decade for np-Mo2C NWs and commercial Mo2C, respectively, are somewhat ambiguous with regard to elucidating the precise HER mechanism at each catalyst modified GCE. One possible conclusion is to eliminate the Volmer-step as the rate determining step, but beyond that only speculation is possible without the intervention of more powerful mechanistic analysis tools. Therefore, the clear perspective now that np-Mo2C NWs have been identified as highly active catalysts is to develop a new suite of techniques that go beyond a routine Tafel analysis based on polarisation curves. Although electron transport (ET) is integral to the HER mechanism, precise analysis of ET at a catalyst modified GCE can be masked by the multiple proton-coupled electron transfer reactions occurring during a typical HER process. To circumvent this issue, recently, Vrubel et al. highlighted the benefits of transitioning from linear sweep voltammetry (LSV)-based analysis to electrochemical impedance spectroscopy (EIS)-based analysis of Tafel slopes.31 They demonstrated the possibility of identifying a slow electron transport process in the HER catalysed by molybdenum sulfides. This method is directly beneficial to the analysis of Mo2C-based electrocatalysts as it is amenable to the study of catalysts of poor electronic conductivity. The latter is a dominant catalyst attribute that may limit the scalability of a catalysts use (i.e. increased resistance to ET in thicker catalyst films) and, additionally, an EIS-based approach should provide less subjective data on the improvements of a catalysts activity by decreases in ET resistance on either physical mixing of the catalyst with carbon materials or direct immobilisation of the catalyst on a carbon support, as discussed vide infra. A plot of log Rct−1vs. overpotential (Fig. S10†) gives a Tafel slope ∼60 mV per decade (η = 70–100 mV) for np-Mo2C NWs from the EIS data. A Tafel slope obtained in this way reflects purely the charge transfer kinetics, in contrast to Tafel slopes obtained from voltammetry data which may include contributions from catalyst resistance.31 Although the Tafel slopes obtained by both methods are comparable, such values do not allow a clear distinction between which of the Heyrovsky or Tafel reactions are rate-limiting, and further mechanistic studies are required in this direction.
logj + a, where j is the current density and b is the Tafel slope), yielding Tafel slopes of ∼53 mV per decade (η = 80–100 mV) and ∼78 mV per decade (η = 180–200 mV) for np-Mo2C NWs and commercial Mo2C, respectively (Fig. 2B). Obtaining a Tafel slope, however, is subjective with moderately different values possible by judicious choice of the regions on the current voltage curves to analyse. Additional complications may arise from the different ways in which the iR-drops are corrected. With this in mind, the Tafel slopes of ∼53 and ∼78 mV per decade for np-Mo2C NWs and commercial Mo2C, respectively, are somewhat ambiguous with regard to elucidating the precise HER mechanism at each catalyst modified GCE. One possible conclusion is to eliminate the Volmer-step as the rate determining step, but beyond that only speculation is possible without the intervention of more powerful mechanistic analysis tools. Therefore, the clear perspective now that np-Mo2C NWs have been identified as highly active catalysts is to develop a new suite of techniques that go beyond a routine Tafel analysis based on polarisation curves. Although electron transport (ET) is integral to the HER mechanism, precise analysis of ET at a catalyst modified GCE can be masked by the multiple proton-coupled electron transfer reactions occurring during a typical HER process. To circumvent this issue, recently, Vrubel et al. highlighted the benefits of transitioning from linear sweep voltammetry (LSV)-based analysis to electrochemical impedance spectroscopy (EIS)-based analysis of Tafel slopes.31 They demonstrated the possibility of identifying a slow electron transport process in the HER catalysed by molybdenum sulfides. This method is directly beneficial to the analysis of Mo2C-based electrocatalysts as it is amenable to the study of catalysts of poor electronic conductivity. The latter is a dominant catalyst attribute that may limit the scalability of a catalysts use (i.e. increased resistance to ET in thicker catalyst films) and, additionally, an EIS-based approach should provide less subjective data on the improvements of a catalysts activity by decreases in ET resistance on either physical mixing of the catalyst with carbon materials or direct immobilisation of the catalyst on a carbon support, as discussed vide infra. A plot of log Rct−1vs. overpotential (Fig. S10†) gives a Tafel slope ∼60 mV per decade (η = 70–100 mV) for np-Mo2C NWs from the EIS data. A Tafel slope obtained in this way reflects purely the charge transfer kinetics, in contrast to Tafel slopes obtained from voltammetry data which may include contributions from catalyst resistance.31 Although the Tafel slopes obtained by both methods are comparable, such values do not allow a clear distinction between which of the Heyrovsky or Tafel reactions are rate-limiting, and further mechanistic studies are required in this direction.
