
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and crystal structure of a series of incommensurately modulated composite oxohalide compounds†
Iwan
Zimmermann
a,
Alexis
Corgnet
a,
Mats
Johnsson
*a and
Sven
Lidin
b
aDepartment of Materials and Environmental Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: mats.johnsson@mmk.su.se; Fax: +46-8-152187; Tel: +46-8-162169
bDepartment of Chemistry, Division of Polymer & Materials Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden
First published on 28th August 2014
Abstract
Transparent, needle-like single crystals of the isostructural compounds [Sb4O7+3δX4][Zn3]1+δ (X = Cl, Br, I) δ ≈ 0.2 were obtained from chemical reactions in evacuated and sealed silica tubes. First, the average structure was solved in P21/n but the model refined poorly and a lowering of the symmetry to the 3 + 1 dimensional space group P21(α0γ)0 gave a significantly better fit to the data. This model used second order positional modulations for all the atoms. Whereas Sb, Cl (Br, I) and most O positions were well behaved, there was a mismatch with Zn that was better described in a sub-cell, thus yielding a composite structure. The composite nature of the structure leads to a charge imbalance that is compensated by oxygen vacancies.
Introduction
A rich structural variety exists among oxides and oxohalides containing antimony because of the flexibility of the coordination of Sb3+. The stereochemically active lone-pair on Sb3+ causes an asymmetric coordination, and the most common coordination polyhedra are [SbO3] trigonal pyramids and [SbO4] see-saws.For ternary Sb–O–X (X = Cl, Br, I) compounds, there is a tendency for the halide ion to take the role of a counter ion rather than forming covalent bonds with antimony. For quaternary compounds also containing a late transition metal, it is most common that antimony only coordinates with oxygen and the transition metal bond to both oxygen and halide ions. This results in the formation of non-bonding volumes in the crystal structure between chalcogen/chalcophile and halogen/halophile parts, where the terminal halogens and lonepairs become next neighbors. The most reasonable explanation for this behavior is that they are excluded from the bonding subvolume of the structure rather than being attracted to each other. The flexibility in the coordination around Sb3+ and its preference to bond only to oxygen led to several types of antimony oxide entities, e.g. small cages resembling cubic Sb2O3 in CuSb2O3Br,1 bigger cages resembling zeolite β-cages in Cu20Sb35O44Cl37,2 layers in CuSbTeO3Cl23 and tubes in Sb8O11X2.4–6 In this work, we present the isostructural compounds [Sb4O7+3δX4][Zn3]1+δ (X = Cl, Br, I), δ ≈ 0.2 with a rather simple columnar average structure that actually proved to be a composite structure, in which the Zn atoms are best described in a sub-cell. Composite structures are relatively rare and are mainly observed among intermetallic compounds and among sulfides and fluorides.7 However, the latter are mostly layer misfit compounds, and there is also a good example of columnar misfit: the “alchemist's gold”, Hg2.86AsF6.8,9 Oxide-based, modulated composite structures have been found in several hexagonal perovskites and related materials such as e.g. Sr1.2872NiO3,10 Sr1+x(CoxMn1−x)O3,11 Ba1+x[(CuxRh1−x)O3]12 and Sr1+x(CuxMn1−x)O3.13 To the best of our knowledge the present compounds are the first oxohalides to show a composite structure.
Experimental
Transparent, needle-like single crystals of the compounds [Sb4O7+3δCl4][Zn3]1+δ, [Sb4O7+3δBr4][Zn3]1+δ and [Sb4O7+3δI4][Zn3]1+δ, δ ≈ 0.2 were obtained from chemical reactions in evacuated and sealed silica tubes. ZnO (ABCR), ZnCl2 (Sigma-Aldrich), ZnBr2 (Sigma-Aldrich), ZnI2 (ABCR) and Sb2O3 (Sigma-Aldrich) were used as starting materials. For the synthesis, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of ZnX2 (X = Cl, Br, or I), ZnO2 and Sb2O3 was subsequently heat treated at 550 °C for 60 h. Single crystals were separated manually from unreacted starting material and crystals of the previously described compound ZnSb2O414 that was observed in minor amounts as by-product. Attempts to synthesize a corresponding oxofluoride phase starting with ZnF2 as a fluorine source failed.
:1:1 mixture of ZnX2 (X = Cl, Br, or I), ZnO2 and Sb2O3 was subsequently heat treated at 550 °C for 60 h. Single crystals were separated manually from unreacted starting material and crystals of the previously described compound ZnSb2O414 that was observed in minor amounts as by-product. Attempts to synthesize a corresponding oxofluoride phase starting with ZnF2 as a fluorine source failed.
Single-crystal X-ray diffraction experiments were carried out on an Oxford Diffraction Xcalibur3 diffractometer equipped with a graphite monochromator. The data collection was at 293 K using MoKα radiation, λ = 0.71073 Å. Data reduction was performed with the software CrysAlis RED, which was also employed for the analytical absorption correction. The crystal structures were solved by charge flipping implemented in SUPERFLIP15 and refined by full matrix least squares on F using the program JANA-2006;16 the W matrix was calculated according to van Smaalen.17,18
The structural drawings are made with the program DIAMOND.19 Powder patterns were collected on a Panalytical X'Pert PRO powder X-ray diffractometer in Bragg-Brentano geometry with Cu-Kα radiation. IR spectra were recorded on a Varian 670-IR FTIR spectrometer in the range 390–4000 cm−1 at ambient temperature. The spectrometer was equipped with an attenuated total reflection (ATR) detection device with a single reflection ATR diamond element. Thermogravimetric studies were performed in a Perkin-Elmer TGA7 unit in nitrogen with a heating rate of 5 °C min−1.
