
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How big is a Cp? Novel cycloheptatrienyl zirconium complexes with tri-, tetra- and pentasubstituted cyclopentadienyl ligands†
Heiko
Bauer
a,
Andreas
Glöckner
b,
Alain C.
Tagne Kuate
b,
Sebastian
Schäfer
c,
Yu
Sun
a,
Matthias
Freytag
b,
Matthias
Tamm
*b,
Marc D.
Walter
*b and
Helmut
Sitzmann
*a
aFachbereich Chemie, Technische Universität Kaiserslautern, Erwin-Schroedinger Strasse, 67663 Kaiserslautern, Germany. E-mail: sitzmann@chemie.uni-kl.de; Fax: +49 631-205-4676; Tel: +49 631-205-4399
bInstitut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: m.tamm@tu-bs.de; Fax: +49 531-391-5309; Tel: +49 531-391-5387
cInstitut für Anorganische Chemie, Karlsruher Institut für Technologie, Engesserstr. 15, 76131 Karlsruhe, Germany
First published on 15th September 2014
Abstract
The new bulky cyclopentadienyl anions 1,2,4-tri(cyclopentyl)cyclopentadienide and 2,3-diisopropyl-1,4-dimethyl-5-trimethylsilyl-cyclopentadienide were prepared. These and the already known 1,2,4-tri(cyclohexyl)-, 1,2,4-tri(isopropyl)-, 2,3-diisopropyl-1,4-dimethyl-, 1,3,4-triisopropyl-2,5-dimethyl-, pentaphenyl-, and p-butylphenyl-tetraphenyl-cyclopentadienide as well as tert-butylindenide were coordinated to the cycloheptatrienylzirconium fragment [(CHT)ZrCl(tmeda)]. The nine zirconium complexes of the [(CHT)Zr(Cp)] type were characterized by elemental analysis and NMR spectroscopy. For five of the sandwich complexes X-ray crystal structure determination could be carried out; structures of the four others were obtained by DFT calculations. The data serve as a basis for cone angle measurements of cyclopentadienyl ligands to evaluate the steric demand of these ligands.
Introduction
In the late forties of the last century the parent cyclopentadienyl ligand was seized by freshly reduced iron from cyclopentadiene vapors at 300 °C1 to form a complex later called ferrocene and initiating a rush for cyclopentadienyl complexes of other metals as well.2–4 About 20 years later, when titanium tetrachloride was reacted with butane isomers and other olefins at 300 °C in an autoclave, unexpected pentamethylcyclopentadienyltitanium trichloride5 was assembled which stimulated experiments with this prototype of polysubstituted cyclopentadienyl ligands.6 Sterically bulky cyclopentadienyl ligands and their metal complexes have been described in review articles.7,8 Among more than 2600 experimentally investigated cyclopentadienylzirconium complexes, only about 30% carry at least one unsubstituted cyclopentadienyl ligand, the remaining 70% exhibit only substituted cyclopentadienyl ligands. For cyclopentadienyl complexes of other metals this fraction may be lower, but cyclopentadienylzirconium complexes are frequently used as catalysts, whose performance can be modified and even tailored with ring substituents.9As early as 1970 the steric bulk of triorganophosphine ligands was quantified by Tolman using a cone angle approach.10 While the number of cyclopentadienyl complexes known in the literature (more than 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700) is in the same order of magnitude as the number of complexes with tertiary phosphine ligands (more than 51000),11 the attention received by one article on the steric bulk of phosphine ligands12 by far exceeds the combined impact of a number of review articles on the quantification of the steric bulk of cyclopentadienyl ligands. The work of Möhring and Coville is focused on the polymerization activity of group IV metallocene derivatives. The authors analyzed a large number of olefin polymerization results and concluded that the catalyst activity is dominated to ca. 80% by the electronic influence of the two cyclopentadienyl, indenyl, or fluorenyl ligands used.13–15 The steric bulk of the cyclopentadienyl ligands in titanocene derivatives was concluded to affect both the angular opening between the two Cp planes corresponding to the access of reactants to the metal center and the angular opening in the plane bisecting the two Cp planes, which limits the angle between the Ti–C(polymer) σ bond and the direction of the olefin approach.16 Janiak et al. concluded, in contrast, that steric effects dominate the catalytic activity of zirconocene derivatives17,18 and find “no influence of … electronic effects …”.18 Different methods of substituted cyclopentadienyl bulk quantification have been outlined in the literature,19 which was later updated in the literature20 where steric bulk measures mostly derived from zirconocene or ferrocene derivatives for 35 cyclopentadienyl ligands with 0–1 substituents and for 8 cyclopentadienyl ligands with two isopropyl or cyclohexyl substituents, two or three tBu groups or 2–5 methyl groups have been listed. Another early approach used force field calculations on 24 iron(II) cyclopentadienyl complexes with a fixed Fe–ring centroid distance of 1.73 Å.21 For most of the bulky cyclopentadienyl ligands with two and more substituents such values are not available. In our research activities bulky alkylcyclopentadienyl ligands play a central role, because they can be used to prevent metal complexes from falling into their preferred energetic sink. For our research a systematic evaluation of steric bulk is therefore quite important.
700) is in the same order of magnitude as the number of complexes with tertiary phosphine ligands (more than 51000),11 the attention received by one article on the steric bulk of phosphine ligands12 by far exceeds the combined impact of a number of review articles on the quantification of the steric bulk of cyclopentadienyl ligands. The work of Möhring and Coville is focused on the polymerization activity of group IV metallocene derivatives. The authors analyzed a large number of olefin polymerization results and concluded that the catalyst activity is dominated to ca. 80% by the electronic influence of the two cyclopentadienyl, indenyl, or fluorenyl ligands used.13–15 The steric bulk of the cyclopentadienyl ligands in titanocene derivatives was concluded to affect both the angular opening between the two Cp planes corresponding to the access of reactants to the metal center and the angular opening in the plane bisecting the two Cp planes, which limits the angle between the Ti–C(polymer) σ bond and the direction of the olefin approach.16 Janiak et al. concluded, in contrast, that steric effects dominate the catalytic activity of zirconocene derivatives17,18 and find “no influence of … electronic effects …”.18 Different methods of substituted cyclopentadienyl bulk quantification have been outlined in the literature,19 which was later updated in the literature20 where steric bulk measures mostly derived from zirconocene or ferrocene derivatives for 35 cyclopentadienyl ligands with 0–1 substituents and for 8 cyclopentadienyl ligands with two isopropyl or cyclohexyl substituents, two or three tBu groups or 2–5 methyl groups have been listed. Another early approach used force field calculations on 24 iron(II) cyclopentadienyl complexes with a fixed Fe–ring centroid distance of 1.73 Å.21 For most of the bulky cyclopentadienyl ligands with two and more substituents such values are not available. In our research activities bulky alkylcyclopentadienyl ligands play a central role, because they can be used to prevent metal complexes from falling into their preferred energetic sink. For our research a systematic evaluation of steric bulk is therefore quite important.
For this reason, we have embarked on an approach, where an average cone angle derived from crystallographic data quantifies the steric bulk of the substituted cyclopentadienyl ligand and additional information on the bulk of the individual substituents is provided by a substituent cone angle adding numerical information on the third dimension.22 The cycloheptatrienyl-zirconium fragment, which turned out to be a very versatile starting compound for an extensive chemistry,23 offers good crystallinity and more than one hemisphere of one of the largest transition metals for very bulky cyclopentadienyl ligands. Thus, these CHT–Zr complexes are the materials of choice for the investigation.
