
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetrametallic lanthanide(III) phosphonate cages: synthetic, structural and magnetic studies†
Karzan H.
Zangana
,
Eufemio Moreno
Pineda
and
Richard E. P.
Winpenny
*
School of Chemistry and Photon Science Institute, The University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: richard.winpenny@manchester.ac.uk
First published on 13th October 2014
Abstract
The synthesis, structures and magnetic properties of a family of lanthanide complexes containing phosphonate ligands are reported. Reaction of hydrated lanthanide nitrate and tbutylphosphonic acid under reflux conditions in iso-butanol, in the presence of pivalic acid as a co-ligand produced five new lanthanide complexes; pyridine (py) was present as a base. The compounds formed are tetrametallic, with the general formula [pyH]4[Ln4(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] where Ln = GdIII, 1; TbIII, 2; DyIII, 3; HoIII, 4 and ErIII, 5. The metal sites within the complexes lie on the vertices of a triangle-based pyramid, with phosphonate ligands on the triangular faces linking the apical Ln site to the Ln sites in the base. Each lanthanide(III) site is eight-coordinate. Magnetic studies of the compounds show a decline in the product χMT with T; modelling the behaviour of 1 shows anti-ferromagnetic exchange between GdIII centres within the triangle with a negligible interaction to the fourth GdIII centre at the apex of the trigonal pyramid.
Introduction
During the past two decades, the synthesis and characterisation of metal phosphonate cages has been an attractive field of synthetic chemistry and molecular magnetism for their aesthetically pleasant structures with interesting magnetic properties,1 including some examples of single molecule magnets (SMMs).2 Other metal phosphonate cages have been studied as possible magnetic refrigerants.3 Phosphonate moieties are interesting functional groups for the construction of molecular cages, as the three tetrahedral oxygen atoms mimic well the zeolite building blocks, providing suitable anchoring sites for a large variety of metal ion centres (including both transition metals and lanthanides).4 It is also possible to influence the reactivity of compounds using the steric bulk of the phosphonate ligands.5Many research groups, notably those of Clearfield and Zubieta have used phosphonate ligands to synthesise a large number of metal phosphonates that possess mainly extended structures 1D-coordination polymer, 2D-layered structured and 3D-pillared structures.6 As a result of these studies a wide range of 3d-phosphonate and 3d–4f phosphonate cages have been synthesised.7 In contrast to transition metal phosphonates, reported molecular assemblies of purely 4f-phosphonates are rare. The first reported lanthanide phosphonates where prepared in 1990 by Mallouk and coworkers;7 cerium and lanthanum phenylphosphonates with composition [Ln(O3PC6H5)(HO3PC6H5)] and a series of alkylphosphonates such as [Ln(O3PMe)(HO3PMe)] were reported. Recently, we have been synthesising molecular cages of lanthanide metal ions with phosphonate ligands with the aim of preparing new cages, optimistically with interesting physical properties.8
Lanthanide phosphonate complexes normally have low solubility and poor crystallinity in water and organic solvents, hence it is generally difficult to obtain single crystals suitable for X-ray structural analysis.9 One of the major reasons for this is that the multisite coordination ability of the phosphonate ligands leads to the formation of insoluble compounds. We and others have proved that synthetically, this problem can be overcome for 3d-metal ions by different strategies, the first example arises from an initial report by Chandrasekhar and Kingsley,10 where a second ligand is added alongside the phosphonate. The co-ligand occupies certain number of coordination sites on the metal ion as a result of which the number of coordination sites that are accessible for the phosphonate ligands decrease and soluble products can be isolated in the form of single crystals. This methodology has been used with a co-ligand to synthesise molecular cages containing manganese,11 iron,12 cobalt,13 nickel,14 copper,15 vanadium.16 A second method for introducing a phosphonate or ligand without causing an immediate precipitate is to react the ligand with a preformed metal carboxylate cage, which was originally reported for iron and manganese and has since been extended by our group and others.17,18
In this work, we extend the co-ligand approach to 4f-ions, reacting simple lanthanide salts LnIII(NO3)3·nH2O (LnIII = Gd, Tb, Dy, Ho and Er) with t-butylphosphonic acid (H2O3PtBu) in the presence of pivalic acid (HO2CtBu) as a co-ligand, giving tetrametallic lanthanide phosphonate cages.
