
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NO-binding in {Ru(NO)2}8-type [Ru(NO)2(PR3)2X]BF4 compounds†
Anna K. E.
Gallien
a,
Dominik
Schaniel
*bc,
Theo
Woike
d and
Peter
Klüfers
*a
aFakultät für Chemie und Pharmazie, Butenandtstraße 5-13, 81377 Munich, Germany. E-mail: kluef@cup.uni-muenchen.de; Fax: +49 (0)89 2180 77407; Tel: +49 (0)89 2180 77404
bUniversité de Lorraine, CRM2, UMR 7036, Vandoeuvre les Nancy, F-54506, France. E-mail: dominik.schaniel@univ-lorraine.fr; Fax: +33 (0)383 68 43 00; Tel: +33 (0)3 83 68 48 70
cCNRS, CRM2, UMR 7036, Vandoeuvre les Nancy, F-54506, France
dTU Dresden, Institut für Strukturphysik, Zellescher Weg 16, 01069 Dresden, Germany. E-mail: theo.woike@tu-dresden.de; Fax: +49 (0)351 463 37048; Tel: +49 (0)351 463 32536
First published on 14th July 2014
Abstract
Two different structure types were found for a series of mononuclear dinitrosyl complexes of the general formula [RuL2(NO)2X]BF4 (L = monodentate phosphane, X = Cl, Br, I). The {Ru(NO)2}8-type target compounds were prepared by the reduction of the respective {RuNO}6 precursors and subsequent oxidative addition of (NO)BF4. About one half of the new compounds share their molecular structure with the hitherto only representative of this class of dinitrosyls, Pierpont and Eisenberg's [RuCl(NO)2(PPh3)2]PF6·C6H6 (Inorg. Chem., 1972, 11, 1088–1094). The Cs-symmetric cations exhibit both a linear and a bent Ru–N–O fragment, in line with a formal 6 + 2 split of the {Ru(NO)2}8 electron sum in the sense of a [RuII(NO+)(1NO−)]2+ bonding. The coordination entity's configuration in this subgroup is described by IUPAC's polyhedral symbol SPY-5. Continuous shape measures (CShM) as defined by Alvarez et al. (Coord. Chem. Rev., 2005, 249, 1693–1708) reveal a uniform deviation from the L–M–L angles expected for SPY-5, in a narrower sense, towards a vacant octahedron (vOC-5). DFT calculations confirmed that Enemark and Feltham's analysis (Coord. Chem. Rev., 1974, 13, 339–406) of the electronic situation of the {Ru(NO)2}8 group remains adequate. The same holds for the second subclass of new compounds the existence of which had been predicted in the same paper by Enemark and Feltham, namely C2v-symmetric, TBPY-5-type cations with two almost equally bonded nitrosyl ligands. In agreement with an 8 + 0 distribution of the relevant electrons, the formal [Ru0(NO+)2]2+ entities are found for L/X couples that donate more electron density on the central metal. Two solid compounds (8a/b, 12a/b) were found in both structures including the special case of the PiPr3/Br couple 12a/b, which led to crystals that contained both structure types in the same solid. Conversely, four compounds showed a single form in the solid but both forms in dichloromethane solution in terms of the solutions’ IR spectra. The irradiation of crystalline 12 with blue laser light resulted in the photoisomerisation of, mainly, the bent 1NO− ligand in terms of low-temperature IR spectroscopy.
Introduction
As nitrogen monoxide is a “non-innocent”, that is, redoxactive, potentially ambident ligand produced endogenously from L-arginine via nitric-oxide-synthase (NOS) catalysis in physiological signal transduction pathways, this simple molecule is one of the most interesting and challenging ligands in bioinorganic and coordination chemistry.1The redox activity is due to the radical character of nitrogen monoxide which enables its participation in metal complexes in four different binding modes exhibiting different M–N–O angles: strongly bent (ca. 120°) as 1NO− in a low-spin complex, weakly bent (ca. 140°) as a neutral 2NO˙ radical in a low-spin complex, (almost) linear as either a 3NO− diradical in a high-spin complex or a formal 1NO+, the “formal” emphasising the high π acidity of the 1NO+ ligand which hardly is a cation due to extensive back-bonding.
The analysis of the electronic structure of an M–N–O moiety is usually based on the spectroscopic and X-ray-crystallographic data of the compound in question. In order to enable a classification of nitrosyl complexes independent of their actual bonding modes, the Enemark–Feltham notation was introduced, wherein the sum of the metal d electrons and the electrons in the π*(NO) orbital(s) is noted.2 In this context, the dinitrosyl compounds of this work are of the {Ru(NO)2}8 type.
In the electronic and structural ground state (GS), the NO ligand of related NO+-type mononitrosyl complexes is κN-bonded. Upon irradiation with light, the metal–nitrosyl bond may be cleaved, resulting in the common photochemical property of NO release, which is used in PDT (photodynamic therapy) to liberate NO in a controlled manner at the target tissue.3,4 As a more intricate type of excitation decay, the potentially ambident NO ligand may be switched into metastable bonding modes instead of dissociating from the metal centre. As a result, two such metastable modes have been detected in solid samples: the κO-bonded, isonitrosyl, MS1 state and the κ2N,O-bonded, side-on, MS2 state (Fig. 1 shows these modes for {RuNO}6 centres).5–7
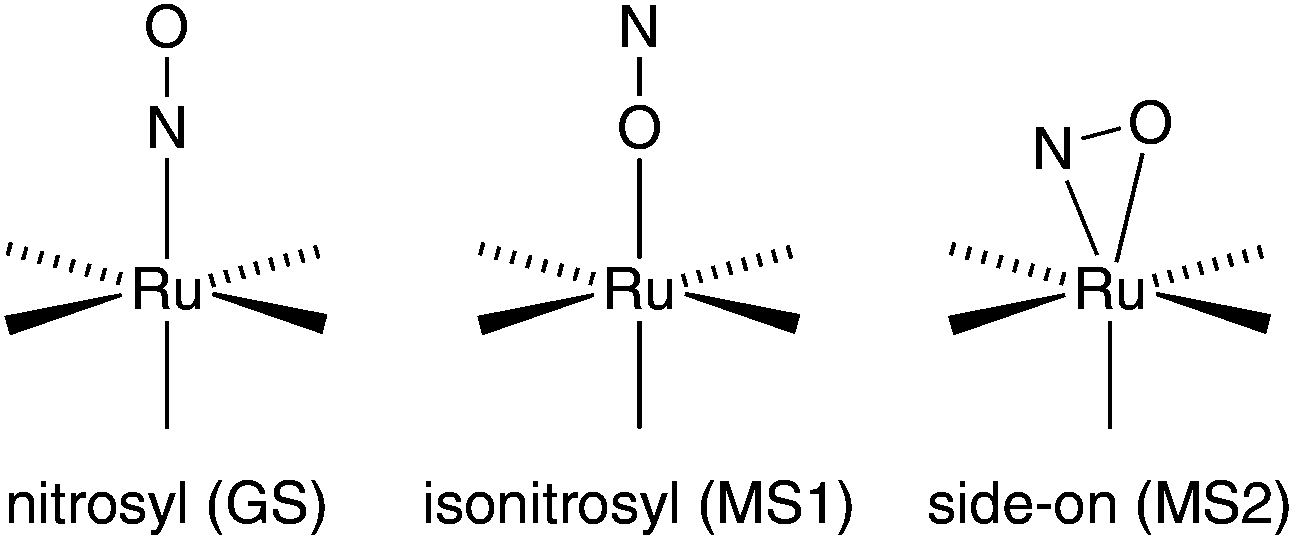 | ||
| Fig. 1 Schematic illustration of the GS, MS1 and MS2 bonding mode in octahedral {RuNO}6 complexes. | ||
This phenomenon is referred to as photoinduced linkage isomerism (PLI). Since the excited states are long-lived below their specific temperature of decay, they can be detected and analysed via low-temperature IR spectroscopy, photocrystallography and DSC (differential scanning calorimetry). PLI has been well studied for {RuNO}6 compounds, in which a single NO+-type ligand is attached linearly to the metal centre. A 1NO−-type nitrosyl has been described as photo-excitable as well, but, until now, for a {PtNO}8 mononitrosyl complex only.8,9 So far, photoinduced phenomena have not been investigated for {Ru(NO)2}8 dinitrosyl compounds.5,10–12 Moreover, ruthenium–dinitrosyl chemistry appears largely underdeveloped in general, particularly in contrast to the chemistry of the homologous dinitrosyl iron compounds (DNICs) which are mostly of the {Fe(NO)2}9 and {Fe(NO)2}10 type.13 (For DNICs, the PLI issue has been addressed as well.14) The mere number of only six published {Ru(NO)2}n-type compounds, four of which are of the {Ru(NO)2}8 type (A–D in Fig. 2), underlines this statement. In terms of structural and spectroscopic parameters, the electronic state of these Ru(NO)2 moieties is characterised by one NO+ and one 1NO− ligand (Fig. 2).
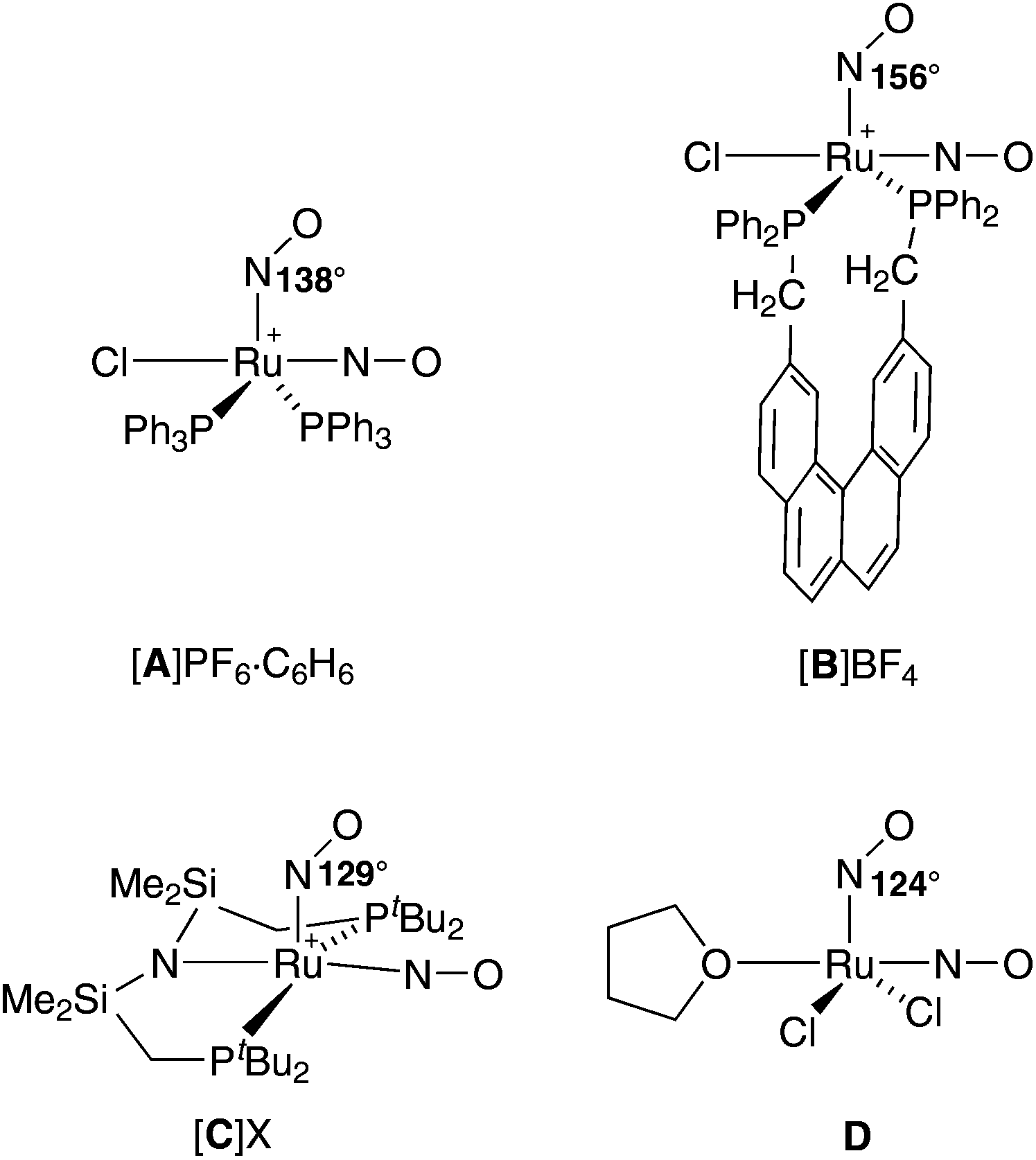 | ||
| Fig. 2 Known {Ru(NO)2}8 dinitrosyls (charges are drawn at the central metal atom); several publications deal with salts of A, including a tetrafluoridoborate with a 14NO/15NO couple.15,17–19 The “X” in [C]X is [Ru(NO)(OH)(NO2)2Cl2].20–22 Ru–N–O angles are given for the bent RuNO moiety. | ||
The issue of NO+/1NO− equilibration in A has attracted considerable interest in the past. On the one hand, in crystals of A, an easily distinguishable NO+/1NO− couple in a square pyramidal complex cation prevails. On the other hand, a dynamic interconversion of the nitrosyl bonding modes within the Ru(NO)2 moiety was detected by 15N NMR spectroscopy. Moreover, the symmetrised intermediate was not a transition state but a well populated intermediate in solution.15 A prototype for the trigonal bipyramidal structure of the intermediate was provided by a related osmium homologue that showed this structure in the solid.16
In this work, we extend the class of halogenido-bis(phosphane)-type {Ru(NO)2}8 compounds by new members with the goal of investigating the photoexcitability of dinitrosyl complexes. Specifically, we address some open questions of nitrosyl bonding and activation: (1) is the NO+/1NO− couple the usual bonding mode in {Ru(NO)2}8 dinitrosyls with the equilibrated form as a less stable intermediate? (2) Are {Ru(NO)2}8 dinitrosyls photoexcitable? (3) If so, which type of nitrosyl ligand, NO+ or NO−, is the switched one? To answer these questions, a series of bis-phosphane complex salts of the formula [Ru(NO)2(PR3)2X]BF4 (X = Cl, Br, I) is presented. This report focuses on the first of the three goals. The issues (2) and (3) are addressed for one of the new compounds to demonstrate the principal qualification of the new dinitrosyl complexes for photophysical research. Photoexcitation studies on other members will be published in a separate work in a more physical context.