Results and discussion
Synthesis of cyclopentadienyl ligands
The synthesis of tricyclopentylcyclopentadiene HCpcpent (2-H) from the parent cyclopentadiene followed a variation24 of the phase-transfer alkylation process25 in a system consisting of aqueous potassium hydroxide solution and an organic phase containing cyclopentadiene, cyclopentyl bromide, and cyclopentadiene alkylation products (Scheme 1).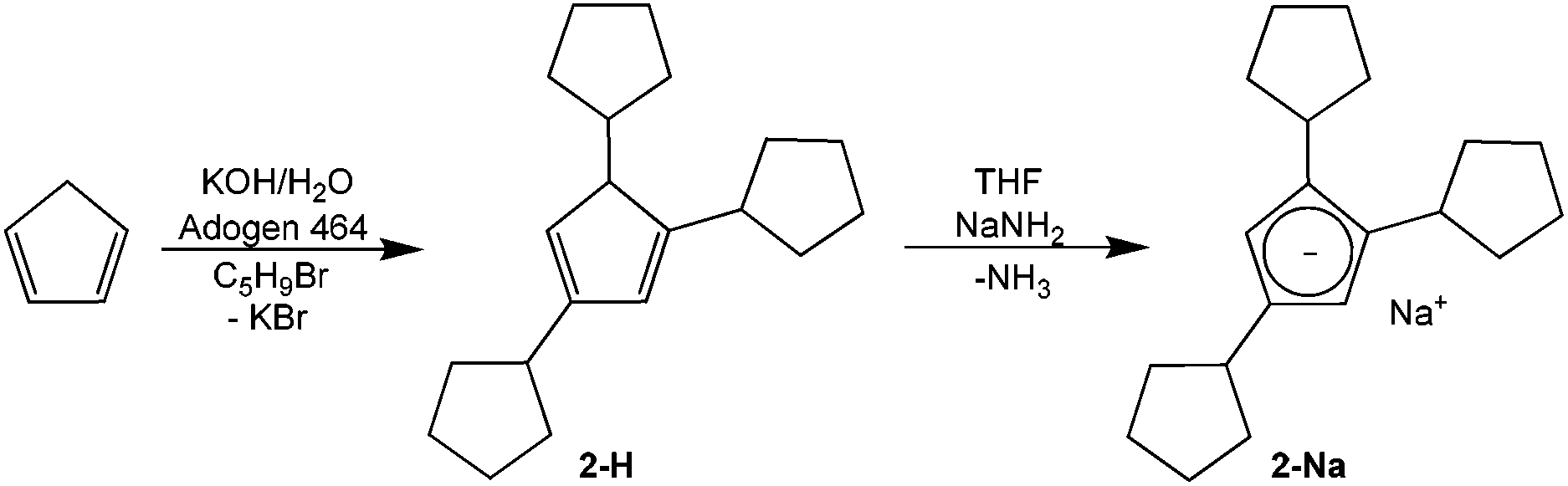 | ||
| Scheme 1 Synthesis of sodium tri(cyclopentyl)cyclopentadienide NaCpcpent (2-Na). | ||
Metalation of the 2-H mixture of isomers with sodium amide gave 46% yield of the sodium salt 2-Na.
For the synthesis of potassium 3,4-diisopropyl-2,5-dimethyl-1-trimethylsilylcyclopentadienide (6-K, Scheme 2) sodium 2,3-diisopropyl-1,4-dimethylcyclopentadienide26 was silylated with chlorotrimethylsilane in tetrahydrofuran to furnish 61% yield of the silylated cyclopentadiene tautomers 6-H, which could be metalated with potassium hydride to the potassium salt 6-K in 77% yield.
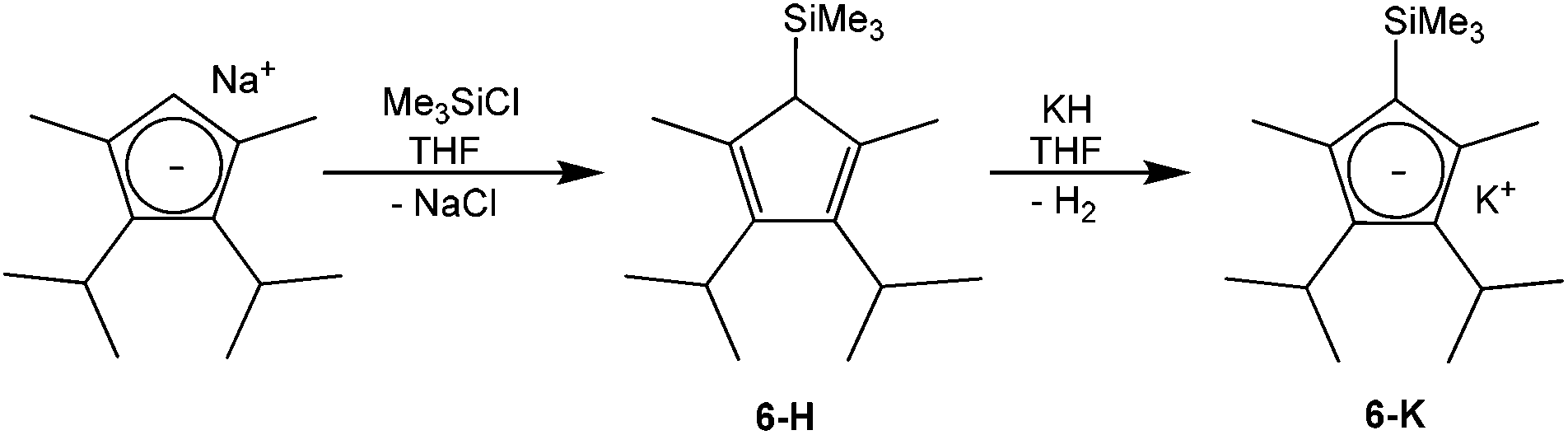 | ||
| Scheme 2 Synthesis of potassium 3,4-diisopropyl-2,5-dimethyl-1-trimethylsilylcyclopentadienide KTMS (6-K). | ||
Literature citations regarding the preparation of other cyclopentadienides used in this study can be found in the experimental part.
Synthesis and characterization of cycloheptatrienyl zirconium complexes 2–10
Salt metathesis reaction between the cycloheptatrienyl zirconium chloride 1 and the substituted cyclopentadienides or the indenide was performed in THF solution at −78 °C (Scheme 3). The changing colour of the reaction mixture turned out to be a very good indicator for the progress of the reaction. Whereas a mixture of the dark blue solution of 1 and the brown solution of the cyclopentadienyl salt displays a grey colour, this turns purple after some minutes or hours, depending on the reaction rate.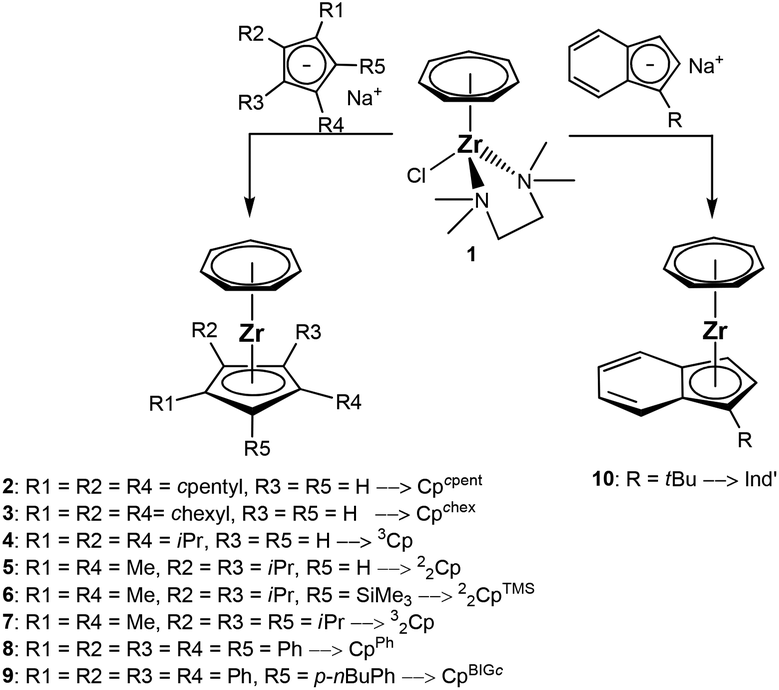 | ||
| Scheme 3 Synthesis of cycloheptatrienyl zirconium complexes 2–10. | ||
The synthesis of the cycloheptatrienyl zirconium complex [(C7H7)Zr{C5H2(C5H9)3-1,2,4}] (2) via a reaction between [(CHT)ZrCl(tmeda)] (1) and 2-Na afforded 53% yield of purple needles. A discussion of proton and 13C NMR spectra of complex 2 can be found in the ESI.†
A crystal structure determination reveals the first structural characterization of the 1,2,4-tricyclopentylcyclopentadienyl ligand in complex 2 (Fig. 1). The three cyclopentyl rings show the expected envelope conformation and are rotated such that the methyne hydrogen atom at the α carbon atom is close to the cyclopentadienyl plane, one of the β carbon atoms is above and the other below the cyclopentadienyl plane. Both β carbon atoms of all three cyclopentyl substituents point towards a CH group of the cyclopentadienyl ligand, the two vicinal cyclopentyl substituents in 1- and 2-positions are turned away from each other. All three C5 envelopes see the cyclopentadienyl moiety in an exo and the α (methyne) hydrogen atom in an endo position as expected from a steric point of view. The Zr–CHTcent distance (1.673 Å) is much smaller than the Zr–Cpcent distance (2.191 Å) because of the larger ring diameter of the cycloheptatrienyl ligand and the steric demand of the cyclopentadienyl ring. Due to the arrangement of the cyclopentyl substituents described before, the CHT ligand is tilted towards the two substituents in 1- and 2-positions (CHTcent–Zr–Cpcent 172.03°).