Experimental section
Starting material
All reagents, metal salts, solvents and ligands were used as purchased without any further purification. Analytical data were obtained by the microanalytical service of the University of Manchester, the data and yields are given in Table 1.| Formula | Yielda | Elemental analysis: calculated (found) | |||||
|---|---|---|---|---|---|---|---|
| C | H | Ln | P | N | |||
| a Calculated based on the lanthanide pivalate starting material. | |||||||
| 1 | [pyH]4[Gd4(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] | 45% | 26.48 (26.22) | 3.86 (3.55) | 30.15 (30.10) | 5.93 (5.89) | 6.71 (6.66) |
| 2 | [pyH]4[Tb4(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] | 39% | 26.40 (26.18) | 3.85 (3.71) | 30.37 (30.25) | 5.92 (5.88) | 6.69 (6.63) |
| 3 | [pyH]4[Dy4(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] | 30% | 26.22 (26.11) | 3.82 (3.79) | 30.84 (30.77) | 5.88 (5.73) | 6.67 (6.61) |
| 4 | [pyH]4[Ho4(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] | 35% | 26.10 (25.98) | 3.80 (3.75) | 31.11 (30.97) | 5.85 (5.80) | 6.61 (6.59) |
| 5 | [pyH]4[Er4(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] | 44% | 25.98 (25.66) | 3.79 (3.71) | 31.46 (31.33) | 5.82 (5.78) | 6.58 (6.51) |
Synthetic method
Pivalic acid (0.8 g, 7.83 mmol), Gd(NO3)3·6H2O (0.55 mmol), t-butyl phosphonic acid (0.07 g, 0.50 mmol) and pyridine (py) (0.5 mL, 6.15 mmol) were dissolved in ibutanol (HOiBu) (15 mL) and the solution was refluxed at 120 °C for 3 hours. The solution was filtered and then allowed to stand undisturbed at room temperature for nine days. Colourless plate-like crystals suitable for single crystal X-ray diffraction of [pyH]4[Gd4III(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] 1 were formed. Similar reactions with LnIII(NO3)3·nH2O, (where LnIII = Tb, Dy, Er and Ho) gave analogous metal cages (Table 1).X-ray data collection and structure solution
X-ray crystallographic measurements for compounds 1–5 were collected on an Agilent SUPERNOVA diffractometer with MoKα radiation (λ = 0.71073 Å). In all cases the selected single crystals were mounted on a tip of a glass pin using Paraton-N oil and placed in a cold nitrogen flow. Structure solution and refinement was performed with the SHELXS-97 package,19a the structures were solved by direct method and completed by iterative cycles of ΔF synthesis and full-matrix least-squares refinement against F2 using program Olex2.19b Crystal data and refinement parameters are given in Table 2. Complete hemispheres data were collected using ϕ and ω scans chosen to give a complete asymmetric unit. All atoms excluding hydrogen were refined anisotropically. Hydrogen atoms were located on the basis of geometrical consideration and treated according to the riding model during refinement with isotropic displacement corresponding to the heavy atom they are linked to. Full crystallographic details can be found in CIF format: CCDC for 1–5, 1021470–1021474.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| a R 1 = ||Fo| − |Fc||/|Fo|, wR2 = [w(|Fo| − |Fc|)2/w|Fo|2]1/2. | |||||
| Chem formula | C51H85O35N11Gd4P4 | C51H85 O35N11Tb4P4 | C46H80O35N10Dy4P4 | C46H80O35N10Ho4P4 | C46H80O35N10Er4P4 |
| fw | 2123.10 | 2129.79 | 2065.01 | 2074.73 | 2084.05 |
| Temp. (K) | 150.01(11) | 150.04(13) | 150.01(10) | 150.02(19) | 150.03(18) |
| Cryst system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| space group | C2/c | C2/c | C2/c | C2/c | C2/c |
| a/Å | 44.8769(7) | 44.8792(6) | 44.8591(11) | 44.7075(5) | 44.7348(10) |
| b/Å | 19.0562(4) | 19.1254(3) | 19.1273(5) | 19.0970(2) | 19.1138(4) |
| c/Å | 24.0257(4) | 24.0145(3) | 23.9486(5) | 23.9771(2) | 23.8851(4) |
| α/° | 90 | 90 | 90 | 90 | 90 |
| β/° | 91.0776(16) | 91.0749(14) | 91.090(2) | 91.2548(9) | 91.3155(18) |
| γ/° | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 20542.7(7) | 20608.8(5) | 20545.1(8) | 20466.1(4) | 20417.6(7) |
| Z | 8 | 8 | 8 | 8 | 8 |
| ρ calcd/g cm−3 | 1.373 | 1.373 | 1.335 | 1.347 | 1.356 |
| μ (Mo Kα)/mm−1 | 2.679 | 2.841 | 3.003 | 3.186 | 3.382 |