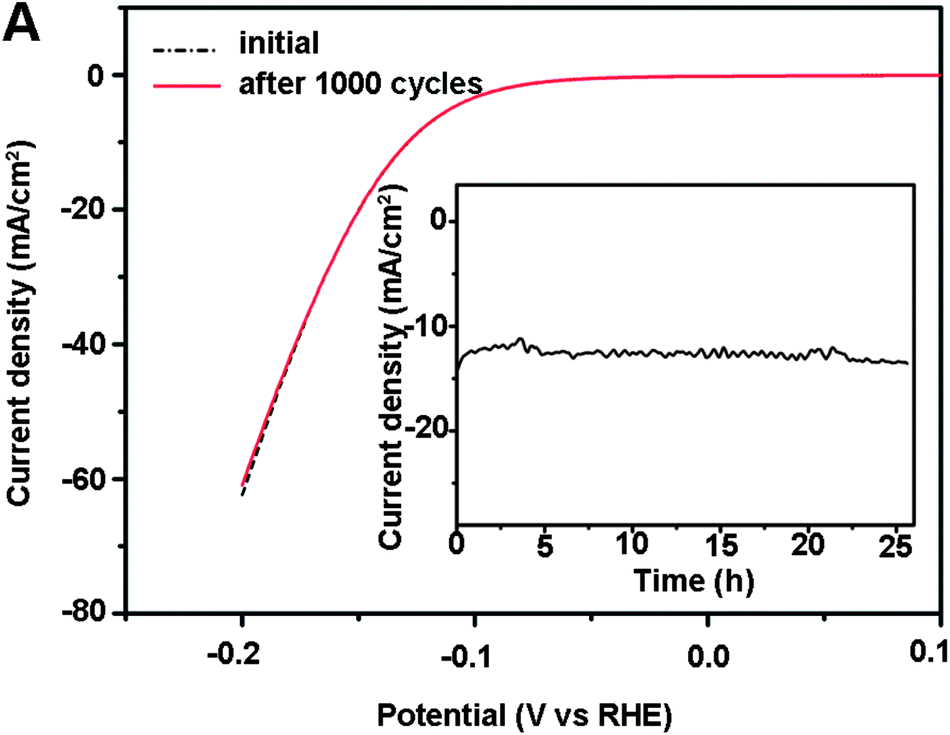
Stability of the catalytic response is evaluated by cycling the np-Mo2C NW-modified GCE continuously for 1000 cycles. At the end of the cycling procedure, the catalyst affords similar i–V curves to the initial cycle with negligible loss of the cathodic current (Fig. 4). The long-term stability of np-Mo2C NWs was also tested by electrolysis at a fixed potential. Fig. 4, inset, shows that the catalytic currents remain at around ca. 12 mA cm−2 over 25 hours demonstrating that np-Mo2C NWs are stable catalysts under the experimental conditions employed for this catalysis system.
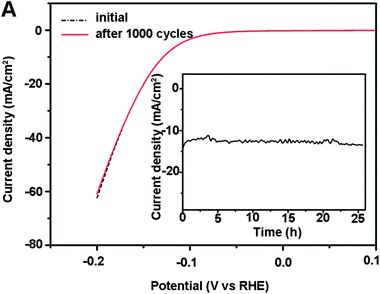 |
| | Fig. 4 Stability of the np-Mo2C NW-modified electrode with an initial LSV polarization curve (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and after 1000 cycles (—) in 0.5 M H2SO4 at a scan rate of 10 mV s−1. The inset shows the time dependence of catalytic currents during electrolysis for np-Mo2C NWs in 0.5 M H2SO4 at η = 130 mV. ) and after 1000 cycles (—) in 0.5 M H2SO4 at a scan rate of 10 mV s−1. The inset shows the time dependence of catalytic currents during electrolysis for np-Mo2C NWs in 0.5 M H2SO4 at η = 130 mV. | |