Results and discussion
Synthesis
The synthesis of the target compounds followed the reaction scheme outlined in Fig. 3. The first step was the coordination of two equivalents of the respective phosphane (Fig. 3) to a {RuNO}6 fragment in a trans-configuration. Depending on the phosphane, the reaction takes place either as a simple ligand substitution (route i) or as a redox reaction with the simultaneous addition of two equivalents of the respective phosphane (route ii). In the next step (iii), the respective {RuNO}n (n = 6, 7, 8) fragment was treated with a zinc/copper alloy to ensure that all {RuNO}n mixtures were uniformly reduced to the {RuNO}8 state. In the last step iv, the second nitrosyl ligand was introduced by the reaction of the {RuNO}8 intermediate with NOBF4 to form the attempted pentacoordinate dinitrosyl complex of the general formula [RuX(NO)2(PR3)2]BF4.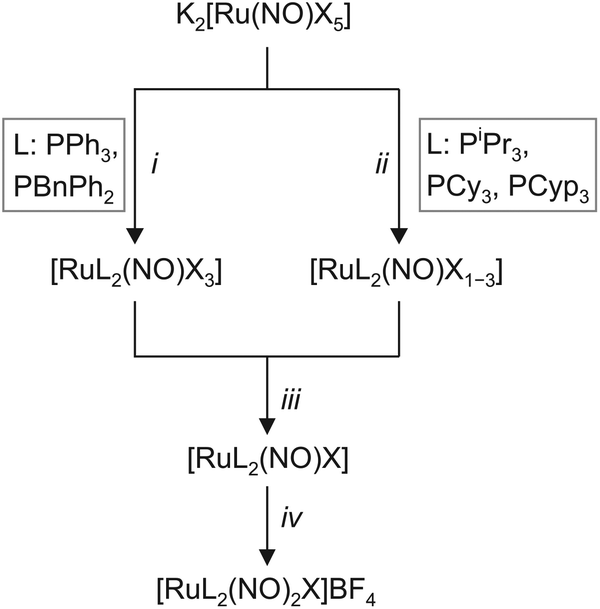 | ||
| Fig. 3 Synthetic route to the [RuL2(NO)2X]BF4 dinitrosyls of this work. The employed phosphanes differ by their electronic and steric properties as given by their Tolman's parameters (ν in cm−1 and cone angle): triphenylphosphane (PPh3) 2068.9, 145°; benzyldiphenylphosphane (PBnPh2) 2068.4, 152°, triisopropylphosphane (PiPr3) 2059.2, 160°; tricyclohexylphosphane (PCy3) 2056.4, 170°; no values were available for tricyclopentylphosphane (PCyp3). Details of the individual steps: i 2.5–3 eq. L, EtOH–H2O; ii 2.5–4 eq. L, EtOH–H2O; iii ZnxCu, toluene; iv (NO)BF4, toluene–EtOH. In the ii step, partial reduction of the Ru species by the more electron rich phosphanes took place prior to the final reduction step iii, hence the [RuL2(NO)X1–3] formulation. Further details are collected in the Experimental section. | ||
Crystalline products, including some as yet unknown iodido and bromido {Ru(NO)2}8 derivatives, were obtained by covering dichloromethane solutions with a layer of diethyl ether or n-pentane. The isolated crystals’ colour depended on the halide, ranging from yellow to reddish brown.
Structural data
Crystal structure analyses revealed two structure types for the monocationic, pentacoordinate coordination entities: first, a vacant octahedron (vOC-5) with linear/bent Ru–N–O couples, and, second, a trigonal bipyramid (TBPY-5) with equilibrated Ru–N–O angles. An overview of the compounds, their numbering and the adopted structure type is given in Fig. 4.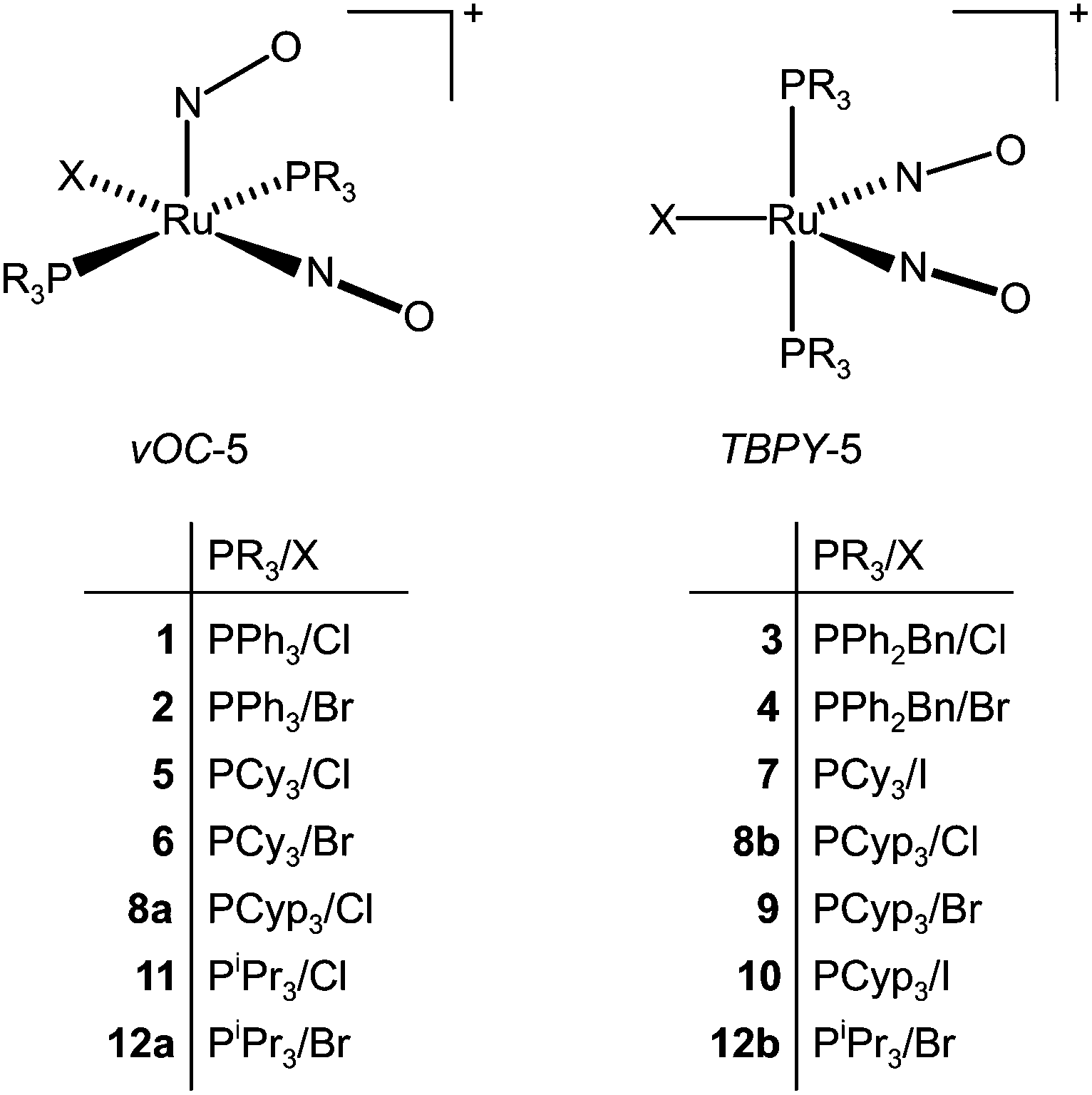 | ||
| Fig. 4 The crystalline compounds of this work and their structure type. The abbreviations TBPY-5 and vOC-5 refer to IUPAC's configuration index and the definitions used by the Alvarez group in the context of Continuous Shape Measures (CShM).23 | ||
