
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unraveling the origins of catalyst degradation in non-heme iron-based alkane oxidation†
Michaela
Grau
,
Andrew
Kyriacou
,
Fernando
Cabedo Martinez
,
Irene M.
de Wispelaere
,
Andrew J. P.
White
and
George J. P.
Britovsek
*
Department of Chemistry, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: g.britovsek@imperial.ac.uk; Fax: +44-(0)20-75945804; Tel: +44-(0)20-75945863
First published on 1st October 2014
Abstract
A series of potentially tetradentate and pentadentate ligands modelled on BPMEN has been prepared and their iron(II) bis(triflate) complexes have been isolated and characterised by spectroscopic and crystallographic techniques (BPMEN = N,N′-bis(pyridylmethyl)ethylenediamine). Changes to the BPMEN ligand have invariably led to complexes with different coordination modes or geometries and with inferior catalytic efficiencies for the oxidation of cyclohexane with H2O2. The reaction of an iron(II) complex containing a pentadentate BPMEN-type ligand with O2 has resulted in ligand degradation via oxidative N-dealkylation and the isolation of a bis(hydroxo)-bridged dinuclear iron(III) complex with a picolinate-type ligand.
Introduction
Catalyst deactivation is a common but often ignored problem in catalyst development, in particular in oxidation catalysis. Oxidative ligand degradation of a homogeneous molecular oxidation catalyst during its lifetime can be a limiting factor for high turnover and the activity of the catalyst is often directly related to its stability under the oxidising reaction conditions.1 In living systems, ligand degradation of oxidation catalysts also occurs in enzymatic systems where both heme and non-heme oxidases have a limited lifetime, but are regenerated in vivo by complex mechanisms.2,3A thorough understanding of the various oxidative ligand degradation processes will be essential for the design and development of more robust oxidation catalysts. In heme-based oxidation catalysts, one mode of deactivation has been shown to involve oxidation of the porphyrin ligand.4–8 While the exact deactivation pathway in the large variety of non-heme metal catalysts is not known at this stage, oxidative degradation of the ligand is probably one of the main causes for catalyst deactivation. Another possible deactivation pathway that has been invoked in a number of non-heme catalyst systems is the formation of inactive dinuclear μ-oxo iron(III) complexes.9,10
Various oxidative ligand degradation pathways have been observed in non-heme iron systems, for example peripheral alkyl and aryl C–H bond oxidations,11–13 and more importantly for amine-based ligands, oxidative N-dealkylation. The cleavage of C–N bonds in metal complexes with polydentate amine ligands via oxidative N-dealkylation is a general problem that has been observed on many occasions.14–16 After initial oxidation of Fe(II) to Fe(III), the C–H bonds adjacent to an amine moiety (CHR–NR′2) (A in Scheme 1) are prone to oxidation resulting in the formation of hemi-aminal (C(OH)R–NR′2) complexes (B).17 Particularly vulnerable to oxidative degradation are methylene or methine protons adjacent to an amine that are also in alpha position to a carbonyl group, for example in amino acids,18–21 or next to an aromatic unit such as phenyl (benzylic),22–25 phenolate,26 or pyridine moieties, as shown in Scheme 1.27–34 Occasionally, the hemi-aminal products are stable enough and can be isolated as O-bound hemi-aminal complexes of type C.35–39 Alternatively, hemi-aminals are prone to dissociation into an aldehyde (or ketone) and a secondary amine (D).40 In many cases where ligand degradation has occurred via oxidative N-dealkylation, degradation products such as aldehydes,23–25,41–44 or ketones30,45 have been isolated. A third possibility is further oxidation of the hemi-aminal intermediate (B) to an amide complex (F). Hydrolysis of this amide intermediate would result in the formation of picolinic acid from pyridylmethylamine moieties, which appears to be a common occurrence, sometimes resulting in the formation of stable metal picolinate complexes (G).42,44,46,47
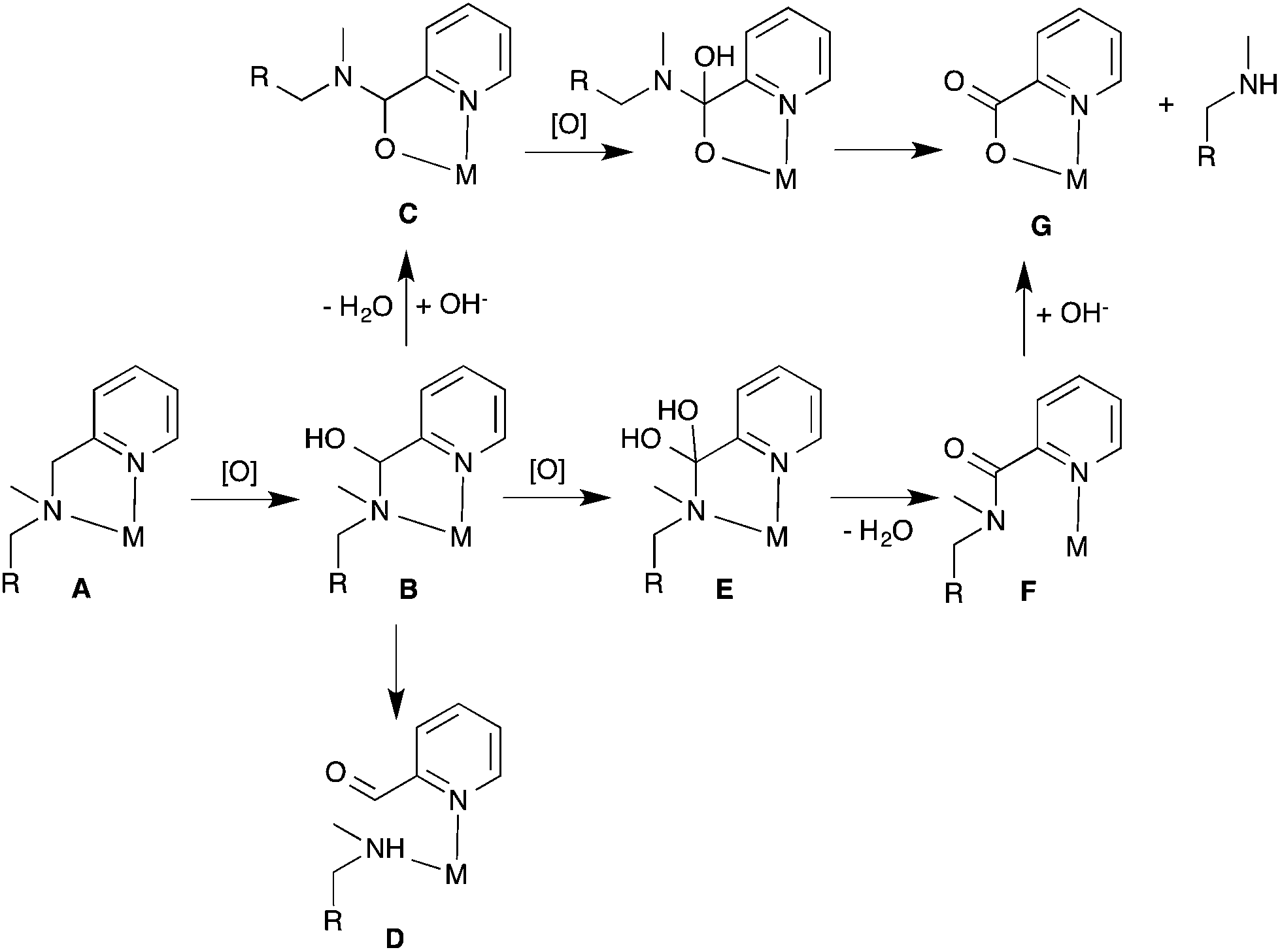 | ||
| Scheme 1 Potential degradation pathways via oxidative N-dealkylation in pyridylmethyl amine complexes ([O] = oxidation). | ||
The prevention of oxidative ligand degradation is an on-going challenge in the design of more efficient oxidation catalysts. In an attempt to prepare more robust biomimetic catalysts for the oxidation of alkanes, we have previously reported various derivatives of the benchmark catalyst [Fe(BPMEN)(OTf)2],48–50 developed by Que and co-workers (BPMEN = N,N′-bis(pyridylmethyl)ethylenediamine).51 The selectivity and stability of [Fe(BPMEN)(CH3CN)2](SbF6)2 is improved by adding electron donating para-methoxy groups to the pyridine moieties of the BPMEN ligand.50,52 Electron donating ligands are preferred because they can support high-valent iron oxo intermediates, which are generally assumed to be the active oxidant in these systems.
Our continuing efforts to understand and improve the stability of non-heme iron-based catalyst have led us to explore three aspects of the BPMEN ligand. Firstly, the effect of N–Me versus N–H substitution in alkane oxidation has been evaluated (ligand 1, Fig. 1). Complexes containing secondary amines are generally prone to oxidative dehydrogenation under oxidising conditions and therefore would be expected to give a poorer performance.53–55 Secondly, methylene (CH2) moieties adjacent to amine donors have been identified as vulnerable to oxidation.17 To avoid such methylene linkages, we have prepared tetradentate ligands with a C(Me)H or a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O linkage between the pyridine and the amine donor (ligands 2–4). Replacing vulnerable C–H bonds with C–Me bonds has previously been used to improve the catalyst stability in other non-heme catalysts,56 and a recent report on a related iron(II) catalyst with C(Ph)H linkages has shown promising results in asymmetric epoxidations.57 In a third series, we have explored the application of linear pentadentate ligands in oxidation catalysis (5–8). An additional donor could result in greater catalyst stability and within this series we have examined the effect of N–H (5) versus N–Me substitution (6), the removal of CH2 linkages (7) and an alternative donor set (8) on the catalytic behaviour in cyclohexane oxidation.
O linkage between the pyridine and the amine donor (ligands 2–4). Replacing vulnerable C–H bonds with C–Me bonds has previously been used to improve the catalyst stability in other non-heme catalysts,56 and a recent report on a related iron(II) catalyst with C(Ph)H linkages has shown promising results in asymmetric epoxidations.57 In a third series, we have explored the application of linear pentadentate ligands in oxidation catalysis (5–8). An additional donor could result in greater catalyst stability and within this series we have examined the effect of N–H (5) versus N–Me substitution (6), the removal of CH2 linkages (7) and an alternative donor set (8) on the catalytic behaviour in cyclohexane oxidation.