Results
An inspection of reciprocal space images from compounds [Sb4O7+3δX4][Zn3]1+δ (X = Cl, Br, I), δ ≈ 0.2 clearly show that the structures are incommensurately modulated with a q vector in the ac-plane of the fundamental monoclinic unit cell. The close-to-perfect orthorhombic pseudo-symmetry of the fundamental unit cell and the presence of two (orthorhombically) symmetry-equivalent q-vectors clearly indicates that pseudo-merohedral twinning according to the orthorhombic-monoclinic symmetry lowering is expected. All the structures contain a set of strong substructure reflections and a set of weaker satellites that appear to be incommensurate with the substructure.The crystallographic information of the three compounds is summarized in Table 1. The value of δ is given by the q vector component along a*. The patterns further share the special feature that first-order satellites dominate for the 1kl reflections, and the second-order satellites dominate for the 2kl reflections, as shown in Fig. 1. This is a strong indication for composite behavior.
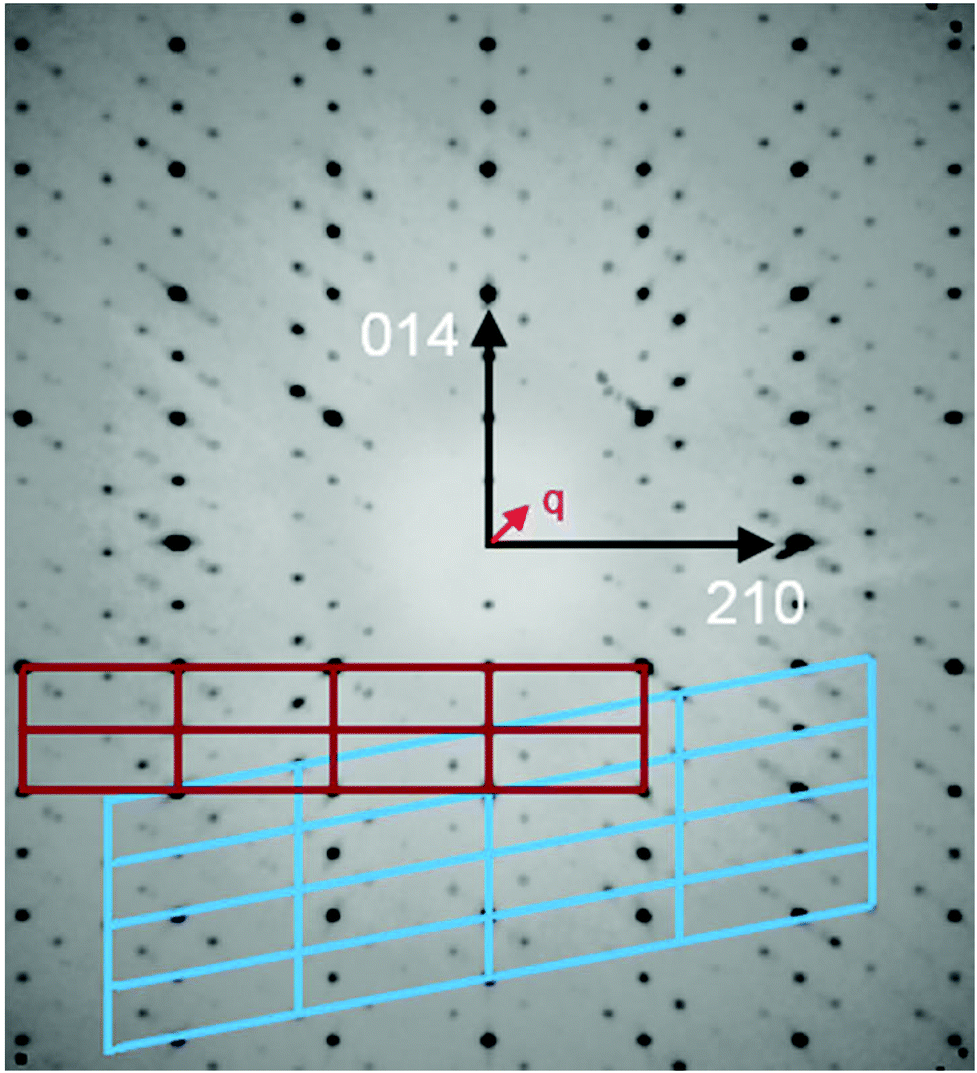 | ||
| Fig. 1 Reconstructed reciprocal lattice section hkl integrated from k = 0 to k = 4 of the iodide compound. Note the relative intensity of first-order satellites for 1kl reflections and second-order satellites for 2kl reflections. Primary and secondary unit cells are indicated in red and blue, respectively. | ||
| Compound | [Sb4O7+3δI4][Zn3]1+δ | [Sb4O7+3δBr4][Zn3]1+δ | [Sb4O7+3δCl4][Zn3]1+δ |
| Space group | P21(α0γ)0 | P21(α0γ)0 | P21(α0γ)0 |
| Symmetry operators | x1, x2, x3, x4 | x1, x2, x3, x4 | x1, x2, x3, x4 |
| −x1, x2 + 0.5, −x3, −x4 | −x1, x2 + 0.5, −x3, −x4 | −x1, x2 + 0.5, −x3, −x4 | |
| a/Å | 4.2710(8) | 4.1307(13) | 4.0634(6) |
| b/Å | 18.508(3) | 17.9042(2) | 17.163(2) |
| c/Å | 10.6741(14) | 10.3021(10) | 10.4699(13) |
| β/° | 90.054(13) | 90.331(6) | 91.024(11) |
| q |

|
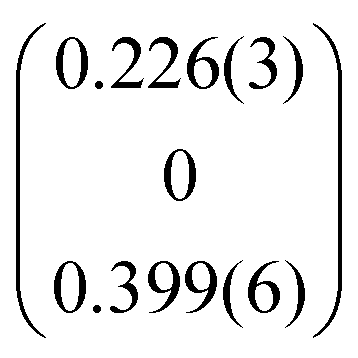
|
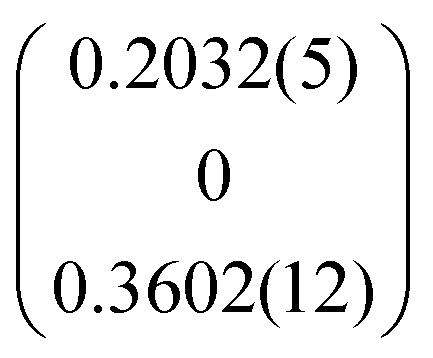
|
| Crystal colour/habit | Colourless/needle | Colourless/needle | Colourless/needle |
| Crystal dimensions | 0.32 × 0.04 × 0.03 | 0.21 × 0.03 × 0.02 | 0.20 × 0.04 × 0.02 |