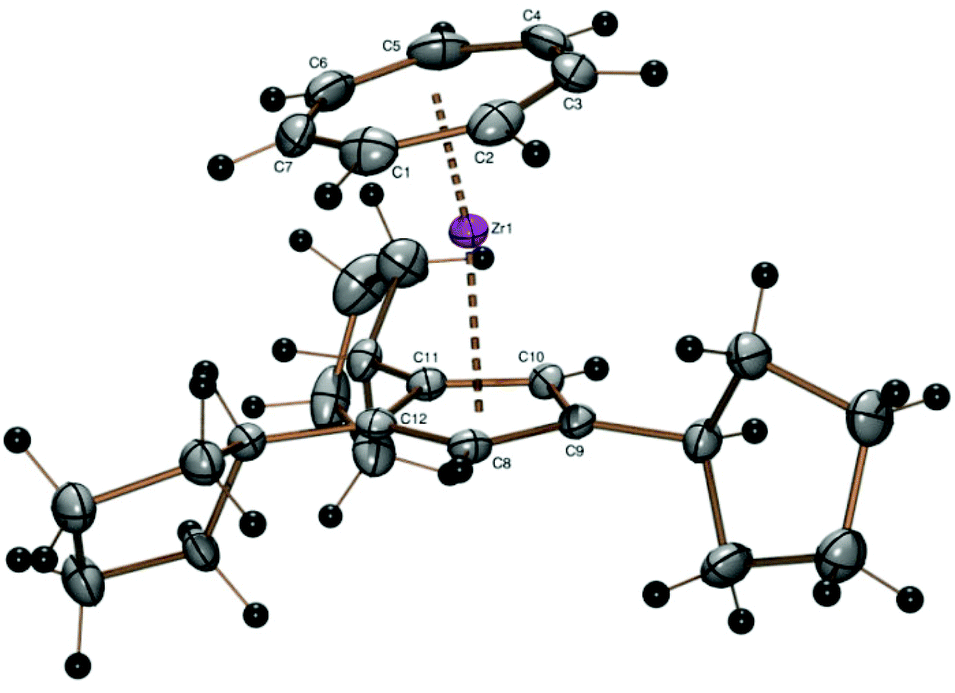 | ||
| Fig. 1 ORTEP plot of the molecular structure of 2. Atomic displacement parameters have been drawn at 50% probability. Selected bond length (Å) and angles (°): Zr–C1 2.327(3), Zr–C2 2.339(3), Zr–C3 2.363(3), Zr–C4 2.348(3), Zr–C5 2.341(4), Zr–C6 2.327(3), Zr–C7, 2.328(4), Zr–C8 2.482(3), Zr–C9 2.515(3), Zr–C10 2.491(3), Zr–C11 2.511(2), Zr–C12 2.496(3), Zr–Cpcent 2.191, Zr–CHTcent 1.673, CHTcent–Zr–Cpcent 172.03. | ||
Other features of the crystallographically determined or theoretically calculated molecular structures of complexes 2–10 are collected in Table 2, details like individual Zr–C distances are listed in the figure caption of Fig. 1.
The tricyclohexylcyclopentadienyl derivative [(C7H7)Zr{C5H2(C6H11)3-1,2,4}] (3) was obtained as purple crystals in 54% yield from 1 and sodium tricyclohexylcyclopentadienide (3-Na)28 by the procedure used also for the synthesis of 2. A discussion of proton and 13C NMR spectra of complex 3 can be found in the ESI.† The structure of 3 was obtained by DFT calculations (Fig. 2), which arrived at a conformation very similar to that discussed for the crystallographically characterized structure of 2.
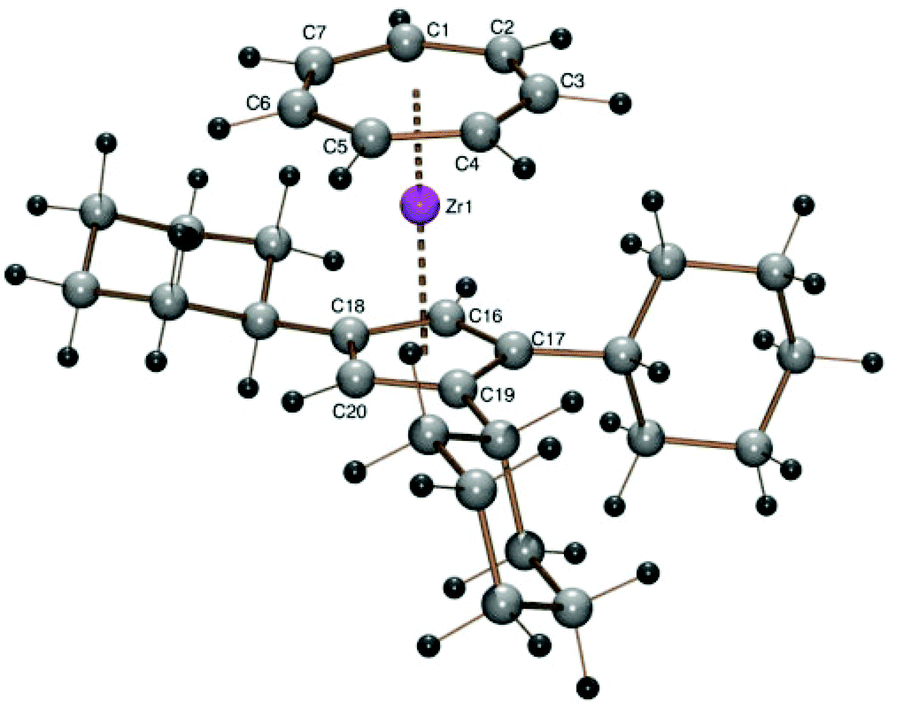 | ||
| Fig. 2 ORTEP plot of the calculated structure of 3. Calculated bond length (Å) and angles (°): Zr–C1 2.352, Zr–C2 2.353, Zr–C3 2.349, Zr–C4 2.347, Zr–C5 2.353, Zr–C6 2.351, Zr–C7 2.353, Zr–C16 2.511, Zr–C17 2.535, Zr–C18 2.523, Zr–C19 2.529, Zr–C20 2.508, Zr–Cpcent 2.213, Zr–CHTcent 1.684, CHTcent–Zr–Cpcent 178.06. | ||
The calculated distances (1.684 Å for CHTcent–Zr and 2.213 Å for Cpcent–Zr) are slightly longer than the values for 2, a tendency, which was found for all calculated structures, irrespective of their steric demand (Table 2). The validity of these calculations had been demonstrated for the tetraisopropylcyclopentadienyl derivative [(C7H7)Zr(C5HiPr4)] before.22 In this case the largest difference between the experimental and the calculated Zr–C distances, for example, was found for Zr–C4 (2.346(4) vs. 2.370 Å). For the other eleven Zr–C distances the deviation was found well below 1%.