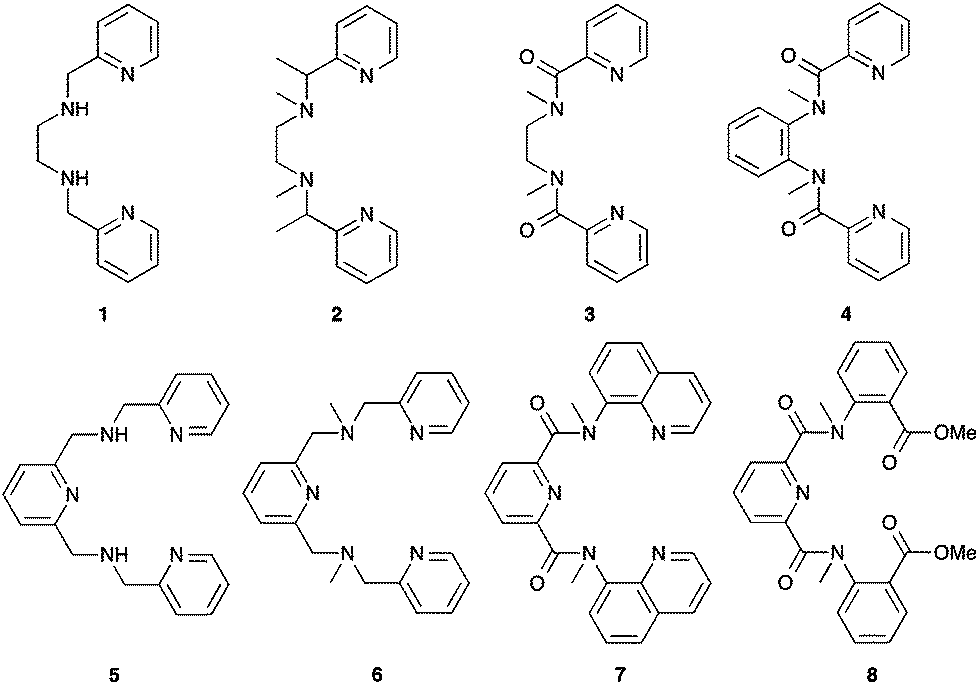 | ||
| Fig. 1 Overview of tetradentate and pentadentate pyridylmethylamine ligands (1–8). | ||
Results and discussion
Synthesis and characterisation of ligands and metal complexes
The N-methylated pyridylamine ligands 2 and 6 were prepared from the non-methylated precursors.58–62 The N-methyl carboxamide ligands 3, 4, 7 and 8 were prepared by methylation of the carboxamides, which were obtained by reacting pyridine dicarboxylic acid or acid chloride with the corresponding amine (see Experimental section). N-methyl carboxamides show restricted rotation around the CO–NMe bond, which can lead to 3 rotamers and complicated NMR spectra.63 The ratio between the three different rotamers was determined as 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3 in the case of ligand 3, as reported previously.64 For the new ligands 4, 7 and 8, the ratio was determined as 10:6:1 for ligand 4 (Fig. S1†) and as 3:2:1 for ligand 8. Only one isomer was observed for ligand 7 (Fig. S2†).
:3:3 in the case of ligand 3, as reported previously.64 For the new ligands 4, 7 and 8, the ratio was determined as 10:6:1 for ligand 4 (Fig. S1†) and as 3:2:1 for ligand 8. Only one isomer was observed for ligand 7 (Fig. S2†).
The iron(II) bis(triflate) complexes were prepared by combining the ligands and [Fe(OTf)2(MeCN)2] in THF as the solvent. The complex [Fe(BPMEN)(OTf)2] has a cis-α coordination mode in the solid state and according to VT-19F NMR studies, there is no fluxional coordination behaviour in CD2Cl2 solution between 233–298 K.50 In acetonitrile solution, the cationic complex [Fe(BPMEN)(CH3CN)2]2+ is formed,51,65 which shows a temperature dependant 5T2→1A1 spin crossover (SC) behaviour with μeff = 1.1μB at 233 K and μeff = 5.1μB at 343 K (Tc = 264 K) in acetonitrile (see (■) Fig. 2).50,66
 | ||
| Fig. 2 Magnetic moment (μeff) versus temperature in CD3CN for complexes [Fe(BPMEN)(OTf)2] (■), [Fe(1)(OTf)2] (▲), [Fe(2)(OTf)2] (●), [Fe(5)(OTf)](OTf) (×), [Fe(6)(OTf)2] (◆). | ||
In contrast, the non-methylated complex [Fe(1)(OTf)2] shows a different behaviour. This complex also forms a dicationic complex [Fe(1)(CH3CN)2]2+ in acetonitrile,67 which was recently crystallographically characterised at 100 K as a low spin complex with cis-α coordination.68 However, we noticed that the signals for this complex in the 1H NMR spectrum in CD3CN are unusually broad at room temperature and more species are observed at higher temperatures (see Fig. S3†). Complex [Fe(1)(CH3CN)2]2+ undergoes SC approximately at room temperature, due to the strong field acetonitrile ligands (see Fig. 2 (▲) and VT-1H NMR spectra in Fig. S4†). For comparison, the complex [Fe(1)(SCN)2] with relatively weak field thiocyanate ligands undergoes 5T2→1A1 relaxation at a very low transition temperature of 70 K,69 and later studies have revealed a very slow spin transition process in the solid state.70 Compared to [Fe(BPMEN)(CH3CN)2]2+, the change in magnetic susceptibility of [Fe(1)(CH3CN)2]2+ is much more gradual with μeff = 2.1μB at 233 K and μeff = 3.9μB at 343 K (Fig. 2). This suggests that [Fe(1)(CH3CN)2]2+ undergoes geometrical changes with temperature, most likely to form other HS complexes with different geometries. Geometrical rearrangements of iron(II) complexes with tetradentate ligands similar to ligand 1 are quite common and we have seen isomerisation between cis-α and other geometries cis-β and trans, for example when the chelate rings size or donor strength has been changed.49,50 Here, the weaker basicity of the N–H versus N–Me donor and the resulting weaker Fe–N bond strength, results probably in a similar rearrangement process. For example the analogous complex with O instead of NH donors forms the trans complex exclusively.50 Noteworthy, cis-β and trans geometries are known for ligand 1 with different metals such as Co(III) and Cr(III).71–74 The activation barriers (ΔH‡) for spin transitions between HS and LS iron(II) complexes can vary in solution between 2 and 34 kJ mol−1 and may become competitive with geometric rearrangements.75,76 Coupling between the spin relaxation process and the geometrical rearrangement of the ligand, as seen in a related system,77 cannot be excluded.
VT-19F NMR analysis of complex [Fe(1)(OTf)2] in CD2Cl2 shows a broad signal at −40 ppm at room temperature, which coalesces at 258 K and reveals multiple species in equilibrium, most likely cis-β and trans isomers in addition to the main cis-α complex (see Fig. S5†). We conclude that the non-methylated complex [Fe(1)(OTf)2] must be conformationally less rigid compared to [Fe(BPMEN)(OTf)2], probably due to the weaker basicity of NH versus NMe donors. The ligand flexibility results in a combination of spin crossover and geometric isomerisation upon changing the temperature, which explains the anomalous magnetic behaviour seen in Fig. 2.
 | (1) |
Ligand 2, with two chiral centres, forms a pair of enantiomers (R,R and S,S) and a meso form (R,S or S,R). Upon coordination to the iron(II) centre, two additional N-chirogenic centres are created at the amino nitrogen atoms. This could result in 16 isomers, but the cis-α coordination mode only supports the [(N)S*,(N)S*] configuration at the central amine donors. This reduces the number of possible isomers to 8 (4 pairs of enantiomers). The four possible isomers with [(N)S,(N)S] configuration at the central amines are shown in Fig. 3. The two meso isomers C are identical, which reduces the number of complexes to three diastereomeric complexes A, B and C in a 1:1:2 ratio.
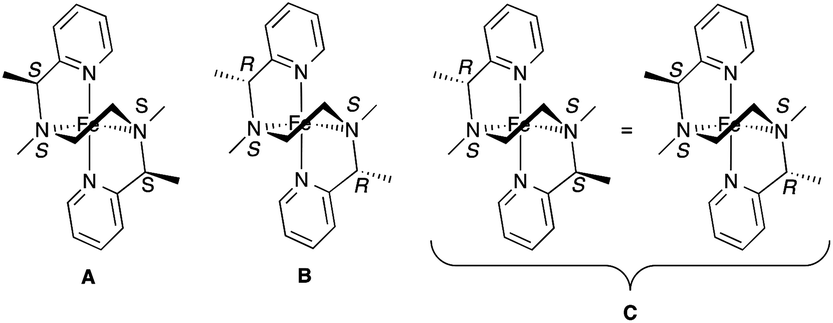 | ||
| Fig. 3 Four of eight possible isomers for complex [Fe(2)(OTf)2] with cis-α geometry (the OTf− ligands have been omitted for clarity). | ||
The presence of diastereomers complicates the 1H NMR spectrum of [Fe(2)(OTf)2] in CD3CN. At 228 K, the spectrum appears to consist of a mixture of two complexes (or groups of complexes), which we presume are A and B on the one hand, and C on the other (see Fig. S6†). One complex is diamagnetic at this temperature, with a chemical shift range between 0 and 10 ppm and the other is partially high spin (HS) with a shift range from −5 to 40 ppm. The latter complex is fully HS at 278 K and follows Curie behaviour upon further temperature increase. The spin crossover temperature for this complex is at Tc ≈ 200 K. The diamagnetic complex shows 9 signals at 228 K and as the temperature is raised, undergoes spin crossover at Tc ≈ 235 K, whereby the chemical shift range increases and the signals become gradually broader. At 288 K, the signals for this complex are extremely broad, due to exchange between coordinated and non-coordinated CD3CN ligands. At the highest temperature of 338 K, the exchange is sufficiently fast for the signals to sharpen again. From 298 K onwards, all complexes are paramagnetic and follow Curie behaviour with further temperature increase. VT 19F NMR spectroscopy (Fig. S8†) and magnetic susceptibility measurements (see (●) in Fig. 2) also indicate a different behaviour compared to [Fe(BPMEN)(OTf)2], indicative of multiple complexes, each with a different spin crossover behaviour and Tc values.
The complexes [Fe(3)(OTf)2] and [Fe(4)(OTf)2] are high spin over the temperature range from 243–343 K, according to VT-1H-NMR studies. The NMR spectra are rather complicated (see for example Fig. S10†) and indicate the presence of more than one species in solution, possibly different isomers due to the different ligand rotamers. IR spectroscopy shows a change in ν(CO) from 1636 and 1648 cm−1 for 3 and 4, respectively, to 1608 and 1610 cm−1 for the corresponding iron(II) complexes, indicating coordination of the carbonyl oxygen, which is not uncommon in picoline amide ligands.27,78 In the case of complex [Fe(4)(OTf)2], X-ray quality crystals were obtained from a THF solution and analysis revealed the formation of a dinuclear complex [Fe(4)(OTf)2]2 with carbonyl oxygen coordination and the ligand binding in a bridging bis(bidentate) mode (see Fig. 4 and 5). The dinuclear complex has adopted a C2 symmetric geometry about an axis that passes through the middle of the Fe2O4N4C8 macrocycle. Each iron centre has a distorted octahedral geometry with cis angles in the range 75.19(5)–102.84(5)°, and is bound to two bidentate N,O donor ligands as well as two triflate groups. The only π–π stacking interaction of note is an intramolecular contact between the N(18) pyridyl ring and its C2 related counterpart with centroid⋯centroid and mean interplanar separations of ca. 3.67 and 3.61 Å, the two rings inclined by ca. 12° (interaction a in Fig. 4).
