
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Down- and up-converting dual-mode YPO4:Yb3+,Tb3+ nanocrystals: synthesis and spectroscopic properties†
Tomasz
Grzyb
*a,
Rafal J.
Wiglusz
b,
Aleksandra
Gruszeczka
a and
Stefan
Lis
a
aAdam Mickiewicz University, Faculty of Chemistry, Department of Rare Earths, Umultowska 89b, 61-614 Poznan, Poland. E-mail: tgrzyb@amu.edu.pl
bInstitute of Low Temperature and Structure Research, Polish Academy of Sciences, P.O. Box 1410, 50-950 Wroclaw, Poland
First published on 15th September 2014
Abstract
Tetragonal YPO4 nanocrystals doped with Yb3+ and Tb3+ ions were synthesized by 900 °C annealing of precursors obtained with a co-precipitation method in the presence of glycerine. These materials exhibited intense green luminescence under ultraviolet excitation and up-conversion emission from the 5D3 and 5D4 Tb3+ excited states after irradiation with near infrared light (λ = 980 nm). The structure and morphology of the products were analysed by recording X-ray diffraction patterns and transmission electron microscopy images. The obtained nanomaterials were single-phased with spherical shaped nanocrystals that had an average size of 18 ± 3 nm. The spectroscopic properties of YPO4:Yb3+,Tb3+ nanocrystals were investigated based on their excitation and emission spectra. The time-resolved luminescence traces were measured, and the luminescence lifetimes of Tb3+ and Yb3+ ions were calculated. The most effective dopant concentrations were determined to be 5% Yb3+ and 15% Tb3+, which exhibited the most intense ultraviolet excited emission and up-conversion. Because the integral intensity was observed to be dependent on the power of the pumping laser, a cooperative energy transfer (CET) mechanism underlying the observed up-conversion was proposed.
Introduction
In recent decades, efforts to synthesize new luminescent nanomaterials activated by lanthanide (Ln3+) ions have been the subject of intense research.1–5 Luminescent Ln3+-doped materials are required for next generation plasma display panels (PDPs), mercury-free fluorescence lamps, fiber amplifiers, forensic sciences and particularly light emitting diodes (LEDs).6–9 However, materials that exhibit up-conversion (UC) properties have attracted special attention due to their potential applications in solving current challenges in cancer diagnosis and therapy as well as in the utilization of solar energy. Therefore, there is growing interest in developing up-conversion nanocrystals for various applications, such as bioimaging, photodynamic therapy and drug delivery.10–14 UC in solar cells has been developed to improve spectral absorption properties of the materials and their efficiencies.15,16Rare earth (RE3+) orthophosphates have been extensively investigated because they are promising hosts for emitting Ln3+ ions.17,18 Their very low solubility in water, high thermal stability and high quantum efficiencies for REPO4-based phosphors are advantageous for the above mentioned applications.19 Up-conversion orthophosphates in the form of nanocrystals have been studied since 2003.20
Lanthanide phosphates occur in two crystal structure forms including xenotime with a tetragonal structure and monazite with a monoclinic structure.21 The xenotime structure is preferred by the heavy lanthanide ions from Tb3+ to Lu3+ with REO8 polyhedron structures. However, the monazite structure is typically observed for the light lanthanides (from La3+ to Gd3+ ions) and larger REO9 polyhedra.21
Up-conversion is a process in which two or more photons from the infrared or near infrared (NIR) range can be converted to photons with a higher energy than the absorbed energy (i.e. typically from the visible or ultraviolet spectral range). The idea of sequential absorption was proposed by Bloembergen in 1959.22 However, the energy transfer process, which is important for Ln3+-doped materials, leading to up-conversion emission was reported by Auzel in 1966.23,24 Since that time, numerous studies have been performed.1,2,25–29 In addition, the mechanisms of up-conversion have been studied, and the main mechanisms responsible for the anti-Stokes emission in Ln3+-doped systems (i.e. ground/excited state absorption (GSA and ESA), energy transfer up-conversion (ETU), photon avalanche (PA) and cooperative energy transfer (CET)) are known.25 However, improvement in the efficiency of UC materials is still needed. In addition, the mechanisms of up-conversion in nanocrystalline systems consisting of Yb3+–Tb3+ ions are still not well understood.
The up-conversion Yb3+/Tb3+ pair of ions has been studied in many micro-materials, such as oxides, fluorides, glasses and polymers.30–41 In addition, the luminescence of the Yb3+/Tb3+ co-doped phosphates has been widely studied in relation to the cooperative energy transfer from two Tb3+ ions to one Yb3+ ion as a quantum cutting system by down-conversion.42–44 However, investigations of down- and up-conversion in co-doped phosphates, especially in the nanocrystalline form, have been limited.45–49 The Tb3+ ion exhibits high quantum efficiencies for luminescence due to the large energy gap between the 7FJ ground state and the 5D4 excited state, which results in the lack of multiphonon relaxation of the 5D4 excited state and the high usage of this ion as a dopant in efficient phosphors. In addition, Yb3+ ions have many advantages as dopants resulting in their wide application as effective sensitizers for NIR radiation. One of the most important advantages is the high oscillator strength of the Yb3+ 2F7/2→2F5/2 transition responsible for the absorption of this ion at approximately 980 nm.
The main goal of the current paper is the evaluation of the physico-chemical properties of YPO4 nanoparticles activated with Tb3+ and Yb3+ ions, with special emphasis on the luminescence properties of the obtained system. The role of the reaction conditions and the influence of the dopant concentration on the final form of the YPO4 particles have been elucidated. The emission properties of the Yb3+ and Tb3+ co-doped system were fully characterized.
Experimental
Materials
All of the reagents were of analytical quality or spectral purity for rare earth oxides. To synthesize the materials, the following reagents were used: Y2O3, Yb2O3 and Tb4O7 were obtained from Stanford Materials (United States, 99.999%), nitric acid (HNO3) was obtained from POCh S.A. (Poland, ultra-pure), ammonium phosphate monobasic ((NH4)H2PO4) was obtained from Sigma-Aldrich (Poland, ReagentPlus®, ≥98.5%) and glycerine was obtained from POCh S.A. (Poland, pure p.a., 99.9%). Rare earth oxides were dissolved in HNO3 to obtain their nitrates. 1 M solutions were used for further synthesis.Synthesis of YPO4:Yb3+,Tb3+ nanocrystals
Yttrium phosphate nanocrystals doped with Yb3+ and Tb3+ ions were prepared using a two-step method. First, the co-precipitation method was used to synthesize phosphates, which were thermally treated at 900 °C. The procedure for obtaining nanocrystals in the first stage is described below. 4 mmol of precipitate was formed in the reaction that was carried out in a beaker containing a 25% solution of glycerine in distilled water. To a beaker containing 100 mL of a glycerine solution, 125% of the stoichiometric amount of NH4H2PO4 was added. Next, the solution was magnetically stirred and heated to 50 °C. Simultaneously, 50 mL of the mixed solution containing Y(NO3)3, Yb(NO3)3 and Tb(NO3)3 was prepared by dissolving calculated volumes of rare earth nitrates in water at a concentration of 1 M, in a 25% solution of glycerine. The following concentrations of dopants ions were chosen: (i) the static molar concentration of Yb3+ was 10% and 5–30% of Tb3+ ions and (ii) the static molar concentration of Tb3+ ions was 15% and varied for Yb3+ ions in the range of 2.5–20% mol. After reaching the required temperature, the solution of RE3+ nitrates was poured into a dropping funnel and slowly added to the NH4H2PO4 solution under magnetic stirring. The reaction was maintained for 30 min at approximately 50 °C. The white precipitate was centrifuged and washed several times with water and ethanol. The as-prepared powders were dried for 24 h at 80 °C in the air and annealed at 900 °C for 2 hours.Characteristics of the YPO4:Yb3+,Tb3+ nanocrystals
Powder diffractograms were recorded on a Bruker AXS D8 Advance diffractometer in Bragg–Brentano geometry with Cu Kα1 radiation in the 2θ range of 6° to 60°. The reference data were obtained from the Inorganic Crystal Structure Database (ICSD). In addition, X-ray diffractograms (XRD) were used in crystallographic data refinement along with the Rietveld method.50,51 Transmission electron microscopy (TEM) images were recorded on a FEI Tecnai G2 20 X-TWIN transmission electron microscope with an accelerating voltage of 200 kV. High-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) images were recorded on a Philips CM-20 Super Twin electron microscope with an accelerating voltage of 200 kV.A Hitachi F-7000 fluorescence spectrophotometer with a 150 W xenon lamp was used for determination of the luminescence properties (excitation and emission) of the samples at room temperature. The excitation and emission spectra were corrected for the instrumental response. For the up-conversion measurements, the solid state 980 nm laser (Dragon Lasers) was used as the excitation source. Emission lifetimes were measured at 545 nm (under excitation at the 978 nm line of a Ti:sapphire laser, LOTIS TII Belarus) using a Jobin-Yvon THR 1000 spectrophotometer (1200 mm−1 holographic grating), a Hamamatsu R928 photomultiplier as a detector and a digital oscilloscope LeCroy WaveSurfer 400 for data collection and on a QuantaMasterTM 40 spectrophotometer equipped with an Opolette 355LD UVDM tunable laser, which had a repetition rate of 20 Hz, as the excitation source and a Hamamatsu R928 photomultiplier as a detector. The decay curve fitting was performed using a single and double decay model with the Origin 9.0 software. The goodness of fit of the time traces was not less than R2 = 0.998. Error bars presented in Fig. 10 and 11 were estimated by three times calculation of lifetime values from the experimental data and further statistical analysis.