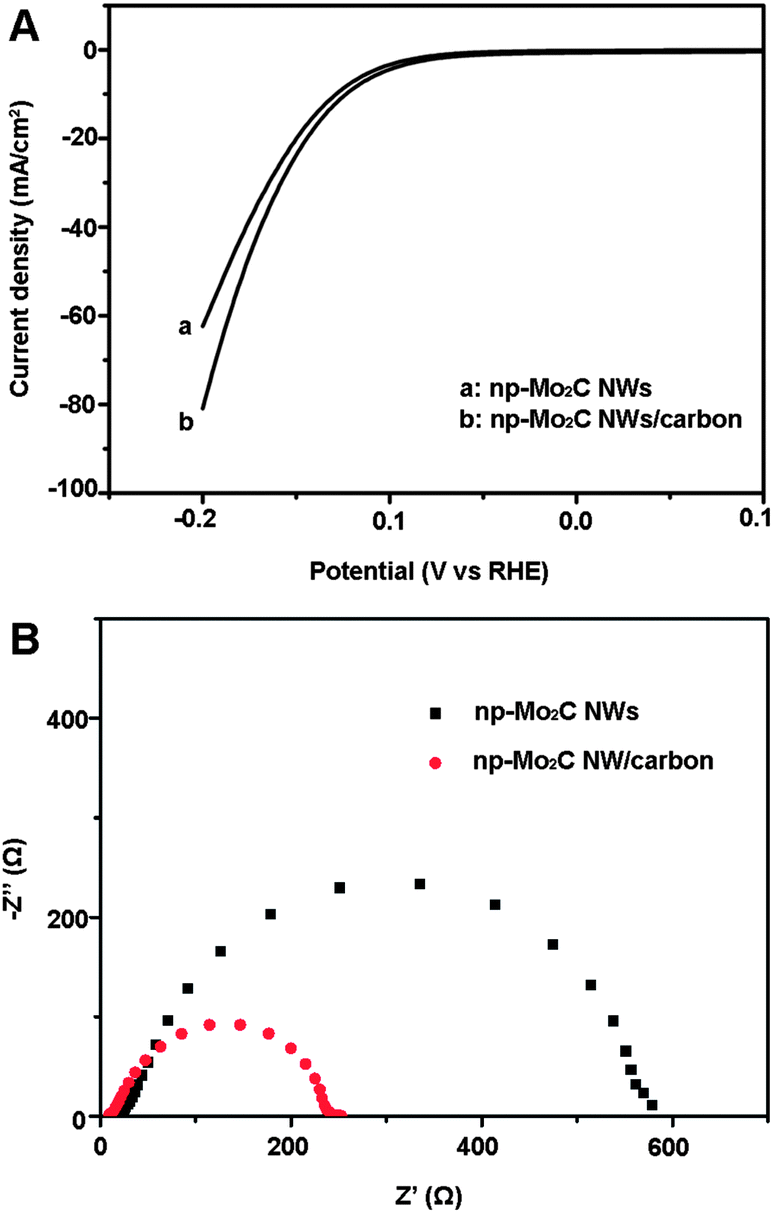
Moreover, when np-Mo2C NWs were physically mixed with commercial Vulcan carbon (1:1 w/w) and used to modify GCEs, an enhanced catalytic performance was observed compared to that of a pure np-Mo2C NW-modified GCE (Fig. 5A) with substantial current densities of ∼80 mA cm−2, at an overpotential of 200 mV, and a Tafel slope of 54 mV per decade (η = 80–100 mV). From the SEM image (Fig. S7†) of np-Mo2C NWs mixed with Vulcan carbon, we can see that the carbon nanoparticles are well-distributed and attached on Mo2C nanowires allowing the conductive carbon to improve electron transfer between the Mo2C-carbon and the underlying electrode. This was further confirmed by electrochemical impedance measurements. The np-Mo2C NW/carbon-modified GCE displays lower impedance than that of an electrode modified with pure np-Mo2C NWs (Fig. 5B). The reduced impedance affords faster electron transfer between the Mo2C nanocrystallite–carbon mixture and the electrode, contributing to the superior kinetics of the HER. It is anticipated that the catalytic performance will be further improved via an in situ formation of nanocomposites of Mo2C on functional carbon based materials.