| R 1 (I > 2σ)(I)a | 0.0319 | 0.0389 | 0.0430 | 0.0361 | 0.0370 |
wR2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
0.0745 | 0.0846 | 0.0841 | 0.0816 | 0.0854 |
Magnetic measurements
Magnetic measurements were performed in temperature ranges 2–300 K, using a Quantum Design MPMS-XL7 SQUID magnetometer equipped with a 7 T magnet. The samples were grounded and placed in a gel capsule. A small amount of eicosane was used to avoid movement of the sample during the measurement. Diamagnetic corrections for the compounds were estimated using Pascal's constants, and the magnetic data were corrected for diamagnetic contribution of the gel capsule, the eicosane and the sample holder. Magnetic data were fitted using the program PHI.20Results and discussion
Synthetic description
To date, just a few examples of lanthanide phosphonates cages have been reported due to the tendency of these systems to form polymeric materials.9 However we, and others, have recently proved that introducing of co-ligands along with aliphatic R groups on the phosphonate can render better solubility allowing their characterisation. This has led to families of {Ln8P6}8a cages and {Ln10P6}8b centred-rings. To illustrate how variation of other reaction conditions influence the product formed, herein we report the synthesis of five new lanthanide phosphonate clusters {Ln4P4}, using the same general procedure as used for synthesising {Ln8P6}, but replacing the base iPrNH2 with pyridine. Using an aromatic base produces a compound of lower nuclearity, containing four Ln(III) metal ions and four phosphonates. Three of the Ln(III) metal ions in the cluster form a μ3-OH centred triangle with the fourth Ln site capping this triangle.Crystal structures
Compounds 1–5 crystallise in the monoclinic space group C2/c and have the same molecular structures (Fig. 1); we describe the structure of [pyH]4[Gd4III(μ3-OH)(O3PtBu)3(HO3PtBu)(O2CtBu)2(NO3)6] 1 as representative. Overall the molecular cage contains four lanthanide metal ions, four phosphonates, six nitrates, one μ3-OH and two pivalates. The metallic core of 1 can be described as a triangular pyramid; the four Gd(III) ions (Gd1, Gd2, Gd3, and Gd4) lie on the vertexes of a triangular pyramid. The three phosphonate ligands sit above the triangular faces linking the base to the apex, and adopt the 3.111 bonding mode (Harris notation).21 The fourth phosphonate is located below the base of a triangular pyramid and bridges the (Gd1⋯Gd3) edge using the 2.11 bonding mode. There are six chelating nitrate ligands in the structure; one nitrate coordinates to each of the vertices in the triangular base and there are three nitrate ligands coordinated to the apex of the triangular pyramid (Gd4). Two of the Gd⋯Gd edges within the triangular base are bridged by 2.11 pivalates.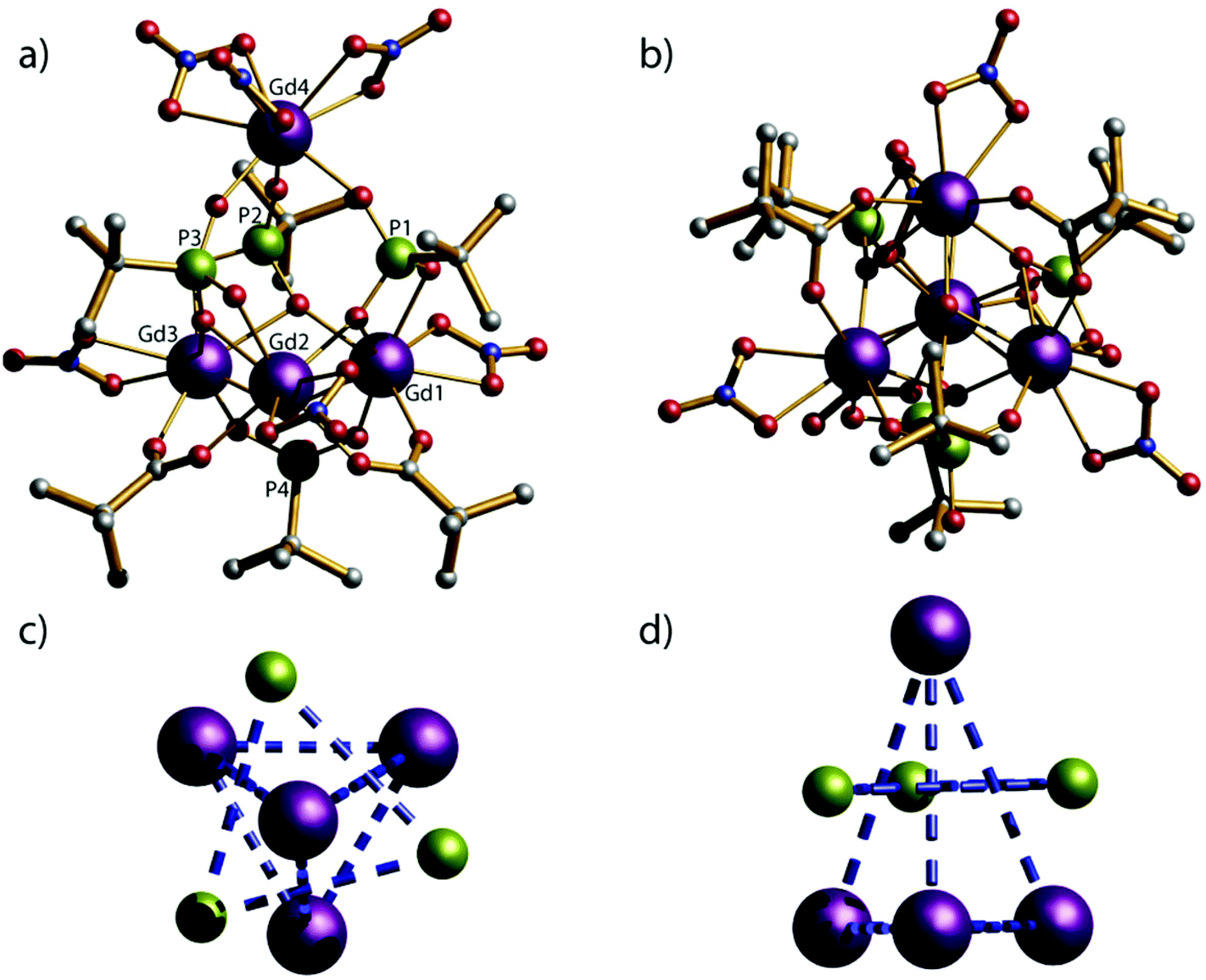 | ||
| Fig. 1 (a) and (b) side and top view of Crystal structure of the {Gd4P4} cluster. Scheme: Ln, purple; P, green; O, red; C, grey; N, cyan; (H omitted for clarity); (c) and (d) top and side view of metal and phosphonate core of the crystal structure of {Gd4P4} cluster. | ||
The distances between Gd1⋯Gd2, Gd2⋯Gd3 and Gd1⋯Gd3 that are forming the edges of the base of the triangular pyramid are 3.8104(6), 3.8151(8) and 3.8642(6) Å respectively. The μ3-OH group is displaced about 0.8230(1) Å out of the plane of the gadolinium metal ions. The Gd–O–Gd angles range from 108.34(11) to 109.19(10)°; these angles are consistent with a μ3-hydroxide and not with a bridging oxide. The distances between the plane of base (Gd1, Gd2 and Gd3) and the apex (Gd4) within the triangular pyramid are exactly 5.1150(1) Å.
Two different geometries are adopted by the GdIII ions in the cluster: whilst Gd2, Gd3 and Gd4 exhibit a biaugmented trigonal prism geometry (C2v) and a Continuous Shape Measure value (CShM) of 2.494, 2.573 and 2.434 respectively; Gd4 adopts a less regular coordination (muffin, Cs, with a CShM value of 2.345).22 There are no significant intermolecular interactions between clusters within the lattice.