Results and discussion
The methods for the synthesis of rare earth orthophosphates have been studied for many years due to interest in their structural properties.52–54 Further studies have been performed to synthesize efficient REPO4-based phosphors and investigate properties of Ln3+ luminescent ions incorporated into the REPO4 structure as host materials.17,18,42,55,56 Recently, many studies of REPO4 synthesized nanomaterials have been reported.45,47,57–59 Most of the developed methods of synthesis of REPO4 powders or nanomaterials are based on the precipitation reaction between the RE3+ and PO43− ions.The co-precipitation method is one of the most widely used methods for nanomaterial synthesis.60–62 This method is based on a chemical exchange reaction resulting in the precipitation of an insoluble inorganic compound. The major advantages of this method are that it is easy and inexpensive. However, some requirements must be met to synthesize nanocrystals with low aggregation, small particle sizes and narrow distributions. In addition, the reaction environment, which typically includes water, can result in a large amount of water being adsorbed on the nanocrystal surface. Therefore, in our reactions, glycerine has been used to inhibit excessive growth of nanoparticles and lower their aggregation.3
Structure and morphology
After the formation of YPO4:Yb3+,Tb3+, the XRD measurements were performed. All of the Bragg reflections in the diffraction patterns were indexed and assigned to the tetragonal crystal system I41/amd space group no. 141 of the YPO4 crystalline phases (see Fig. 1). No additional secondary phase, impurities or amorphous forms were observed, which confirmed the structural purity of the obtained compounds. This result confirmed that high lanthanide doping levels are achievable in the YPO4 host material without altering the structure of the host under the given synthesis conditions.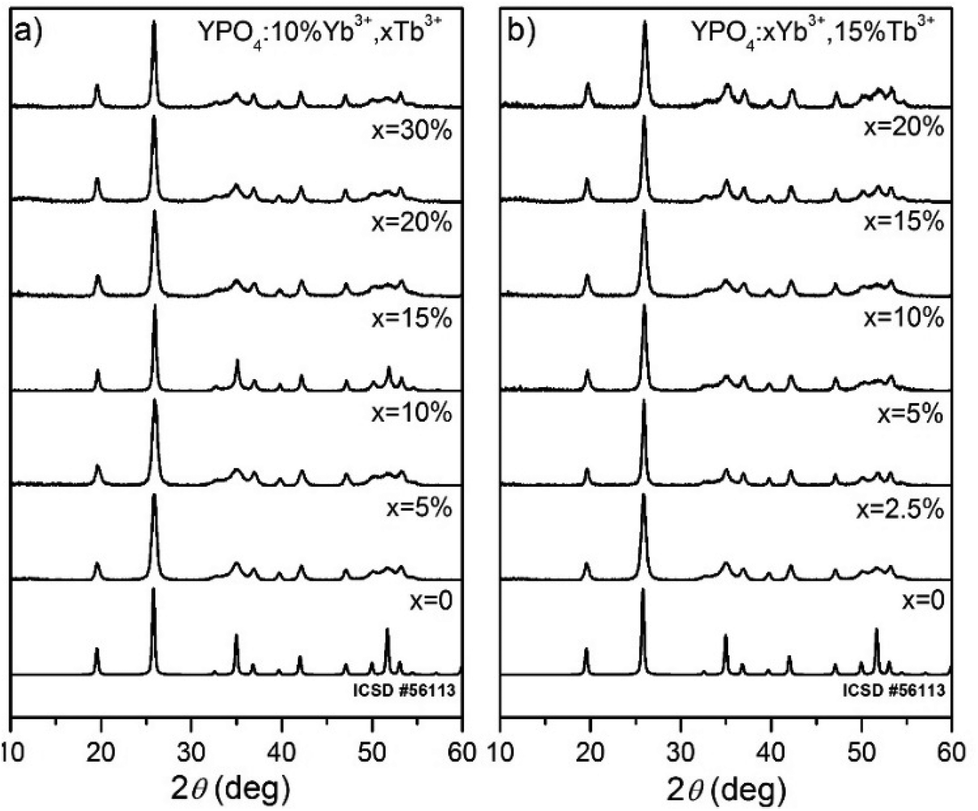 | ||
| Fig. 1 XRD diffraction patterns of (a) 10% Yb3+, x% Tb3+ doped YPO4 and (b) x% Yb3+, 15% Tb3+ doped YPO4 nanocrystals annealed at 900 °C for 2 h. | ||
To determine the structural changes and calculate the cell parameters of the synthesized YPO4 co-doped with Yb3+ and Tb3+ ions, Rietveld analysis has been used.51 The structural refinement method has several advantages over conventional quantitative analysis methods. This method uses a whole pattern-fitting algorithm, and all of the lines are explicitly considered. Therefore, even severely overlapped lines are typically not a problem. The Rietveld refinement was performed based on YPO4 crystals with a tetragonal structure using the crystallographic information file (CIF) no. 56113. Fig. 2 shows a comparison of the calculated cell volumes with the reference data. The crystallographic data and structural refinement indicated some changes in the structural parameters with the dopant concentrations.
 | ||
| Fig. 2 Dependence of the crystal cell volumes of the YPO4:Yb3+,Tb3+ nanocrystals on the dopant ion concentration. | ||
The calculated cell volumes increased as the concentration of Tb3+ ions increased. For the Yb3+ doping, there was a decrease in the cell volumes as the concentration of this ion increased.
The significant difference between the unit cell parameters of the YPO4 single crystal and those of the YPO4 nanoparticles without dopant ions, which possess larger unit cell parameter values, is caused by the so-called grain size effect.63,64
In this phenomenon, a reduction in the particle size contributes to the creation of negative pressure on the crystal lattice, which causes expansion of the lattice cell volume. In the YPO4 nanocrystals that are co-doped with Yb3+ and Tb3+, two effects play a role in causing differences in the cell volumes between the reference and synthesized materials. The first effect is due to the above mentioned grain size effect. The second effect is related to the differences in the ionic radii of the Y3+, Yb3+ and Tb3+ ions (for the coordination number, CN = 8, r(Y3+) = 1.019 Å, r(Tb3+) = 1.040 Å, r(Yb3+) = 0.985 Å). Substitution of the Y3+ ions in the crystallographic structure by the smaller Yb3+ and bigger Tb3+ ions could result in contraction or expansion of the dimensions of the crystal cell.