To illustrate the structural principles, the cations in crystals of 1, 3, 8a/b, 9, 10, and 12a/b are depicted in Fig. 5–12. Significant bond distances and angles are collected in Table 1. Crystallographic data for all compounds as well as drawings and metrical parameters for 2, 4, 5–7 and 11 are collected in the ESI.†
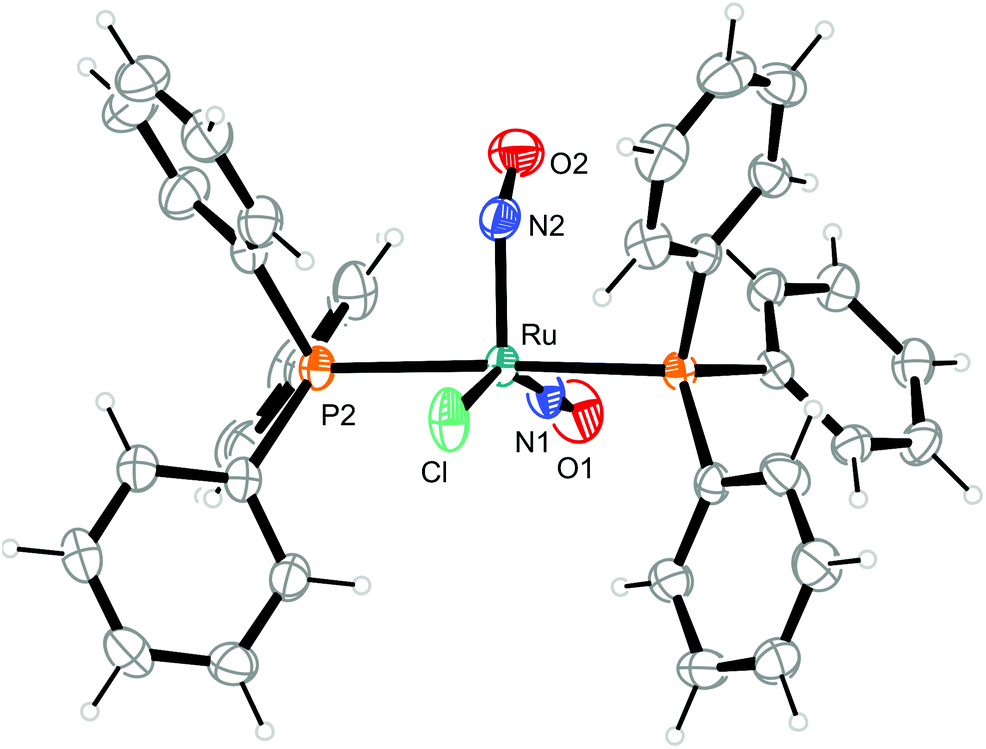 | ||
| Fig. 5 Structure (50% ellipsoids) of the vOC-5-type complex cation in crystals of [RuCl(NO)2(PPh3)2]BF4 (1). | ||
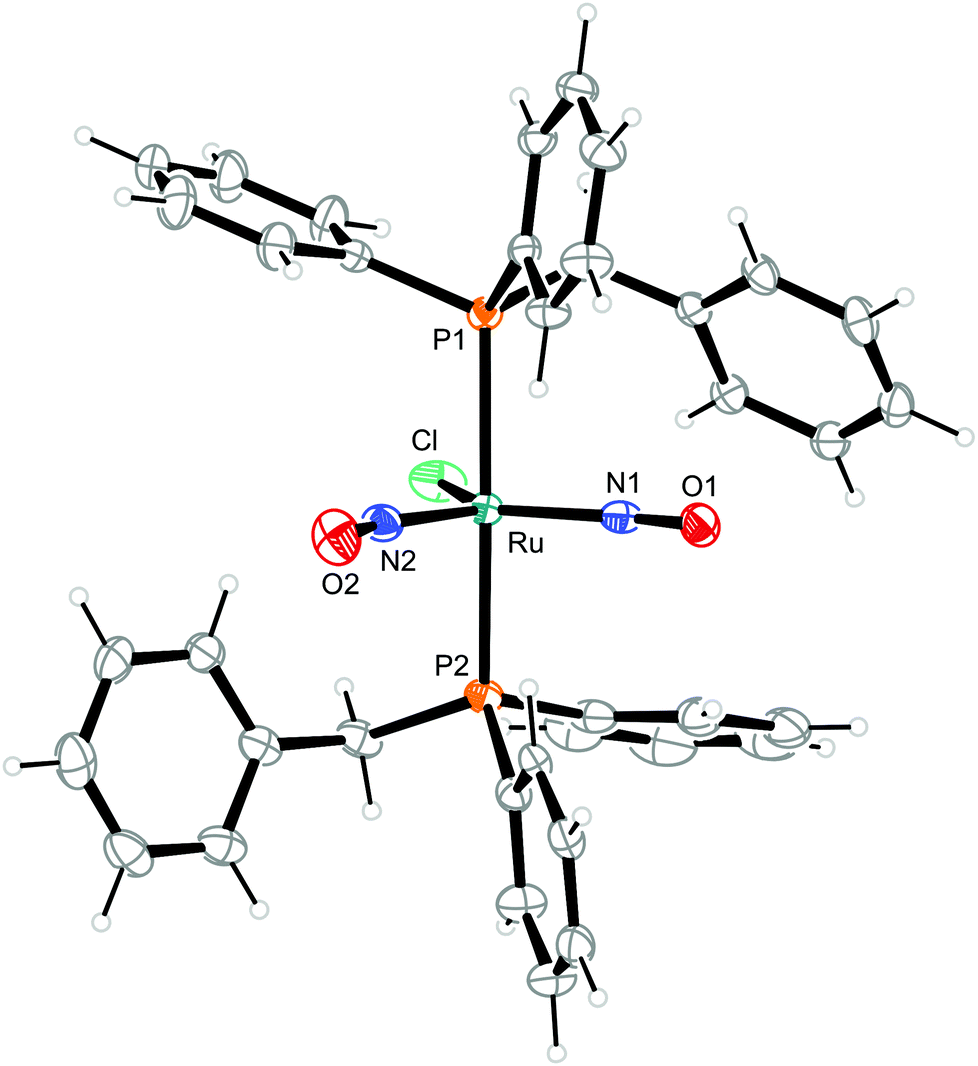 | ||
| Fig. 6 Structure (50% ellipsoids) of the TBPY-5-type complex cation [RuCl(NO)2(PPh2Bn)2]+ in crystals of 3. | ||
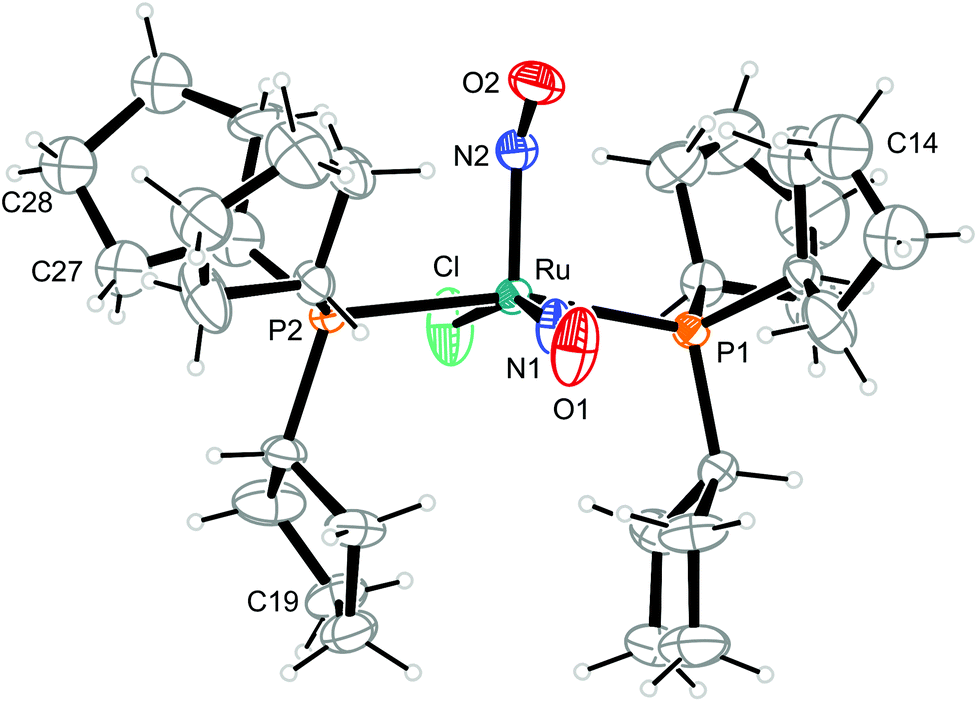 | ||
| Fig. 7 Structure of the vOC-5-type complex cation [RuCl(NO)2(PCyp3)2]+ in crystals of 8a. The thermal ellipsoids are drawn at 40% probability level. Some carbon atoms are disordered. The minor parts of C14 (44%), C19 (42%) and C27 and C28 (47%) are not shown. C13–15 and C26–29 were refined isotropically. | ||
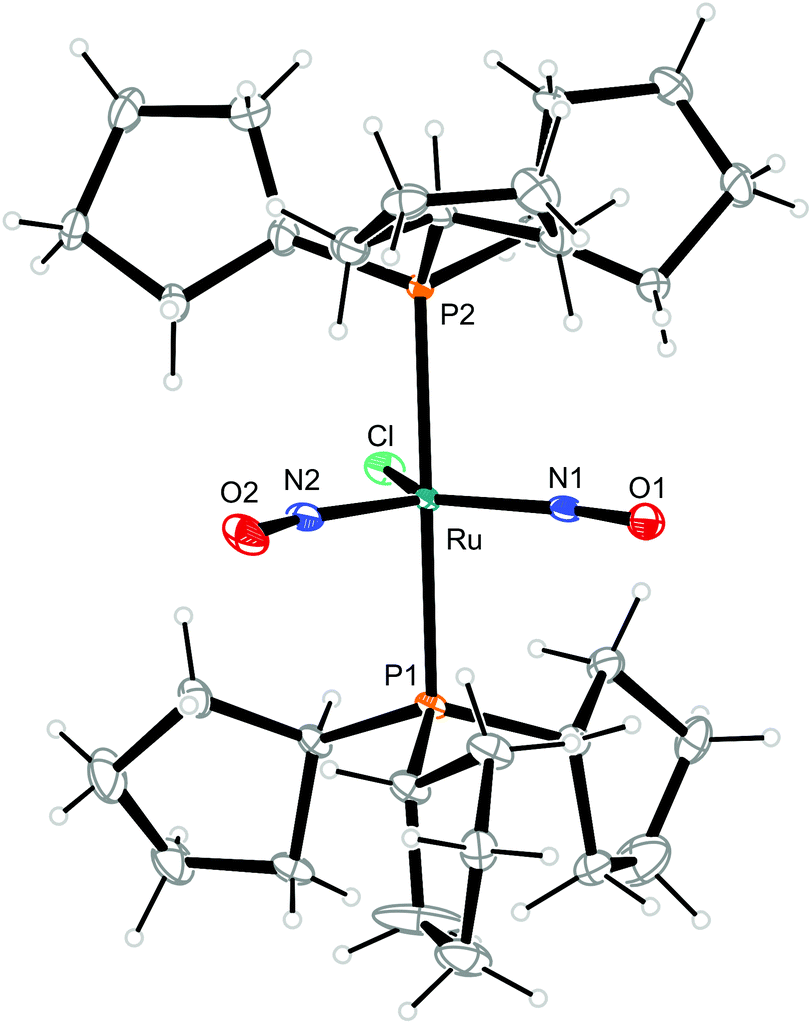 | ||
| Fig. 8 Structure (50% ellipsoids) of the TBPY-5-type complex cation [RuCl(NO)2(PCyp3)2]+ in crystals of 8b. | ||
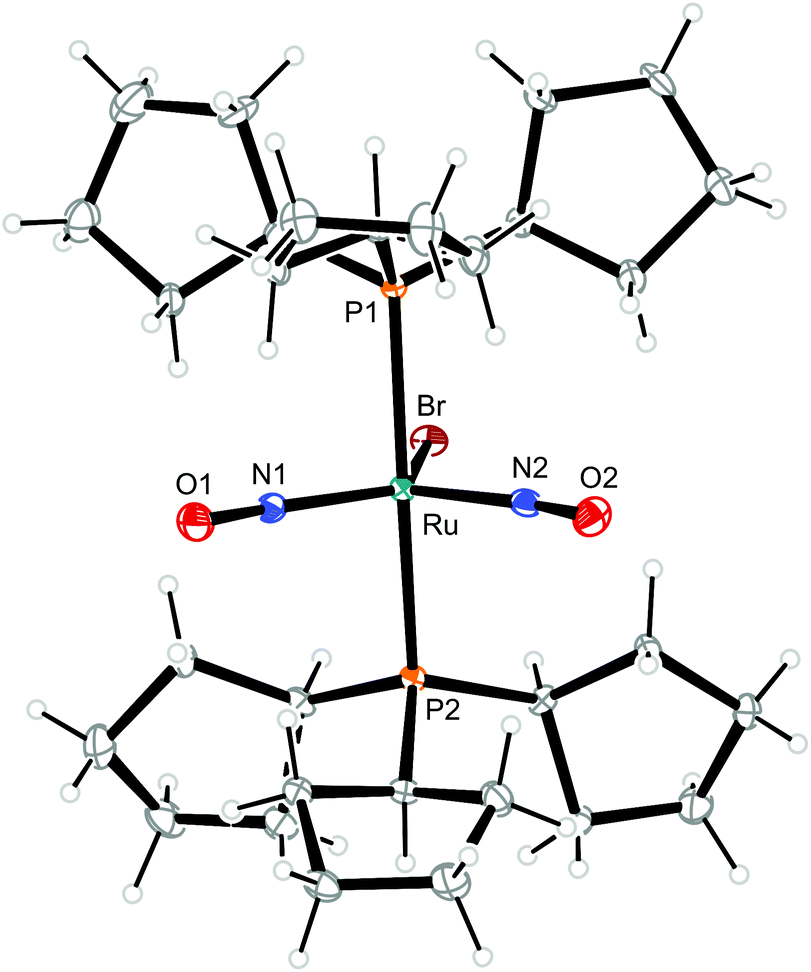 | ||
| Fig. 9 Structure (50% ellipsoids) of the TBPY-5-type complex cation [RuBr(NO)2(PCyp3)2]+ in crystals of 9. | ||
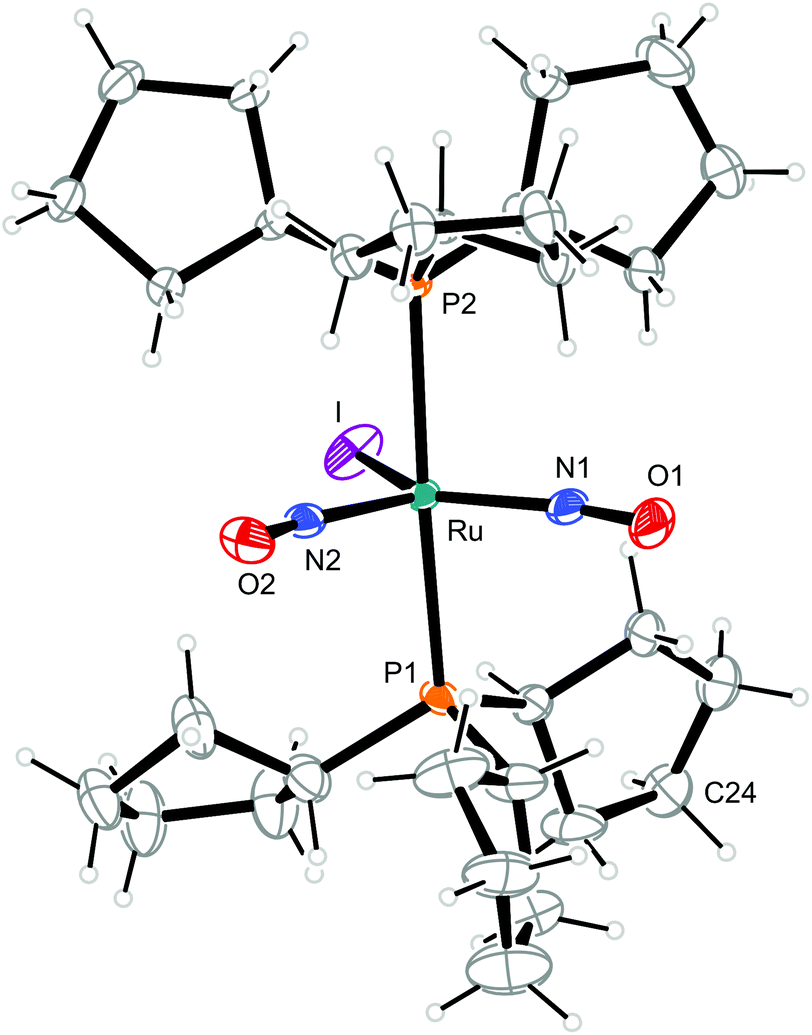 | ||
| Fig. 10 Structure (50% ellipsoids) of the TBPY-5-type complex cation [RuI(NO)2(PCyp3)2]+ in crystals of 10. C24 is disordered, the minor part (34%) is not shown. | ||
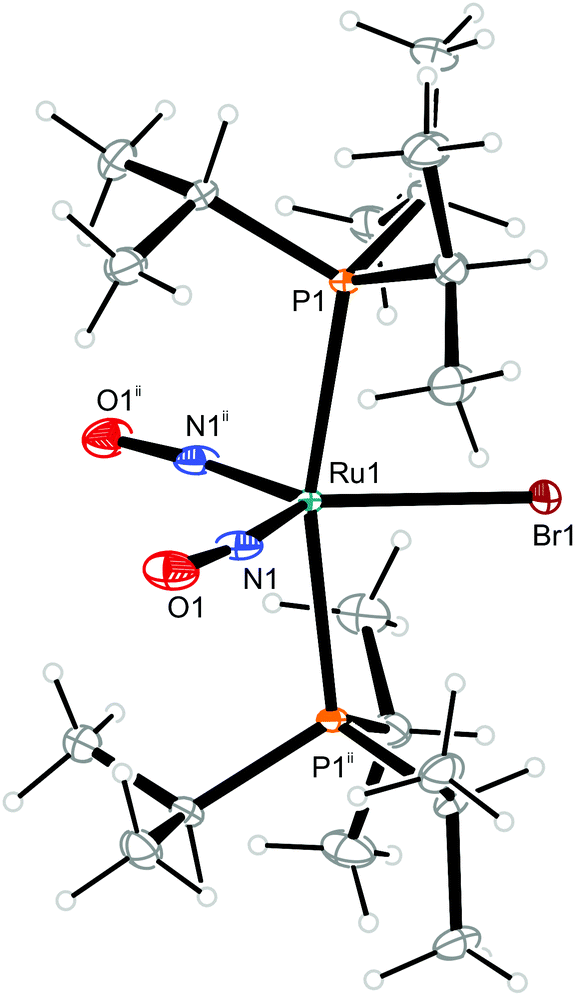 | ||
| Fig. 11 Structure (50% ellipsoids) of the TBPY-5-type conformer 12b of the complex cation [RuBr(NO)2(PiPr3)2]+ in crystals of 12. Symmetry code: ii −x + 1, y, −z + 1/2. | ||
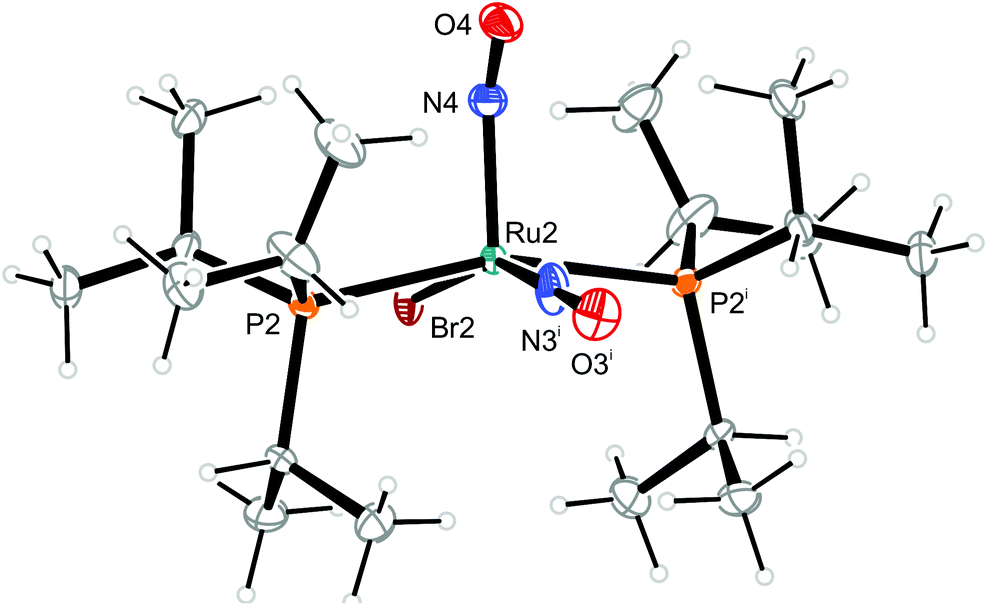 | ||
| Fig. 12 Structure (50% ellipsoids) of the vOC-5-type conformer 12a of the complex cation [RuBr(NO)2(PiPr3)2]+ in crystals of 12. Symmetry code: i −x, y, −z + 1/2. The linear NO group and the Br ligand are disordered in such a way as to be superimposed onto each other. The bent NO group is also disordered in such a way to ensure that the O4 atom is always inclined to the linear NO group. This kind of disorder corresponds to crystallographic mm2 site symmetry of the cations. | ||
| PR3/X | Ru–N1–O1/° | Ru–N2–O2/° | ΔRu–N–O/° | Ru–N1/Å | Ru–N2/Å | ΔRu–N/Å | ||
|---|---|---|---|---|---|---|---|---|
| exp. | exp. | exp. | calc. | exp. | exp. | exp. | ||
| 1 | PPh3/Cl | 178.9(2) | 134.8(2) | 44.1 | 39.1 | 1.746(2) | 1.872(2) | 0.126 |
| 2 | PPh3/Br | 175.3(3) | 143.1(2) | 32.2 | 35.1 | 1.753(2) | 1.854(2) | 0.101 |
| 3 | PPh2Bn/Cl | 167.2(2) | 164.0(2) | 3.2 | 5.5 | 1.781(2) | 1.785(2) | 0.004 |
| 4 | PPh2Bn/Br | 168.3(3) | 162.4(3) | 5.9 | 7.0 | 1.776(3) | 1.781(3) | 0.005 |
| 5 | PCy3/Cl | 179.9(3) | 136.5(2) | 43.4 | 39.1 | 1.739(3) | 1.870(3) | 0.131 |