 | ||
| Fig. 4 The crystal structure of the C2-symmetric complex [Fe(4)(OTf)2]2. Selected bond lengths (Å) and angles (°); Fe(1)–N(1) 2.1674(15), Fe(1)–O(7) 2.1769(13), Fe(1)–O(31) 2.0948(13), Fe(1)–O(41) 2.1100(14), Fe(1)–O(16A) 2.0964(11), Fe(1)–N(18A) 2.1632(14), N(1)–Fe(1)–O(7) 75.19(5), N(1)–Fe(1)–O(31) 102.84(5), N(1)–Fe(1)–O(41) 88.85(6), N(1)–Fe(1)–O(16A) 86.60(5), N(1)–Fe(1)–N(18A) 161.27(5), O(7)–Fe(1)–O(31) 88.21(5), O(7)–Fe(1)–O(41) 161.14(5), O(7)–Fe(1)–O(16A) 94.02(5), O(7)–Fe(1)–N(18A) 99.13(5), O(31)–Fe(1)–O(41) 85.60(6), O(31)–Fe(1)–O(16A) 170.56(5), O(31)–Fe(1)–N(18A) 94.70(5), O(41)–Fe(1)–O(16A) 94.98(5), O(41)–Fe(1)–N(18A) 99.12(6), O(16A)–Fe(1)–N(18A) 75.90(5). | ||
 | ||
| Fig. 5 Molecular structures of iron(II) bis(triflate) complexes of ligands 3 and 4. | ||
We postulate a similar coordination for ligand 3 in complex [Fe(3)(OTf)2]2 (see Fig. 5). Coordination via the carbonyl oxygen donor is clearly preferred over coordination by the weakly basic amide nitrogen donors. In acetonitrile, formation of acetonitrile-coordinated complexes occurs and the triflate anions are not coordinated to the metal centre in the temperature range from 233–343 K, as shown by a single peak between −70 and −80 ppm in the VT-19F NMR spectra (see Fig. S9 and S11†). Only a few related dinuclear iron complexes have been reported with non-methylated pyridyl amide and pyridyl ester ligands.79–81 Coordination via the carbonyl oxygen donors was also seen in a palladium(II) complex containing ligand 3,82 which is the only previously reported structure of a metal complex containing 3.
The six-coordinate octahedral complex [Fe(5)(OTf)](OTf) is the major species at low temperature, as indicated by two signals in the 19F NMR spectrum, one for coordinated triflate at 35 ppm and one for non-coordinated triflate anions at −68 ppm at 203 K (Fig. 6). As the temperature is raised, the rate of exchange between coordinated and non-coordinated triflate anions will become faster, but there is also a shift in the equilibrium from a six-coordinate complex [Fe(5)(OTf)]+ to a five-coordinate complex [Fe(5)]2+, as illustrated in eqn (2). Both complexes are high spin, as can be seen from the VT 1H NMR spectra in CD2Cl2 (Fig. S12†). The two iron(II) complexes are similar to the previously reported six- and five-coordinate zinc(II) complexes with the same pentadentate ligand, [Zn(5)Cl]+ and [Zn(5)]2+.60
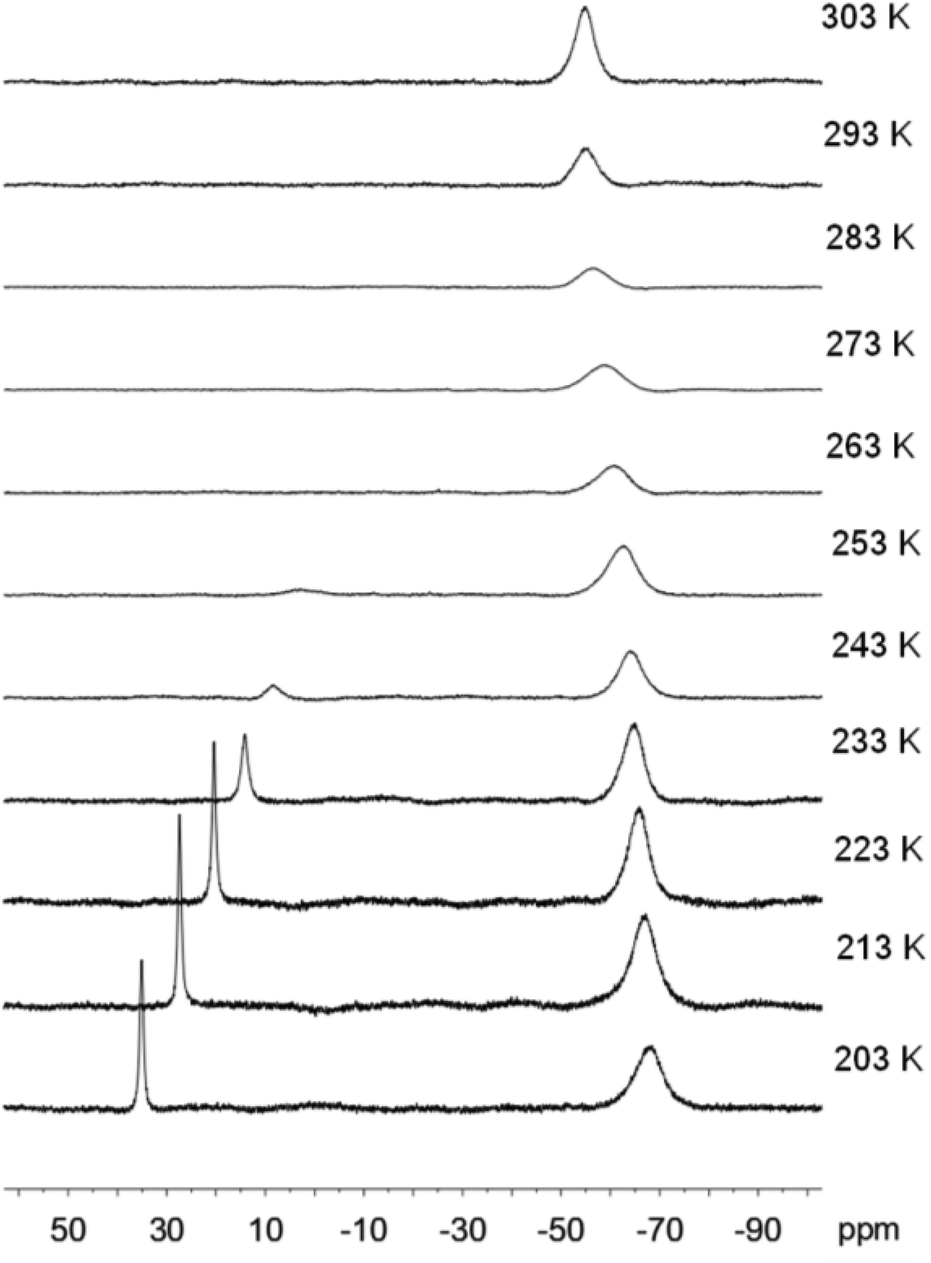 | ||
| Fig. 6 VT-19F NMR spectra of [Fe(5)(OTf)](OTf) in CD2Cl2 from 203 to 303 K. | ||
In acetonitrile, the octahedral iron(II) complex [Fe(5)(CH3CN)]2+ is formed with non-coordinating triflate anions (see VT 19F NMR in Fig. S13†). At low temperature this is a low spin complex, which shows SC behaviour as the temperature is increased (see Fig. 7). The HS [Fe(5)(CH3CN)]2+ complex is more labile and the equilibrium will shift between the six-coordinate complex [Fe(5)(CH3CN)]2+ and a five-coordinate complex [Fe(5)]2+, resulting in an anomalous magnetic behaviour similar to complex [Fe(1)(CH3CN)2]2+, as shown by the magnetic moment measurements in Fig. 2(×).
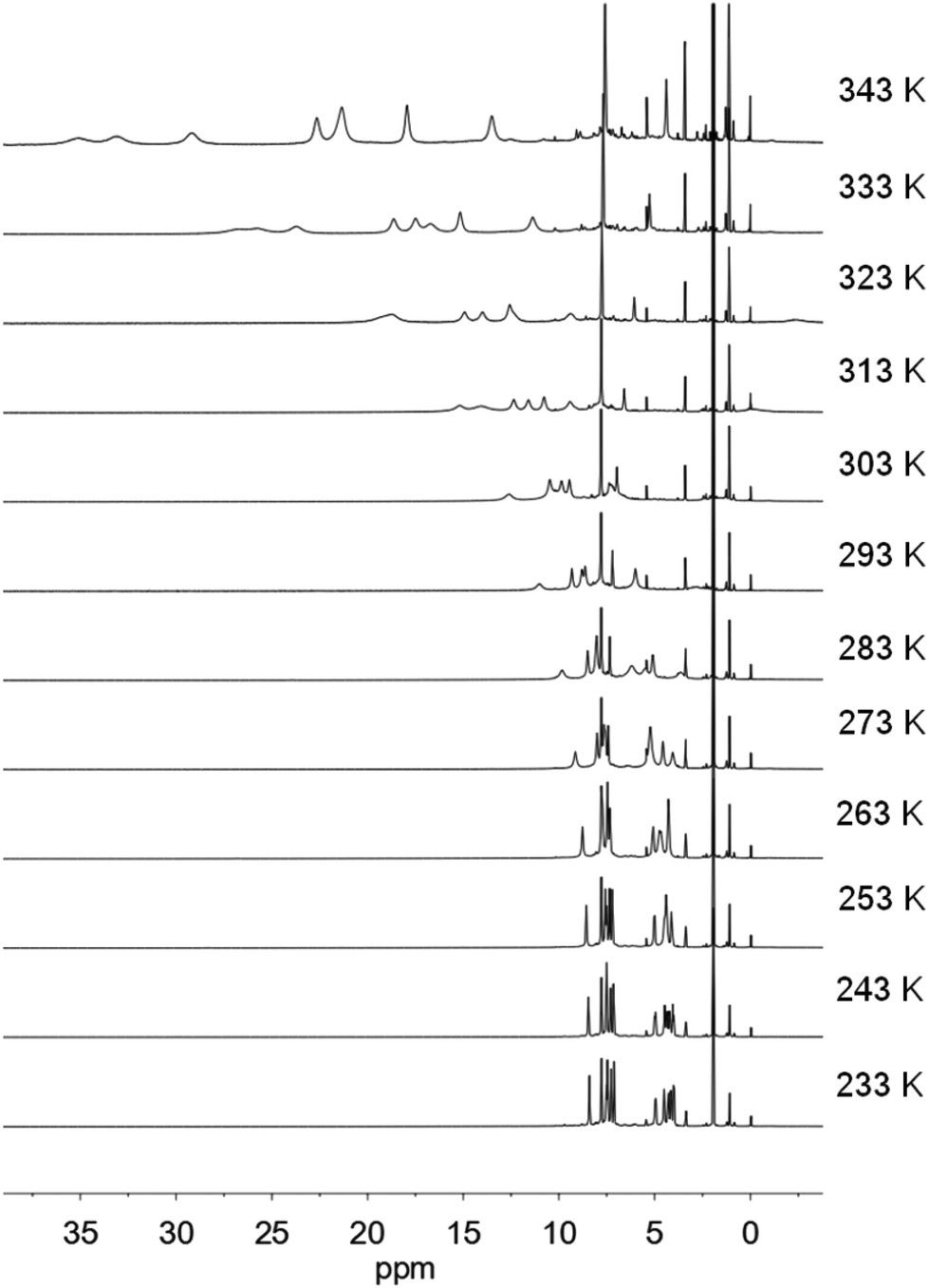 | ||
| Fig. 7 VT-1H NMR spectra of [Fe(5)(OTf)](OTf) in CD3CN. | ||
We have previously reported the remarkable coordination behaviour of complex [Fe(6)(OTf)2], which is distinctly different from [Fe(5)(OTf)](OTf). In non-coordinating solvents such as DCM, an equilibrium is observed between a seven-coordinate complex [Fe(6)(OTf)2] with pentagonal bipyramidal coordination and a five-coordinate trigonal bipyramidal complex [Fe(6)](OTf)2 with two non-coordinating triflate anions (see eqn (3)).62 In acetonitrile, a seven-coordinate [Fe(6)(CH3CN)2]2+ complex is formed, which is in equilibrium with the five-coordinate complex [Fe(6)]2+. These seven- and five-coordinate complexes are all high spin, as shown by their magnetic moment measurements in Fig. 2(◆), which appears to be the preferred spin state for this complex rather than forming an octahedral LS complex [Fe(6)(CH3CN)]2+.