| No. of reflections all/[I > 3σ(I)] | 16749/4073 |
26084/6011 |
22220/3537 |
| Goodness of fit S | 1.51 | 2.02 | 1.39 |
| R factors [I > 3σ(I)] | R 1 = 0.0729 (wR2 = 0.0711) | R 1 = 0.0982 (wR2 = 0.1015) | R 1 = 0.0714 (wR2 = 0.0690) |
| Equivalents | R int = 0.0701 | R int = 0.1192 | R int = 0.1486 |
| Main reflections | R = 0.0556 (2355) | R = 0.0808 (3201) | R = 0.0592 (2302) |
| First-order satellites | R = 0.1180 (1610) | R = 0.1297 (2316) | R = 0.1103 (1003) |
| Second-order satellites | R = 0.1751 (108) | R = 0.1920 (494) | R = 0.1580 (232) |
| Residual e− density (min/max)/Å−3 | 4.23/−4.84 | 13.86/−9.56 | 9.13/−9.04 |
Structural solutions using charge flipping in superspace (superflip) of the compounds was relatively straight-forward. A first structural solution was carried out in the centro-symmetric space group P21/n but this solution refined rather poorly. Lowering the symmetry to Pn did little to improve the situation, whereas a model in the 3 + 1 dimensional space group P21(α0γ)0 provided a considerably better fit to the data. This model used second-order positional modulations for all the atoms. The automatic procedure produced excellent starting positions and modulations for Sb and halides, whereas the modulations for Zn turned out to be very large. Oxygen positions were only localized to some extent. The amplitude of the modulation of Zn positions was particularly large along the a direction, and while this could be modeled as a saw-tooth type positional modulation, it was clear that a composite structure model would be more suitable (see Fig. 2). After moving all the Zn positions to the secondary unit cell using the transformation matrix expressed as
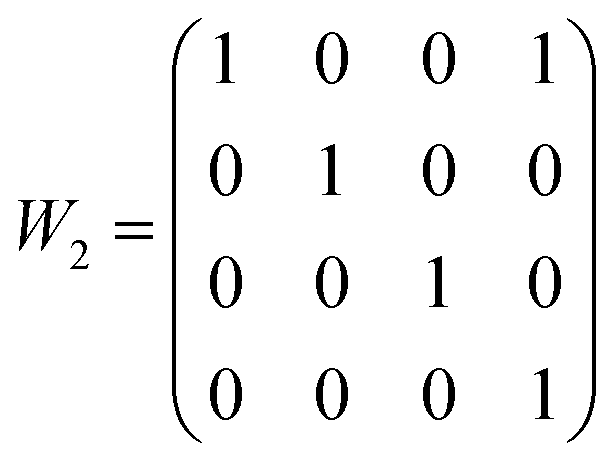
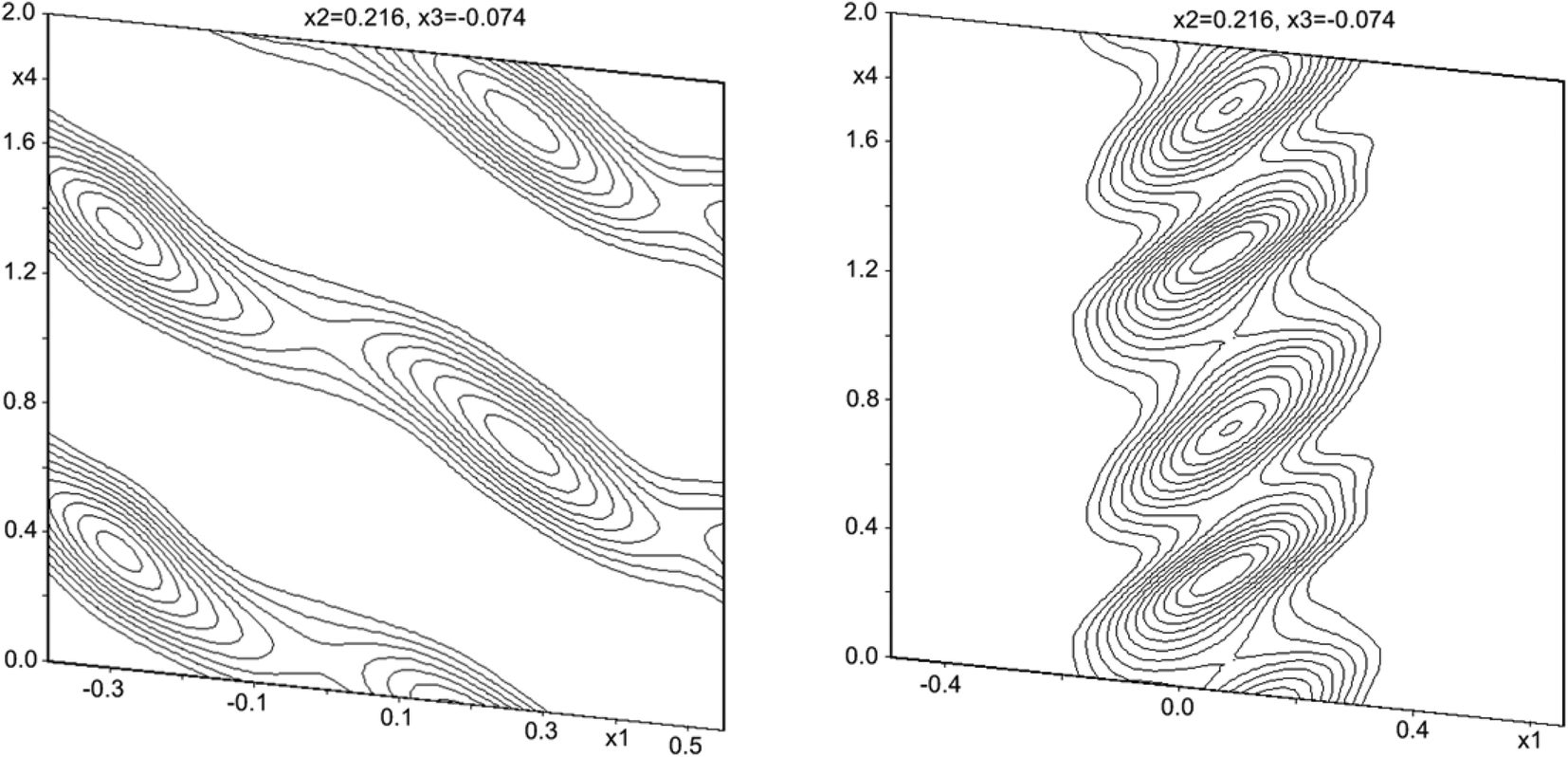 | ||
| Fig. 2 Electron density map of Zn1 in the primary (left) and the secondary unit cells (right). | ||