From sodium 1,2,4-triisopropylcyclopentadienide (3Cp) and the zirconium starting compound 1 the triisopropylcyclopentadienyl derivative [(C7H7)Zr{C5H2(i-C3H7)3-1,2,4}] (4) was obtained as purple crystals in 60% yield. The spectroscopic properties of the 1,2,4-triisopropylcyclopentadienyl ligand have been discussed before.27 Since the crystals were found unsuitable for X-ray diffraction, a structure was obtained by DFT calculations (Fig. 3). The calculation shows the expected structure of the complex. The isopropyl substituent in the 4-position is oriented in a way that one methyl carbon atom is close to the ring plane and the methyne proton points towards the zirconium atom to reduce steric strength. Resulting from this orientation, the cycloheptatrienyl ring is tilted towards the 4-position of the five-membered ring (CHTcent–Zr–Cpcent 174.20°).
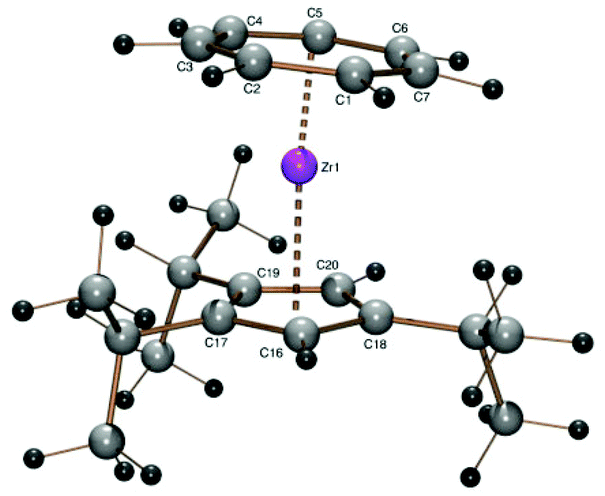 | ||
| Fig. 3 ORTEP plot of the calculated structure of 4. Calculated bond length (Å) and angles (°): Zr–C1 2.349, Zr–C2 2.360, Zr–C3 2.356, Zr–C4 2.346, Zr–C5 2.344, Zr–C6 2.339, Zr–C7 2.342, Zr–C16 2.508, Zr–C17 2.522, Zr–C18 2.515, Zr–C19 2.524, Zr–C20 2.505, Zr–Cpcent 2.205, Zr–CHTcent 1.679, CHTcent–Zr–Cpcent 174.20. | ||
Even with the almost insoluble sodium 2,3-diisopropyl-1,4-dimethylcyclopentadienide26 (22Cp) the reaction with the blue solution of 1 proceeded smoothly and afforded the sublimable sandwich complex [(C7H7)Zr{C5H(i-C3H7)2-2,3-(CH3)2-1,4}] (5) as purple crystals in 76% yield. 1H and 13C NMR spectra exhibit the signals expected for pairs of symmetry-equivalent methyl and isopropyl substituents (the latter with two diastereotopic methyl positions, cf. lit.27) as well as the CH signals for both ring ligands. The violet prisms of 5 obtained by sublimation at about 120 °C in a sealed glass tube at 10−3 mbar gave X-ray diffraction data of excellent quality (wR2 value 0.0473, all data, Table 1) (Fig. 4).
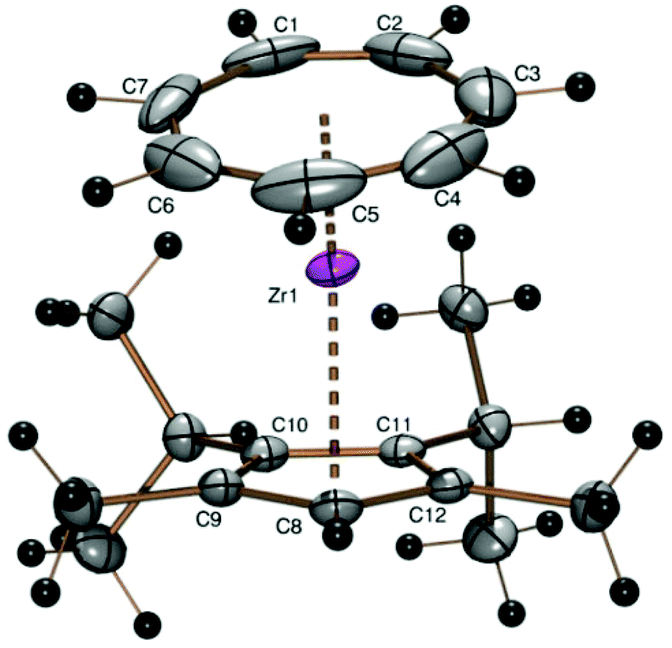 | ||
| Fig. 4 ORTEP plot of the molecular structure of 5. Atomic displacement parameters have been drawn at 50% probability. Selected bond length (Å) and angles (°): Zr–C1 2.349(2), Zr–C2 2.352(2), Zr–C3 2.345(2), Zr–C4 2.321(2), Zr–C5 2.310(2), Zr–C6 2.310(2), Zr–C7 2.325(2), Zr–C8 2.4714(16), Zr–C9 2.4928(17), Zr–C10 2.2.5358(16), Zr–C11 2.5164(16), Zr–C12 2.4998(16), Zr–Cpcent 2.192, Zr–CHTcent 1.677, CHTcent–Zr–Cpcent 167.98. | ||
| Complex | 2 | 5 | 6 | 8 | 10 |
|---|---|---|---|---|---|
| Formula | C27H36Zr | C20H28Zr | C23H36SiZr | C46H40OZr | C20H22Zr |
| Fw (g mol−1) | 451.78 | 359.64 | 431.83 | 700.00 | 353.60 |
| Crystal size (mm3) | 0.19 × 0.12 × 0.04 | 0.12 × 0.12 × 0.04 | 0.25 × 0.24 × 0.06 | 0.20 × 0.25 × 0.04 | 0.23 × 0.17 × 0.10 |
| Color, habit | Violet, plate | Violet, prism | Violet, plate | Violet, block | |
| Space group | Pna21 | Pbca | Cmca |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| Lattice parameters | |||||
| a (Å) | 20.8280(2) | 13.8451(1) | 14.3913(2) | 9.9259(6) | 12.2202(2) |
| b (Å) | 11.2237(1) | 15.7176(1) | 14.1131(2) | 10.2193(6) | 11.4037(2) |
| c (Å) | 18.7759(2) | 31.7801(3) | 21.5608(3) | 17.2817(9) | 11.7719(2) |
| α (°) | 90 | 90 | 90 | 91.290(4) | 90 |
| β (°) | 90 | 90 | 90 | 106.486(5) | 100.431(2) |
| γ (°) | 90 | 90 | 90 | 91.193 | 90 |
| Volume (Å3) | 4389.19(7) | 6915.72(9) | 4379.13(11) | 1679.81(17) | 1613.37(5) |
| Z | 8 | 16 | 8 | 2 | 4 |
| T (K) | 150(2) | 150(2) | 150(2) | 100(2) | 150(2) |
| ρ calc. (g cm−3) | 1.367 | 1.382 | 1.310 | 1.384 | 1.456 |
| μ (mm−1) | 4.147 | 5.117 | 4.633 | 2.949 | 5.482 |
| Transmission factors | 0.44798–1.00000 | 0.39483–1.00000 | 0.26508–1.00000 | 0.63893–1.00000 | 0.53445–1.00000 |
| Theta limits (°) | 4.25–62.78 | 4.924–62.66 | 4.10–62.68 | 4.33–75.93 | 3.68–62.62 |
| Total reflections | 17862 |
56677 |
14566 |
72660 |
9749 |
| Unique reflections | 6228 | 5527 | 1838 | 6975 | 2565 |
| Absorption correction | Semi-empirical | Semi-empirical | Semi-empirical | Semi-empirical | Semi-empirical |
| Structure solution | Direct methods | Direct methods | Direct methods | Direct methods | Direct methods |
| Program used | SIR92 | SIR92 | SIR92 | SIR92 | SIR92 |
| Refinement | SHELXL-97 | SHELXL-97 | SHELXL-97 | SHELXL-97 | SHELXL-97 |
| Data/restraints/parameters | 6228/106/597 | 5517/42/455 | 1838/8/130 | 6975/0/461 | 2565/36/229 |
| R 1, wR2 (I > 2σ(I)) | 0.0231, 0.0594 | 0.0182, 0.0466 | 0.0290, 0.0794 | 0.0358, 0.0900 | 0.0208, 0.0535 |
| R 1, wR2 (all data) | 0.0237, 0.0599 | 0.0208, 0.0473 | 0.0300, 0.0800 | 0.0371, 0.0914 | 0.0220, 0.0540 |
| GooF (all data) | 1.074 | 1.042 | 1.145 | 1.044 | 1.136 |
| Δρ (e Å−3) | 0.483, −0.251 | 0.266, −0.271 | 0.894, −0.393 | 0.850, −0.974 | 0.289, −0471 |
Due to the sterically demanding isopropyl substituents in 2- and 3-positions, the cycloheptatrienyl ligand is tilted towards the substitution gap of the cyclopentadienyl ring (167.98°). The Zr–CHTcent distance of 1.677 Å and the Cpcent–Zr distance of 2.192 Å are in the same range as for other complexes of the [(CHT)Zr(Cp)] type.