 | (2) |
 | (3) |
Iron(II) complexes of the potentially pentadentate ligands 7 and 8 are paramagnetic (see Fig. S14 and S16†) and contain coordinated carbonyl donors according to IR analysis. The ν(CO) signal of ligand 7 decreases from 1647 to 1603 cm−1 upon coordination. Similar changes were observed in a related nickel(II) complex of a pyridine dicarboxamide ligand with coordinated carbonyl oxygens.83 Attempts to react the non-methylated precursor of ligand 7 with iron(II) bis(triflate) were unsuccessful, probably due to the poor solubility of this precursor. The 19F NMR spectrum of [Fe(7)(OTf)2] in CD3CN indicates non-coordinating triflate anions (Fig. S15†) and the UV-vis spectrum in CH3CN shows a relatively weak MLCT absorption at 425 nm (ε = 260 M−1 cm−1). X-ray analysis of the iron(II) bis(triflate) complex of ligand 8 revealed a tridentate coordination of the ligand via the carbonyl oxygen donors with an additional THF ligand to complete the octahedral coordination in [Fe(8)(OTf)2(thf)] (see Fig. 8). The iron centre has a severely distorted octahedral geometry with cis angles in the range 74.00(5)–117.74(16)°, and the metal atom lies ca. 0.74 Å out of the N(1)-pyridyl ring plane. Noteworthy intermolecular interactions are a pair of F⋯π contacts across independent centres of symmetry. The F(51) triflate fluorine atom in one molecule approaches the N(1) pyridyl ring in a Ci-related counterpart (F⋯π 3.59 Å), whilst F(41) approaches the N(8)-bound aryl ring across a different centre of symmetry (F⋯π 3.51 Å), resulting in an extended chain of molecules (interactions a and b respectively in Fig. S21; see the ESI†). Tridentate coordination of pyridine dicarboxamide ligands via the carbonyl oxygens appears to be the dominant coordination mode for nickel, copper and cobalt complexes,84,85 and based on the available data on the complex with ligand 7, we postulate a similar coordination mode in this case.
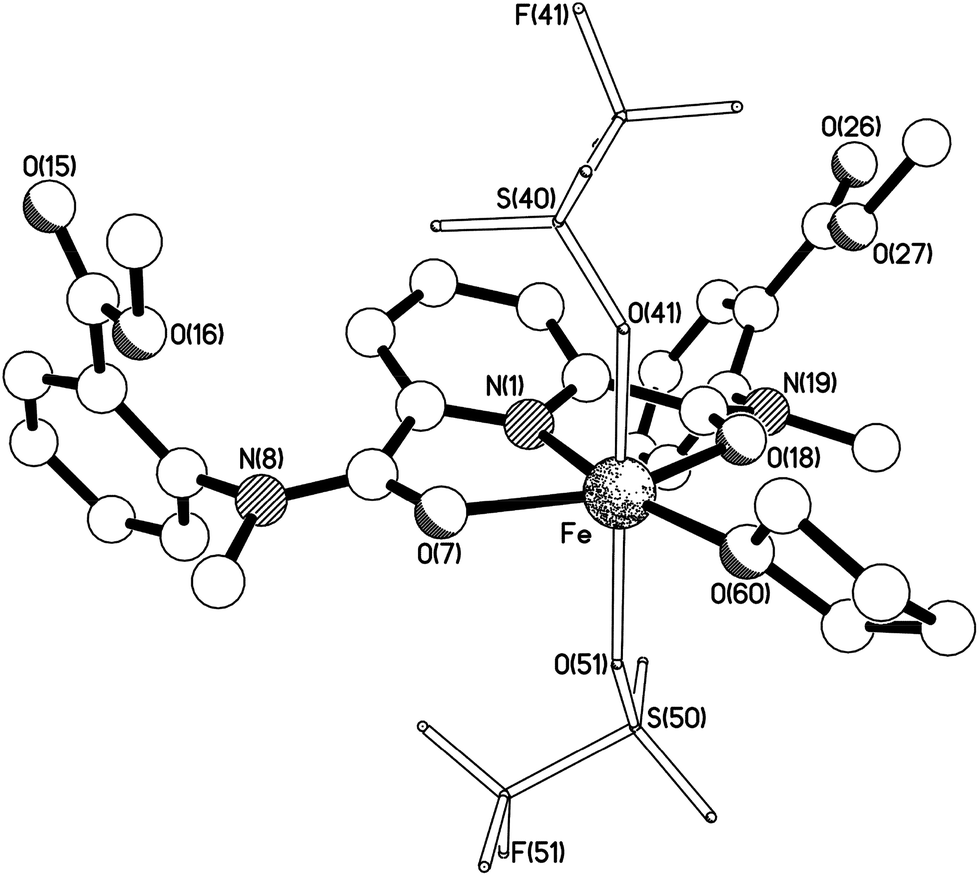 | ||
| Fig. 8 The crystal structure of [Fe(8)(OTf)2(thf)]. Selected bond lengths (Å) and angles (°); Fe–N(1) 2.1259(14), Fe–O(7) 2.1139(12), Fe–O(18) 2.1178(13), Fe–O(41) 2.147(2), Fe–O(51) 2.1081(15), Fe–O(60) 2.091(3), N(1)–Fe–O(7) 74.00(5), N(1)–Fe–O(18) 74.69(5), N(1)–Fe–O(41) 93.66(9), N(1)–Fe–O(51) 98.35(6), N(1)–Fe–O(60) 168.17(16), O(7)–Fe–O(18) 148.67(5), O(7)–Fe–O(41) 89.61(10), O(7)–Fe–O(51) 87.25(6), O(7)–Fe–O(60) 117.74(16), O(18)–Fe–O(41) 93.51(10), O(18)–Fe–O(51) 96.14(6), O(18)–Fe–O(60) 93.59(16), O(41)–Fe–O(51) 166.22(9), O(41)–Fe–O(60) 85.32(14), O(51)–Fe–O(60) 84.29(12). | ||
Several conclusions can be drawn from these structural studies. Firstly, the BPMEN ligand appears to have a unique ability to provide a very rigid ligand framework with a cis-α coordination mode and a strong binding of the iron centre. Any flexibility in the ligand framework or weakening of the N-donor strength and a consequent weakening of the metal–ligand interaction (e.g. ligand 1) can lead to changes in the coordination behaviour (cis-β and trans isomers). Secondly, the addition of a pyridine donor in the pentadentate ligands 5 and 6 does not improve the strength of the metal–ligand interaction and disfavours the formation of octahedral complexes. Thirdly, if a pyridylmethylene amine unit is changed to a pyridyl carboxamide unit (ligands 3, 4, 7 and 8), the amide N-donor will be too weak to coordinate effectively and the complexes rearrange to a preferred coordination of another donor.
Catalytic oxidation of cyclohexane
The catalytic properties of the iron(II) bis(triflate) complexes containing ligands 1–8 for the oxidation of cyclohexane with H2O2 have been evaluated (eqn (4)). | (4) |
The oxidation reactions were carried out under our standard conditions in order to compare the results with previously reported data (see ESI†).48,49 Hydrogen peroxide solution (10 equiv. or 100 equiv.) was added to an acetonitrile solution containing the catalyst (1 equiv.) and cyclohexane (1000 equiv.). A large excess of substrate was used to minimize over-oxidation of cyclohexanol (A) to cyclohexanone (K). The addition of dilute H2O2 was carried out slowly using a syringe pump, in order to minimise H2O2 decomposition. The yields are based on the amount of oxidant (H2O2) converted into oxygenated products. All the individual catalytic runs were performed at least twice.
Catalytic experiments were carried out initially using 10 equiv. of H2O2. The amount of cyclohexanol (A) and cyclohexanone (K) are measured by GC and the percentage conversion of H2O2 into oxidised products (A + K) for the different catalysts is shown in Table 1. The iron bis(triflate) complex [Fe(BPMEN)(OTf)2] is used as a benchmark against which the other catalysts are compared. We have previously reported that this catalyst, when using 10 equiv. of H2O2, converts 65% of the added H2O2 into oxygenated products, with a large ratio of cyclohexanol to cyclohexanone (A/K ratio) of 9.86 These results are consistent with those reported previously by Que and co-workers for the complex [Fe(BPMEN)(CH3CN)2](ClO4)2.87 The addition of more H2O2 (100 equiv.) results in a lower percentage conversion and a lower A/K ratio. The results obtained with [Fe(OTf)2(CH3CN)2] have been added for comparison (run 8), which shows only low conversions and A/K ratios, indicative of Fenton-type behaviour.