The TEM images (see Fig. 3a) of the as-prepared sample indicate that after the co-precipitation reaction, nanofibers with a thickness of less than 5 nm are formed. Their length is difficult to estimate due to the web that is formed. The sample sintered at 900 °C for 2 h, which is shown in Fig. 3d–f, has considerably different morphology with nanocrystals that are rounded with an average size of 18 ± 3 nm. These crystal sizes are typical for powders prepared by the co-precipitation method and annealed at a higher temperature. High temperate also causes aggregation of crystallites.
 | ||
| Fig. 3 TEM and SAED images of the as-prepared (a–c) and annealed (d–f) YPO4:5%Yb3+,15%Tb3+ nanocrystals (annealed at 900 °C for 2 hours). | ||
Spectroscopic properties
The luminescence of the YPO4:Yb3+,Tb3+ nanomaterials exhibits a dual nature. These nanomaterials can be excited by ultraviolet (UV) radiation due to the f–d and f–f electronic transitions within the Tb3+ ions or by NIR radiation due to Yb3+ absorption and energy transfer between dopants ions. Both excitation methods yield efficient green luminescence of the Tb3+ ions. The presence of Yb3+ and Tb3+ dopant ions has a strong influence on the observed luminescence properties because these ions are responsible for dual mode excitation and they can exchange energy, transfer charge and undergo other processes due to their complex spectroscopic nature and interactions between the ions. The luminescence excitation spectra were measured in the range of 200–400 nm and are shown in Fig. 4. These spectra are characterized by broad excitation peaks located at higher energies and a series of narrower peaks at lower energies. The broad excitation bands at shorter wavelengths (with maxima at approximately 225 nm in each sample) are associated with the 4f8→4f75d1 transition in the Tb3+ ions, and the peaks located above 250 nm originated from the f–f electronic transitions characteristic of Tb3+ containing materials. By applying excitation with sufficiently energetic photons, electrons from the 4f electronic levels of Tb3+ ions can be promoted to the excited 5d shell. This results in excited states with two configurations as follows: a high-spin 9DJ state and a 7DJ low-spin state.65 According to Hund's rule, the 9DJ states have a lower energy than the 7DJ states. However, the 7FJ→7DJ transitions are spin-allowed and connected with the excitation band of the transition, which has a maximum at approximately 235 nm.66,67 Normally, the spin-forbidden 7FJ→9DJ transitions can be observed in the range of 250–280 nm, and all of the studied samples exhibited very weak intensities.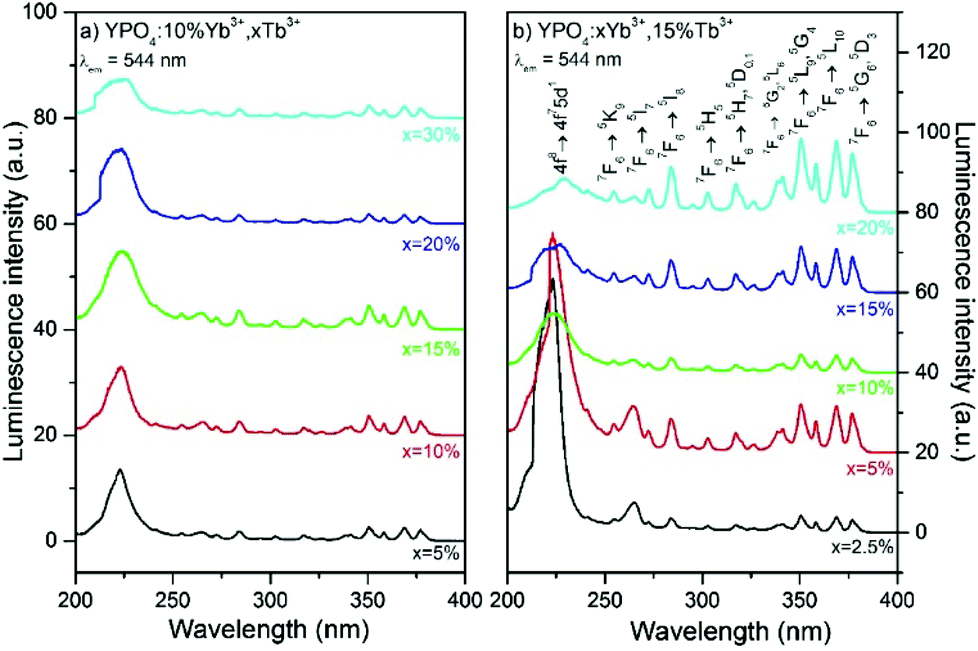 | ||
| Fig. 4 Excitation spectra of (a) YPO4 doped by 10% Yb3+, x% Tb3+ and (b) YPO4 doped by x% Yb3+, 15% Tb3+. | ||
The intensity of the 4f8→4f75d1 transition band is sensitive to the concentration of the Yb3+ ions, as shown in Fig. 4b.
An increase in the amount of Yb3+ ions lowers the probability of the excitation of Tb3+ into the 4f75d1 state and reduces the intensity of the excitation band associated with this process. A combination of ions, such as Tb3+, has a tendency to be oxidized by the Yb3+ ion, which can be easily reduced to generate charge transfer states (CTSs). Indeed, in the Yb3+ and Tb3+ co-doped materials, the Tb3+→Yb3+ charge transfer has been observed.33,68 After absorption of UV photons, energy from the Tb3+ ions that was excited into the 4f75d1 state is transferred to the Yb3+ ions via emission at NIR wavelengths.
This process requires the existence of an intermediate Tb4+–Yb2+ CTS.68 The Tb4+–Yb2+ CTS has an energy higher than the 4f75d1 state of the Tb3+ ions, but thermally induced crossing between states is possible. In addition, the Tb3+→Yb3+ energy transfer to the CTS is 10–102 times faster than the relaxation of Tb3+ from the 4f75d1 state to the 5D4 excited state. Therefore, this charge transfer based mechanism explains the observed changes in the excitation spectra and quenching of the 4f8→4f75d1 excitation band.
Fig. 5 and 6 show the luminescence spectra which were measured using UV (366 nm) or NIR (980 nm) excitation, of the Yb3+ and Tb3+ co-doped YPO4 nanocrystals. The spectra in Fig. 5 exhibit characteristic luminescence bands associated with the f–f electronic transitions of Tb3+ ions. These transitions are due to radiative relaxation from the Tb3+ 5D4 energy level to the 7FJ components of the ground state. Emission from higher levels (i.e., 5G6 and 5D3) was not observed in the samples due to cross-relaxation processes (CR) associated with the Tb3+ ions.69,70 This process depopulated the 5D3 state of the Tb3+ ion and transferred the energy difference between the 5D3 and 5D4 states to the neighbouring Tb3+ ion. This energy difference match the energy gap between 7F6 and 7F0 energy levels of the Tb3+ ion.70,71 The emission peaks at 489, 544, 588 and 622 nm can be assigned to the 5D4→7F6,5,4,3 transitions, respectively, and are responsible for the green luminescence under UV irradiation of the prepared materials.