| 6 | PCy3/Br | 177.3(6) | 139.3(6) | 38.0 | 37.8 | 1.756(6) | 1.846(6) | 0.090 |
| 7a | PCy3/I | 170.9(8) | 165.9(8) | 5.0 | 4.7 | 1.773(8) | 1.789(9) | 0.016 |
| 7b | PCy3/I | 170.3(9) | 163.1(8) | 7.2 | 4.7 | 1.791(9) | 1.790(7) | 0.001 |
| 8a | PCyp3/Cl | 176.9(4) | 137.0(4) | 39.9 | 39.7 | 1.758(3) | 1.850(4) | 0.092 |
| 8b | PCyp3/Cl | 168.1(2) | 164.4(2) | 3.7 | 9.7 | 1.775(2) | 1.783(2) | 0.008 |
| 9 | PCyp3/Br | 168.7(3) | 166.6(3) | 2.1 | 5.4 | 1.779(3) | 1.780(3) | 0.001 |
| 10 | PCyp3/I | 168.9(4) | 165.5(4) | 3.4 | 5.3 | 1.779(4) | 1.787(4) | 0.008 |
| 11 | PiPr3/Cl | 178.0(9) | 149.2(8) | 28.8 | 38.3 | 1.740(8) | 1.831(8) | 0.091 |
| 12a | PiPr3/Br | 177(1) | 150.8(3) | 26.2 | 41.2 | 1.72(1) | 1.860(5) | 0.140 |
| 12b | PiPr3/Br | 165.34(3) | 165.34(3) | 0.0 | 0.5 | 1.785(3) | 1.785(3) | 0.000 |
The members of the first group resemble Pierpont and Eisenberg's prototypic dinitrosyl A (Fig. 2). They share the structural property of well separated bent/linear 1NO−/NO+ nitrosyl bonding.17 In terms of the Enemark–Feltham notation, a ruthenium(II) centre contributes its six 4d electrons to the {Ru(NO)2}8 formula, an NO+ ligand none and a 1NO− ligand two electrons. This description also applies to 1, 2, 5, 6, 8a, 11, and 12a. Geometrically, the structure type is characterised by a considerable difference of the two Ru–N–O angles (projection of the data points of Fig. 13 on the ordinate; note that a reliable separation of the two groups succeeds by the combination of ΔRuNO as a structural and Δν(NO) as a spectroscopic parameter [Fig. 13]). In terms of continuous shape measures (CShM), the complex cations are best described as vacant octahedra (vOC-5; in a vacant octahedron, the central atom is closer to the basal plane than it is in the SPY-5 conformation in the sense of ref. 23; the applicable IUPAC recommendation does not note this difference).24 If Addison's τ5 parameter was used to assign the conformation, the square pyramid resulted (sqp; using τ5 categories, there is no difference between vOC-5 and SPY-5; hence, in this work, the CShM terminology is used).25 The bent nitrosyl ligand forms the apex of the vacant octahedron and the trans-configured phosphanes together with the halide ligand and the linear NO group comprises the basal plane (Table 1). The mean P–Ru–P angle deviates from linearity by 14.5° since the phosphorus atoms are bent away from the bent NO group. The two NO ligands are clearly distinct from each other, not only in terms of the Ru–N–O-angle difference which ranges from 26.2° to 44.1°, but also in terms of the Ru–N bond length difference (0.090 to 0.140 Å, Table 1). The oxygen atom of the bent NO group points towards the linear nitrosyl ligand. The maximum symmetry for these compounds is that of point group Cs.