Magnetic description
The magnetic properties of 1–5 were investigated by solid-state magnetic susceptibility (where χM stands for molar magnetic susceptibility) measurements in the 2–300 K range in a 0.1 Tesla (T) DC field. The resulting data for complexes 1–5 are shown as χMT versus T plot in Fig. 2 and 3, respectively. In each case the room temperature value for χMT is equivalent to that for four non-interacting metal ions of the appropriate type. For compounds 2–5χMT decreases steadily upon cooling down to ca. 90 K (Fig. 2), below which they drop at 2 K, respectively. Such behaviour is typical for the individual metal ions concerned, and is probably due to depopulation of Stark levels within the individual metal sites. While magnetic exchange between the Ln sites in these compounds cannot be ruled out, there is certainly no evidence for such exchange from these measurements. Magnetisation (M) versus applied magnetic field were performed at temperatures of 2 and 3 K in the field range of 0–7 T. For 1M saturates at 27.7 emu K mol−1 at 7 T at 2 K, as expected for four non-interacting GdIII metal ions with S = 7/2 and gGd = 1.99 (Fig. 3). For compounds 2–5, there is a gradual increase in M vs. H curve with increasing field, which reach 20.8, 22.7, 23.7, 21.5 μB at 7 T at 2 K without reaching saturation.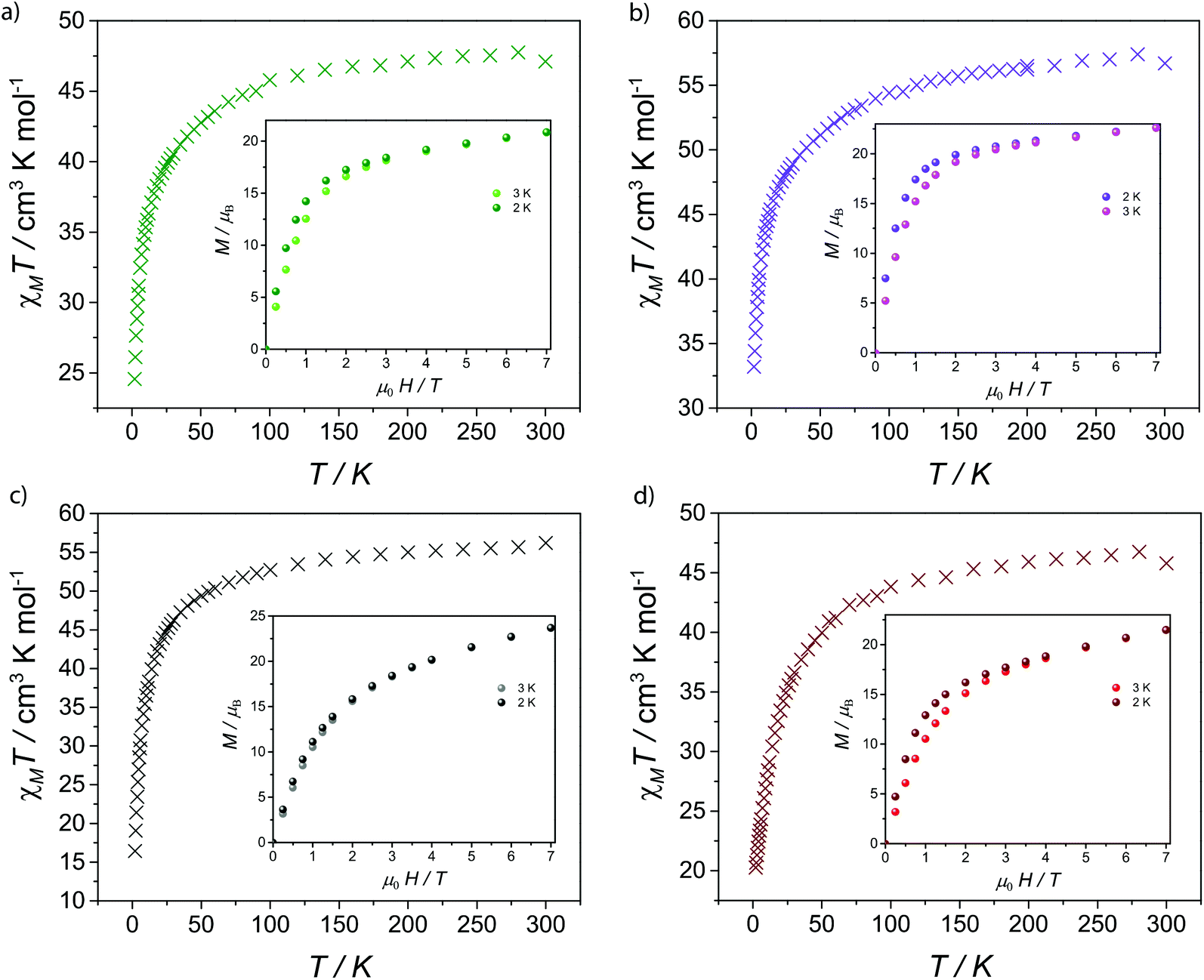 | ||