| Run | Catalyst | Equiv. H2O2 | A + Kb (%) | A/Kc |
|---|---|---|---|---|
| a Catalytic conditions: see ESI. b Total percentage yield of cyclohexanol (A) + cyclohexanone (K), expressed as moles of product per mole of H2O2. c Ratio of moles of cyclohexanol (A) to moles of cyclohexanone (K). d Data taken from ref. 49. e Data taken from ref. 89. | ||||
| 1d | [Fe(BPMEN)(OTf)2] | 10 | 65 | 9.5 |
| 2d | [Fe(BPMEN)(OTf)2] | 100 | 48 | 2.5 |
| 2 | [Fe(1)(OTf)2] | 10 | 23 | 1.4 |
| 2 | [Fe(1)(OTf)2] | 100 | 4 | 2.4 |
| 3 | [Fe(2)(OTf)2] | 10 | 37 | 9.6 |
| 4 | [Fe(5)(OTf)2] | 10 | 22 | 4.5 |
| 5 | [Fe(6)(OTf)2] | 10 | 25 | 2.0 |
| 6 | [Fe(7)(OTf)2] | 10 | 5 | 1.8 |
| 7 | [Fe(8)(OTf)2] | 10 | 19 | 2.1 |
| 8e | [Fe(OTf)2(CH3CN)2] | 10 | 4 | 1.6 |
The non-methylated complex [Fe(1)(OTf)2] shows a lower catalytic conversion and a smaller A/K ratio. This complex was also evaluated using 100 equiv. H2O2, showing comparable results to those reported by Shteinman an co-workers, who reported a ratio of A/K = 2 using 140 equiv. H2O2.67 The lower activity of this complex compared to the benchmark catalyst is tentatively ascribed to the ease of ligand degradation in the case of secondary amines. Complex [Fe(2)(OTf)2] gave the best conversion and A/K ratio within the series, although still lower compared to [Fe(BPMEN)(OTf)2]. Complex [Fe(2)(OTf)2] exists as a mixture of isomers (see Fig. 3) that are likely to have different individual catalytic oxidation activity. Evaluation of the catalytic activity of the individual isomers would require the synthesis of enantiomerically pure ligands and complexes. The complexes [Fe(3)(OTf)2] and [Fe(4)(OTf)2] with H2O2 showed no conversion under these conditions. The pentadentate complexes [Fe(5)(OTf)2] and [Fe(6)(OTf)2] showed a comparable but moderate conversion and A/K ratios, indicating that the two cis-labile sites are not essential for catalytic oxidation activity in these complexes. Relatively low conversions and A/K ratios were observed with complexes [Fe(7)(OTf)2] and [Fe(8)(OTf)2], where the pyridyl diamides are coordinated as tridentate ligands. Previous experiments using iron(II) complexes with tridentate ligands have shown similar results for the oxidation of cyclohexane.88
Catalyst decomposition
Previous studies and the structural analysis carried out here for complexes [Fe(1)(OTf)2] and [Fe(2)(OTf)2] have shown that changes to the BPMEN ligand framework generally lead to a change in ligand flexibility, such that different coordination modes (cis-β and trans) become accessible. An increase in ligand flexibility results in complexes that show inferior catalytic activity in alkane oxidation.48,49,86 As a result of these studies, it has become increasingly clear that catalyst stability, under the harsh oxidising conditions required to oxidise alkanes, is a major factor that determines the catalytic efficiency of a given catalyst. One possible deactivation pathway that has been invoked in a number of related non-heme catalyst systems is the formation of inactive dinuclear μ-oxo iron(III) complexes.9,10 However, certain dinuclear μ-oxo iron(III) complexes are active alkane hydroxylation catalysts,90–92 which suggests that dinuclear μ-oxo iron(III) complexes can be in equilibrium with mononuclear iron(III) hydroxo complexes (probably together with other dinuclear μ-oxo/μ-hydroxo intermediates).93–96 The formation of dinuclear complexes may be minimized by steric congestion around the metal centre.10,91,97,98 Ligand rigidity, a strong ligand field and low chemical reactivity of the ligand appear to be critically important for the stability and lifetime of non-heme catalysts. | (5) |
When complex [Fe(6)(OTf)2] was exposed to air and moisture in acetonitrile, oxidative ligand degradation occurred and an isolable complex could be obtained, which was characterised by X-ray diffraction. A dinuclear μ2-(OH)2-iron(III) complex [Fe(6′)(OH)]2(OTf)2 was obtained (see eqn (5) and Fig. 9) with a centre of symmetry in the middle of the Fe2O2 ring. The unique iron centre has a distorted octahedral geometry with cis angles in the range 73.70(8)–109.25(8)°, and is bound to one tetradentate N,N′,N′′,O donor ligand as well as to two bridging hydroxo ligands. The Fe–O–Fe bridges are symmetric [Fe(1)–O(20) 1.9664(17), Fe(1A)–O(20) 1.9700(17)] and subtend an angle of 103.40(8)° at the oxygen. The triflate anions sit in the clefts formed by the pyridyl rings on each iron centre with O⋯centroid separations of ca. 3.07 and 3.68 Å (interactions a and b respectively in Fig. S23†).
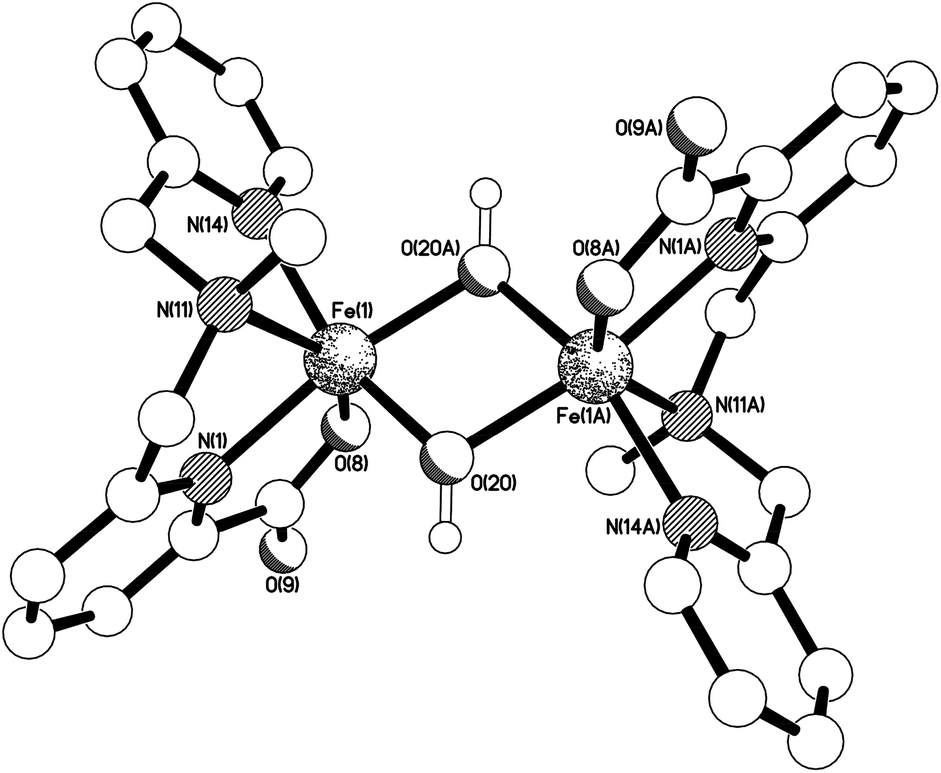 | ||
| Fig. 9 The structure of the Ci-symmetric di-cation present in the crystal of [Fe(6′)(OH)]2(OTf)2. Selected bond lengths (Å) and angles (°); Fe(1)–N(1) 2.085(2), Fe(1)–O(8) 1.9728(19), Fe(1)–N(11) 2.247(2), Fe(1)–N(14) 2.111(2), Fe(1)–O(20) 1.9664(17), Fe(1)–O(20A) 1.9700(17), N(1)–Fe(1)–O(8) 77.75(8), N(1)–Fe(1)–N(11) 73.70(8), N(1)–Fe(1)–N(14) 102.22(9), N(1)–Fe(1)–O(20) 93.18(8), N(1)–Fe(1)–O(20A) 169.40(8), O(8)–Fe(1)–N(11) 147.17(8), O(8)–Fe(1)–N(14) 93.70(9), O(8)–Fe(1)–O(20) 103.97(8), O(8)–Fe(1)–O(20A) 101.79(8), N(11)–Fe(1)–N(14) 77.08(8), N(11)–Fe(1)–O(20) 93.55(8), N(11)–Fe(1)–O(20A) 109.25(8), N(14)–Fe(1)–O(20) 158.72(9), N(14)–Fe(1)–O(20A) 88.38(8), O(20)–Fe(1)–O(20A) 76.60(8), Fe(1)–O(20)–Fe(1A) 103.40(8). | ||
The pentadentate ligand 6 has undergone oxidative C–N cleavage to give a picolinate-type ligand 6′, presumable together with (N-methyl)-2-aminomethyl pyridine as the by-product. The iron(II) centre has been oxidised, most likely by oxygen which in turn is reduced and in the presence of moisture results in the formation of hydroxide anions, as observed in complex [Fe(6′)(OH)]2(OTf)2. The oxidation of pyridylmethylamine moieties appears to be a common occurrence, sometimes resulting in the formation of stable metal picolinate complexes.42,44,46,47 Ligand degradation reactions via oxidative N-dealkylation was recently observed during catalytic toluene oxidation with H2O2 with a related iron complex featuring the BPMCN ligand, an analogue of BPMEN with a cyclohexyl backbone.34
The mechanism by which oxidative ligand degradation occurs in pyridylamine complexes such as [Fe(6)(OTf)2] with oxidants is not yet understood, but the results obtained here can be explained according to the general mechanism shown in Scheme 1. Oxidation of a methylene unit in ligand 6 results in a hemi-aminal complex (B), which can react further or rearrange to an O-bound hemi-aminal complex C. Further oxidation of the tertiary C–H bond in N-bound hemi-aminal complex B results in the formation of complex (E), which will dehydrate to an amide complex (F). As we have seen here for complexes [Fe(4)(OTf)2] and [Fe(8)(OTf)2], the weak basicity of amide nitrogen donors will likely result in a rearrangement due de-coordination of the nitrogen donor. Hydrolysis of the amide complex would give a picolinate complex G. In the case of complex [Fe(6)(OTf)2], this results in the picolinate ligand 6′ (see eqn (5)). An alternative pathway involves rearrangement of the N-bound hemi-aminal complex B to an O-bound complex of type C. Related O-bound hemi-aminal complexes have been isolated and characterised on several occasions.35–39 Further oxidation and C–N cleavage would also result in the picolinate complex G.
In conclusion, ligand degradation in non-heme oxidation catalysts is an important factor that affects catalyst stability and lifetime under the oxidising reaction conditions. In order to investigate potential ligand degradation pathways and to improve catalyst stability, we have prepared a series of iron(II) complexes with tetradentate and pentadentate pyridylamine-type ligands 1–8. Compared to [Fe(BPMEN)(OTf)2], all complexes have shown lower activities as catalysts for the oxidation of cyclohexane with H2O2 as the oxidant. The BPMEN ligand ensures a strong coordination environment with a cis-α geometry at the iron centre, stabilises intermediates in various oxidation states along the catalytic oxidation cycle and undergoes negligible oxidative ligand degradation. The NH donors in [Fe(1)(OTf)2] provide a weaker ligand field resulting in a conformationally less rigid complex with different geometrical high spin isomers (cis-β and trans in addition to cis-α). Secondary amines are also believed to be more vulnerable to oxidative degradation. A mixture of isomers was obtained in the case of [Fe(2)(OTf)2], probably with different catalytic activities. Amide donors in ligands 3 and 4 result in dinuclear oxygen-bound complexes with no catalytic activity. Pentadentate ligands in [Fe(5)(OTf)2] and [Fe(6)(OTf)2] provide moderate catalytic activity, despite the absence of two cis-labile sites, where ligands 7 and 8 were found to coordinate as tridentate ligands and showed only low catalytic activities. The reaction of complex [Fe(6)(OTf)2] with O2 has shown that ligand degradation can occur via oxidative N-dealkylation, based on the isolation of an iron(III) complex [Fe(6′)(OH)]2(OTf)2 with a picolinate-type ligand 6′. We are continuing our efforts to develop robust non-heme iron-based catalysts for the selective oxidation of alkanes.