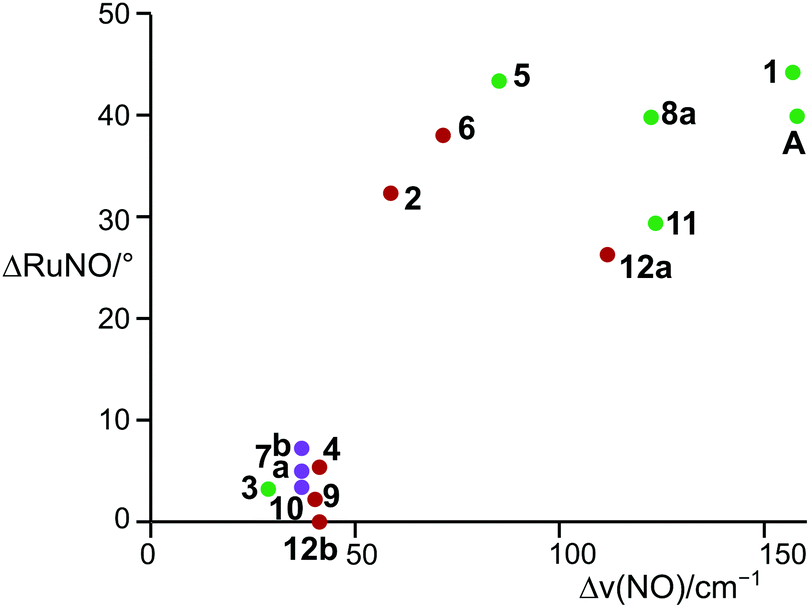 | ||
| Fig. 13 Adopted structures (bottom left group: TBPY-5, top right data points: vOC-5) depending on the Ru–N–O angle and the ν(NO) stretching frequency. Colour code: purple (X = I), reddish brown (X = Br), green (X = Cl). | ||
The members of the second, the TBPY-5 group, are well separated in terms of metrical parameters. Hence, the structure of the cations in 3, 4, 7, 8b, 9–10, and 12b resembles that of the nitrosyl-equilibrated intermediate described in the introduction. The structure type is best described as a trigonal bipyramid (TBPY-5) in terms of CShM (and τ5) values (CShM values are collected in the ESI†). The trans-arranged phosphane ligands form the apexes of the bipyramid, the halide together with the two NO groups form the trigonal plane. Again, the P–Ru–P angle deviates from linearity by 14.0° on average. The phosphorus atoms are bent away from the NO groups, the nitrosyl ligands themselves are slightly bent (13.5° in average) in a cisoid fashion. The equilibration of the two NO ligands is mirrored in the angles and the distances of the Ru(NO)2 moieties, the Ru–N–O angle difference ranging from 0.0 to 7.2°, the Ru–N distances differing by 0.016 Å in maximum. The trigonal-bipyramidal conformation had been as yet unknown for pentacoordinate {Ru(NO)2}8 compounds with simple monodentate co-ligands. The maximum symmetry reachable in these structures refers to point group C2v, which is the crystallographic symmetry of 12b, in which the two NO ligands are indistinguishable from each other.
Table 1 gives an overview of the metrical data from the X-ray analyses. Two compounds, 8 and 12, were found in both conformations. For 8, crystals of the vOC-5 form 8a were accompanied by a minor polymorph, the TBPY-5 conformer 8b. In the case of 12, both conformers, the vOC-5 form 12a and the TBPY-5 conformer 12b co-crystallise in the same solid.
The result of the continuous-shape-measures analysis is shown in Fig. 14. The assignment of a particular compound to one of the groups succeeded satisfactorily on a mere structural basis with a single exception: the data point for the Br/PPh3 derivative 2 is found close to the TBPY-5 group but is assigned a vOC-5-type complex in a combined structural/spectroscopic view (Fig. 13).
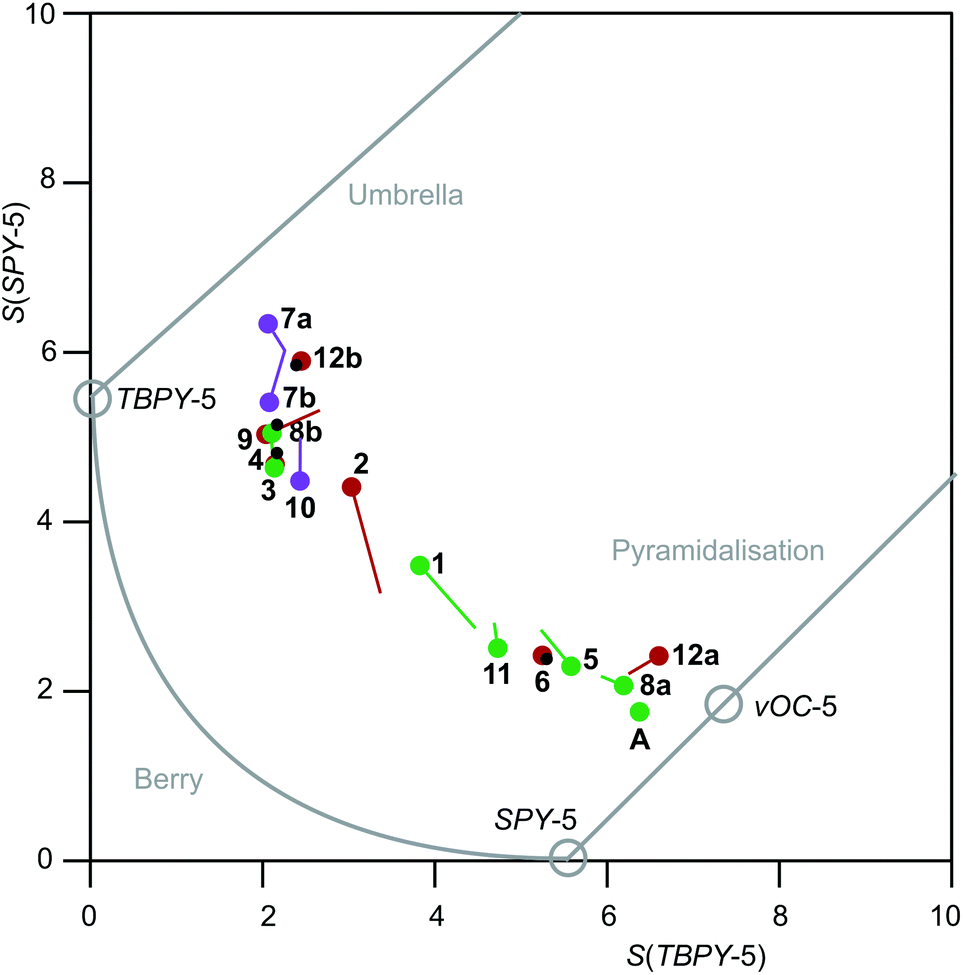 | ||
| Fig. 14 Shape map for pentacoordinate complex ions [Ru(NO)2(PR3)2X]+. The graph was drawn by using Fig. 6 of ref. 23 as a template (from which the commentary printed in grey was transferred as well). Purple (X = I), reddish brown (X = Br) and green (X = Cl) circles are drawn at the experimental values; lines of the respective colour end at the DFT-calculated values; small black circles within larger coloured circles indicate that the DFT value gave a data point within the experimental value's circle radius. | ||
IR spectroscopy
The structural differences between the vOC-5 and the TBPY-5 group are mirrored in the excitation-energy difference of the symmetrically and asymmetrically coupled N–O stretches. In the case of vOC-5, the Δν(NO) values show a large difference (71–157 cm−1, solid state). Considerably smaller differences (28–41 cm−1, solid state) characterise the TBPY-5 group (for all ranges and mean values, 2, which was correctly assigned in the combined map of Fig. 13, was not included). IR spectra of crystals of 12, which contained the vOC-5 and TBPY-5 conformers in equal parts, provided us with useful information for the interpretation of solution spectra: for a vOC-5 conformer, the symmetrically coupled vibration is dominated by the linearly bonded ligand (see the DFT part below) and reaches the highest excitation energy. The symmetrically coupled vibration of the TBPY-5 conformer is observed at a somewhat lower excitation energy. However, the two symmetrically coupled stretches may appear so close in the spectrum that they may overlap, at least at higher temperatures (Fig. 15). The asymmetrically coupled nitrosyl stretches are well resolved with the lower frequency for the vOC-5 conformer whose lower-energy stretch is dominated by the 1NO− ligand. As a result, three to four bands are observed in the case of compounds that exhibit both conformations, the couple of TBPY-5 bands (“b” in Fig. 15) being nested in the band couple of the vOC-5 form (“a” in Fig. 15).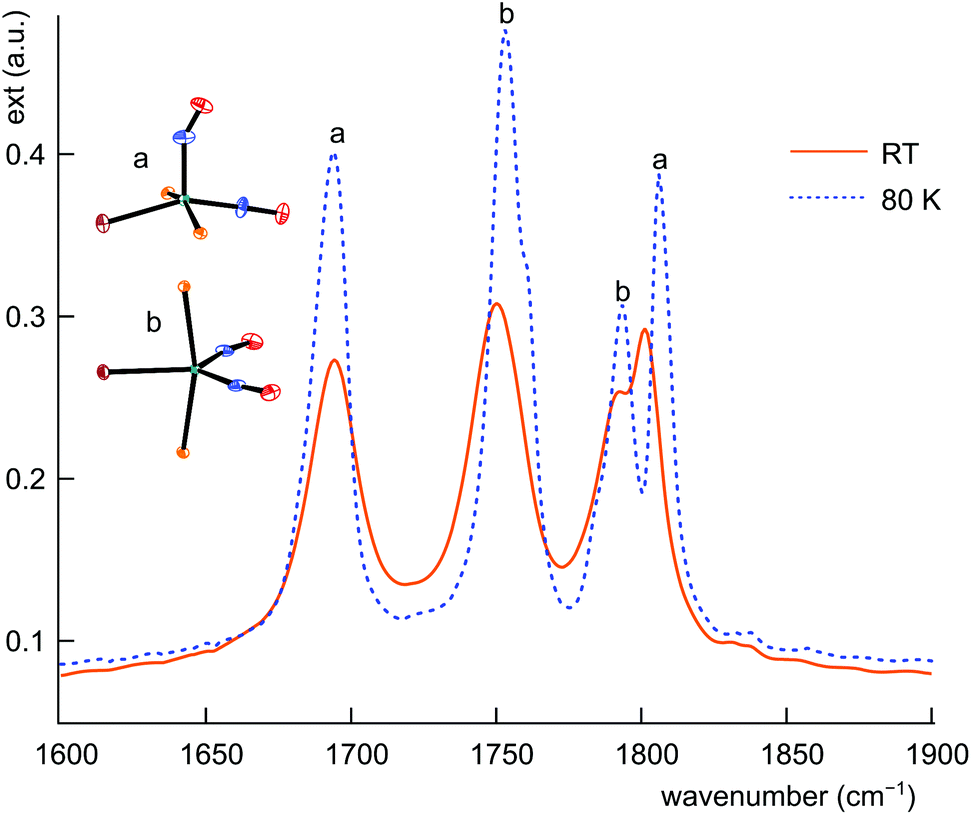 | ||
| Fig. 15 IR spectrum of the ground state of 12a/b in the ν(NO) range at RT (solid line) and 80 K (dashed line). | ||
The clear-cut difference between the asymmetrically coupled stretches allows for the interpretation of the whole body of data of Table 2. The dissolution of crystals of 12 in dichloromethane results in the transformation of the vOC-5 conformer to the TBPY-5 form as the only conformer in solution. In the case of 1, 6, 8, and 11, dissolution in CH2Cl2 induces the formation of a mixture of the conformers. The full set of bands was resolved for 8, whereas the coincidence of the symmetrically coupled stretches was observed for 1, 6, and 11. (For a solution of A in methanol, four bands had been observed.15)
| PR3/X | ν(NO)sym,asym/cm−1 | ν(NO)sym,asym/cm−1 | ν(NO)sym,asym/cm−1 | Δν(NO)/cm−1 | Conformation | |
|---|---|---|---|---|---|---|
| Solid | DFT | CH2Cl2 | Solid/calc. | (Solid) | ||
| a TBPY-5/vOC-5 conformational mixture in solution; cf.ref. 15. b Coincidence of the symmetric N–O stretches of the two conformations at the maximum of the three values. c No local minimum found for the TBPY-5 conformer in the DFT calculation both with and without COSMO approach to solvation. d Local minimum for the TBPY-5 conformer found for the COSMO(CH2Cl2) calculation; calculated frequencies/cm−1 (TBPY-5 values in italics): 6 1791, 1780/1745/1691, 8a/b 1800/1795/1766/1688. e Frequencies measured on a Nicolet 5700 FTIR device for the sake of its better spectral resolution (2 cm−1). f Transformation of the vOC-5 conformer to TBPY-5 on dissolution. | ||||||
| 1 | PPh3/Cl | 1842/1685 | 1830/1724 | 1823/1776/1720a,b,c | 157/106 | vOC-5 |
| 2 | PPh3/Br | 1824/1765 | 1819/1736 | 1813/1778 | 59/83 | vOC-5 |
| 3 | PPh2Bn/Cl | 1799/1771 | 1811/1788 | 1818/1776 | 28/23 | TBPY-5 |
| 4 | PPh2Bn/Br | 1817/1776 | 1809/1787 | 1815/1778 | 41/22 | TBPY-5 |
| 5 | PCy3/Cl | 1789/1704 | 1806/1709 | 1812/1706 | 85/97 | vOC-5 |
| 6 | PCy3/Br | 1785/1714 | 1799/1709 | 1800/1760/1716a,b,d | 71/90 | vOC-5 |
| 7 | PCy3/I | 1788/1751 | 1791/1768 | 1797/1765 | 37/23 | TBPY-5 |
| 8a | PCyp3/Cl | 1805/1681 | 1809/1710 | 1834/1797/1754/1710a,d | 124/99 | vOC-5 |
| 9 | PCyp3/Br | 1810/1770 | 1809/1783 | 1795/1759 | 40/26 | TBPY-5 |
| 10 | PCyp3/I | 1809/1772 | 1793/1769 | 1794/1759 | 37/24 | TBPY-5 |
| 11 | PiPr3/Cl | 1808/1682 | 1815/1717 | 1809/1759/1714a,b,c | 126/98 | vOC-5 |
| 12a | PiPr3/Br | 1806/1694e | 1809/1710 | 1802/1765f | 112/99 | vOC-5 |
| 12b | PiPr3/Br | 1794/1753e | 1799/1776 | 1802/1765 | 41/23 | TBPY-5 |
The combination of the crystallographical and the spectroscopical analysis allows for a reliable assignment of a conformation in question. Fig. 13 shows two clearly resolved fields of existence of the conformers, a narrow field for the TBPY-5 group and a broader one for the vOC-5 conformer.