The compounds [Sb4O7+3δX4][Zn3]1+δ, (X = Cl, Br, I), δ ≈ 0.2 are isostructural, and may be described by the behavior of the iodide compound without loss of generality. The Br compound produced crystals of relatively low quality, and for these convergence was difficult to achieve. The problem was solved by restricting all oxygen thermal displacement parameters to be equal. The non-stoichiometry originates from the composite nature of the structures. The primary unit cell is the locus of the Sb4O8I4 sub-structure, whereas the secondary unit cell given by the relationship a2* = a1* + q, b2* = b1*, c2* = c1* yielding a = 3.4797 Å, b = 18.508 Å, c = 10.7768 Å, β = 97.915°, q = (α0γ), α = 0.1852, γ = 0.3451 contains Zn only. This model refines very well yielding R1 values better than 6% for main reflections and better than 12% and 18% for first- and second-order satellites, respectively. The number of Sb positions in the primary cell is 4, whereas the number of Zn positions in the secondary cell is 3. However, because of the metric relationship between the two unit cells, the relative amount Sb:Zn is not 4:3, but rather 4:3.682; that is, because the Zn content refers to a smaller cell, it must be multiplied by the ratio of the a-axes of the two cells, 4.271/3.4797. This still leaves the structure unbalanced with respect to charge, for which there are several possible remedies. The answer might be found in the partial replacement of O by OH, the oxidation of Sb(III) to Sb(V) or the under-occupancy of either oxygen or iodine. The oxidation of Sb is unlikely because of the absence of oxidizing agents in the synthesis and the local coordination of Sb that clearly indicates the trivalent state throughout the structure. Similarly, the absence of iodide should be very noticeable as negative residual electron density. The most likely charge compensation is therefore either partial occupancies on oxygen and/or the presence of hydrogen in the structure. The latter can hardly constitute the sole contribution to charge balance because this would require a rather substantial moisture content in the reaction mixture. Given that the title compounds are the main product of the reaction, the required hydrogen content is [Sb4O7.38(OH)0.62I4][Zn3]1.23, rather more than expected from only contaminant water. This leaves oxygen vacancies as the only plausible main contributor to balance the charge of the non-stoichiometry.