| Fig. 2 Molar magnetic susceptibility (χMT) vs. T plot for 2–5 under 1 kG dc field and molar magnetization (M) as a function of applied magnetic field (H) at 2 and 3 K (inset) for (a) 2, (b) 3, (c) 4 and (d) 5. | ||
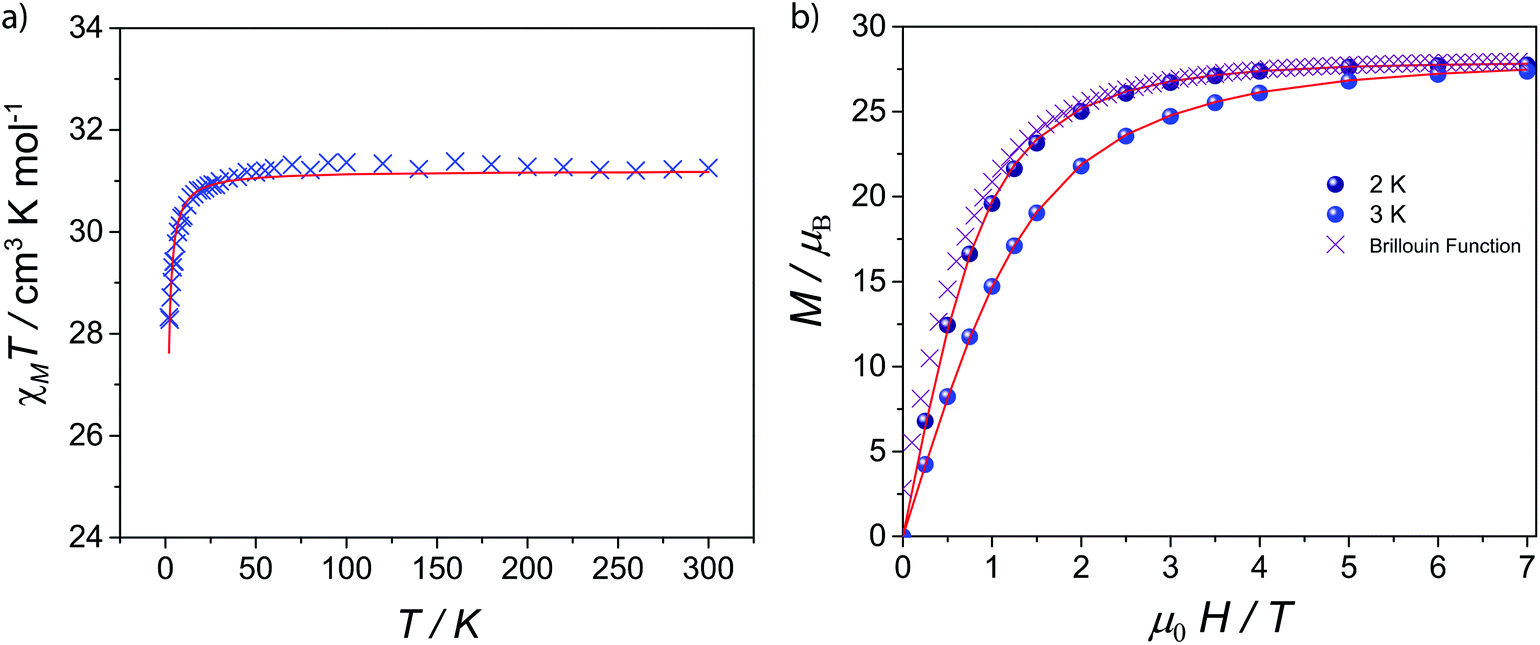 | ||
| Fig. 3 (a) Molar magnetic susceptibility (χMT) vs. T and simulation plots for 1 under 1 kG dc field; (b) Molar magnetization (M) as a function of applied magnetic field (H) at 2 and 3 K for 1. | ||
As GdIII is an isotropic metal ion this allowed us to simulate the magnetic data of complex 1. Structurally there is little difference between the Gd⋯Gd edges of the triangles (see above); however the Gd⋯Gd contacts between the apical Gd and those in the base are between 5.544(1)–5.604(1) Å. Furthermore, any intermolecular interaction between would be rather small since the closest Gd⋯Gd intercluster distance is 9.8665(6) Å. Based on this structure, we have fitted simultaneously χMT(T) and M(H), using the program PHI,20 using as a model an equilateral Gd3, with no exchange interaction to the fourth Gd, using the Hamiltonian (1).
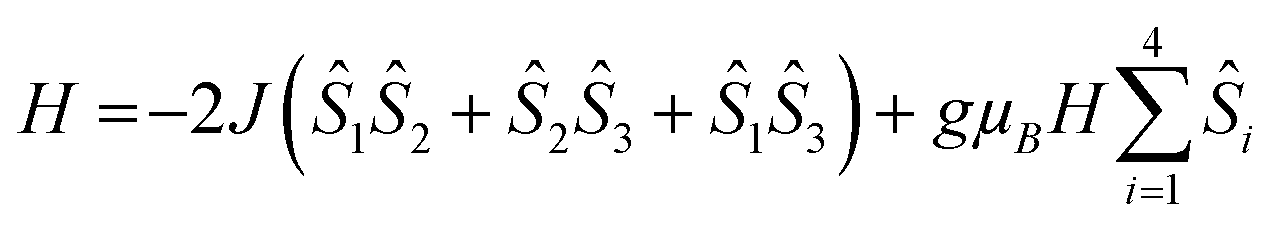 | (1) |
AC magnetic susceptibility measurements are a suitable method to judge whether a molecule exhibits a slow magnetisation relaxation rate, which is an essential property of a molecules that might be a single-molecule magnet (SMM). Such measurements were carried out between 10 and 1.8 K with and without applied DC field, however no out-of-phase component was obtained for any of the clusters.