DFT calculations
Both the metrical as well as the spectroscopical data are satisfyingly reproduced by calculation in the framework of density functional theory (compare the “calc.” entries in Tables 1 and 2). In Fig. 14, the computational results are sketched in as well. The resemblance of experimental and calculated values is mostly satisfactory. Larger deviations ensued for the PPh3/Cl (1) and the PPh3/Br (2) couples only. The τ5 values from calculation and experiment show a maximum deviation of 0.10, found for 2.In terms of frequencies, the measured values in the solid and liquid states are in relatively good agreement with those calculated as well. If the calculated frequencies and the frequencies measured in solid samples are compared, it is obvious that the asymmetric vibration tends to be predicted a bit too high, the mean deviation being +12.9 cm−1 [Δν(NO) = ν(NO)calcd − ν(NO)exp.], whereas the symmetric vibration mode is predicted more reliably (Δν(NO) = +1.8 cm−1). It should be noted that all DFT-derived frequencies are listed without applying a correction factor.
A slightly better agreement between calculated and observed frequencies was found for the liquid state values that are devoid of contributions from specific intermolecular interactions. Specifically, the uncorrected asymmetric vibration was predicted slightly too high (Δν(NO) = 2.5 cm−1), and the symmetric vibration mode was predicted slightly too low (Δν(NO) = −1.83 cm−1).
For 8, both the vOC-5 and the TBPY-5 forms were found in the solid state, the vOC-5 conformer 8a as the major form, with an admixture of a few crystals of 8b (hence, no solid-state IR data are given for 8b in Table 2). On attempts to model both conformers using the standard procedure (no solvent model applied), only 8a was found as a minimum structure, whereas 8b converged into 8a on refinement. However, a local minimum was obtained for 8b also by the application of a COSMO model with the parameters of dichloromethane. 8a also remained a minimum structure in the COSMO calculations, the energetic difference of both conformers being rather small (1.4 kJ mol−1 instability of 8b), in line with the common occurrence of both conformers in solution spectra.
For 12, both conformations were local minima on the conformational hypersurface in the standard as well as the COSMO-DFT treatment with the vOC-5 conformer the global minimum, and the TBPY-5 at some 3 kJ mol−1 less stable for 12.
Though two crystallographically confirmed conformers were found for 8 and 12 only, IR spectra of solutions of the vOC-5 species 1, 6, and 11 revealed the existence of their TBPY-5 counterparts. However, with or without the application of the COSMO solvent model, we did not succeed in locating a minimum structure for a TBPY-5 isomer with the specified methods and basis sets for 1 and 11. For 6, however, both the COSMO and the pure gas-phase approach led to local minima for the two conformations. As expected for the IR result, the stability of both conformers was practically equal (TBPY-5 instable by 1.1 and 1.4 kJ mol−1 for the gas-phase and the COSMO refinement, respectively).
The distribution of the conformers is summarised in Table 3. Dinitrosyl complexes with the vOC-5 structure are primarily found on the top left, compounds which adopt both structures in the middle, and compounds adopting the TBPY-5 structure bottom right.
| PPh3 | PiPr3 | PCy3 | PCyp3 | |
|---|---|---|---|---|
| Cl | vOC-5 | vOC-5 | vOC-5 | vOC-5 + TBPY-5 |
| +TBPY-5 | +TBPY-5 | |||
| Br | TBPY-5 | vOC-5 + TBPY-5 | vOC-5 | TBPY-5 |
| TBPY-5 only | +TBPY-5 | |||
| I | TBPY-5 | TBPY-5 | TBPY-5 |
Bonding in the [RuX(NO)2(PR3)2]+ cations
The argumentation used in the following attempt to rationalise the experimental results agrees with qualitative molecular-orbital considerations published by Enemark and Feltham and by Hoffmann and Rossi some four decades ago.2,26 As shown before, the observed conformation is obviously correlated to the coordination entity's electronic situation. Thus, A, 1, 2, 5, 6, 8a, 11 and 12a, all of which adopt the vOC-5 conformation, constitute a group of less electron-rich species in terms of the kind of halide and Tolman's parameter of the phosphane. 3, 4, 7, 8b–10 and 12b adopt the TBPY-5 structure in agreement with the higher electron supply of the heavier halides and/or the more electron-donating phosphanes.The fact that a square-planar conformer with one bent nitrosyl, and a trigonal bipyramidal conformer with more or less linear nitrosyls are the two alternatives for five-coordinate {M(NO)2}8 species had been recognised by Enemark and Feltham prior to the discovery of a first representative of the TPBY-5 type.2 Their analysis starts with the presentation of the four combinations of the NO-π* orbitals in a cis-M(NO)2 fragment. Fig. 16 shows, for the point group C2v, these four ligand group orbitals which represent the nitrosyl ligands’ contribution to the frontier orbital range. As a starting point for the discussion, an idealised conformation was considered with a 90° N–M–N angle and linear M–N–O fragments. In this situation, the a1, a2 and b2 orbitals are metal–ligand-bonding by their interaction with the x2 − z2, xy and yz metal d-orbitals, respectively. Moreover, these three molecular orbitals remain metal–ligand bonding in the course of distorting the depicted starting conformation and thus are filled for all conformations in question with six of the eight electrons of the {M(NO)2}8 moiety. The ligand b1 orbital, however, is orthogonal to the metal d orbital of the same symmetry (xz) in the virtual starting conformation. To remove this loss of stability, Enemark and Feltham considered, depending on the overall electron supply, two possible distortions that provide a metal–ligand bonding molecular orbital also for the remaining two electrons. Fig. 17, re-drawn from ref. 2, shows the two considered distortional paths that stabilise this electron pair. From the metal's viewpoint, the right part of Fig. 17 treats the electron-rich variant with all eight electrons metal centered. Here, the b1 electron pair is positioned in the metal xz orbital [Fig. 17(c)]. To make the orthogonal metal–ligand contact of Fig. 17(c) metal–ligand-bonded, the N–M–N angle increases [Fig. 17(d)] and some degree of bonding overlap is achieved (the mean N–Ru–N angle for the TBPY-5 species of this work is 117.4°). As a result, the electron-rich TBPY-5 complexes reported herein are interpreted as d8-ruthenium(0) centres with two formal NO+ ligands.
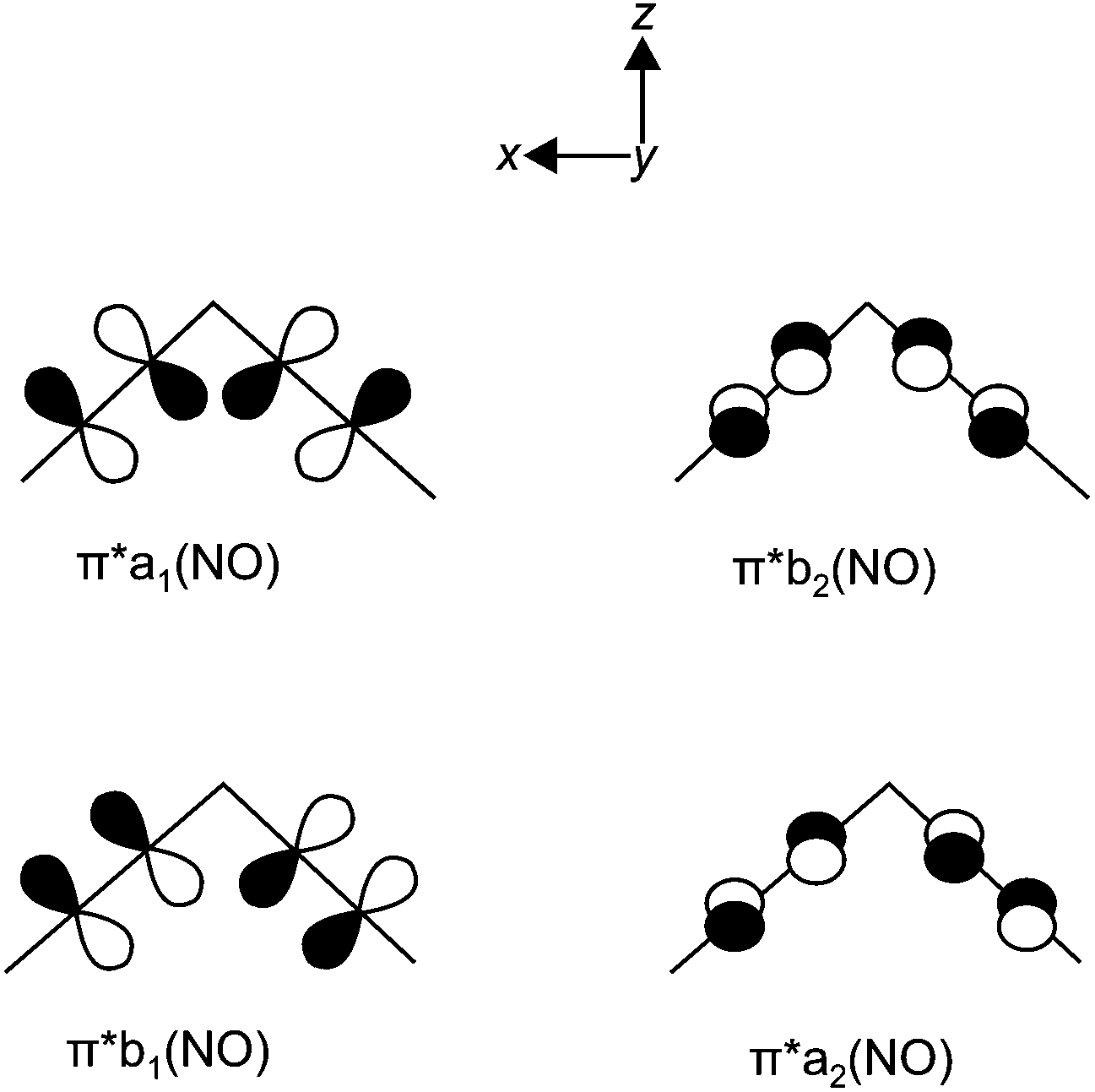 | ||
| Fig. 16 The ligand group orbitals in C2v symmetry derived from the π* orbitals of the two NO ligands. Adapted from ref. 2. | ||
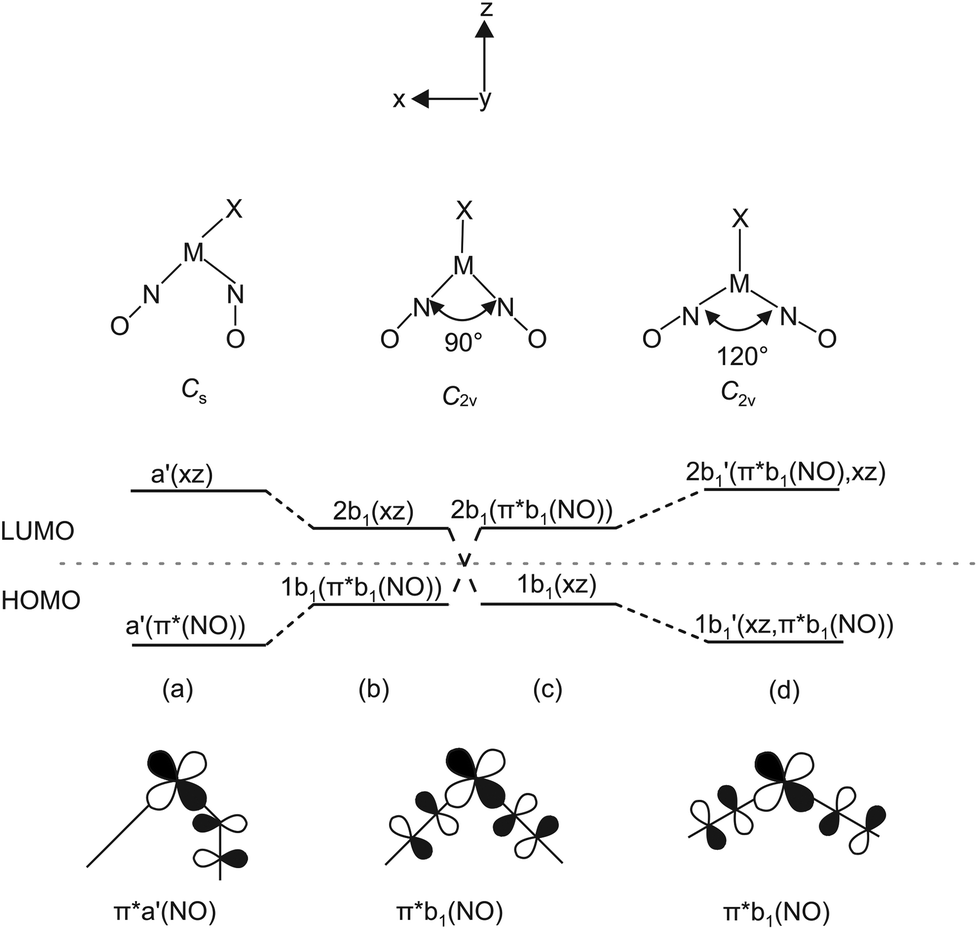 | ||
| Fig. 17 Left: Correlation diagram showing the proposed behaviour of the 1b1 and 2b1 molecular orbitals in five-coordinate {M(NO)2}8 complexes with a (1b1)2 electron configuration. Scheme (b) has π*b1(NO) lower in energy than dxz and leads to structure (a). Scheme (c) has dxz lower in energy than π*b1(NO) and leads to (d). Adapted from ref. 2 Bottom: The relevant HOMO which is metal–ligand non-bonding at an N–M–N angle of 90°. | ||
In the metal-electron-poor variant, Enemark and Feltham placed the two electrons in question in the ligand-centered b1 orbital [Fig. 17(b)]. The system relaxes by a bend of one of the nitrosyl ligands, thus lowering the symmetry to Cs [Fig. 17(a)]. Metal–ligand bonding is achieved via the overlap of the respective orbital of a formal 1NO− ligand and the metal-d(xz) orbital with local σ symmetry. Hence, in the more electron-poor complexes of this work, d6-ruthenium(II) centres are bonded to a NO+/1NO− nitrosyl couple.