It was found that bond valence sum calculations were quite a useful tool in this quest. While the BVS for all the Sb positions vary, the BVS of Sb1 stands out as singular, reaching a maximum value of well above four. A closer inspection reveals that the high value of this BVS is caused by a short Sb1–O5 distance over part of a range of the internal coordinate, describing the propagation of the structure along the direction of the q-vector. Introducing occupational modulation on position O5 yields an improvement of the fit of the model to the data and reduces the offending BVS value to one more on par with those of the other Sb positions. The reduced occupancy of O5 also brings the compound closer to an overall charge balance. A somewhat less problematic but still high BVS is found for Sb3, and herein, the cause is a short distance to O6. Following the procedure for O5, an occupational modulation was introduced for O6, which led to further improvement and a change in composition to slightly below the ideal value [Sb4O7.69X4][Zn3]1.23. In the final model, the sum of the occupancies for positions O5 and O6 was set at 1.69 to avoid overcompensating for Zn deficiency. Unsurprisingly, the agreement between model and data is quite insensitive to such small changes in oxygen content.
The structure is composed of columns centered by three Zn positions, with the central being coordinated exclusively by oxygen, whereas the two outer columns also bind to Cl/Br/I. The oxygen atoms are further connected to Sb atoms belonging to Sb–O ladders. The structure looks rather simple when viewed along the a direction (see Fig. 3). In a perpendicular view, the complexity becomes more pronounced. The mismatch between Zn and the rest of the structure is most easily seen if the Sb–O ladders are removed from the column (see Fig. 4). The coordination around the three Zn atoms is clarified with t-plots (see Fig. 5) to show how the coordination number varies, depending on the modulation. The composite nature of the structure leads to a charge imbalance that is compensated by oxygen vacancies, which are located in one of the Sb–O ladders (see Fig. 6). The Sb–O ladders are strongly reminiscent of the ones in Sb8O11X2.5,6 The electron density maps show that the atomic modulation functions capture the behavior of each atomic position well (see ESI†).