Discussion
The core of compounds 1–5 is related to those found in 3d-metal phosphonate cages (Fig. 4).12a,b An Fe(III) phosphonate cage {Fe4P4} has been reported which contains a μ3-O centred triangle of Fe(III) sites capped by a fourth FeIII ion. The μ3-O Fe(III) triangle is clearly related to the oxo-centred {Fe3} carboxylate triangle precursor. As in 1–5 three phosphonate units sit on the faces of the trigonal pyramid that link the base to the apex. All Fe(III) metal ion centres are six coordinated. The distance between the plane of base (Fe1, Fe2 and Fe3) and the apex (Fe4) is 4.25(1) Å while the Fe⋯Fe distances within the triangular base of the tetrahedron are between 3.34 and 3.37 Å. A similar manganese phosphonate cage {Mn4P4} involves the bulky tritylphosphonic acid (TPA) as ligand; this manganese cage is a mixed valent with two MnII and two MnIII sites. In this compound all four faces of the triangle pyramid are occupied by TPA ligands and the oxo-centred Mn triangle is much more distorted with Mn⋯Mn distances within of 3.19 and 3.61 Å; the Mn⋯Mn distances to the apical Mn site remain longer at 3.88(1) Å. In these 3d-cages, the phosphonates present 3.111 coordination mode and each metal ion centre is coordinated to a pyridine molecule to complete its coordination sphere. In the 4f-phosphonate compounds 1–5 reported here there is a μ3-OH centred triangle, not oxide centred, and the phosphonates show two diverse binding modes. There is no pyridine binding to 4f metals, with the coordination of the 4f-centres completed by chelating nitrates.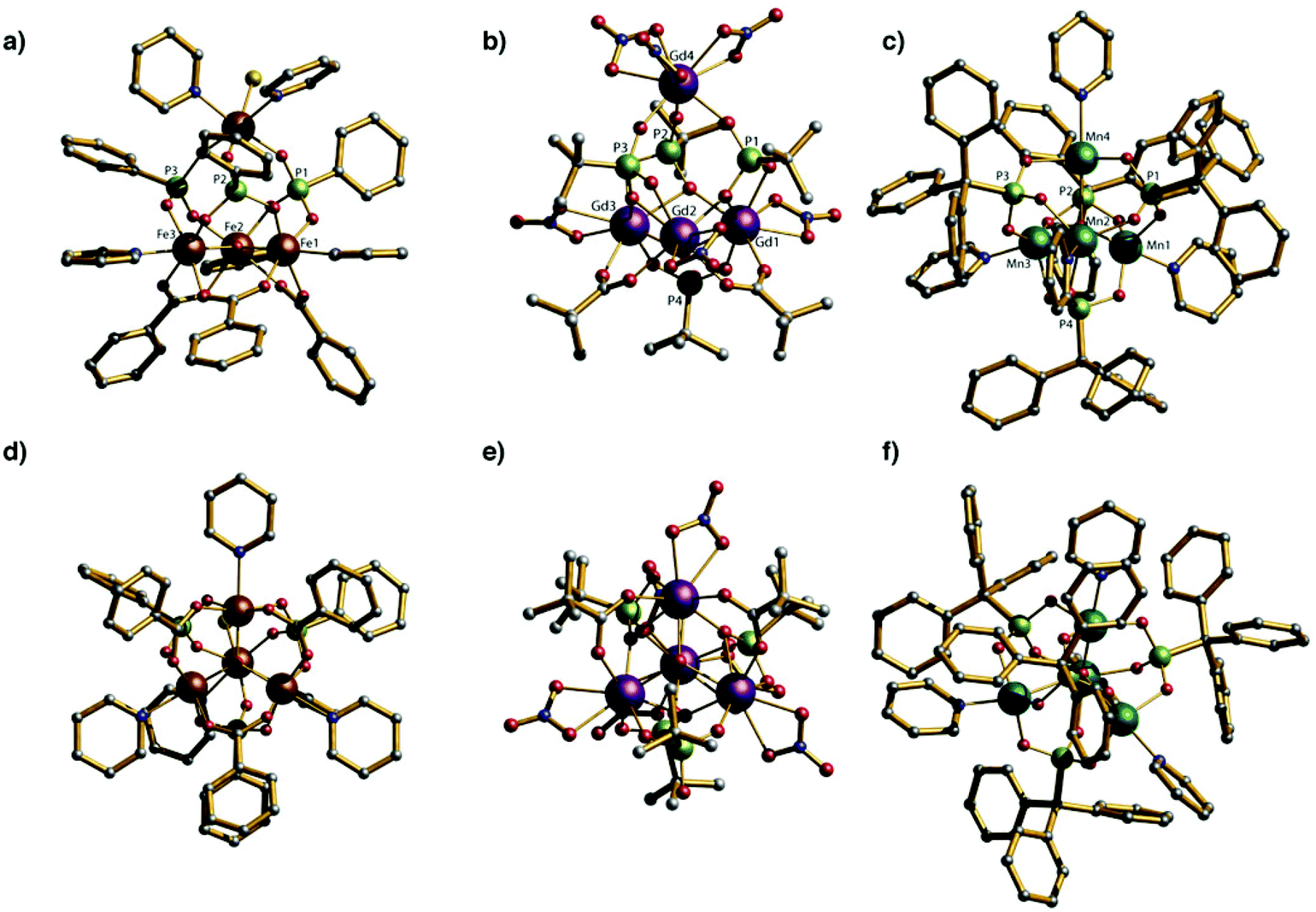 | ||
| Fig. 4 (a) and (d) side and top view of crystal structure of the {Fe4P4} cluster; (b) and (e) side and top view of Crystal structure of the {Ln4P4} cluster; (c) and (f) side and top view of Crystal structure of the {Mn4P4} cluster; Scheme: Ln, purple; Fe, brown; Mn, cyan; P, green; O, red; C, grey; N, cyan; (H omitted for clarity). | ||
Conclusions
A new group of 4f-phosphonate tetranuclear clusters have been synthesised from the reactions of a simple Ln(III) salt with t-butyl phosphonic acid. These products represent new additions to what is still a very small family of such molecular 4f-phosphonate species. We have previously reported that reaction of lanthanide nitrates with phosphonates in the presence of aliphatic base like isopropylamine produce a series of {Ln8} clusters.20 Herein, our original goal of synthetic program was to explore whether same strategy with aromatic base could be used to assemble Ln(III) ions into large combinations with new structures and magnetic properties. It was observed that the reaction progress well in the presence of aromatic base, as the base promotes the reaction by deprotonation of the phosphonic acid ligand. So, in the present reaction system the identity of the cluster isolated is controlled by the nature of the base used, each single component of the reaction matrix has a strong effect on the final product, and hence on the magnetic behaviour. The magnetic susceptibility data for all complexes display dominant antiferromagnetic interactions. Hopefully further efforts in this area will lead to more magnetically interesting compounds.Acknowledgements