The steric bulk of the 22Cp ligand can be significantly enlarged by replacement of the only hydrogen atom connected to the five-membered ring by a fifth substituent. The trimethylsilyl derivative [(C7H7)Zr{C5(SiMe3)(i-C3H7)2-3,4-(CH3)2-2,5}] (6) could be obtained in 39% yield from the corresponding potassium 3,4-diisopropyl-2,5-dimethyl-1-trimethylsilylcyclo-pentadienide (K22CpTMS) and the zirconium starting compound 1. A discussion of proton and 13C NMR spectra of complex 6 can be found in the ESI.†
The crystal structure of the purple orthorhombic blocks obtained by sublimation of 6 in a sealed glass tube under vacuum at 120 °C shows the protruding silyl group (ring C–Si bond length 1.883 Å, Si–CH3 1.852(5) and 1.860(3) Å) and the almost parallel orientation of the ring ligands (Tables 1 and 2 and Fig. 5). The trimethylsilyl substituent is bent 3.78° out of the five-membered ring plane such as to reduce steric hindrance.
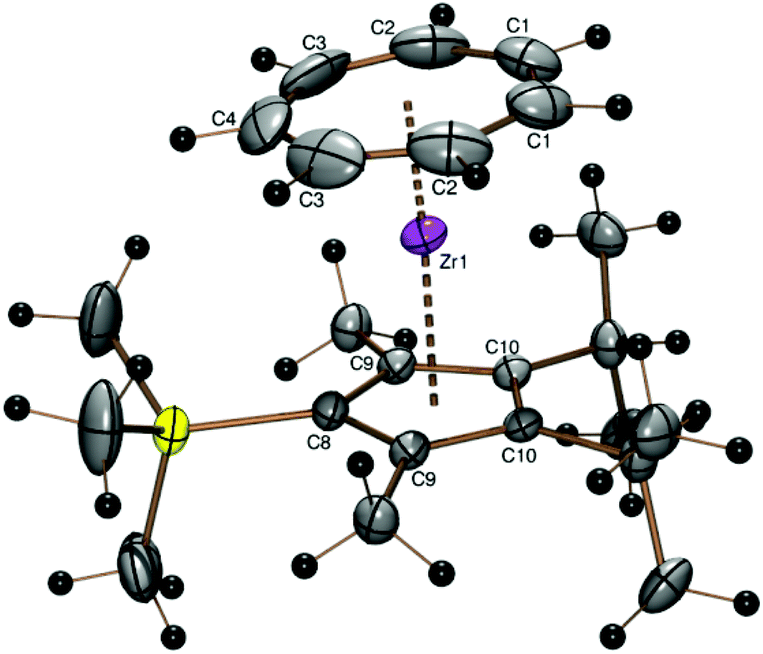 | ||
| Fig. 5 ORTEP plot of the molecular structure of 6. Atomic displacement parameters have been drawn at 50% probability. Selected bond length (Å) and angles (°): Zr–C1 2.356(3), Zr–C2 2.345(3), Zr–C3 2.322(3), Zr–C4 2.308(4), Zr–C8 2.488(3), Zr–C9 2.502(2), Zr–C10 2.5121(19), Zr–Cpcent 2.187, Zr–CHTcent 1.673, CHTcent–Zr–Cpcent 176.75. | ||
Equipped with a third isopropyl group, the 3,5-dimethyl-1,2,4-triisopropylcyclopentadienyl (32Cp) ligand could also be introduced using 1 and sodium 3,5-dimethyl-1,2,4-triisopropylcyclopentadienide (7-Na)28 to form the sandwich complex [(C7H7)Zr{C5(i-C3H7)3-1,2,4-(CH3)2}] (7) as a purple oil, which could not be crystallized, but analyzed quite well (Experimental part). A discussion of proton and 13C NMR spectra of complex 6 can be found in the ESI.† A structure model had to be calculated by DFT methods (Fig. 6 and Table 2) which shows a slightly bent sandwich structure with an interplanar angle of 8.6°.
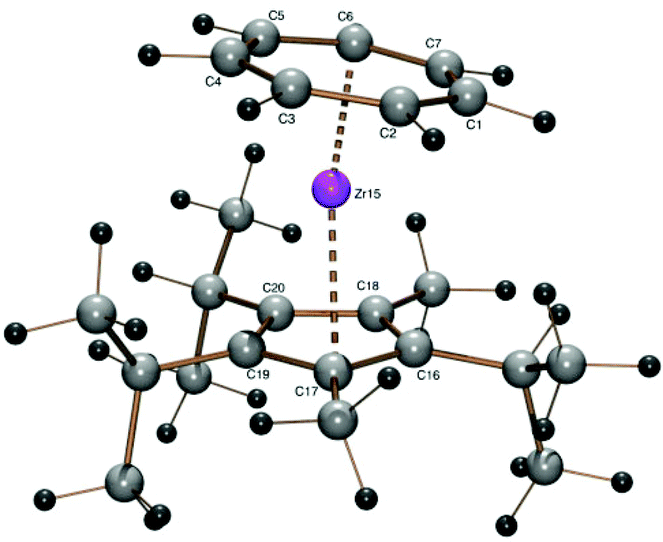 | ||
| Fig. 6 ORTEP plot of the calculated structure of 7. Calculated bond length (Å) and angles (°): Zr–C1 2.340, Zr–C2 2.349, Zr–C3 2.359, Zr–C4 2.366, Zr–C5 2.358, Zr–C6 2.346, Zr–C7 2.338, Zr–C16 2.518, Zr–C17 2.521, Zr–C18 2.515, Zr–C19 2.517, Zr–C20 2.506, Zr–Cpcent 2.202, Zr–CHTcent 1.683, CHTcent–Zr–Cpcent 172.05. | ||
The reaction of sodium pentaphenylcyclopentadienide with 1 was successful and afforded the sandwich complex [(C7H7)Zr(C5Ph5)] (8) as a purple solid, which is insoluble in hexane or toluene, sparingly soluble in tetrahydrofuran and moderately soluble in dichloromethane. 8 could be crystallized from saturated solutions in tetrahydrofuran by slow evaporation at ambient temperature. This result is interesting because the corresponding pentaisopropylcyclopentadienyl complex could not be obtained with lithium pentaisopropylcyclopentadienide.22 Another attempt at the synthesis of [(C7H7)Zr(C5(i-C3H7)5)] using potassium pentaisopropylcyclopentadienide was also unsuccessful. Thus, it can be supposed that the larger cone angle of the pentaisopropylcyclopentadienide in comparison with the pentaphenylcyclopentadienide is responsible for the failure of 1 to react, not the reactivity of the alkali cation (Fig. 7).