Experimental section
Starting materials
The following ligands and starting materials have been prepared following literature procedures: N,N′-dimethyl-bis(2-pyridylmethyl)ethylene diamine (BPMEN),89N,N′-bis(2-pyridylmethyl)ethylene diamine (1),59N,N′-dimethyl-bis(2-pyridyl-1-ethyl)ethylene diamine (2),99N,N′-dimethyl-N,N′-bis(2-pyridinecarboxamide)-1,2-ethane (3),64,100 2,6-bis[(2-pyridylmethyl)aminomethyl]-pyridine (5) and 2,6-bis[(N-methyl(2-pyridylmethyl)-amino)methyl]pyridine (6),60,61,101N,N′-bis(2-pyridinecarboxamide)-1,2-benzene,100 2-N,6-N-bis(quinolin-8-yl)pyridine-2,6-dicarboxamide.102 The synthesis of the iron complexes [Fe(BPMEN)(OTf)2] and [Fe(6)(OTf)2] has been reported previously.62,89Synthesis of ligands
:10)) gave pure 4 as beige solid (0.84 g, 48%). 1H-NMR (CDCl3, 400 MHz) (three stereoisomers in a 10:6:1 ratio) δ (ppm): 3.33 (s, 3/17 of 6H, CH3), 3.44 (s, 1/17 of 6H, CH3), 3.56 (s, 3/17 of 6H, CH3), 3.67 (s, 10/17 of 6H, CH3), 6.64 (m, 2.8/28 of 12H, Ph-H), 6.81 (m, 2.8/28 of 12H, Ph-H), 7.02 (br, 0.95/28 of 12H, arom-H), 7.17 (m, 4.5/28 of 12H, Py-H), 7.32 (br, 0.5/28 of 12H, arom-H), 7.35–7.43 (m, 1.9/28 of 12H, arom-H), 7.5 (br, 0.5/28 of 12H, arom-H), 7.68 (br, 1/28 of 12H, arom-H), 7.75 (t, 3.6/28 of 12H, 3JHH = 7.8 Hz, Py-H), 7.85 (m, 1.7/28 of 12H, arom-H), 7.90 (m, 3.2/28 of 12H, Py-H), 8.15 (d, 2.8/28 of 12H, 3JHH = 4.36 Hz, Py-H), 8.22 (br, 0.75/28 of 12H, arom-H), 8.68 (br, 1/28 of 12H, arom-H); 13C-NMR (CDCl3, 400 MHz): (Three stereoisomers in a 10:6:1 ratio) δ (ppm) = 35 (CH3), 125 (Py-CH), 125 (Py-CH), 127 (Ph-CH), 129 (Ph-CH), 136 (Py-CH), 140 (Ph-CR), 148 (Py-CH), 154 (Py-C), 168 (CO); IR: ν (cm−1): 1648 (CO stretching), 1599 (phenyl), 1586 and 1568 (pyridine), 751 (ortho-disubstituted); ESI-MS: m/z (%): 715 (49) [M2 + Na]+, 369 (30) [M + Na]+, 347 (100) [M]+.
O), 153 (quat. C), 151 (2-Qu-CH), 143 (quat. C), 141 (quat. C), 137 (meta-Py-CH), 129 (para-Py-CH or other Qu-CH), 129 (para-Py-CH or other Qu-CH) 128 (7-QuH), 127 (para-Py-CH or other Qu-CH), 123 (quat. C), 122 (para-Py-CH or other Qu-CH) 38 (NCH3); ESI-MS: m/z (%) = 448 (98) [M + H]+, 470 (100) [M + Na]+; IR: ν (cm−1) 1647; Anal. Calcd for C27H21N5O2: C 72.47, H 4.73, N 15.65, Found: C 71.62, H 4.81, N 15.35.
(b) 2,6-Bis[(methylanthranilate)carboxamide]pyridine (700.0 mg, 1.5 mmol) was dissolved in abs. THF and NaH (110.0 mg, 4.6 mmol) were added. After stirring the suspension for 4 h, MeI (0.3 mL, 4.8 mmol) was added and the suspension was stirred at room temperature overnight. After removal of all volatiles at reduced pressure, the residue was taken up in chloroform and saturated aqueous sodium hydrogencarbonate. After the phases were separated the aqueous phase was extracted three times with chloroform. The organic phase was dried over MgSO4, filtered and the solvent removed under reduced pressure. The product 8 was obtained as viscous yellow oil (430 mg, 62%). 1H NMR (CDCl3, 400 MHz) (due to the presence of three rotamers in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :2:1 ratio, a large number of signals appear at room temperature): δ (ppm) = 7.97 (d, J = 8.2 Hz, 0.4H), 7.85–7.64 (m, 2.7H), 7.60 (t, J = 8.0 Hz, 0.6H), 7.51 (d, J = 7.8 Hz, 0.4H), 7.44–7.17 (m, 5.8H), 7.12 (d, J = 7.8 Hz, 0.4H), 6.84 (d, J = 8.0 Hz, 0.4H), 6.61 (d, J = 8.0 Hz, 0.2H), 6.52 (t, J = 8.0 Hz, 0.2H), 3.79 (m, 6H), 3.44 (m, 1.5H), 3.29 (m, 3H), 2.89 (m, 1.5H). 13C NMR (CDCl3, 101 MHz): δ (ppm) = 167.8, 167.4, 167.4, 165.9, 165.8, 165.7, 165.6, 152.6, 152.5, 152.2, 151.9, 144.6, 144.0, 143.4, 137.5, 136.3, 134.6, 133.7, 133.1, 133.0, 132.6, 131.5, 131.4, 131.2, 131.1, 130.7, 130.3, 130.1, 128.7, 128.2, 127.8, 127.6, 127.5, 127.4, 124.7, 123.9, 123.7, 123.5, 114.3, 110.7, 52.5, 52.4, 52.2, 51.4, 40.2, 38.7, 38.0, 37.9, 29.7, 29.5. ESI-MS: m/z = 484 [M + Na]+, 462 [M + H]+.
:2:1 ratio, a large number of signals appear at room temperature): δ (ppm) = 7.97 (d, J = 8.2 Hz, 0.4H), 7.85–7.64 (m, 2.7H), 7.60 (t, J = 8.0 Hz, 0.6H), 7.51 (d, J = 7.8 Hz, 0.4H), 7.44–7.17 (m, 5.8H), 7.12 (d, J = 7.8 Hz, 0.4H), 6.84 (d, J = 8.0 Hz, 0.4H), 6.61 (d, J = 8.0 Hz, 0.2H), 6.52 (t, J = 8.0 Hz, 0.2H), 3.79 (m, 6H), 3.44 (m, 1.5H), 3.29 (m, 3H), 2.89 (m, 1.5H). 13C NMR (CDCl3, 101 MHz): δ (ppm) = 167.8, 167.4, 167.4, 165.9, 165.8, 165.7, 165.6, 152.6, 152.5, 152.2, 151.9, 144.6, 144.0, 143.4, 137.5, 136.3, 134.6, 133.7, 133.1, 133.0, 132.6, 131.5, 131.4, 131.2, 131.1, 130.7, 130.3, 130.1, 128.7, 128.2, 127.8, 127.6, 127.5, 127.4, 124.7, 123.9, 123.7, 123.5, 114.3, 110.7, 52.5, 52.4, 52.2, 51.4, 40.2, 38.7, 38.0, 37.9, 29.7, 29.5. ESI-MS: m/z = 484 [M + Na]+, 462 [M + H]+.
[Fe(1)(OTf)2]: grey-green solid. 90% yield. 1H NMR (CD3CN, 400 MHz, 298 K, all peaks appear as broad singlets): δ (ppm) = 17.75, 14.87, 13.92, 10.11, 9.83, 6.42, 3.70, 0.66. 19F NMR (CD2Cl2) δ −42. MS (FAB, m/z (%)): 447 (100) [(M − OTf)+], 296 (2) [(M − 2OTf)+]. μeff (CD3CN, 298 K) = 2.8 BM. UV-Vis (CH3CN, 298 K): λ(ε) (nm, M−1 cm−1): 375 (4100), 533 (200).
[Fe(2)(OTf)2]: yellow solid. 85% yield. Mixture of diastereomers: 1H NMR (CD3CN) δ 132.6, 73.6, 70.4, 63.3, 59.3, 48.6, 42.2, 41.2, 26.5, 24.4, 18.6, 10.3. 19F NMR (CD3CN) δ −78.6 (ν1/2 = 900 Hz). 19F NMR (CD2Cl2) δ −25.6. MS (+FAB, m/z (%)): 503 (100) [(M − OTf)+]. μeff (CD3CN) = 4.39μB. UV-Vis (CH3CN, 298 K): λ(ε) (nm, M−1 cm−1): 376 (3600), 515 (30); Anal. Calcd for C20H26F6FeN4O6S2: C, 36.82; H, 4.02; N, 8.59. Found: C, 36.87; H, 3.96; N, 8.59.
[Fe(3)(OTf)2]: yellow solid (1.03 g, 94%). 1H-NMR (CD3CN, 400 MHz, 298 K) δ (ppm): −1.01, 7.21, 10.25, 18.54, 23.62, 48.96, 66.30, 74.04; 19F-NMR (CD3CN, 376 MHz, 298 K) δ (ppm): −65.4; UV-Vis (CH3CN, 298 K): λ(ε) (nm, M−1 cm−1): 217 (12000), 264 (9500), 414 (750); IR: ν (cm−1) 1608 (CO), 1590 and 1572 (pyridine); LSIMS: m/z (%): 1155 (2.5) [M2 − OTf]+, 503 (71.3) [M − OTf]+. Anal. Calcd for C18H18F6FeN4O8S2: C, 33.14; H, 2.78; N, 8.59. Found: C, 33.29; H, 2.88; N, 8.46.
[Fe(4)(OTf)]: orange solid (0.83 g, 82%). 1H-NMR (CD3CN, 400 MHz, 298 K): ∼45 signals indicative of multiple species; 19F-NMR (CD3CN, 376 MHz): δ (ppm): −59.44. UV-Vis (CH3CN): λ(ε) (nm, M−1 cm−1) = 217(12000), 262(6800), 416(500); IR: ν (cm−1): 1610 (CO), 1585 and 1565 (pyridine). LSIMS: m/z (%): 1251 (20.9) [M2 − OTf]+, 551 (100) [M − OTf]+. Anal. calcd for C22H18F6FeN4O8S2: C, 37.73; H, 2.59; N, 8.00. Found: C, 37.81; H, 2.49; N, 7.94. Crystals suitable for X-ray analysis were grown from an acetonitrile solution layered with diethyl ether at room temperature. Crystal data for [Fe(4)(OTf)2]2: C44H36F12Fe2N8O16S4·2MeCN, M = 1482.86, rhombohedral, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c (no. 167), a = b = 25.4174(4), c = 51.8599(7) Å, V = 29015.1(8) Å3, Z = 18 (C2 symmetry), Dc = 1.528 g cm−3, μ(Mo-Kα) = 0.684 mm−1, T = 173 K, orange/red needles, Oxford Diffraction Xcalibur 3 diffractometer; 9380 independent measured reflections (Rint = 0.0293), F2 refinement,103R1(obs) = 0.0396, wR2(all) = 0.1164, 6515 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 63°], 390 parameters. CCDC 995554.
c (no. 167), a = b = 25.4174(4), c = 51.8599(7) Å, V = 29015.1(8) Å3, Z = 18 (C2 symmetry), Dc = 1.528 g cm−3, μ(Mo-Kα) = 0.684 mm−1, T = 173 K, orange/red needles, Oxford Diffraction Xcalibur 3 diffractometer; 9380 independent measured reflections (Rint = 0.0293), F2 refinement,103R1(obs) = 0.0396, wR2(all) = 0.1164, 6515 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 63°], 390 parameters. CCDC 995554.