KZ thanks the KRG-Scholarship program in “Human Capacity Development (HCDP)”. EMP thanks the Panamanian agency SENACYT-IFARHU. REPW thanks the Royal Society for a Wolfson Merit Award. We thank the EPSRC (UK) for funding for an X-ray diffractometer (grant number EP/K039547/1).References
- (a) S. K. Langley, M. Helliwell, R. Sessoli, P. Rosa, W. Wernsdorfer and R. E. P. Winpenny, Chem. Commun., 2005, 5029 RSC; (b) E. I. Tolis, L. P. Engelhardt, P. V. Mason, G. Rajaraman, K. Kindo, M. Luban, A. Matsuo, H. Nojiri, J. Raftery, C. Schroder, G. A. Timco, F. Tuna, W. Wernsdorfer and R. E. P. Winpenny, Chem. – Eur. J., 2006, 12, 8961 CrossRef CAS PubMed; (c) S. K. Langley, M. Helliwell, R. Sessoli, S. J. Teat and R. E. P. Winpenny, Inorg. Chem., 2008, 47, 497 CrossRef CAS PubMed; (d) J.-T. Li, Y.-S. Ma, S.-G. Li, D.-K. Cao, Y.-Z. Li, Y. Song and L.-M. Zheng, Dalton Trans., 2009, 5029 RSC; (e) M. Wang, C. Ma, H. Wen and C. Chen, Dalton Trans., 2009, 994 RSC.
- (a) Y.-Z. Zheng, M. Evangelisti, F. Tuna and R. E. P. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057 CrossRef CAS PubMed; (b) V. Baskar, K. Gopal, M. Helliwell, F. Tuna, W. Wernsdorfer and R. E. P. Winpenny, Dalton Trans., 2010, 39, 4747 RSC.
- (a) G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430 RSC; (b) A. Clearfeld, Curr. Opin. Solid State Mater. Sci., 1996, 1, 268 CrossRef.
- (a) A. Clearfield, Curr. Opin. Solid State Mater. Sci., 2002, 6, 495 CrossRef CAS; (b) E. M. Pineda, F. Tuna, R. G. Pritchard and A. C. Regan, Chem. Commun., 2013, 49, 3522 RSC; (c) M. Thompson, Chem. Mater., 1994, 6, 1168 CrossRef CAS; (d) A. Subbiah, D. Pyle, A. Rowland, J. Huang, R. A. Narayanan, P. Thiyagarajan, J. Zon and A. Clearfield, J. Am. Chem. Soc., 2005, 127, 10826 CrossRef CAS PubMed.
- (a) V. Baskar, K. Gopal, M. Helliwell, F. Tuna, W. Wernsdorfer and R. E. P. Winpenny, Dalton Trans., 2010, 39, 4747 RSC; (b) Y.-Z. Zheng, M. Evangelisti and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 3692 CrossRef CAS PubMed; (c) Y.-Z. Zheng, E. M. Pineda, M. Helliwell, M. Evangelisti and R. E. P. Winpenny, Chem. – Eur. J., 2012, 18, 4161 CrossRef CAS PubMed; (d) E. M. Pineda, F. Tuna, Y.-Z. Zheng, S. J. Teat, R. E. P. Winpenny, J. Schnack and E. J. L. McInnes, Inorg. Chem., 2014, 53, 3032 CrossRef CAS PubMed; (e) M. I. Khan and J. Zubieta, Prog. Inorg. Chem., 1995, 43, 1 CrossRef CAS; (f) V. Chandrasekhar and S. Kingsley, Angew. Chem., Int. Ed., 2000, 39, 2320 CrossRef CAS.
- (a) J.-G. Mao, Chem. Rev., 2007, 251, 1493 CAS; (b) S.-M. Ying and J.-G. Mao, Cryst. Growth Des., 2006, 6, 964 CrossRef CAS; (c) S.-F. Tang, J.-L. Song, X.-L. Li and J.-G. Mao, Cryst. Growth Des., 2007, 7, 360 CrossRef CAS; (d) Y.-Q. Guo, S.-F. Tang, B.-P. Yang and J.-G. Mao, J. Solid State Chem., 2008, 181, 2713 CrossRef CAS PubMed.
- G. Cao, V. M. Lynch, J. S. Swinnea and T. E. Mallouk, Inorg. Chem., 1990, 29, 2112 CrossRef CAS.
- (a) K. H. Zangana, E. M. Pineda, J. Schnack and R. E. P. Winpenny, Dalton Trans., 2013, 42, 14045 RSC; (b) K. H. Zangana, E. M. Pineda, E. J. L. McInnes, J. Schnack and R. E. P. Winpenny, Chem. Commun., 2014, 50, 1438 RSC.