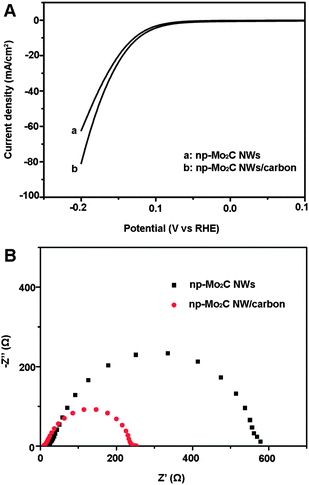 |
| | Fig. 5 (A) Polarization curves of np-Mo2C NWs and physically mixed with Vulcan carbon (1:1 w/w); scan rate: 2 mV s−1. (B) AC impedance spectra of np-Mo2C NWs and np-Mo2C NWs/carbon modified GCEs at η = 90 mV. The np-Mo2C NWs/carbon exhibited smaller Faradic impedance. | |
Conclusion
In summary, nanoporous Mo2C nanowires have been successfully demonstrated as highly active and stable catalysts towards hydrogen evolution reaction. The catalyst was prepared by simply calcining a MoOx/amine hybrid precursor with a sub-nanosized periodic structure. The special features of np-Mo2C NWs, an enriched nanoporosity by being composed of nanosized crystallites, and a resulting large active surface area, greatly accelerate the interfacial electrochemical reaction and contribute to their potent HER activity. Future perspectives include further augmenting the exceptional catalytic activity of Mo2C by in situ formation of Mo2C nanocomposites on functional matrices. Such highly catalytic, abundant and inexpensive materials hold great promise for a variety of applications in the fields of energy conversion and storage.
Experimental
Reagents
Ammonium heptamolybdate ((NH4)6Mo7O24·4H2O), aniline, commercial Mo2C, Vulcan carbon and Nafion® perfluorinated ion-exchange resin solution (5% w/w) were purchased from Sigma-Aldrich. Other reagents were obtained from the Shanghai Chemical Plant.
Synthesis of Mo2C
The nanoporous molybdenum carbide nanowires (np-Mo2C NWs) were prepared according to our reported method22 with modifications. Typically, 2.48 g of ammonium heptamolybdate and 3.20 g of aniline were added to 40 mL of distilled water and aqueous HCl (1 M) was added drop-wise, with magnetic stirring at room temperature, until a white precipitate was obtained at pH 4–5. After stirring at 50 °C for 6 hours, the product was filtered, washed with ethanol and dried at 50 °C for a further 10 hours. After expelling air for 4 hours at room temperature using argon, the products obtained above were calcined at 725 °C for 5 hours in an argon flow and, finally, stored in a vacuum desiccator. The molybdenum carbide nanoflake and bulky foam products were prepared by a similar process to that of np-Mo2C NWs except changing the pH to 1.0 and 2.0, respectively. The morphology of the Mo8O26 (C6H8N)4·2H2O precursor formed at pH 2.0 is bulky and, hence, the bulky foam morphology of the resultant Mo2C obtained after calcination.23
Characterization
TEM and SEM images were obtained on JEOL JEM-2011 and Philips XL 30 instruments, respectively. XRD patterns were taken on a Rigaku D/MAX 2000 diffractometer using Cu Kα radiation (λ = 1.54056 Å). N2 sorption isotherms were collected on a Micromeritics ASAP 2010 adsorption analyzer at −196 °C (77 K). BET specific surface areas were calculated from adsorption data in a relative pressure range from 0.071 to 0.20. The pore size distribution was derived from the adsorption branch of the isotherms.
Electrochemical measurements
The np-Mo2C NWs (4 mg) were dispersed in 1 mL of solution composed of 4:1 (v/v) distilled water and ethanol containing Nafion (80 μL, 5% wt) by sonication to form a homogeneous slurry. Then, 5 μL of the slurry was loaded onto the surface of a home-made glassy carbon electrode (GCE, 3.5 mm in diameter) with a catalyst loading of 0.21 mg cm−2. The np-Mo2C NW-modified GCE was then dried at room temperature. For comparison, GCEs were also modified with commercial Mo2C, bulky Mo2C, flaky Mo2C and physical mixtures of np-Mo2C NW and Vulcan carbon (1:1 w/w). Each modified GCE was loaded with the same amount of catalyst. All electrochemical studies were performed using a CHI660 potentiostat (CH Instruments, China) in a standard three-electrode setup with a modified glassy carbon working electrode, a saturated calomel electrode as a reference, and platinum foil as a counter electrode. The electrocatalytic activity of Mo2C towards HER was examined by obtaining polarization curves using linear sweep voltammetry (LSV) at a scan rate of 2 mV s−1 at room temperature in 0.5 M H2SO4 solutions. The time dependency of catalytic currents during electrolysis for np-Mo2C NWs was tested in 0.5 M H2SO4 at η = 130 mV after equilibrium. Electrochemical impedance spectroscopic (EIS) measurements were carried out using an AutoLab in 0.5 M H2SO4 at different potentials in the frequency range 10−2 to 106 Hz with a single modulated AC potential of 10 mV. Experimental EIS data were analysed and fitted with the software NOVA 1.7.