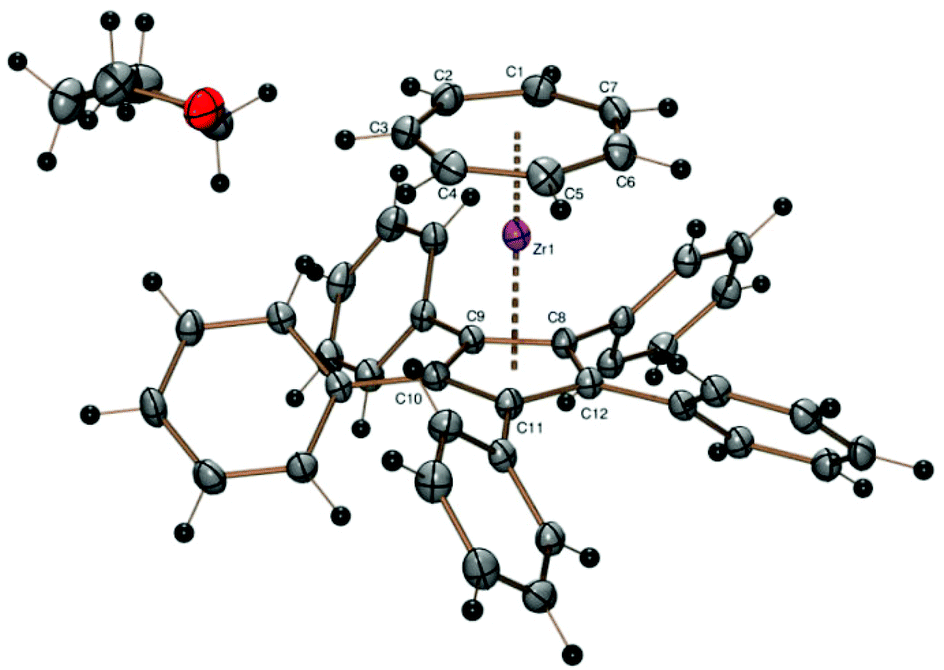 | ||
| Fig. 7 ORTEP plot of the molecular structure of 8. Atomic displacement parameters have been drawn at 50% probability. Selected bond length (Å) and angles (°): Zr–C1 2.339(2), Zr–C2 2.333(2), Zr–C3 2.338(2), Zr–C4 2.342(2), Zr–C5 2.335(2), Zr–C6 2.334(2), Zr–C7 2.339(2), Zr–C8 2.534(2), Zr–C9 2.536(2), Zr–C10 2.526(2), Zr–C11 2.528(2), Zr–C12 2.536(2), Zr–Cpcent 2.220, Zr–CHTcent 1.666, CHTcent–Zr–Cpcent 178.9. | ||
Through slow evaporation of a saturated THF solution of 8, crystals suitable for X-ray diffraction could be obtained.
During preparation and handling of pentaphenylcyclopentadiene (8-H)29 single crystals of this starting compound suitable for X-ray diffraction were obtained. The data for crystal structure determination were measured at room temperature and solved in P21/n with two independent molecules in the unit cell. In the literature29 there exists another structure of 1,2,3,4,5-pentaphenylcyclopentadiene with one independent molecule in a cell with almost the same cell constants b = 6.267(1) Å, c = 24.517(8) Å, β = 93.49(2)° as the structure determined in our laboratory with a = 24.3208(8) Å, b = 6.2097(2) Å, β = 93.649(3)°, except for c (previously 15.954(4) Å, we found c = 31.4383(10) Å). Careful analyses of both structures provided no indication of any obvious mistake in the structure determination. A possible explanation would be a slow phase transformation without any destructive effects on the crystal upon cooling from room temperature to −173 °C.
The introduction of a butyl chain in the para position of one of the phenyl rings of the pentaphenylcyclopentadienyl ligand was accomplished as described for the tert-butyl derivative.30 Indeed the purple complex [(C7H7)Zr{C5Ph4(C6H4nBu-4)}] (9) obtained from the potassium salt of 4-butylphenyl-tetraphenylcyclopentadiene and 1 displays better solubility and could be extracted with toluene from the product mixture. After the toluene solution was taken to dryness, the purple residue could be washed with pentane because complex 9 is almost insoluble in pentane. Crystallization of 9 was not successful from saturated toluene solutions at low temperatures. The product could only be obtained as a fine, purple powder. Thus, a crystal structure of 9 could not be obtained and the structure had to be calculated with DFT methods. This calculation reveals a Zr–CHTcent distance of 1.679 Å and a Zr–Cpcent distance of 2.233 Å which are both slightly larger than the corresponding values for the pentaphenyl derivative 8. Since one para-nBu substituent is not expected to enhance the steric demand of the pentaphenylcyclopentadienyl ligand, the higher values for the centroid–Zr distances might result from the calculation method, as mentioned before (Fig. 8).
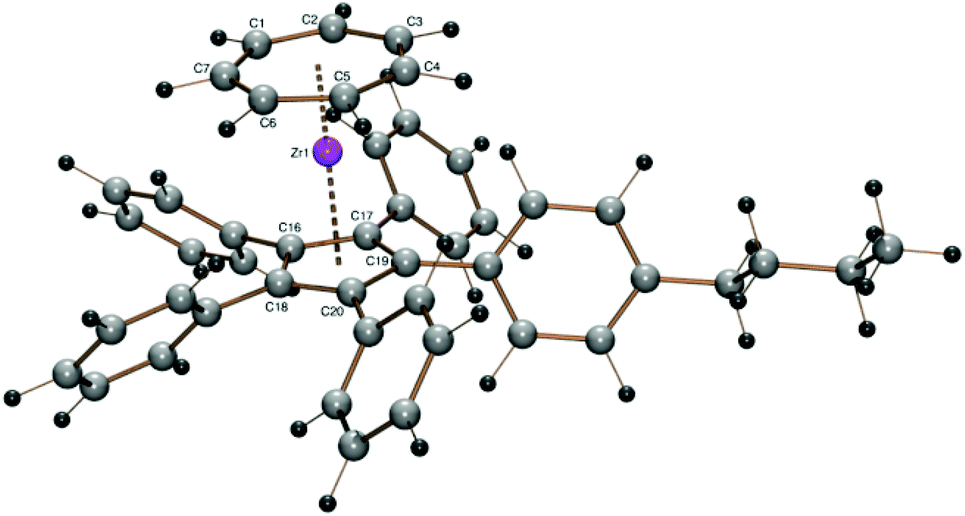 | ||
| Fig. 8 ORTEP plot of the calculated structure of 9. Calculated bond length (Å) and angles (°): Zr–C1 2.345, Zr–C2 2.351, Zr–C3 2.348, Zr–C4 2.346, Zr–C5 2.349, Zr–C6 2.347, Zr–C7 2.342, Zr–C16 2.541, Zr–C17 2.542, Zr–C18 2.544, Zr–C19 2.542, Zr–C20 2.541, Zr–Cpcent 2.233, Zr–CHTcent 1.679, CHTcent–Zr–Cpcent 179.43. | ||
While the cycloheptatrienylzirconium complex of the 1,3-di(tert-butyl)indenyl ligand as well as of the unsubstituted indenyl ligand have been investigated previously,22 the corresponding mono(tert-butyl) derivative has been prepared from 1 and sodium 1-tert-butylindenide and could be crystallized as purple needles from pentane in 47% yield. A discussion of proton spectra of complex 10 can be found in the ESI.† The chiral molecules crystallize as a racemate in the space group P21/c, where the enantiomers are symmetry-related by a crystallographic inversion center (Fig. 9).