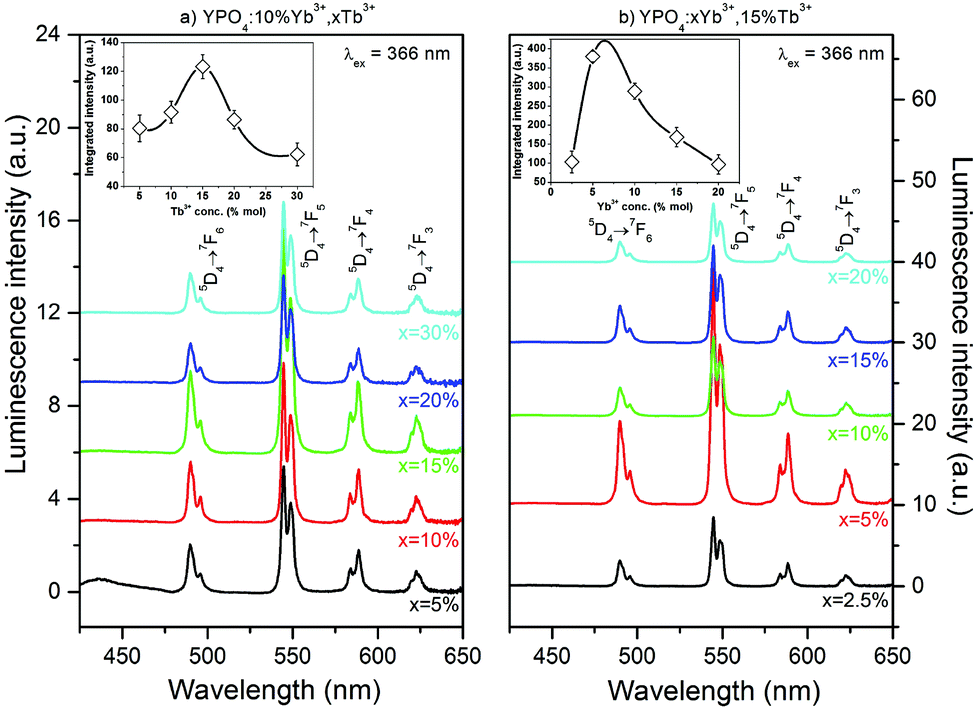 | ||
| Fig. 5 Emission spectra of (a) YPO4 doped with 10% Yb3+, x% Tb3+ and (b) YPO4 doped with x% Yb3+, 15% Tb3+; dependence on Tb3+ (a) and Yb3+ (b) concentrations and dependence of integral luminescence intensity on the Tb3+ (a) and Yb3+ (b) concentrations (shown in insets). | ||
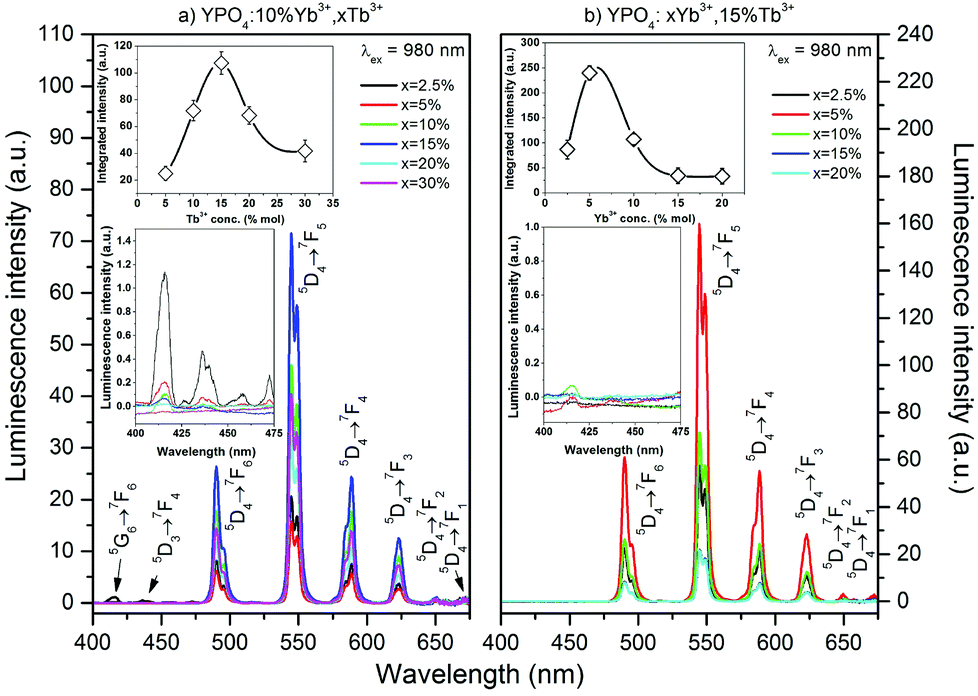 | ||
| Fig. 6 Dependence of the up-conversion luminescence spectra of YPO4:Yb3+,Tb3+ nanocrystals on the concentrations of Tb3+ (a) and Yb3+ (b); insets: dependence of integral intensity on the concentrations of Tb3+ (a) and Yb3+ (b) and emission in the 400–475 nm range. | ||
The insets shown in Fig. 5 show the integral luminescence intensity of the Tb3+ ions as a function of the concentration of (a) Tb3+ or (b) Yb3+ ions. The sample that exhibited the most intense luminescence was doped with 5% of Yb3+ and 15% of Tb3+ ions. A concentration of Tb3+ ions higher than 15% caused quenching of the green luminescence. In addition, a concentration of Yb3+ ions higher than 5% decreased the Tb3+ luminescence intensity. In materials doped with Ln3+ ions, non-radiative processes, such as multiphonon decay, quenching by impurities or energy migration among active ions, are typically responsible for luminescence quenching. For Tb3+ ions, multiphonon relaxation can be neglected due to the high energy gap between the 5D3 and 5D4 or 5D4 and 7FJ levels, which require at least 5 or 15 phonons, respectively, to depopulate these excited states (YPO4 lattice phonon energy is approximately 1080 cm−1).72 The observed emission quenching as the concentration of Tb3+ ions increased may be due to the energy transfer between two Tb3+ ions in the 5D4 excited state. As the result of this process, one ion is excited into upper lying levels. However, the second ion becomes depopulated. This process can also be explained by the slight increase in the luminescence intensity of the measured emission decays (see ESI†).73
In fact, the observed increase in the luminescence intensity as the amount of Yb3+ ions increased in the sample is important. According to the literature, the Yb3+ ions strongly quench the emission of the Tb3+ ions due to cooperative energy transfer from the Tb3+ ion in its 5D4 excited state to two Yb3+ ions.49,68 This effect can be observed in the luminescence intensity when the Yb3+ concentration was higher than 10% or as the shortening of Tb3+ luminescence lifetimes in the whole range of Yb3+ concentrations (see Fig. 9). Similar results for the changes in the luminescence intensity as the amount Yb3+ ions increased have also been previously reported.68,74 However, the cause of this unexpected phenomenon was not explained and remains unclear. The mechanism of this anomaly may be connected to the expansion of the crystal cell volume after co-doping with Yb3+ ions. Cooperative energy transfer is effective only over very short distances between interacting ions.75 Therefore, an increase in the cell volume decreases the efficiency of CET between Tb3+ and Yb3+ ions. If other processes, such as phonon assisted energy transfer (PET), quench the Tb3+ ions luminescence, another explanation of the observed initial increase in Tb3+ luminescence intensity is a backward energy transfer from neighbouring Yb3+ ions that also pump the 5D4 excited state of the Tb3+ ion.
The up-conversion emission spectra of the co-doped YPO4 thermally treated at 900 °C were observed after excitation at 980 nm, and the results are shown in Fig. 6. The spectra are similar to those obtained by UV excitation. The maximum green emission was recorded for the sample doped with 15% Tb3+ and 5% Yb3+ ions. The insets (see Fig. 6) show the integral luminescence intensity as a function of the concentration of Tb3+ or Yb3+ ions. The estimated optimal concentrations of Yb3+ and Tb3+ ions demonstrate that a relatively large portion of the Y3+ ions in the host material must be replaced, especially by the acceptor (i.e., Tb3+ ions). This fact is a direct result of the relatively low probability of CET and small distances between interacting ions, which are effective for this process.75 In addition to the emission from the 5D4 excited state, small peaks associated with the transitions from the 5G6 and 5D3 states were observed and are shown in the insets in Fig. 6.
The UC emission in the Yb3+/Tb3+ system is rare, and there are only a few reports regarding up-conversion from excited states higher than 5D4.30,38,75–78 The only way to produce Tb3+ ions in the 5D3 or 5G6 excited state is via a three photon process. The Tb3+ ion in the populated 5D4 level can be further excited up to the 5D1 level by one photon through the ESA or by energy transfer from a neighbouring Yb3+ ion. In addition, cooperative energy transfer from three Yb3+ ions is possible.78 The power dependence of the Tb3+ emission (see Fig. 7) confirms these assumptions. In our previous studies of Yb3+/Tb3+ co-doped LaPO4 and GdPO4 hosts, we did not detect emission from the 5D3 state of Tb3+ ions, which may be due to the different crystal system in YPO4.48,49
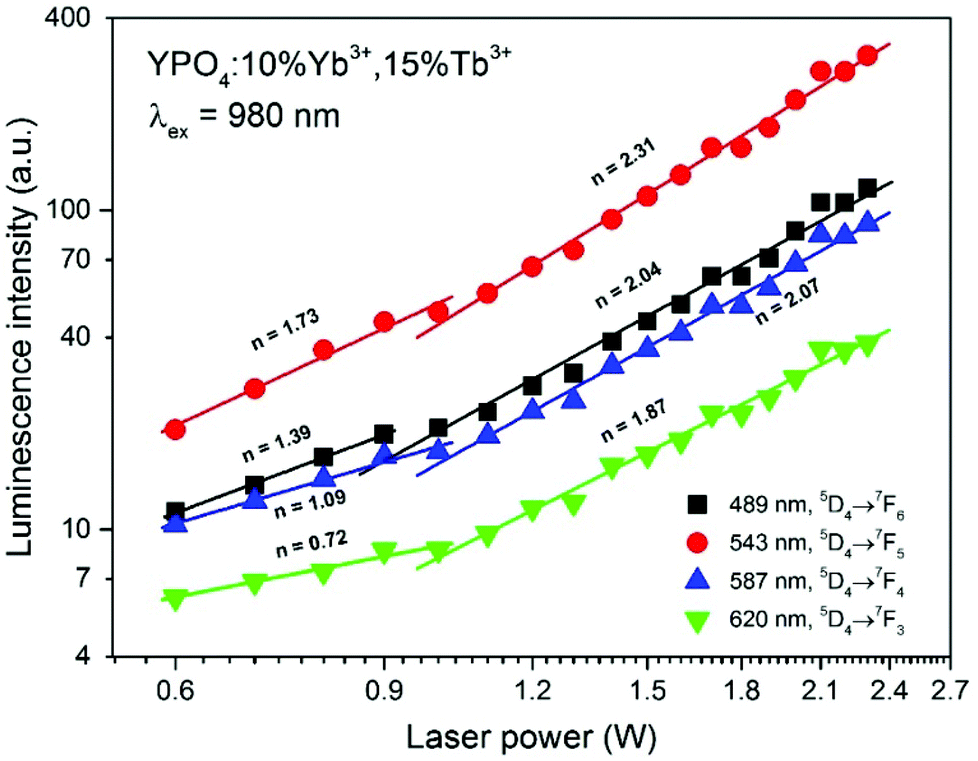 | ||
| Fig. 7 Integral emission intensity of the YPO4:10%Yb3+,15%Tb3+ sample as a function of the pumping power of the NIR laser (980 nm). | ||
The dependence of the integral luminescence intensity on the pumping laser power at 980 nm can provide insight into the number of photons involved in the excitation process. The relationship between the UC intensity IUC and the pumping laser intensity IP is given by the following equation:
| IUC = α(IP)n | (1) |
The power dependence of the luminescence intensity shown in Fig. 7 is complex. The differences between the slopes calculated for each transition of the Tb3+ ions indicate that the up-conversion mechanism involves additional processes, which results in divergence between the obtained values. In addition, when the power of the pumping laser was more than 1 W, the energy transfer mechanism changed, and the number of photons involved in this process was higher. According to the conditions required to match the differences between the Tb3+ and Yb3+ energy levels, cooperative energy transfer has been previously proposed in many studies.30,31,35–37,45,79,80 The most likely process involves a simultaneous interaction of two excited Yb3+ ions with one Tb3+, which results in excitation of the Tb3+ ions into a 5D4 state. However, at low temperatures, GSA and ESA processes also lead to UC of Tb3+ ions after Yb3+ excitation.35,36 The proposed scheme of the energy transfer mechanism between Yb3+ and Tb3+ ions in the YPO4 matrix is presented in Fig. 8.