[Fe(5)(OTf)](OTf): purple solid, 58% yield. 1H NMR (CD3CN, 400 MHz, 298 K, all peaks appear as broad singlets): δ (ppm) = 10.91, 9.25, 8.72, 8.57, 8.20, 7.80, 7.45, 7.33, 7.22, 5.05, 4.69, 4.59, 2.94, 2.50. 19F NMR (CD3CN, 376 MHz, broad singlet): δ (ppm) = −77.76. UV/Vis (CH3CN): λ(ε) (nm, M−1 cm−1): 546 (1000), 381 (7400), 283 (5200). μeff (CD3CN, 298 K) = 1.74 BM. LSIMS m/z = 524 [M − OTf]+. Anal. Calcd (found) for C21H21F6FeN5O6S2: %C 37.46 (37.51), %H 3.14 (3.05), %N 10.40 (10.35).
[Fe(7)(OTf)2]: red solid. Yield: 146 mg (74%); 1H-NMR (400 MHz, CD3CN, 298 K): peaks are overlapping and too broad to assign; 19F-NMR (376 MHz, CD3CN): δ (ppm) −70 (s, 3F, −OSO2CF3). IR: ν (cm−1) 2983, 1603, 1566, 1498, 1400, 1318, 1267, 1213, 1148, 1026, 924, 878, 837, 800, 766, 737, 688. UV-Vis (CH3CN): λ(ε) (nm, M−1 cm−1): 296 (3000), 425 (260). LSIMS: m/z (%) = 652 (10) [M − OTf]+.
[Fe(8)(thf)(OTf)2]: red solid, 81% yield. 1H NMR (CD2Cl2, 400 MHz, 298 K, all peaks appear as broad singlets): δ (ppm) = 67.11, 65.51, 30.53, 28.47, 26.78, 8.56, 8.42, 8.00, 7.87, 7.68, 6.81, 6.44, 6.06, 5.55, 5.33, 5.10, 4.07, 3.79. 19F NMR (CD2Cl2, 376 MHz, broad singlet): δ (ppm) = −29. LSIMS m/z = 666 [M − OTf]+. Anal. Calcd (found) for C21H19F6FeN3O6S4: %C 42.00 (41.92), %H 3.41 (3.35), %N 4.74 (4.44). UV/Vis (CH3CN): λ(ε) (nm, M−1 cm−1): 279 (4200). Crystals suitable for X-ray analysis were grown from a tetrahydrofuran solution at room temperature. Crystal data for [Fe(8)(OTf)2(thf)]: C31H31F6FeN3O13S2, M = 887.56, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 12.1402(4), b = 12.1595(4), c = 13.5926(3) Å, α = 81.615(2), β = 70.378(3), γ = 88.442(3)°, V = 1869.29(10) Å3, Z = 2, Dc = 1.577 g cm−3, μ(Mo-Kα) = 0.612 mm−1, T = 173 K, orange/red blocks, Oxford Diffraction Xcalibur 3 diffractometer; 12149 independent measured reflections (Rint = 0.0184), F2 refinement,103R1(obs) = 0.0473, wR2(all) = 0.1310, 9276 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 65°], 581 parameters. CCDC 995555.
(no. 2), a = 12.1402(4), b = 12.1595(4), c = 13.5926(3) Å, α = 81.615(2), β = 70.378(3), γ = 88.442(3)°, V = 1869.29(10) Å3, Z = 2, Dc = 1.577 g cm−3, μ(Mo-Kα) = 0.612 mm−1, T = 173 K, orange/red blocks, Oxford Diffraction Xcalibur 3 diffractometer; 12149 independent measured reflections (Rint = 0.0184), F2 refinement,103R1(obs) = 0.0473, wR2(all) = 0.1310, 9276 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 65°], 581 parameters. CCDC 995555.
[Fe(6′)(OH)]2(OTf)2: brown crystals were obtained from a solution of [Fe(6)(OTf)2] in acetonitrile, upon exposure to air for several days. Crystal data for [Fe(6′)(OH)]2(OTf)2: C28H30Fe2N6O6·2(CF3SO3)·2MeCN, M = 1038.53, triclinic, P (no. 2), a = 9.0685(5), b = 9.9914(4), c = 13.1697(5) Å, α = 104.096(3), β = 109.381(4), γ = 98.362(4)°, V = 1057.85(9) Å3, Z = 1 (Ci symmetry), Dc = 1.630 g cm−3, μ(Cu-Kα) = 7.283 mm−1, T = 173 K, brown plates, Oxford Diffraction Xcalibur PX Ultra diffractometer; 4080 independent measured reflections (Rint = 0.0273), F2 refinement,103R1(obs) = 0.0417, wR2(all) = 0.1193, 3607 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 145°], 327 parameters. CCDC 995556.
Acknowledgements
We are grateful to the Department of Chemistry for funding and to the IDEA League program for a bursary to IdW.References
- T. J. Collins, Acc. Chem. Res., 1994, 27, 279–385 CrossRef CAS.
- E. Nagababu and J. M. Rifkind, Antioxid. Redox Signaling, 2004, 6, 967–978 CrossRef CAS PubMed.
- T. Matsui, M. Unno and M. Ikeda-Saito, Acc. Chem. Res., 2010, 43, 240–247 CrossRef CAS PubMed.
- I. D. Cunningham, T. N. Danks, J. N. Hay, I. Hamerton and S. Gunathilagan, Tetrahedron, 2001, 57, 6847–6853 CrossRef CAS.
- I. D. Cunningham, T. N. Danks, J. N. Hay, I. Hamerton, S. Gunathilagan and C. Janczak, J. Mol. Catal., 2002, 185, 25–31 CrossRef CAS.
- I. D. Cunningham, T. N. Danks, K. T. A. O'Connell and P. W. Scott, J. Chem. Soc., Perkin Trans. 2, 1999, 2133–2139 RSC.
- A. C. Serra, E. C. Marçalo and A. M. d. A. Rocha Gonsalves, J. Mol. Catal., 2004, 215, 17–21 CrossRef CAS PubMed.
- N. A. Stephenson and A. T. Bell, J. Am. Chem. Soc., 2005, 127, 8635–8643 CrossRef CAS PubMed.
- N. A. Vermeulen, M. S. Chen and M. C. White, Tetrahedron, 2009, 65, 3078–3084 CrossRef CAS PubMed.
- L. Gómez, I. Garcia-Bosch, A. Company, J. Benet-Buchholz, A. Polo, X. Sala, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2009, 48, 5720–5723 CrossRef PubMed.
- S. J. Lange, H. Miyake and L. Que Jr., J. Am. Chem. Soc., 1999, 121, 6330–6331 CrossRef CAS.
- Y. Mekmouche, S. Ménage, C. Toia-Duboc, M. Fontecave, J.-B. Galey, C. Lebrun and J. Pécaut, Angew. Chem., Int. Ed., 2001, 40, 949–952 CrossRef CAS.
- A. Nielsen, F. B. Larsen, A. D. Bond and C. J. McKenzie, Angew. Chem., Int. Ed., 2006, 45, 1602–1606 CrossRef CAS PubMed.
- A. M. Calafat and L. G. Marzilli, Inorg. Chem., 1993, 32, 2906–2911 CrossRef CAS.
- M. Yashiro, T. Mori, M. Sekiguchi, S. Yoshikawa and S. Shiraishi, J. Chem. Soc., Chem. Commun., 1992, 1167–1168 RSC.
- B. Sonnberger, P. Hühn, A. Waßerburger and F. Wasgestian, Inorg. Chim. Acta, 1992, 196, 65–71 CrossRef CAS.
- M. R. Bukowski, S. Zhu, K. D. Koehntop, W. W. Brennessel and L. Que Jr., J. Biol. Inorg. Chem., 2004, 9, 39–48 CrossRef CAS PubMed.
- M. Yamaguchi, M. Saburi and S. Yoshikawa, J. Am. Chem. Soc., 1984, 106, 8293–8295 CrossRef CAS.
- K. Jitsukawa, T. Yamamoto, T. Atsumi, H. Masuda and H. Einaga, J. Chem. Soc., Chem. Commun., 1994, 2335–2336 RSC.
- M.-C. Rodríguez, F. Lambert, I. Morgenstern-Badarau, M. Cesario, J. Guilhem, B. Keita and L. Nadjo, Inorg. Chem., 1997, 36, 3525–3531 CrossRef PubMed.
- R. M. Hartshorn, J. Chem. Soc., Dalton Trans., 2002, 3214–3218 RSC.
- I. Sanyal, M. Mahroof-Tahir, M. S. Nasir, P. Ghosh, B. I. Cohen, Y. Gultneh, R. W. Cruse, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, Inorg. Chem., 1992, 31, 4322–4332 CrossRef CAS.
- D. Lee and S. J. Lippard, J. Am. Chem. Soc., 2001, 123, 4611–4612 CrossRef CAS.
- D. Lee and S. J. Lippard, Inorg. Chem., 2002, 41, 827–837 CrossRef CAS PubMed.
- E. C. Carson and S. J. Lippard, J. Inorg. Biochem., 2006, 100, 1109–1117 CrossRef CAS PubMed.
- A. Böttcher, H. Elias, E.-G. Jäger, H. Langfelderova, M. Mazur, L. Müller, H. Paulus, P. Pelikan, M. Rudolph and M. Valko, Inorg. Chem., 1993, 32, 4131–4138 CrossRef.
- S. Zhu, W. W. Brennessel, R. G. Harrison and L. Que Jr., Inorg. Chim. Acta, 2002, 337, 32–38 CrossRef CAS.
- J. M. Rowland, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2001, 41, 2754–2760 CrossRef PubMed.
- M. Renz, C. Hemmert, B. Donnadieu and B. Meunier, Chem. Commun., 1998, 1635–1636 RSC.
- M. Renz, C. Hemmert, H. Gornitzka and B. Meunier, New J. Chem., 1999, 23, 773–776 RSC.
- C. Hemmert, M. Renz, H. Gornitzka, S. Soulet and B. Meunier, Chem. – Eur. J., 1999, 5, 1766–1774 CrossRef CAS.
- M. Ostermeier, C. Limberg, B. Ziemer and V. Karunakaran, Angew. Chem., Int. Ed., 2007, 46, 5329–5331 CrossRef CAS PubMed.
- H. Kooijman, S. Tanase, E. Bouwman, J. Reedijk and A. L. Spek, Acta. Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m510–m512 Search PubMed.
- M. Canals, R. Gonzalez-Olmos, M. Costas and A. Company, Environ. Sci. Technol., 2013, 47, 9918–9927 CrossRef CAS PubMed.
- N. Arulsamy and D. J. Hodgson, Inorg. Chem., 1994, 33, 4531–4536 CrossRef CAS.