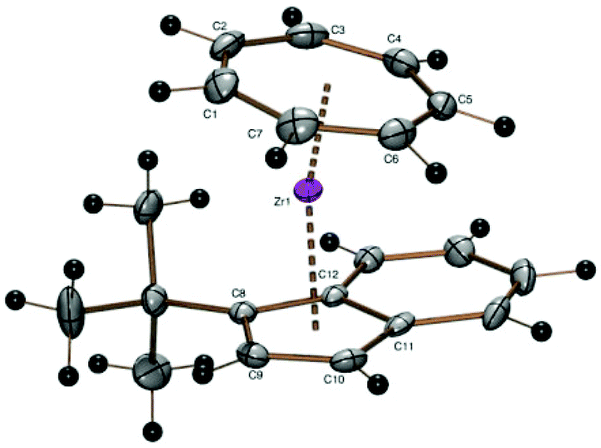 | ||
| Fig. 9 ORTEP plot of the molecular structure of 10. Atomic displacement parameters have been drawn at 50% probability. Selected bond length (Å) and angles (°): Zr–C1 2.345(2), Zr–C2 2.354(2), Zr–C3 2.349(2), Zr–C4 2.319(2), Zr–C5 2.312(2), Zr–C6 2.319(2), Zr–C7 2.334(2), Zr–C8 2.507(2), Zr–C9 2.492(2), Zr–C10 2.494, Zr–C11 2.5335(19), Zr–C12 2.544(2), Zr–Cpcent 2.202, Zr–CHTcent 1.661, CHTcent–Zr–Cpcent 167.55. | ||
As expected, the CHT ligand is tilted towards the unsubstituted side of the indenyl ligand (CHTcent–Zr–Cpcent 167.55°).
Cone angle measurement
From crystallographically determined and theoretically calculated structural data, for each complex two cone angles were derived as outlined in an earlier publication.22 The angle Θ is the cone angle of the cyclopentadienyl ligand with the zirconium atom as the apex and the center of the outermost hydrogen of the substituents (with the maximum cone angle θi) as the limit (Scheme 4, top). The second angle Ω is defined as the cone angle of the substituents of the Cp ligand and thus the steric demand of the ligand in the z-dimension (Scheme 4, bottom). | ||
| Scheme 4 Measurement of the angles θi and ωi leading to Θ (top) and Ω (bottom). The large sphere represents the metal and the small spheres represent the methyl hydrogens. The relevant group is not necessarily directly bonded to the ring, nor does it necessarily lie in the ring plane. For each substituent the outermost methyl hydrogen, corresponding to the largest θi value, is chosen. | ||
The cone angles Θ and Ω of the complexes 2–10 reported in this manuscript as well as those reported earlier22 are listed in Table 3. In comparison with the triisopropylcyclopentadienyl ligand (3Cp) the tricyclopentylcyclopentadienyl ligand occupies an almost identical, but slightly smaller cone angle Θ of 131.7° vs. 132.6°. This tendency is more pronounced, when the cone angles of one isopropyl group Ω (89.1°) and the cyclopentyl group (86.1°) at the cyclopentadienyl ligand are compared. The reason for the slim appearance of the cyclopentyl group is ring strain visible in the smaller Cβ–Cα–Cβ angles of the cyclopentyl substituent (typically between 101.5° and 102.5° in complex 2) compared to the same angle for the isopropyl substituents of complexes 5 and 6 (between 110.5° and 111.5°). The cone angle Θ of the tricyclohexylcyclopentadienyl ligand (134.8°) is larger than that of the tricyclopentylcyclopentadienyl ligand (131.7°) and even slightly larger than that of the triisopropylcyclopentadienyl ligand (132.6°). For the 22Cp ligand, the cone angle Θ is comparable even to Cpchex (Table 3), but the ring tilt in 5 is larger than that in the tris(secondary alkyl)cyclopentadienyl complexes 2–4 (Table 2) because the steric bulk of the 22Cp ligand is concentrated on one edge of the five-membered ring and rather small on the other side of the ring.
| Complex | Indenyl | Θ | Ω |
|---|---|---|---|
| a Calculated for a given substituent by measuring the maximum cone angle of each group in the α-C of the substituent with the ipso-C atom as the apex, the averaged. In the case of iPr and chexyl the C–H proton was included in the calculation. b Two independent molecules in the asymmetric unit. c See ref. 22 for details. d Data are based on the calculated structure. | |||
| C9H7 | 102.6 | 0 | |
| 10 | Ind′ | 119.0 | 101.0 |
| , | Ind′′ | 130.7/131.8 | 101.3/100.2 |
| , | Indchex | 131.0/131.7 | 84.9/84.2 |
The calculated cone angle Θ of the 32Cp ligand (150.2°) is practically identical to that of 22CpTMS (150.3°).
The cone angle Θ of 167.4° for the pentaisopropylcyclopentadienyl ligand is significantly larger than the 157.4° angle for the pentaphenylcyclopentadienide. This could be an explanation for the failure of 1 to react; another alkali cation does not make a difference.
The calculated cone angle Θ (163.8°) of the monobutyl derivative of pentaphenylcyclopentadienide (CpPh) is significantly larger than the value found for the pentaphenyl derivative 8 (157.4°), which is due to the conformation of the ligand and varies with the rotational orientation of the phenyl ring planes relative to the cyclopentadienyl plane. This cone angle should be viewed with caution. There is no obvious reason why one butyl group in the para position should significantly increase the steric bulk the pentaphenylcyclopentadienyl ligand presents to the central atom. The cone angle Θ of the 1-tert-butylindenyl ligand was determined to be 119° and is in the expected range between indenyl (102.6°) and 1,3-di(tert-butyl)indenyl (ca. 131°, see Table 3).
Comparison of the values for the cyclopentadienyl ligands listed in the table above results in the following order of bulk:
C5H5 < C5H4CH3 < C9H7 ≈ C5H4Si(CH3)3 ≈ C5H4(allyl) < Cp′′ < Ind′ < C5(CH3)5 < 22Cp < Cpcpent ≈ 3Cp ≈ Cpchex < 22CpTMS < 32Cp < Ind′′ ≈ Indchex ≈ Cp′′′ < CpPh < 4Cp < CpBIGc < 5Cp
If the cone angle of the cycloheptatrienyl ligand is calculated according to the equation Θ = (Θ1 + Θ2 + Θ3 + Θ4 + Θ5 + Θ6 + Θ7) × 2/7, values of 117–118° are obtained, which place the steric demand of the CHT ligand between those of 1,3-di(tert-butyl)cyclopentadienyl and 1-tert-butylindenyl ligands.