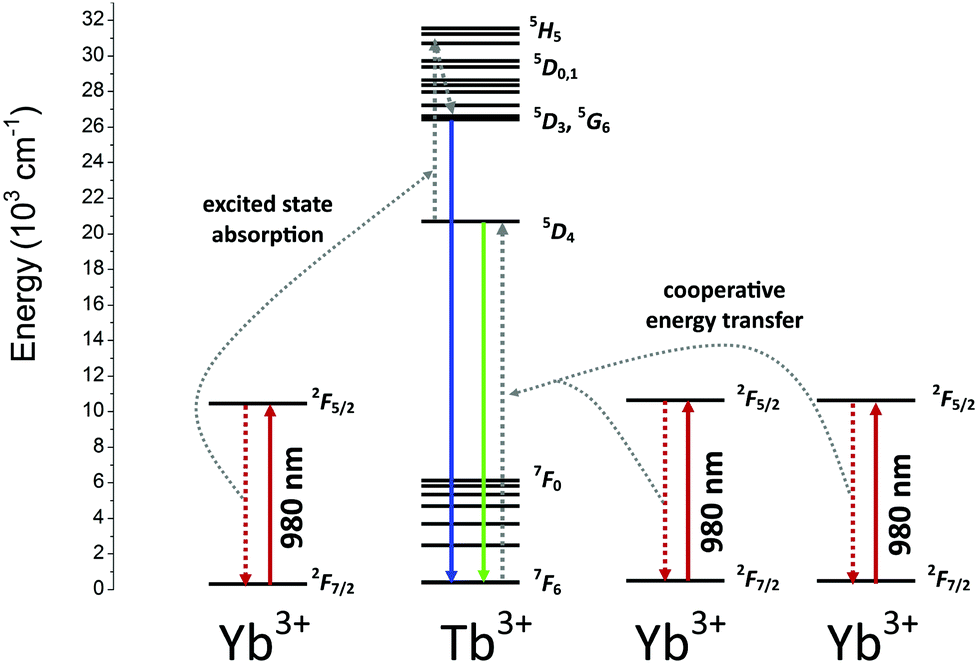 | ||
| Fig. 8 The proposed scheme of energetic processes occurring in the YPO4:Yb3+,Tb3+ samples. | ||
In general, the up-conversion in the Yb3+ and Tb3+ doped YPO4 samples can be treated as a two photon process. The observed deviations from the theoretical value can be explained as described below. The cross-relaxation (CR) that populates the 5D4 state from the 5D3 excited state of the Tb3+ ion is the most likely cause of more than 2 photons being involved in the observed UC. When the energies of the excitation source are less than 1 W, competitive processes occur and lead to quenching of the Tb3+ emission.
The energy transfer (ET) between the Yb3+ and Tb3+ ions is responsible for the relatively large increase in time observed in the luminescence decay of Tb3+ ions after excitation at 940 nm (see Fig. 9). A comparison of the luminescence decay of Yb3+ and Tb3+ after excitation at 940 nm indicated ET processes. During the time when emission from the Yb3+ ions disappeared, an increase in the luminescence of Tb3+ ions, which are the energy acceptors, was observed.
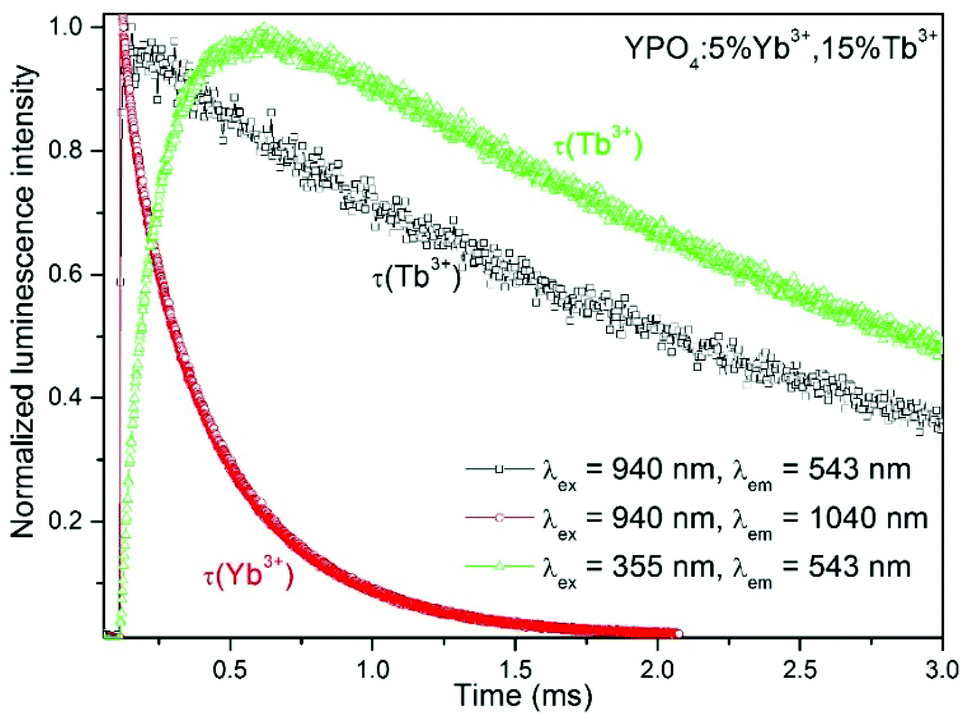 | ||
| Fig. 9 Luminescence decays of the YPO4:5%Yb3+,15%Tb3+ nanocrystals. | ||
The luminescence decays of the Tb3+ and Yb3+ ions in all of the samples measured using UV and NIR laser light (355 nm and 940 nm) are reported in the ESI.† The recorded decays were used to calculate luminescence lifetimes, which are compared in Fig. 10. The shapes of the measured Tb3+ and Yb3+ luminescence decay curves (λex = 355 nm or 940 nm) were non-exponential due to the quenching processes. However, the decrease in the lifetime may still be approximated using an exponential function. The single exponential model for the lifetime value calculations of the Tb3+ and Yb3+ ion (Fig. 10a, b, d and e) luminescence was as follows:
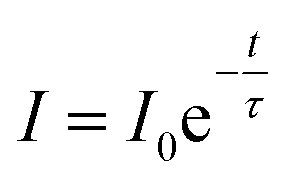 | (2) |
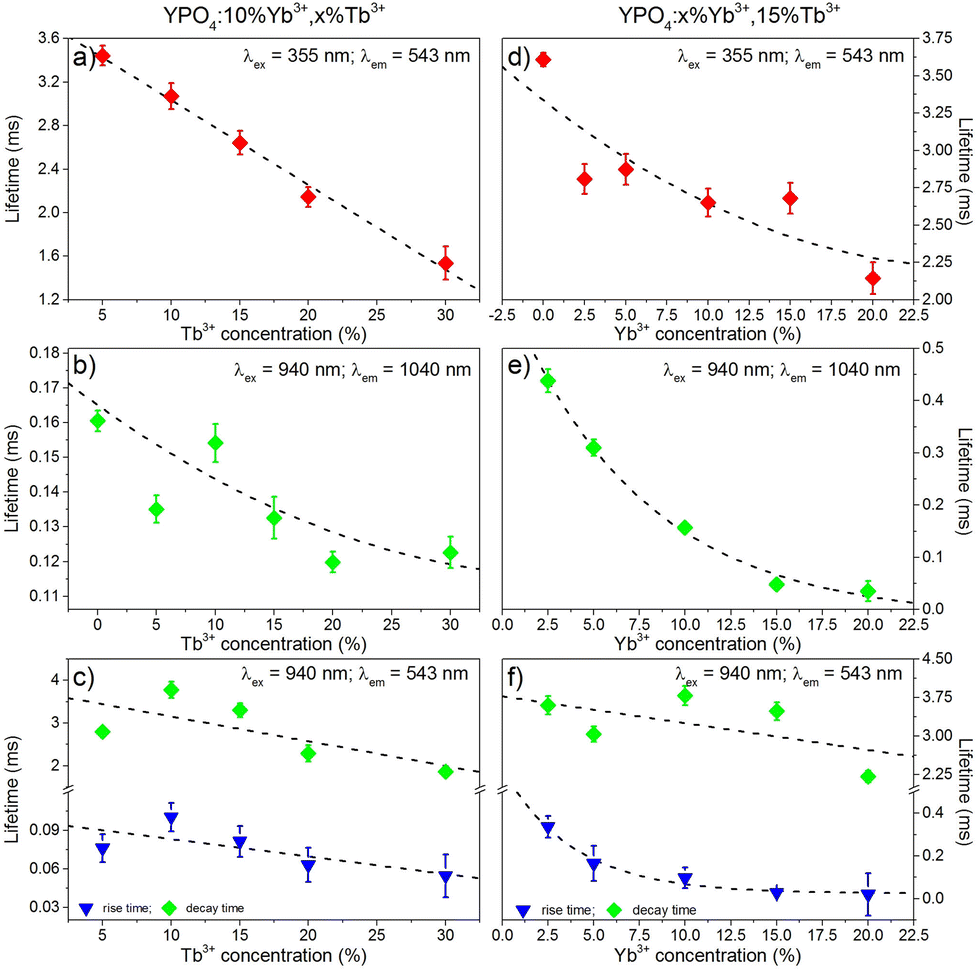 | ||
| Fig. 10 Luminescence lifetimes of the Tb3+ (a, c, d, f) and Yb3+ (b, e) ions in the YPO4 nanocrystals, which are as dependent on the concentration of Tb3+ (a, b, c) or Yb3+ (d, e, f). | ||
Luminescence decays of Tb3+ ions, which result from up-conversion (λex = 940 nm), exhibited rise times in all of the samples. Therefore, to calculate the luminescence lifetimes and rise times, the following equation was used:
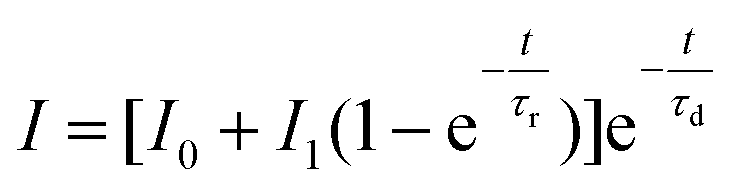 | (3) |