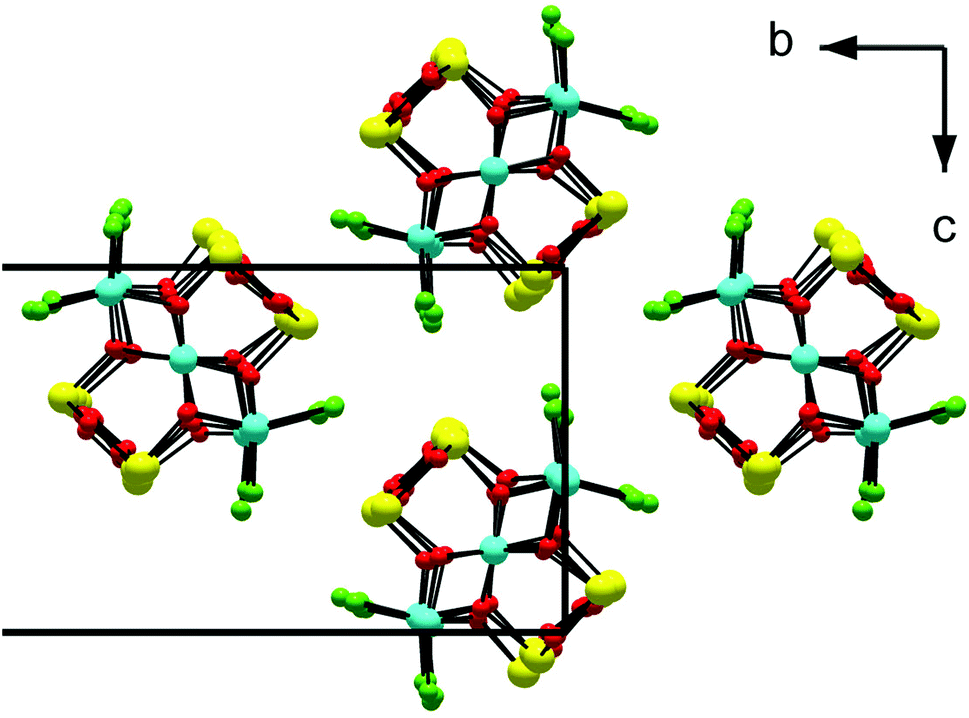 | ||
| Fig. 3 Structure of single columns of SbZn1−yO2−yI viewed along a. Zinc (blue) is coordinated entirely by oxygen (red) or by oxygen and iodine (green), whereas antimony forms oxygen-connected ladders. Note the exposure of Sb to the surroundings of the column, a typical trait of lone-pair behavior. | ||
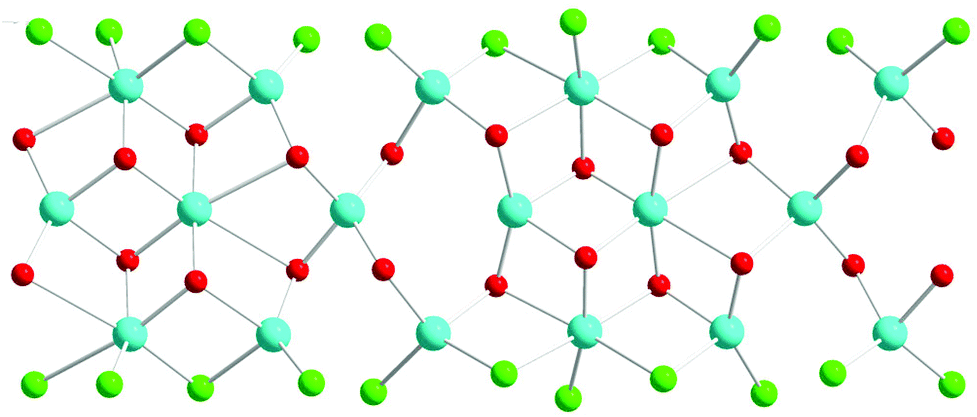 | ||
| Fig. 4 Coeur-du-filé of a single column of the structure, displaying the misfit between on one hand Zn(blue) and on the other hand oxygen (red) and I (green). The a direction is horizontal. | ||
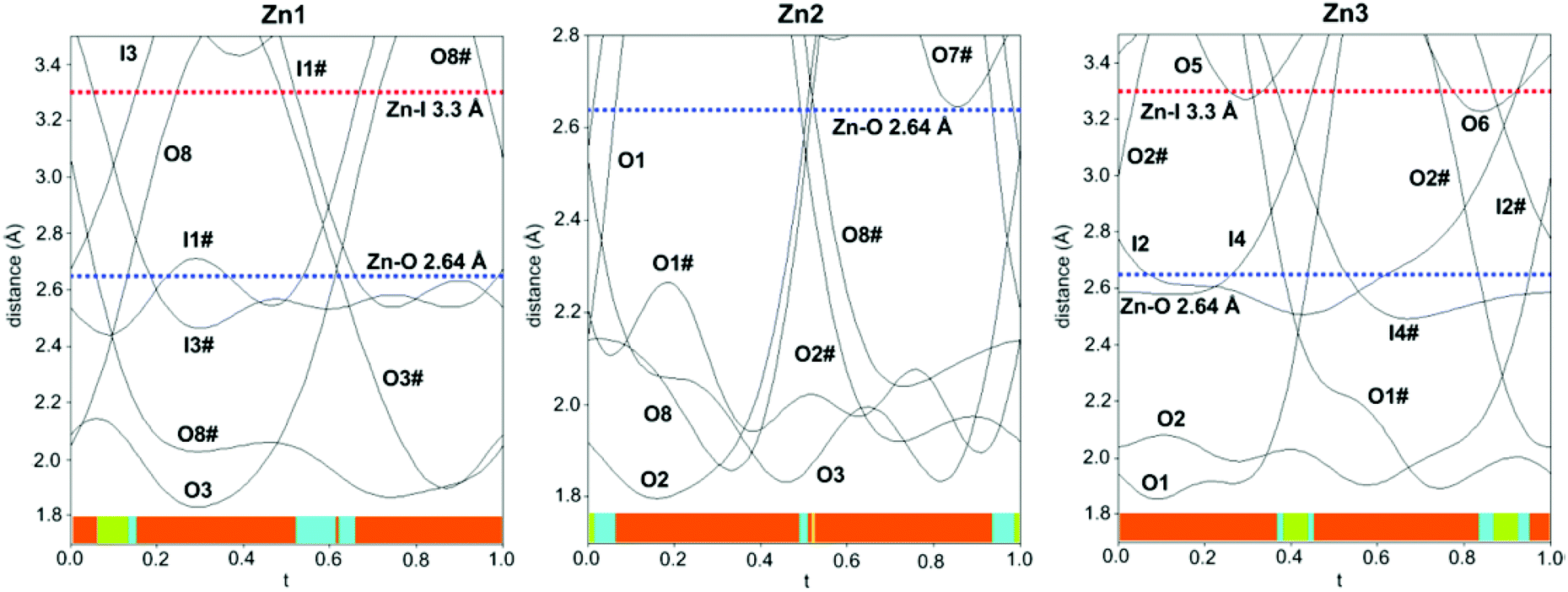 | ||
| Fig. 5 Variation in the coordination around the three crystallographically different Zn-atoms in the form of t-plots. The maximum Zn−O and Zn−I distances to be considered for covalent bonding are indicated with dotted lines. The coordination number is outlined with colored bars; yellow = 3-coordination, red = 4-coordination, blue = 5-coordination, and green = 6-coordination. # sign implies symmetry-equivalent atoms. | ||
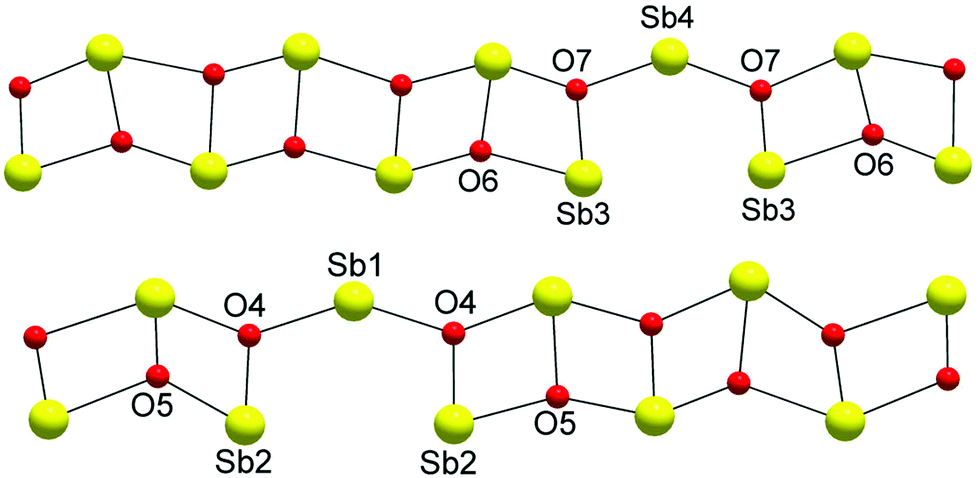 | ||
| Fig. 6 Two types of Sb–O ladders. Note that oxygen vacancies balance the charge deficiency. In one ladder, there are O6 vacancies, and in the other, O5 vacancies. | ||
Almost phase-pure material (see powder diffractogram in ESI†) could be synthesized for the iodine compound, which was used to collect an IR spectrum and to study thermal decomposition. Fig. 7a shows the transmission IR spectrum for [Sb4O7+3δI4][Zn3]1+δ in the range 4000 to 400 cm−1. Intense peaks are observed in the low-energy region, which are most probably due to Sb–O and Zn–O vibrations. The absence of peaks in the high-energy region confirms the absence of hydroxyl groups, which would give rise to strong signals. Thermogravimetry (TG) data of [Sb4O7+3δI4][Zn3]1+δ were recorded from 30–900 °C in a nitrogen atmosphere with a heating rate of 5 °C min−1. The TG curve in