Enemark and Feltham's qualitative discussion is supported by our DFT calculations. Fig. 18 shows the result for the Cs-symmetric vOC-5 conformer 12a and the C2v-symmetric TBPY-5 conformer 12b. Both conformers present energetic minima that assure metal–ligand bonds for all four electron pairs of the {Ru(NO)2}8 configuration, even for the least stable b1/a′ pair.
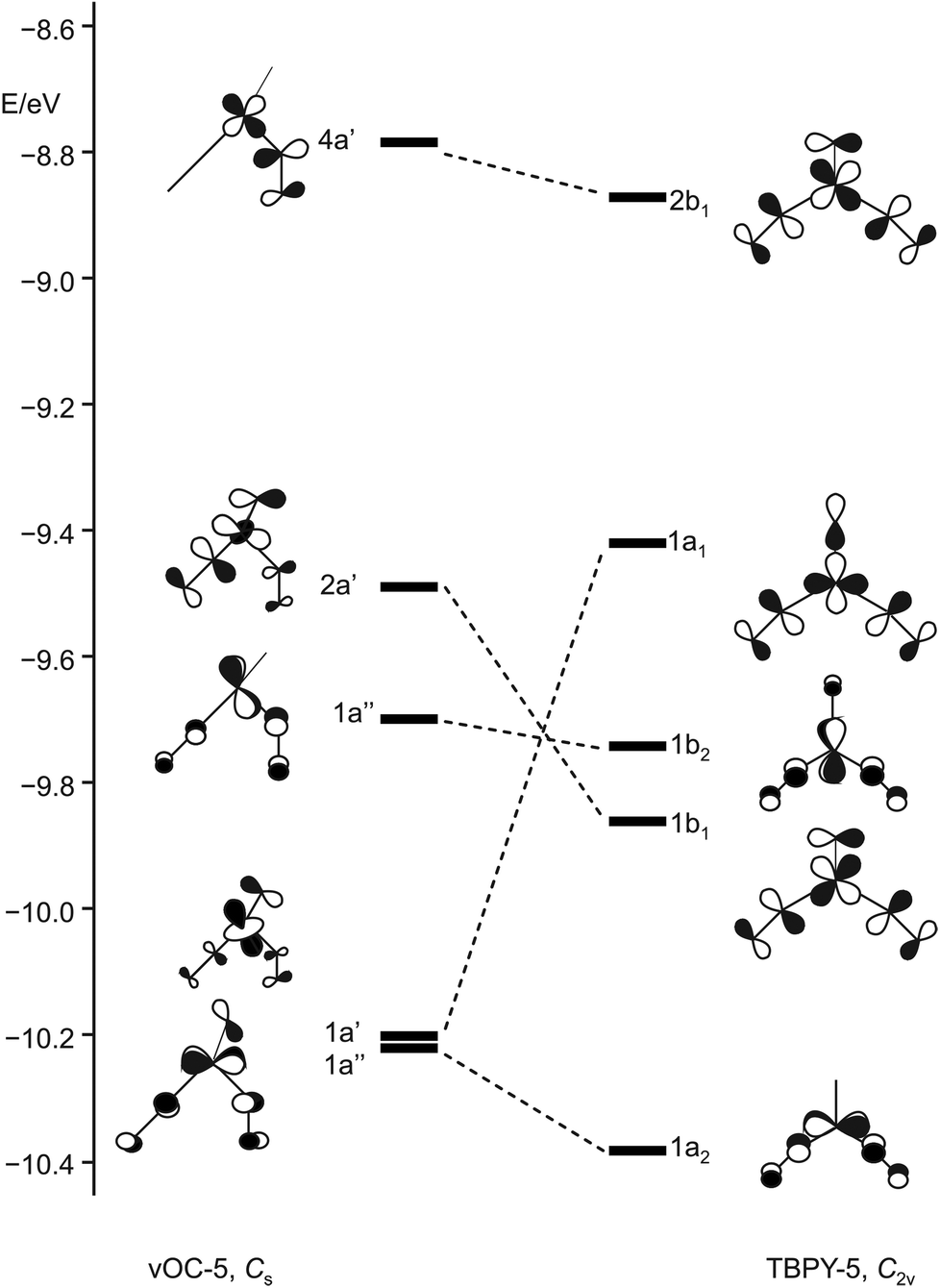 | ||
| Fig. 18 Schematic representation of selected frontier orbitals for the C2v (TBPY-5) and the Cs (vOC-5) conformer of 12 calculated by a DFT-based method. | ||
Photophysical investigation of [RuBr(NO)2(PiPr3)2]BF4 (12)
Dinitrosyl complexes promise to be particularly interesting objects for photophysical research on the linkage isomer issue since they provide a larger number of excitation and decay pathways than mononitrosyl compounds – if they are excitable at all. To check the principal suitability of {Ru(NO)2}8 compounds for the formation of metastable linkage isomers via photoexcitation, we chose 12 since both conformers 12a and 12b are present at an equal ratio in crystals of the tetrafluoridoborate.To test under which conditions the population of metastable isomers is achievable, 12 was irradiated with laser light of the wavelengths 405, 445 and 476 nm at 80 K. The maximal photo-excitation was reached at 405 nm. Fig. 19 shows an infrared spectrum in the ν(NO) range of the ground state and the photo-excited state at 80 K. Upon illumination in the blue spectral range, the two conformers reacted differently with the vOC-5 conformer showing the more distinct change. Hence, the two NO stretches of 12a (1806 and 1694 cm−1 in the ground state) were affected most and lost about 70% of their intensity. New absorption bands appeared at 1649 and 1821 cm−1, −45 cm−1 relative to the asymmetrically coupled stretch, and +15 cm−1 relative to the symmetrical stretching mode (blue spectrum in Fig. 19). The ground-state bands of the TBPY-5 isomer 12b were much less affected on irradiation. A decay of about 30% is apparent but no new bands of matching intensity were observed. The photo-switching is reversible: irradiation with red light (660 nm) restored most of the original spectrum (Fig. 19), complete restoration of the initial state was observed on warming the samples (Fig. 20). Since the asymmetrically coupled, 1NO−-dominated absorption band of 12a experienced the largest shift, we see this ligand of the four different nitrosyls of 12 as the switched one. The smaller shift of the linearly bonded NO ligand of 12a would thus simply mirror the altered bonding situation within the bent Ru–N–O moiety in the photoinduced isomer of 12a.
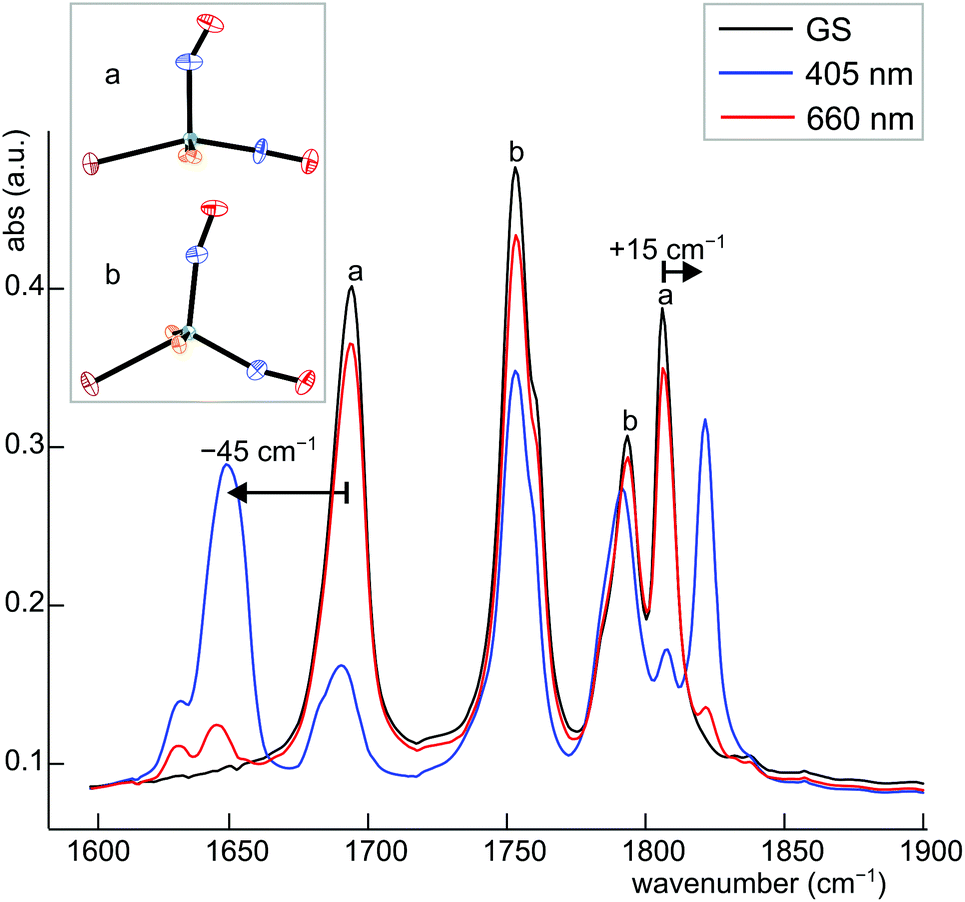 | ||
| Fig. 19 IR spectrum of 12a/b in the ν(NO) range before (black line) and after population (405 nm, blue line) and depopulation (606 nm, red line) by irradiation with light of the appropriate wavelength at 80 K. | ||
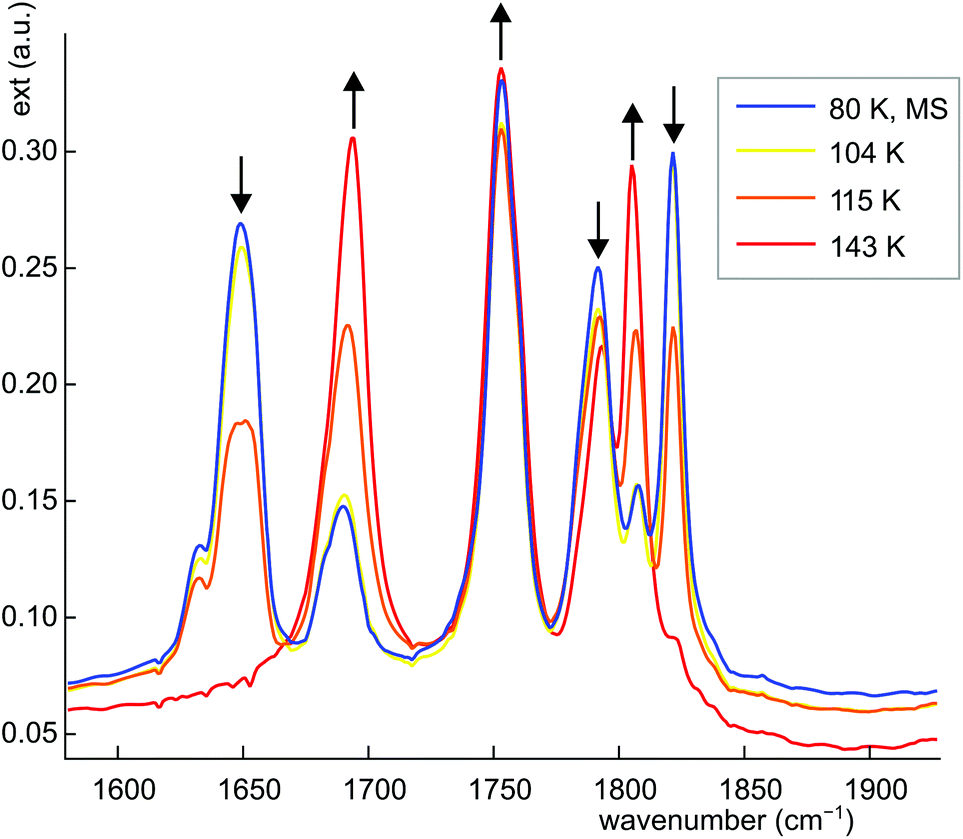 | ||
| Fig. 20 IR spectrum of 12a/b in the ν(NO) range after population at 80 K and subsequent thermal depopulation by gradual heating (yellow, orange and red line), the arrows indicate the change upon warming. | ||
No attempts were made in this report to investigate the characteristics of the metastable state in detail. Work on this issue is in progress and will be reported in due course.