The decay times were dependent on the dopant concentration and became shorter with increasing amounts of Tb3+ or Yb3+ ions in the YPO4 host for each excitation wavelength used. In more highly doped samples, the distance between the donor and acceptor ions is shorter, and therefore, the energy transfer occurs faster. In addition, phenomena such as cross-relaxation and cooperative energy transfer become more effective. The calculated values of the luminescence lifetimes of the Tb3+ ions are typical for this ion for UV (355 nm) and NIR (940 nm) excitations and are in the range of 1.5–3.8 ms. The use of UV light for excitation caused much more effective quenching as the amounts of Tb3+ or Yb3+ ions increased compared to NIR radiation, and this result was especially noticeable in samples doped with 30% Tb3+ ions (Fig. 10a, c, d and f). The highly energetic UV photons generate larger numbers of quenching centres than those generated from NIR radiation. In addition, the lifetimes of Yb3+ were typical for this ion and are in the range of 0.12–0.48 ms. The effect of the increasing Tb3+ ion concentration on the luminescence lifetimes of the Yb3+ ions (Fig. 10b) is in agreement with the proposed energy transfer from Yb3+ to Tb3+ ions. The shortened lifetimes associated with an increased amount of Tb3+ ions confirm the effectiveness of this process. However, the relatively simple electronic structure of the Yb3+ ions is not conducive for concentration quenching, and this effect is visible in Fig. 10b where a shortening in the Yb3+ lifetimes occurs when the concentration of this ion was increased due to the presence of Yb2+ ions or by radiation trapping caused by the presence of resonant 2F7/2↔2F5/2 transitions between Yb3+ ions.81
The calculated values of the rise times of Tb3+ luminescence (Fig. 10c and f) are comparable to the lifetimes of Yb3+ ions when we take into account the proposed CET up-conversion mechanism. The longest rise times were calculated for samples doped with 10% Tb3+ ions in the series of samples shown in Fig. 10c or 2.5% Yb3+, which is shown in Fig. 10f.
The efficiency of the cooperative energy transfer between the Yb3+ and Tb3+ ions can be estimated based on the measured luminescence decays. The method of calculation has been previously described.31 In short, the following equation can be used to calculate the cooperative energy transfer efficiency (ηCET) for different Tb3+ ion concentrations:
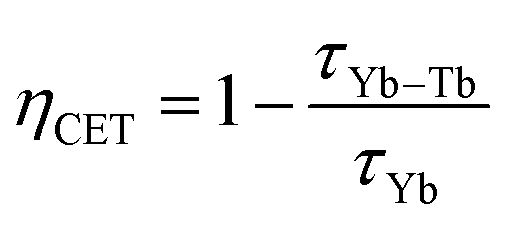 | (4) |
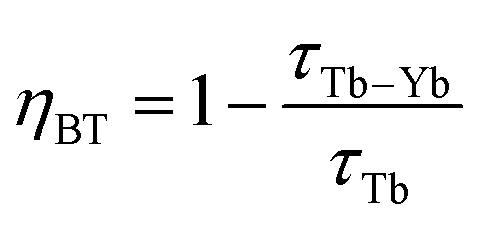 | (5) |
The calculated values of both parameters (i.e., ηCET and ηBT) are plotted in Fig. 11. The energy transfer efficiency increased as the amount of Tb3+ ions in the YPO4 host increased. Therefore the highest up-conversion was observed for the largest concentrations used. However, the backward energy transfers are also very effective in the studied system and become high when Yb3+ concentrations exceed 10%. Therefore, these materials can also be promising down-conversion systems.