Fig. 7b shows that the compound is stable up to 400 °C and decomposes shortly thereafter in a single step by releasing iodine as I2(g). This is in good agreement with the total weight loss of about 37%. A powder diffraction pattern of the decomposition product shows a mixture of the oxides ZnSb2O6 and Zn2.33Sb0.67O4, which implies that antimony has been oxidized to Sb(V).
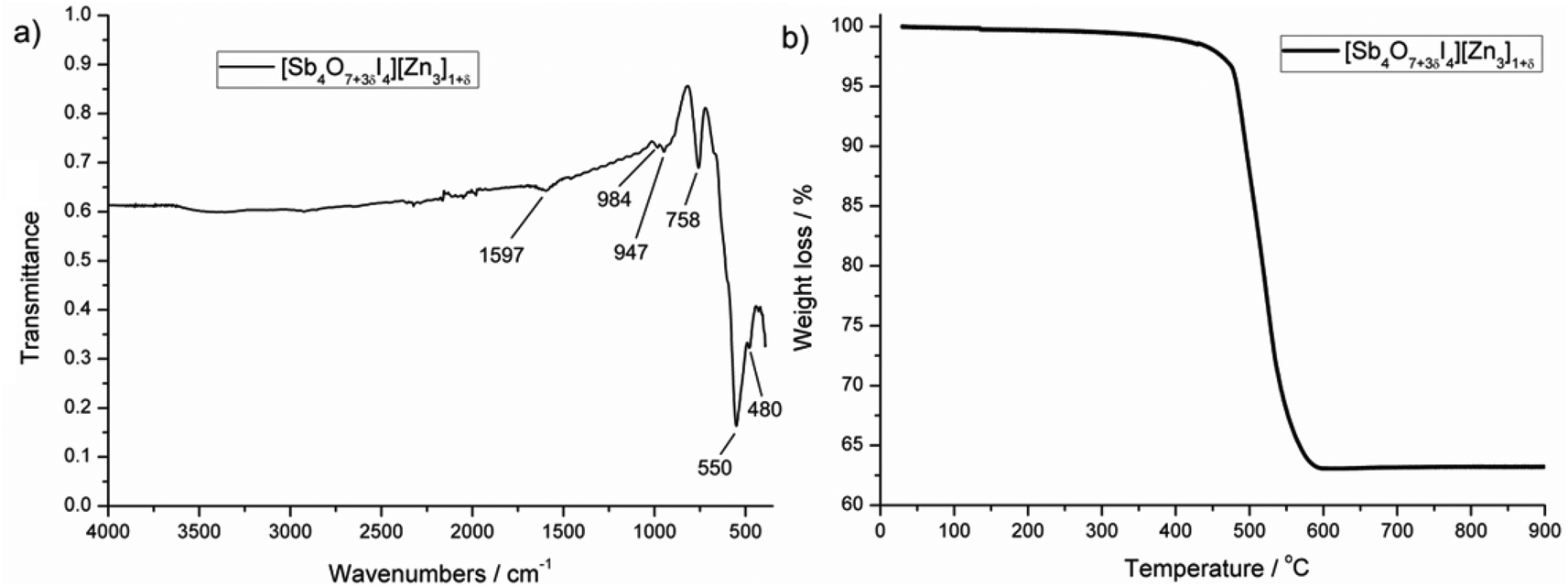 | ||
| Fig. 7 (a) FT-IR spectrum and (b) TG curve of [Sb4O7+3δI4][Zn3]1+δ. | ||
Conclusions
The new isostructural compounds [Sb4O7+3δX4][Zn3]1+δ, (X = Cl, Br, I), δ ≈ 0.2 were synthesized via chemical reactions in evacuated and sealed silica tubes. The crystal structure was found to be incommensurately modulated and was refined in the monoclinic 3 + 1 dimensional space group P21(α0γ)0 having a q vector in the ac-plane. The close-to-perfect orthorhombic pseudo-symmetry of the fundamental unit cell and the presence of two (orthorhombically) symmetry-equivalent q-vectors results in pseudo merohedral twinning. The structure consists of one-dimensional columns having the Zn atoms in the center to which ladders of antimony oxides are attached. The domination of first-order and second-order satellites for 1kl and 2kl reflections, respectively, strongly indicate a composite behavior and that all the Zn atoms were described in a second composite unit cell. The overall charge balance was obtained by introducing oxygen vacancies in the antimony ladders, which is supported by bond valance sum calculations and by the IR measurements that do not show any signs of hydroxyl groups being present. From TG, one can observe the decomposition of the iodide phase in a single step above 400 °C by releasing I2(g) and oxidising antimony to Sb(V).Acknowledgements
The Swedish Research Council is acknowledged for financial support.References
- Z. Mayerová, M. Johnsson and S. Lidin, J. Solid State Chem., 2005, 178, 3471–3475 CrossRef PubMed.
- Z. Mayerová, M. Johnsson and S. Lidin, Angew. Chem., Int. Ed., 2006, 45, 5602–5606 CrossRef PubMed.
- R. Becker, M. Johnsson, R. K. Kremer and P. Lemmens, Solid State Sci., 2003, 5, 1411–1416 CrossRef CAS PubMed.