Conclusion
The cycloheptatrienylzirconium fragment accommodates cyclopentadienyl ligands as bulky as tetraisopropylcyclopentadienyl or pentaarylcyclopentadienyl without steric interactions with the cycloheptatrienyl ligand. Up to now sixteen derivatives of the [(CHT)Zr(Cp)] type with different cyclopentadienyl or indenyl ligands coordinated to the cycloheptatrienylzirconium fragment crystal structures have been obtained and used for data compilation.The cone angle Θ of the substituted cyclopentadienyl ligand alone in some cases leads to unrealistic conclusions. Tetraisopropylcyclopentadienide, for instance, is bulkier than triisopropyl-dimethylcyclopentadienide, but the cone angles are reverted. Similarly, diisopropyl-dimethylcyclopentadienide, triisopropylcyclopentadienide, and tri(tert-butyl)cyclopenta-dienide exhibit very similar cone angles Θ for the ligand, but the cone angles Ω of the substituents are different. It is therefore necessary to take both values into account when comparing different cyclopentadienyl derivatives. The cone angles found experimentally for the pentaphenylcyclopentadienyl ligand and by DFT calculations for its close relative CpBIG with just one n-butyl group added in the para position of one phenyl ring differing by ca. 6° are due to different torsion angles of the substituents. This discrepancy may arise from crystal packing forces or may be sensitive to the basis sets used for the calculations.
Experimental section
General
All preparations and spectroscopic manipulations of air and moisture sensitive compounds were carried out under an atmosphere of nitrogen or argon, using Schlenk line techniques and a glove box (MBraun company, Garching) filled with argon. Solvents were rigorously dried and deoxygenated by distillation under nitrogen with molten potassium (tetrahydrofuran) or sodium/potassium alloy (pentane, benzene-d6) and distilled prior to use. Bruker Avance 400 and Avance 600 spectrometers were used to obtain NMR spectra which were referenced to the residual solvent signal (C6D6: 7.16 ppm). The chemical shifts δ are given in ppm and the coupling constants J are given in Hz. Elemental analyses were carried out in the analytical laboratory of the chemical department of the TU Kaiserslautern, using a Vario Micro Tube of Elementar Analysentechnik/Hanau. Crystal structures were obtained by XRD measurements on an Oxford Diffraction Gemini Ultra diffractometer.The sodium salts NaCpchex,32 Na3Cp,27 Na22Cp,26 Na32Cp28 and NaCpPh31 as well as (C7H7)Zr(Cl)(tmeda)33 were prepared according to literature procedures; a sample of NaCpBIGc was kindly provided by Professor S. Harder.
DFT calculations
For the DFT calculations of the zirconium complexes a B3LYP functional of the Gaussian09, Revision C.0134 was used. A triple zeta basis (6-311G**)35 for the main group elements (C, H) was combined with a double zeta basis (Stuttgart RSC 1997 ECP)36 for zirconium, which also includes the effective core potential.Synthetic work
Acknowledgements
H. S. is grateful to Professor Scherer for many years of friendly support. The authors thank Professor S. Harder for providing a sample of the sodium salt NaCpBIGc.Notes and references
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632–635 RSC.
- P. Laszlo and R. Hoffmann, Angew. Chem., 2000, 112, 127–128 ( Angew. Chem., Int. Ed. , 2000 , 39 , 123–124 ) CrossRef.
- G. Wilkinson, J. Organomet. Chem., 1975, 100, 273–278 CrossRef CAS.
- E. O. Fischer and W. Pfab, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1952, 7, 377–379 Search PubMed.
- H. Röhl, E. Lange, T. Gößl and G. Roth, Angew. Chem., Int. Ed. Engl., 1962, 74, 155 Search PubMed.
- R. B. King and A. Efraty, J. Am. Chem. Soc., 1972, 94, 3773–3779 CrossRef CAS.
- C. Janiak and H. Schumann, Adv. Organomet. Chem., 1991, 33, 291–393 CrossRef CAS.
- J. Okuda, Top. Curr. Chem., 1991, 160, 97–145 CrossRef.
- W. Kaminsky, Rapra Rev. Rep., 2000, 10, 1–136 Search PubMed.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956–2965 CrossRef CAS.
- Literature search was carried out in August 2013, only compounds with experimental properties have been counted.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS . This article received more than 3700 citations, is still earning more than one hundred citations per year and has been referred to in many textbooks on inorganic chemistry.
- P. C. Möhring and N. J. Coville, J. Organomet. Chem., 1994, 479, 1–29 CrossRef.
- P. C. Möhring and N. J. Coville, J. Mol. Catal., 1992, 77, 41–50 CrossRef.
- P. C. Möhring and N. J. Coville, J. Mol. Catal., 1995, 96, 181–195 CrossRef.
- P. C. Möhring, M. Vlachakis, N. E. Grimmer and N. J. Coville, J. Organomet. Chem., 1994, 483, 159–166 CrossRef.
- C. Janiak, U. Versteeg, K. C. H. Lange, R. Weimann and E. Hahn, J. Organomet. Chem., 1995, 501, 219–234 CrossRef CAS.
- C. Janiak, K. C. H. Lange, U. Versteeg, D. Lentz and P. H. M. Budzelaar, Chem. Ber., 1996, 129, 1517–1529 CrossRef CAS PubMed.
- D. White and N. J. Coville, Adv. Organomet. Chem., 1994, 36, 95–158 CrossRef CAS.
- P. C. Möhring and N. J. Coville, Coord. Chem. Rev., 2006, 250, 18–35 CrossRef PubMed.
- N. J. Coville, M. S. Loonat, D. White and L. Carlton, Organometallics, 1992, 11, 1082–1090 CrossRef CAS.
- A. Glöckner, H. Bauer, M. Maekawa, T. Bannenberg, C. G. Daniliuc, P. G. Jones, Y. Sun, H. Sitzmann, M. Tamm and M. Walter, Dalton Trans., 2012, 41, 6614–6624 RSC.
- A. Glöckner and M. Tamm, Chem. Soc. Rev., 2013, 42, 128–142 RSC.
- D. Weismann, D. Saurenz, R. Boese, D. Bläser, G. Wolmershäuser, Y. Sun and H. Sitzmann, Organometallics, 2011, 30, 6351–6364 CrossRef CAS.
- C. G. Venier and E. W. Casserly, J. Am. Chem. Soc., 1990, 112, 2808–2809 CrossRef CAS.
- S. Schäfer, H. Bauer, J. Becker, Y. Sun and H. Sitzmann, Eur. J. Inorg. Chem., 2013, 5694–5700 CrossRef PubMed.
- H. Sitzmann, J. Organomet. Chem., 1988, 354, 203–214 CrossRef CAS.
- V. Quindt, D. Saurenz, O. Schmitt, M. Schär, T. Dezember, G. Wolmershäuser and H. Sitzmann, J. Organomet. Chem., 1999, 579, 376–384 CrossRef CAS.
- L. D. Field, T. W. Hambley, C. M. Lindall and A. F. Masters, Inorg. Chem., 1992, 31, 2366–2370 CrossRef CAS.
- H. Schumann, C. Janiak and H. Khani, J. Organomet. Chem., 1987, 330, 347–355 CrossRef CAS.
- R. Zhang, M. Tsutsui and D. E. Bergbreiter, J. Organomet. Chem., 1982, 229, 109–112 CrossRef CAS.
- J. A. Burman, M. L. Hays, D. J. Burkey, P. S. Tanner and T. P. Hanusa, J. Organomet. Chem., 1994, 479, 135–139 CrossRef CAS.
- A. Glöckner, T. Bannenberg, M. Tamm, A. M. Arif and R. D. Ernst, Organometallics, 2009, 28, 5866–5876 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Menucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylow, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09 (Revision C.01), Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS PubMed.
- (a) A. Bergner, M. Dolg, W. Kuechle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS; (b) M. Kaupp, P. v. R. Schleyer, H. Stoll, H. Preuss and R. M. Pitzer, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the electronic structure calculations, X-ray data, the determination of Θ and Ω, and discussions of NMR spectra of five complexes. CCDC 1012756–1012760. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01581a |
| This journal is © The Royal Society of Chemistry 2014 |