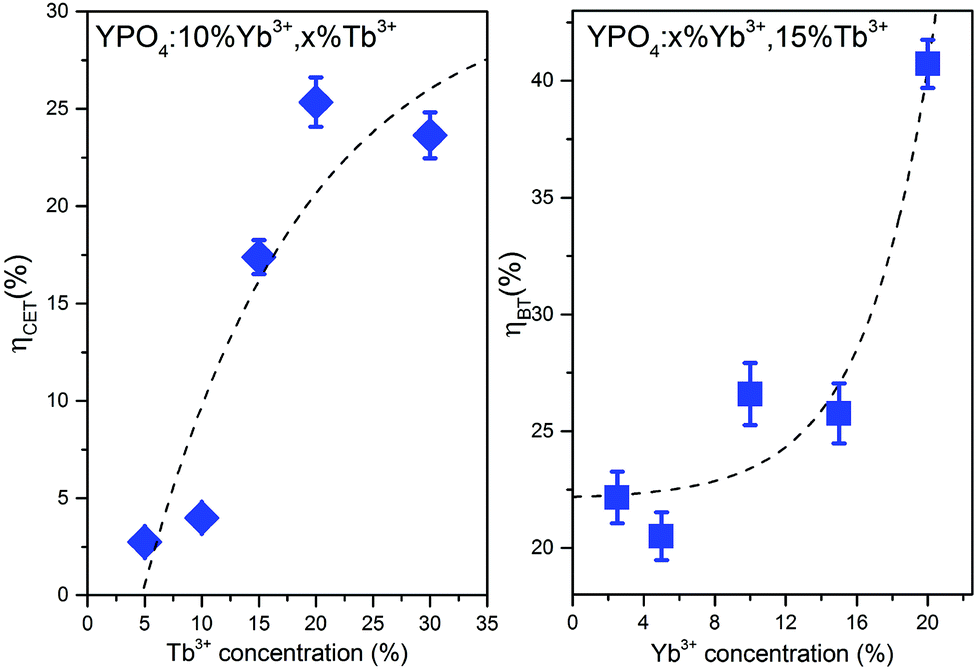 | ||
| Fig. 11 Cooperative energy transfer efficiency (ηCET) and backward energy transfer efficiency (ηBT) as functions of the Tb3+ or Yb3+ concentrations. | ||
Conclusions
Rare earth co-doped YPO4 nanocrystals can be synthesized using a co-precipitation method. To obtain efficient UV-excited and up-conversion luminescence, annealing of the as-prepared tetragonal nanocrystals at 900 °C is necessary. As a result of the thermal treatment, the obtained materials consisting of nanofibers were converted to spherical nanocrystals. The XRD studies confirmed that the obtained materials were a single phase with a tetragonal crystal structure in the I41/amd space group.The observed luminescence exhibited a dual mode nature. The prepared materials could be excited by UV or NIR radiation yielding intense green luminescence. The spectroscopic properties were analysed using both UV and NIR wavelengths as excitation sources. The excitation spectra indicated the presence of a charge transfer Tb4+→Yb2+ state in the samples. The best dopant concentrations that resulted in the highest luminescence upon UV light (365 nm) or NIR (980 nm) excitation were 5% Yb3+ ions and 15% Tb3+ ions. In addition to the typical Tb3+ ion emission from the 5D4 excited state, emission bands associated with the transitions from the 5G6 and 5D3 states were observed due to up-conversion. The effects of the presence of other quenching processes, such as cross-relaxation between Tb3+ ions, multiphonon quenching and backward energy transfer from Yb3+ to Tb3+ ions were determined. The dependence of the integral up-conversion luminescence intensity on the pumping laser power confirmed that at least two photons participated in the excitation of the Tb3+ ions. In addition, evidence of a three photon process was observed (i.e., increasing slope of the curves above 2). The recorded emission decays exhibited a relatively long rise in the luminescence intensity after the laser pulse, which confirmed that an energy transfer occurred between the Yb3+ and Tb3+ ions. The obtained results present a route for the synthesis of YPO4 nanocrystals doped with Yb3+ and Tb3+ ions showing efficient UV-excited luminescence, as well as up-conversion under NIR radiation.
Acknowledgements
Funding for this research was provided by the National Science Centre (grant no. DEC-2011/03/D/ST5/05701).References
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- D. Vennerberg and Z. Lin, Sci. Adv. Mater., 2011, 3, 26–40 CrossRef PubMed.
- T. Grzyb, M. Runowski, A. Szczeszak and S. Lis, J. Phys. Chem. C, 2012, 116, 17188–17196 CAS.
- T. Grzyb, A. Szczeszak, J. Rozowska, J. Legendziewicz and S. Lis, J. Phys. Chem. C, 2012, 116, 3219–3226 CAS.
- R. Pazik, R. Tekoriute, S. Håkansson, R. Wiglusz, W. Strek, G. a Seisenbaeva, Y. K. Gun'ko and V. G. Kessler, Chemistry, 2009, 15, 6820–6826 CrossRef CAS PubMed.
- M. a. M. Lucena, G. F. de Sá, M. O. Rodrigues, S. Alves, M. Talhavini and I. T. Weber, Anal. Methods, 2013, 5, 705 RSC.
- K. Pavani, J. S. Kumar, T. Sasikala, B. C. Jamalaiah, H. J. Seo and L. R. Moorthy, Mater. Chem. Phys., 2011, 129, 292–295 CrossRef CAS PubMed.
- C. C. Lin and R.-S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268–1277 CrossRef CAS.
- V. Bedekar, D. P. D. Dutta, M. Mohapatra, S. V. Godbole, R. Ghildiyal and A. K. Tyagi, Nanotechnology, 2009, 125707, 125707–125714 CrossRef PubMed.
- S. Wang, J. Feng, S. Song and H. Zhang, CrystEngComm, 2013, 15, 7142 RSC.
- Y. Il Park, H. M. Kim, J. H. Kim, K. C. Moon, B. Yoo, K. T. Lee, N. Lee, Y. Choi, W. Park, D. Ling, K. Na, W. K. Moon, S. H. Choi, H. S. Park, S.-Y. Yoon, Y. D. Suh, S. H. Lee and T. Hyeon, Adv. Mater., 2012, 24, 5755–5761 CrossRef PubMed.
- D. Tu, Y. Liu, H. Zhu and X. Chen, Chem. – Eur. J., 2013, 19, 5516–5527 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- L.-D. Sun, Y.-F. Wang and C.-H. Yan, Acc. Chem. Res., 2014, 47, 1001–1009 CrossRef CAS PubMed.
- W. G. van Sark, J. de Wild, J. K. Rath, A. Meijerink and R. E. Schropp, Nanoscale Res. Lett., 2013, 8, 81 CrossRef PubMed.
- B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081–11095 RSC.
- C. Brecher, J. Chem. Phys., 1968, 49, 3303 CrossRef CAS PubMed.
- S. Erdei, F. W. Ainger, D. Ravichandran, W. B. White and L. E. Cross, Mater. Lett., 1997, 30, 389–393 CrossRef CAS.
- C.-H. Yan, Z.-G. Yan, Y.-P. Du, J. Shen, C. Zhang and W. Feng, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneider, J.-C. G. Bünzli and V. K. Percharsky, Elsevier, 2011, vol. 41, pp. 275–472 Search PubMed.
- S. Heer, O. Lehmann, M. Haase and H.-U. H.-U. Güdel, Angew. Chem., Int. Ed., 2003, 42, 3179–3182 CrossRef CAS PubMed.
- A. N. M. Yunxiang Ni and John M. Hughes, Am. Mineral., 1995, 80, 21–26 Search PubMed.
- N. Bloembergen, Phys. Rev. Lett., 1959, 2, 84–85 CrossRef CAS.
- F. Auzel, C. R. Acad. Sci. Paris B, 1966, 263, 819–821 Search PubMed.
- F. Auzel, C. R. Acad. Sci. Paris B, 1966, 262, 1016–1019 Search PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- W. Feng, C. Han and F. Li, Adv. Mater., 2013, 1–17 Search PubMed.
- A. Gnach and A. Bednarkiewicz, Nano Today, 2012, 7, 532–563 CrossRef CAS PubMed.
- M. Lin, Y. Zhao, S. Wang, M. Liu, Z. Duan, Y. Chen, F. Li, F. Xu and T. Lu, Biotechnol. Adv., 2012, 30, 1551–1561 CrossRef CAS PubMed.
- R. Scheps, Prog. Quantum Electron., 1996, 20, 271–358 CrossRef CAS.
- L. de S. Menezes, G. S. Maciel, C. B. de Araújo and Y. Messaddeq, J. Appl. Phys., 2003, 94, 863 CrossRef CAS PubMed.
- Y. Arai, T. Yamashidta, T. Suzuki and Y. Ohishi, J. Appl. Phys., 2009, 105, 83105–83111 CrossRef PubMed.
- B. Lai, J. Wang and Q. Su, Appl. Phys. B, 2009, 98, 41–47 CrossRef PubMed.
- X. Liu, S. Ye, Y. Qiao, G. Dong, B. Zhu, D. Chen, G. Lakshminarayana and J. Qiu, Appl. Phys. B, 2009, 96, 51–55 CrossRef CAS.