Acknowledgements
This work was supported by National Key Basic Reseach Program of China (2013CB934101), NSFC (20925517, 21073041), SKLEAC (201101) and Swiss Science Foundation grant “Solar Fuels” (200 021-134 745). Dr Heron Vrubel, EPFL Switzerland, is thanked for helpful discussions.
References
- J. W. Sun, D. K. Zhong and D. R. Gamelin, Energy Environ. Sci., 2010, 3, 1252–1261 CAS.
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2002, 27, 731–740 CrossRef CAS.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- D. E. Bartak, B. Kazee, K. Shimazu and T. Kuwana, Anal. Chem., 1986, 58, 2756–2761 CrossRef CAS.
- P. Millet, F. Andolfatto and R. Durand, Int. J. Hydrogen Energy, 1996, 21, 87–93 CrossRef CAS.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- N. Krstajic, V. Jovic, L. Gajickrstajic, B. Jovic, A. Antozzi and G. Martelli, Int. J. Hydrogen Energy, 2008, 33, 3676–3687 CrossRef CAS PubMed.
- M.-R. Gao, Z.-Y. Lin, T.-T. Zhuang, J. Jiang, Y.-F. Xu, Y.-R. Zheng and S.-H. Yu, J. Mater. Chem., 2012, 22, 13662 RSC.
- Z. Wu, B. Fang, A. Bonakdarpour, A. Sun, D. P. Wilkinson and D. Wang, Appl. Catal., B, 2012, 125, 59–66 CrossRef CAS PubMed.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- S. Wirth, F. Harnisch, M. Weinmann and U. Schröder, Appl. Catal., B, 2012, 126, 225–230 CrossRef CAS PubMed.
- D. V. Esposito, S. T. Hunt, Y. C. Kimmel and J. G. Chen, J. Am. Chem. Soc., 2012, 134, 3025–3033 CrossRef CAS PubMed.
- C. Ma, J. Sheng, N. Brandon, C. Zhang and G. Li, Int. J. Hydrogen Energy, 2007, 32, 2824–2829 CrossRef CAS PubMed.
- Y. Wang, S. Song, V. Maragou, P. K. Shen and P. Tsiakaras, Appl. Catal., B, 2009, 89, 223–228 CrossRef CAS PubMed.
- D. Ham, R. Ganesan and J. Lee, Int. J. Hydrogen Energy, 2008, 33, 6865–6872 CrossRef CAS PubMed.
- M. C. Weidman, D. V. Esposito, Y.-C. Hsu and J. G. Chen, J. Power Sources, 2012, 202, 11–17 CrossRef CAS PubMed.
- H. Vrubel and X. L. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- W. F. Chen, C. H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. T. Muckerman, Y. Zhu and R. R. Adzic, Energy Environ. Sci., 2013, 6, 943–951 CAS.
- Q. Gao, C. Zhang, S. Xie, W. Hua, Y. Zhang, N. Ren, H. Xu and Y. Tang, Chem. Mater., 2009, 21, 5560–5562 CrossRef CAS.
- Q. Gao, S. Wang, H. Fang, J. Weng, Y. Zhang, J. Mao and Y. Tang, J. Mater. Chem., 2012, 22, 4709–4715 RSC.
- J. Kubisztal, A. Budniok and A. Lasia, Int. J. Hydrogen Energy, 2007, 32, 1211–1218 CrossRef CAS PubMed.
- M. Jafarian, O. Azizi, F. Gobal and M. G. Mahjani, Int. J. Hydrogen Energy, 2007, 32, 1686–1693 CrossRef CAS PubMed.
- R. K. Shervedani and A. R. Madram, Electrochim. Acta, 2007, 53, 426–433 CrossRef CAS PubMed.
- E. Navvaro-Flores and Z. Chong, J. Mol. Catal. A: Chem., 2005, 226, 179–197 CrossRef PubMed.
- R. Solmaz and G. Kardas, Int. J. Hydrogen Energy, 2011, 36, 12079–12087 CrossRef CAS PubMed.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- J. O. M. Bockris and E. C. Potter, J. Electrochem. Soc., 1952, 99, 169–186 CrossRef CAS PubMed.
- H. Vrubel, T. Moehl, M. Gratzel and X. Hu, Chem. Commun., 2013, 49, 8985–8987 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ee42441c |
| ‡ The authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.