- S. Menchetti, C. Sabeli and R. Trosti-Rerroni, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1506–1510 CrossRef.
- Z. Mayerová, M. Johnsson and S. Lidin, Solid State Sci., 2006, 8, 849–854 CrossRef PubMed.
- S. Lidin, M. Johnsson and Z. Hugonin, Solid State Sci., 2009, 11, 1198–1205 CrossRef CAS PubMed.
- J. Sun, S. Lee and J. Lin, Chem. – Asian J., 2007, 2, 1204–1229 CrossRef CAS PubMed.
- B. D. Cutforth, Preparation, Structural Characterization and Electrical Properties of Some Polyatomic Cations of Mercury, Open Access Dissertations and Theses, Paper 3741, McMaster University, Canada, 1975 Search PubMed.
- I. D. Brown, B. D. Cutforth, C. G. Davies, R. J. Gillespie, P. R. Ireland and J. E. Vekris, Can. J. Chem., 2011, 52, 791–793 CrossRef.
- M. Evain, F. Boucher, O. Gourdon, V. Petricek, M. Dusek and P. Bezdicka, Chem. Mater., 1998, 10, 3068–3076 CrossRef CAS.
- N. A. Jordan, P. D. Battle, S. van Smaalen and M. Wunschel, Chem. Mater., 2003, 15, 4262–4267 CrossRef CAS.
- M. Zakhour-Nakhl, J. B. Claridge, J. Darriet, F. Weill, H.-C. zur Loye and J. M. Perez-Mato, J. Am. Chem. Soc., 2000, 122, 1618–1623 CrossRef CAS.
- A. E. Abed, E. Gaudin, H.-C. zur Loye and J. Darriet, Solid State Sci., 2003, 5, 59–71 CrossRef.
- S. Stahl, Ark. Kemi, Mineral. Geol., 1943, 17, 1–7 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, JANA-2006, Institute for Physics AVCR, Praha, Czech Republic, 2006.
- S. van Smaalen, Phys. Rev., 1991, B43, 11330–11341 Search PubMed.
- S. van Smaalen, Crystallogr. Rev., 1995, 4, 79–202 CrossRef CAS.
- K. Brandenburg, DIAMOND, Release 2.1e, Crystal impact GbR, Bonn, Germany, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further details on crystal structural investigations can be obtained from the Fachinformationszentrum Karlsruhe, Abt. PROKA, 76344 Eggenstein-Leopoldshafen, Germany (fax +49-7247-808-666; E-mail: crysdata@fiz-karlsruhe.de) on quoting the following depository numbers: CSD 427521 for [Sb4O7+3δCl4][Zn3]1+δ, CSD 427522 for [Sb4O7+3δBr4][Zn3]1+δ, and CSD 427523 for [Sb4O7+3δI4][Zn3]1+δ.. See DOI: 10.1039/c4dt02084g |
| This journal is © The Royal Society of Chemistry 2014 |