- M. a. Noginov, P. Venkateswarlu and M. Mahdi, J. Opt. Soc. Am. B, 1996, 13, 735 CrossRef CAS.
- G. Salley, R. Valiente and H. Güdel, Phys. Rev. B: Condens. Matter, 2003, 67, 1–9 CrossRef.
- G. M. Salley, R. Valiente and H. U. Guedel, J. Lumin., 2001, 94–95, 305–309 CrossRef.
- V. Scarnera, B. Richards, A. Jha, G. Jose and C. Stacey, Opt. Mater., 2010, 33, 159–163 CrossRef CAS PubMed.
- C. H. Yang, Y. X. Pan, Q. Y. Zhang and Z. H. Jiang, J. Fluoresc., 2007, 17, 500–504 CrossRef CAS PubMed.
- Z. Yang, D. Yan, Z. Song, D. Zhou, X. Yu, Y. Yang, Z. Yin, L. Yan, R. Wang, H. Wu and J. Qiu, J. Lumin., 2012, 132, 1550–1552 CrossRef CAS PubMed.
- K. Zhu, Z. Yang, D. Yan, Z. Song, D. Zhou, R. Wang and J. Qiu, J. Sol-Gel Sci. Technol., 2012, 62, 149–152 CrossRef CAS PubMed.
- K. Prorok, A. Gnach, A. Bednarkiewicz and W. Stręk, J. Lumin., 2013, 140, 103–109 CrossRef CAS PubMed.
- P. Vergeer, T. Vlugt, M. Kox, M. den Hertog, J. van der Eerden and A. Meijerink, Phys. Rev. B: Condens. Matter, 2005, 71, 1–11 CrossRef.
- L. Li, X. Wei, Y. Chen, C. Guo and M. Yin, J. Rare Earths, 2012, 30, 197–201 CrossRef CAS.
- Y.-T. An, C. Labbé, J. Cardin, M. Morales and F. Gourbilleau, Adv. Opt. Mater., 2013, 1, 855–862 CrossRef.
- M. L. Debasu, D. Ananias, S. L. C. Pinho, C. F. G. C. Geraldes, L. D. Carlos and J. Rocha, Nanoscale, 2012, 4, 5154–5162 RSC.
- W. Ren, G. Tian, L. Zhou, W. Yin, L. Yan, S. Jin, Y. Zu, S. Li, Z. Gu and Y. Zhao, Nanoscale, 2012, 4, 3754–3760 RSC.
- M. L. Debasu, D. Ananias, J. Rocha, O. L. Malta and L. D. Carlos, Phys. Chem. Chem. Phys., 2013, 15, 15565–15571 RSC.
- T. Grzyb, A. Gruszeczka, R. J. Wiglusz, Z. Śniadecki, B. Idzikowski and S. Lis, J. Mater. Chem., 2012, 22, 22989–22997 RSC.
- T. Grzyb, A. Gruszeczka, R. J. Wiglusz and S. Lis, J. Mater. Chem. C, 2013, 1, 5410–5418 RSC.
- D. Bish and J. Post, Am. Mineral., 1993, 78, 932–940 CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- A. Hezel and S. D. Ross, J. Inorg. Nucl. Chem., 1967, 29, 2085–2089 CrossRef CAS.
- G. M. Begun, G. W. Beall, L. A. Boatner and W. J. Gregor, J. Raman Spectrosc., 1981, 11, 273–278 CrossRef CAS.
- D. F. Mullica, D. A. Grossie and L. A. Boatner, Inorg. Chim. Acta, 1985, 109, 105–110 CrossRef CAS.
- H. Meyssamy, K. Riwotzki, A. Kornowski, S. Naused and M. Haase, Adv. Mater., 1999, 11, 840–844 CrossRef CAS.
- K. Riwotzki, H. Meyssamy, A. Kornowski and M. Haase, J. Phys. Chem. B, 2000, 104, 2824–2828 CrossRef CAS.
- N. K. Sahu, R. S. Ningthoujam and D. Bahadur, J. Appl. Phys., 2012, 112, 014306 CrossRef PubMed.
- J. Cybińska, C. Lorbeer and A.-V. Mudring, J. Mater. Chem., 2012, 22, 9505 RSC.
- T. Shimizu and T. Isobe, IOP Conf. Ser. Mater. Sci. Eng., 2011, 18, 032021 CrossRef.
- J. Wang, Y. Xu, M. Hojamberdiev, Y. Cui, H. Liu and G. Zhu, J. Alloys Compd., 2009, 479, 772–776 CrossRef CAS PubMed.
- P. P. Fedorov, M. N. Mayakova, S. V. Kuznetsov, V. V. Voronov, R. P. Ermakov, K. S. Samarina, A. I. Popov and V. V. Osiko, Mater. Res. Bull., 2012, 47, 1794–1799 CrossRef CAS PubMed.
- P. P. Fedorov, A. a. Luginina, S. V. Kuznetsov and V. V. Osiko, J. Fluorine Chem., 2011, 132, 1012–1039 CrossRef CAS PubMed.
- R. J. Wiglusz, R. Pazik, A. Lukowiak and W. Strek, Inorg. Chem., 2011, 300–301 Search PubMed.
- P. Ayyub, V. Palkar, S. Chattopadhyay and M. Multani, Phys. Rev. B: Condens. Matter, 1995, 51, 6135–6138 CrossRef CAS.
- L. Ning, C. Mak and P. Tanner, Phys. Rev. B: Condens. Matter, 2005, 72, 085127 CrossRef.
- L. Yang, L. Zhou, X. Chen, X. Liu, P. Hua, Y. Shi, X. Yue, Z. Tang and Y. Huang, J. Alloys Compd., 2010, 509, 3866–3871 CrossRef PubMed.
- R. J. Wiglusz and T. Grzyb, Opt. Mater., 2011, 33, 1506–1513 CrossRef CAS PubMed.
- J.-L. Yuan, X.-Y. Zeng, J.-T. Zhao, Z.-J. Zhang, H.-H. Chen and X.-X. Yang, J. Phys. D: Appl. Phys., 2008, 41, 105406 CrossRef.
- P. C. Ricci, M. Salis, R. Corpino, C. M. Carbonaro, E. Fortin and A. Anedda, J. Appl. Phys., 2010, 108, 43512–43517 CrossRef PubMed.
- A. D. Sontakke, K. Biswas and K. Annapurna, J. Lumin., 2009, 129, 1347–1355 CrossRef CAS PubMed.
- P. Berdowski, M. J. J. Lammers and G. Blasse, Chem. Phys. Lett., 1985, 113, 387–390 CrossRef CAS.
- A. K. Parchur, A. I. Prasad, S. B. Rai, R. Tewari, R. K. Sahu, G. S. Okram, R. A. Singh and R. S. Ningthoujam, AIP Adv., 2012, 2, 032119 CrossRef PubMed.
- T. Grzyb and S. Lis, Inorg. Chem., 2011, 50, 8112–8120 CrossRef CAS PubMed.
- X. Y. Huang, D. C. Yu and Q. Y. Zhang, J. Appl. Phys., 2009, 106, 113521 CrossRef PubMed.
- T. Yamashita and Y. Ohishi, J. Non-Cryst. Solids, 2008, 354, 1883–1890 CrossRef CAS PubMed.
- R. Martín-Rodríguez, R. Valiente, S. Polizzi, M. Bettinelli, A. Speghini and F. Piccinelli, J. Phys. Chem. C, 2009, 113, 12195–12200 Search PubMed.
- J. Adam, N. Duhamel-Henry and J. Allain, J. Non-Cryst. Solids, 1997, 213–214, 245–250 CrossRef.
- L. Huang, T. Yamashita, R. Jose, Y. Arai, T. Suzuki and Y. Ohishi, Appl. Phys. Lett., 2007, 90, 131116–131119 CrossRef PubMed.
- I. a. a. Terra, L. J. Borrero-González, L. a. O. Nunes, M. P. Belançon, J. H. Rohling, M. L. Baesso and O. L. Malta, J. Appl. Phys., 2011, 110, 083108 CrossRef PubMed.
- W. Stręk, A. Bednarkiewicz and P. J. Dereń, J. Lumin., 2001, 92, 229–235 CrossRef.
- G. Boulon, J. Alloys Compd., 2008, 451, 1–11 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Luminescence decays of the YPO4:Tb3+,Yb3+ nanocrystals. See DOI: 10.1039/c4dt02234c |
| This journal is © The Royal Society of Chemistry 2014 |