- (a) J.-G. Mao, Coord. Chem. Rev., 2007, 251, 1493 CrossRef CAS PubMed; (b) S.-M. Ying and J.-G. Mao, Cryst. Growth Des., 2006, 6, 964 CrossRef CAS; (c) S.-F. Tang, J.-L. Song, X.-L. Li and J.-G. Mao, Cryst. Growth Des., 2007, 7, 360 CrossRef CAS; (d) Y.-Q. Guo, S.-F. Tang, B.-P. Yang and J.-G. Mao, J. Solid State Chem., 2008, 181, 2713 CrossRef CAS PubMed.
- V. Chandrasekhar and S. Kingsley, Angew. Chem., Int. Ed., 2000, 39, 2320 CrossRef CAS.
- (a) S. Maheswaran, G. Chastanet, S. J. Teat, T. Mallah, R. Sessoli, W. Wernsdorfer and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2005, 44, 5044 CrossRef CAS PubMed; (b) M. Shanmugam, G. Chastanet, T. Mallah, R. Sessoli, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Chem. – Eur. J., 2006, 12, 8777 CrossRef CAS PubMed.
- (a) E. I. Tolis, M. Helliwell, S. Langley, J. Raftery and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 3804 CrossRef CAS PubMed; (b) S. Konar and A. Clearfield, Inorg. Chem., 2008, 47, 3492 CrossRef CAS PubMed.
- (a) S. Langley, M. Helliwell, R. Sessoli, S. J. Teat and R. E. P. Winpenny, Inorg. Chem., 2008, 47, 497 CrossRef CAS PubMed; (b) S. Langley, M. Helliwell, R. Sessoli, S. J. Teat and R. E. P. Winpenny, Dalton Trans., 2009, 3102 RSC; (c) S. K. Langley, M. Helliwell, S. J. Teat and R. E. P. Winpenny, Dalton Trans., 2012, 41, 12807 RSC.
- (a) B. A. Breeze, M. Shanmugam, F. Tuna and R. E. P. Winpenny, Chem. Commun., 2007, 5185 RSC; (b) S. K. Langley, M. Helliwell, S. J. Teat and R. E. P. Winpenny, Inorg. Chem., 2014, 53, 1128 CrossRef CAS PubMed.
- (a) V. Chandrasekhar and S. Kingsley, Angew. Chem., Int. Ed., 2000, 39, 6175 Search PubMed; (b) V. Chandrasekhar, L. Nagarajan, K. Gopal, V. Baskar and P. Kögerler, Dalton Trans., 2005, 3143 RSC; (c) V. Chandrasekhar, L. Nagarajan, R. Clérac, S. Ghosh, T. Senapati and S. Verma, Inorg. Chem., 2008, 47, 5347 CrossRef CAS PubMed.
- (a) S. Khanra, M. Kloth, H. Mansaray, C. A. Muryn, F. Tuna, E. C. Sañudo, M. Helliwell, E. J. L. McInnes and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2007, 46, 5568 CrossRef CAS PubMed; (b) S. Khanra, L. Batchelor, M. Helliwell, F. Tuna, E. J. L. McInnes and R. E. P. Winpenny, J. Mol. Struct., 2008, 890, 157 CrossRef CAS PubMed; (c) S. Khanra, R. Shaw, M. Helliwell, F. Tuna, C. A. Muryn, E. J. L. McInnes and R. E. P. Winpenny, Materials, 2010, 3, 232 CrossRef CAS PubMed.
- (a) K. Brechin, R. A. Coxall, A. Parkin, S. Parsons, P. A. Tasker and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2001, 40, 2700 CrossRef; (b) E. I. Tolis, M. Helliwell, S. Langley, J. Raftery and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 3804 CrossRef CAS PubMed; (c) E. I. Tolis, L. P. Engelhardt, P. V. Mason, G. Rajaraman, K. Kindo, M. Luban, A. Matsuo, H. Nojiri, J. Raftery, C. Schroder, G. A. Timco, F. Tuna, W. Wernsdorfer and R. E. P. Winpenny, Chem. – Eur. J., 2006, 12, 8961 CrossRef CAS PubMed; (d) M. Maheswaran, G. Chastanet, S. J. Teat, T. Mallah, R. Sessoli, W. Wernsdorfer and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2005, 44, 5044 CrossRef PubMed; (e) M. Shanmugam, G. Chastanet, T. Mallah, R. Sessoli, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Chem. – Eur. J., 2006, 12, 8777 CrossRef CAS PubMed.
- (a) I. G. Fomina, M. A. Kiskin, A. G. Martynov, G. G. Aleksandrov, Z. V. Dobrokhotova, Y. G. Gorbunova, Y. G. Shvedenkov, A. Y. Tsivadze and V. M. Novotortsev, Zh. Neorg. Khim., 2004, 49, 1463 Search PubMed; (b) T. A. Zoan, N. P. Kuzmina, S. N. Frolovskaya, A. N. Rykov, N. D. Mitrofanova, S. I. Troyanov, S. I. Pisarevsky, A. P. Martynenko and L. I. Korenev, J. Alloys Compd., 1995, 225, 396 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed; (b) O. V. Dolomanov, L. J. Bourthis, R. L. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
- Harris notation describes the binding mode as [X.Y1Y2...Yn] where X is the overall number of metal bond by the whole ligand, and each value of Y refers to the number of metal atoms attached to the different donor atoms. See ESI† and R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, Dalton Trans., 2000, 14, 2349 RSC.
- (a) S. Alvarez, Dalton Trans., 2005, 2209 RSC; (b) S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cif files, further synthetic details, structural and magnetic plots. CCDC 1021470–1021474. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02630f |
| This journal is © The Royal Society of Chemistry 2014 |