- V. M. Ugalde-Saldívar, M. E. Sosa-Torres, L. Ortiz-Frade, S. Bernès and H. Höpfl, J. Chem. Soc., Dalton Trans., 2001, 3099–3107 RSC.
- Z. Liu, H. Kooijman, A. L. Spek and E. Bouwman, Prog. Org. Coat., 2007, 60, 343–349 CrossRef CAS PubMed.
- J. P. Saucedo-Vázquez, V. M. Ugalde-Saldívar, A. R. Toscano, P. M. H. Kroneck and M. E. Sosa-Torres, Inorg. Chem., 2009, 48, 1214–1222 CrossRef PubMed.
- S. K. Padhi, R. Sahu and V. Manivannan, Inorg. Chim. Acta, 2011, 367, 57–63 CrossRef CAS PubMed.
- L. You, S. R. Long, V. M. Lynch and E. V. Anslyn, Chem. – Eur. J., 2011, 17, 11017–11023 CrossRef CAS PubMed.
- P. Comba, S. Kuwata, G. Linti, H. Pritzkow, M. Tarnai and H. Wadepohl, Chem. Commun., 2006, 2074–2076 RSC.
- S. Groni, P. Dorlet, G. Blain, S. Bourcier, R. Guillot and E. Anxolabéhère-Mallart, Inorg. Chem., 2008, 47, 3166–3172 CrossRef CAS PubMed.
- A. Thibon, J.-F. Bartoli, S. Bourcier and F. Banse, Dalton Trans., 2009, 9587–9594 RSC.
- D. Pijper, P. Saisaha, J. W. de Boer, R. Hoen, C. Smit, A. Meetsma, R. Hage, R. P. van Summeren, P. L. Alsters, B. L. Feringa and W. R. Browne, Dalton Trans., 2010, 39, 10375–10381 RSC.
- S. Mahapatra, V. G. Young Jr., S. Kaderli, A. D. Zuberbühler and W. B. Tolman, Angew. Chem., Int. Ed. Engl., 1997, 36, 130–133 CrossRef CAS.
- M. S. Vad, A. Nielsen, A. Lennartson, A. D. Bond, J. E. McGrady and C. J. McKenzie, Dalton Trans., 2011, 40, 10698–10707 RSC.
- D. G. Lonnon, D. C. Craig and S. B. Colbran, Inorg. Chem. Commun., 2003, 6, 1351–1353 CrossRef CAS PubMed.
- J. England, G. J. P. Britovsek, N. Rabadia and A. J. P. White, Inorg. Chem., 2007, 46, 3752–3767 CrossRef CAS PubMed.
- J. England, C. R. Davis, M. Banaru, A. J. P. White and G. J. P. Britovsek, Adv. Synth. Catal., 2008, 350, 883–897 CrossRef CAS.
- J. England, R. Gondhia, L. Bigorra-Lopez, A. R. Petersen, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2009, 5319–5334 RSC.
- K. Chen and L. Que Jr., Chem. Commun., 1999, 1375–1376 RSC.
- G. Xue, D. Wang, R. De Hont, A. T. Fiedler, X. Shan, E. Münck and L. Que Jr., Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20713–20718 CrossRef CAS PubMed.
- M. Goto, M. Takeshita, N. Kanda, T. Sakai and V. L. Goedken, Inorg. Chem., 1985, 24, 582–587 CrossRef CAS.
- V. L. Goedken and D. H. Busch, J. Am. Chem. Soc., 1972, 94, 7355–7340 CrossRef CAS.
- G. J. Christian, A. Llobet and F. Maseras, Inorg. Chem., 2010, 49, 5977–5985 CrossRef CAS PubMed.
- C. Hemmert, A. P. Maestrin, M. Renz, H. Gornitzka and B. Meunier, C. R. Acad. Sci., Ser. C, 2000, 3, 735–741 CrossRef CAS.
- M. Wu, C.-X. Miao, S. Wang, X. Hu, C. Xia, F. E. Kühn and W. Sun, Adv. Synth. Catal., 2011, 353, 3014–3022 CrossRef CAS.
- T. M. Suzuki, T. Kimura and J. Fujita, Bull. Chem. Soc. Jpn., 1980, 53, 77–81 CrossRef CAS.
- H. A. Goodwin and F. Lions, J. Am. Chem. Soc., 1960, 82, 5013–5023 CrossRef CAS.
- T. Darbre, C. Dubs, E. Rusanov and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 2002, 3284–3291 CrossRef CAS.
- G. R. Newkome, V. K. Gupta, F. R. Fronczek and S. Pappalardo, Inorg. Chem., 1984, 23, 2400–2408 CrossRef CAS.
- M. Grau, J. England, R. Torres Martin de Rosales, H. S. Rzepa, A. J. P. White and G. J. P. Britovsek, Inorg. Chem., 2013, 52, 11867–11874 CrossRef CAS PubMed.
- I. Okamoto, M. Terashima, H. Masu, M. Nabeta, K. Ono, N. Morita, K. Katagiri, I. Azumaya and O. Tamura, Tetrahedron, 2011, 67, 8536–8543 CrossRef CAS PubMed.
- A. V. Malkov, L. Gouriou, G. C. Lloyd-Jones, I. Stary, V. Langer, P. Spoor, V. Vinader and P. Kocovsky, Chem. – Eur. J., 2006, 12, 6910–6929 CrossRef CAS PubMed.
- M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194–7195 CrossRef CAS.
- K. P. Bryliakov, E. A. Duban and E. P. Talsi, Eur. J. Inorg. Chem., 2005, 72–76 CrossRef CAS.
- E. A. Turitsyna, O. N. Gritsenko and A. A. Shteinman, Kinet. Catal., 2007, 48, 53–59 CrossRef CAS.
- S. Kundu, E. Matito, S. Walleck, F. F. Pfaff, F. Heims, B. Rábay, J. M. Luis, A. Company, B. Braun, T. Glaser and K. Ray, Chem. – Eur. J., 2012, 18, 2787–2791 CrossRef CAS PubMed.
- H. Toftlund, E. Pedersen and S. Yde-Andersen, Acta Chem. Scand., 1984, 38A, 693–697 CrossRef PubMed.
- J.-F. Létard, S. Asthana, H. J. Shepherd, P. Guionneau, A. E. Goeta, N. Suemura, R. Ishikawa and S. Kaizaki, Chem. – Eur. J., 2012, 18, 5924–5934 CrossRef PubMed.
- J. G. Gibson and E. D. McKenzie, J. Chem. Soc., 1971,(A), 1666–1683 RSC.
- S. Utsuno, A. Hayashi, S. Kondo and M. Utsumi, Chem. Lett., 1979, 351–352 CrossRef CAS.
- L. Xu, I. A. Setyawati, J. Pierreroy, M. Pink, V. G. Young Jr., B. O. Patrick, S. J. Rettig and C. Orvig, Inorg. Chem., 2000, 39, 5958–5963 CrossRef CAS.
- M. A. Heinrichs, D. J. Hodgson, K. Michelsen and E. Pedersen, Inorg. Chem., 1984, 23, 3174–3180 CrossRef CAS.
- J. J. McGarvey, I. Lawthers, K. Heremans and H. Toftlund, Inorg. Chem., 1990, 29, 252–256 CrossRef CAS.
- J. K. Beattie, Adv. Inorg. Chem., 1988, 32, 1–53 CrossRef CAS.
- S. Schenker, P. C. Stein, J. A. Wolny, C. Brady, J. J. McGarvey, H. Toftlund and A. Hauser, Inorg. Chem., 2001, 40, 134–139 CrossRef CAS.
- W. Jacob and R. Mukherjee, Inorg. Chim. Acta, 2006, 359, 4565–4573 CrossRef CAS PubMed.
- L. Yang, R.-N. Wei, R. Li, X.-G. Zhou and J.-L. Zou, J. Mol. Catal. A: Chem., 2007, 266, 284–289 CrossRef CAS PubMed.
- E. A. Turitsyna and A. A. Shteinman, Russ. Chem. Bull., 2011, 60, 2094–2099 CrossRef CAS PubMed.
- J. J. Kodanko and S. J. Lippard, Inorg. Chim. Acta, 2008, 361, 894–900 CrossRef CAS PubMed.
- M. W. Mulqi, F. S. Stephens and R. S. Vagg, Inorg. Chim. Acta, 1982, 63, 197–207 CrossRef CAS.
- J. G. H. du Preez and B. J. A. M. van Brecht, Inorg. Chim. Acta, 1989, 162, 49–56 CrossRef CAS.
- P. Kapoor, A. P. S. Pannu, M. Sharma, M. S. Hundal and R. Kapoor, J. Coord. Chem., 2010, 63, 3635–3647 CrossRef CAS.
- P. Kapoor, A. P. S. Pannu, M. Sharma, G. Hundal, R. Kapoor and M. S. Hundal, J. Coord. Chem., 2011, 64, 256–271 CrossRef CAS.
- G. J. P. Britovsek, J. England and A. J. P. White, Dalton Trans., 2006, 1399–1408 RSC.
- K. Chen and L. Que Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed.
- G. J. P. Britovsek, J. England, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Dalton Trans., 2005, 945–955 RSC.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed.
- K. Chen and L. Que Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed.
- Y. Meckmouche, S. Menage, C. Toia-Duboc, M. Fontecave, J. B. Galey, C. Lebrun and J. Pecaut, Angew. Chem., Int. Ed., 2001, 40, 949 CrossRef.
- K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que Jr., J. Am. Chem. Soc., 2002, 124, 3026–3035 CrossRef CAS PubMed.
- D. M. Kurtz Jr., Chem. Rev., 1990, 90, 585–606 CrossRef.
- S. Poussereau, G. Blondin, M. Cesario, J. Guilhem, G. Chottard, F. Gonnet and J.-J. Girerd, Inorg. Chem., 1998, 37, 3127–3132 CrossRef CAS.
- S. Taktak, S. V. Kryatov and E. V. Rybak-Akimova, Inorg. Chem., 2004, 43, 7196–7209 CrossRef CAS PubMed.
- R. Hazell, K. B. Jensen, C. J. McKenzie and H. Toftlund, J. Chem. Soc., Dalton Trans., 1995, 707 RSC.
- L. Gómez, M. Canta, D. Font, I. Prat, X. Ribas and M. Costas, J. Org. Chem., 2013, 78, 1421–1433 CrossRef PubMed.
- P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef CAS PubMed.
- R. P. Halliday, W. J. Kinnard and J. P. Buckley, J. Pharm. Sci., 1964, 53, 19–23 CrossRef CAS.
- D. J. Barnes, R. L. Chapman, R. S. Vagg and E. C. Watton, J. Chem. Eng. Data, 1978, 23, 349–350 CrossRef CAS.
- D. W. Gruenwedel, Inorg. Chem., 1968, 7, 495–501 CrossRef CAS.
- K. Hiratani and K. Taguchi, Bull. Chem. Soc. Jpn., 1990, 63, 3331–3333 CrossRef CAS.
- (a) SHELXTL, Bruker AXS, Madison, WI Search PubMed; (b) SHELX-97, G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format and experimental details regarding the synthesis and characterisation of the ligands and metal complexes, including spectroscopic details. CCDC 995554–995556. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02067g |
| This journal is © The Royal Society of Chemistry 2014 |