Conclusions
Eleven compounds of the {Ru(NO)2}8 type were synthesised by slightly modified procedures adopted from Townsend and Ibers.27,28 The dinitrosyl complexes of the general formula [RuX(NO)2(PR3)2]BF4 (X = Cl, Br, I; PR3 = PPh3, PPh2Bn, PCy3, PCyp3, PiPr3) were characterised by X-ray diffraction and spectroscopic methods (IR, NMR), mass spectrometry and elemental analysis. X-ray studies of the dinitrosyl compounds reveal that they adopt two different structures: one which was already known for {Ru(NO)2}8 compounds, and another one which has, so far, been unknown for ruthenium dinitrosyls but has been found for an osmium homologue. The known structure is of the vacant-octahedron type (vOC-5) in terms of continuous shape measures (CShM);23 the maximum possible symmetry is Cs. It shows two distinct bonding modes for the two NO ligands, a formal NO+ and a formal 1NO− group. Animations of the calculated ν(NO) vibrations reveal that, of the two N–O stretches, the higher-energy symmetrically coupled vibration is dominated by the linearly coordinated nitrosyl and the lower-energy asymmetrically coupled vibration by the bent-coordinated nitrosyl ligand. The extent of vibrational coupling is higher in the second group of complex cations which adopt a trigonal bipyramidal structure (TBPY-5) in terms of CShM values; the maximum possible symmetry is C2v. The coordination entities show an equal bonding situation for the two NO ligands. This structure, predicted in a review article by Enemark and Feltham for five-coordinate {M(NO)2}8 compounds of third-row transition metals and good π-accepting ligands X, is known for M = Re, Mn and X = Cl, CO, and, more closely to the compounds of this work, for the osmium homologue of 1, [OsCl(NO)2(PPh3)2]BF4 (it might be noted that Enemark and Feltham's bonding analysis was correct but their prediction regarding the occurrence of the TBPY-5 form was not).16 Animations of the calculated ν(NO) vibrations of the compounds with this structure reveal, as expected, equal contributions of both N–O stretches to the symmetrically and the asymmetrically coupled vibrations at higher and lower excitation energy, respectively.All structures were verified by DFT calculation, both in terms of structural data and vibrational frequencies. The actually developed structure type depends, primarily, on the nature of X, and, secondarily, on the substituent R of the phosphane. Both structure types for the same compound were found experimentally for the complex cations of [RuBr(NO)2(PiPr3)2]BF4 (12) and [RuCl(NO)2(PCyp3)2]BF4 (8) in the solid state, and, in dichloromethane solution, for 1, 6, 8, and 11 (as a peculiarity, both conformers of 12 are transformed to the TBPY-5 form 12b on dissolution in CH2Cl2).
The frontier orbitals of the two conformers of 12 were compared with the predictions made by Enemark and Feltham for square planar and trigonal bipyramidal pentacoordinate {M(NO)2}8 compounds.2 Although TBPY-5 compounds of this type were not known at that time, and the predictions were, thus, based only on symmetry and overlap criteria, the energetic order as well as the type of orbitals involved are, to a great extent, consistent with the DFT results.
Generally, the vOC-5 structure is found preferentially for the chlorido species, whereas the iodido compounds are the realm of the TBPY-5 type. Thus, we conclude that the adopted structure is primarily dominated by the halogenido ligand and, secondarily, by Tolman's electronic factor of the phosphane ligand (Table 3, Fig. 14). Obviously, the occurrence of any of the two conformers mirrors the electronic supply by the halogenide and the phosphane. In a simplifying view, the TBPY-5 class is consistent with the electron-rich situation of d8-Ru0 centres that bind two NO+ ligands. Less electron-rich complexes switch to a bent coordination of one nitrosyl ligand. These, in the ideal case, Cs-symmetrical compounds can be regarded as derivatives of d6-RuII centres coordinating to one NO+ and one 1NO− ligand.
An orienting investigation on the photo-excitability was conducted for 12 for which both conformers are present within the same crystal structure at equal parts. As a preliminary result, a high degree of population of the metastable, 1NO−-switched state was found for the vOC-5 conformer on irradiation with blue light.
After having demonstrated the photo-excitability of {Ru(NO)2}8 compounds, photo-physical investigations including photo-crystallographic experiments as well as quantum-chemical calculations of the metastable states are currently in progress and will be reported in a separate work.
Experimental
Materials
RuCl3·xH2O was purchased by Alfa Aesar or ABCR. Benzyldiphenylphosphane, tricyclohexylphosphane and tri-isopropylphosphane were used as supplied by ABCR. HBr (48%), ZnnCu and triphenylphosphane were purchased by ACROS Organics. Tricyclopentylphosphane, NOBF4 and toluene (kept over molecular sieve) were purchased by Sigma-Aldrich. KNO2 and HCl (37%) were used as supplied by Fluka. Diethyl ether, ethanol and HI (57%) were purchased from Merck. 1 M HCl was used as supplied by AppliChem. Ethanol was dried over molecular sieve and degassed. Water used for the preparation of the phosphane-containing mononitrosyl compounds, and toluene were also degassed. K2[RuCl5(NO)], K2[RuBr5(NO)], K2[RuI5(NO)], [RuCl3(NO)(PPh3)2] were prepared according to the literature.19,29,30 [RuCl(NO)2(PPh3)2]BF4 was prepared by a slightly modified literature procedure. The synthesis as well as the consecutive reaction of the precursor compounds of the general formula [RuX1–3(NO)(PR3)2] were performed using standard Schlenk technique, since the compounds are air-sensitive, unless X = 3. The {RuNO}6 as well as the {Ru(NO)2}8 compounds are air-stable.General information
Standard procedures are specified in the ESI.†Computational details
All calculations were performed with the ORCA 3.0 program package. Structures were optimised with the Becke-Perdew density (BP) method and the Ahlrichs-type basis set tzvp for all atoms except ruthenium and iodine. Found stationary points were confirmed with subsequent frequency analyses on the corresponding level of theory, except that no ECPs were used. Instead, scalar relativistic all-electron calculations were performed for ruthenium and iodine.31 Continuous shape measures were calculated with the programme SHAPE.23,32IR spectroscopy at low temperature
Measurements of IR spectra at low temperatures were performed using a Nicolet 5700 FTIR spectrometer. The powdered samples were mixed with KBr and pressed into pellets. The KBr pellets were mounted on a copper cold finger using silver paste for good thermal contact. The samples were cooled to 85 K in a liquid nitrogen cryostat. KBr windows allowed the irradiation of the sample with laser light and absorption measurements down to 390 cm−1. Irradiation was performed with lasers as described above.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 mixture of dichloromethane–ethanol–n-hexane (12 mL) and dried in vacuo. Yield: 1.26 g, 1.59 mmol, 89.3%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 19.6. Selected IR bands νmax/cm−1: 1850 (s, NO), 1599 (vw), 1495 (w), 1482 (w), 1453 (w), 1433 (m), 1408 (w), 1330 (w), 1185 (w), 1143 (w), 1094 (w), 1096 (w), 1030 (w), 1000 (w), 914 (w), 831 (m), 773 (m), 752 (m), 740 (s), 697 (s). MS-FAB (NBA): m/z calcd 754.1 for [RuCl2(NO)(PPh2Bn)2]+ = [M − Cl]+, found 754.0. Elemental analysis (%): calc. C 57.77, H 4.34, Cl 13.46, N 1.77; found: C 58.05, H 4.44, Cl 13.13, N 1.64.
:1, 10 mL) and heated under reflux for 30 minutes. In the course of the reaction a yellow-orange solid precipitated which, after cooling to room temperature, was filtered off and washed with a mixture of ethanol–dichloromethane–n-hexane (1:1:2). The product was freed from all volatile components in vacuo. Yield: 0.610 g, 0.661 mmol, 94.3%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 12.9. Selected IR bands νmax/cm−1: 1847 (s, NO), 1494 (vw), 1453 (vw), 1432 (w), 1408 (vw), 1143 (vw), 1099 (vw), 1069 (vw), 1030 (vw), 830 (m), 772 (m), 750 (m), 739 (s), 697 (vs). MS-FAB (NBA): m/z calcd 844.0 for [RuBr2(NO)(PPh2Bn)2]+ = [M − Br]+, found 844.0. Elemental analysis (%): calc. C 49.43, H 3.71, N 1.52; found: C 50.58, H 3.83, N 1.30.
:1, 60 mL), was added to a solution of tricyclohexylphosphane (2.06 g, 7.35 mmol) in hot ethanol (55 mL). The reaction mixture was kept under refluxing conditions for 4 hours. The resulting solid was collected by filtration and dried in vacuo. Yield: 1.67 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 38.8, 25.2, 17.6. Selected IR bands νmax/cm−1: 2921 (vs), 2846 (vs), 1826 (vs), 1802 (w), 1712 (vs), 1442 (s), 1264 (m), 1195 (m), 1173 (s), 1127 (w), 1002 (s), 899 (m), 847 (s), 734 (m). MS-FAB (NBA): m/z calcd for C36H66ClNOP2Ru = [M]+ 762.3045, found 762.3013; [M − Cl]+ 727.3359, found 727.3315.
:1, 70 mL), was added to a solution of tricyclohexylphosphane (1.15 g, 4.11 mmol) in hot ethanol (50 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting green solid was collected by filtration, washed with ethanol–dichloromethane–n-hexane (1:1:2, 28 mL) and dried in vacuo. Yield: 1.00 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 73.7, 36.5, 24.8, 16.2, 9.3. Selected IR bands νmax/cm−1: 2922 (vs), 2846 (m), 1825 (w), 1802 (m), 1752 (w), 1709 (s), 1442 (m), 1265 (vw), 1172 (m), 1127 (vw), 1001 (m), 886 (w), 846 (m), 732 (m). MS-FAB (NBA): m/z calcd for C36H66BrNOP2Ru = [M]+ 852.2023, found 852.2046; [M − Br]+ 773.2888, found 773.2847.
:1, 24 mL), was added to a solution of tricyclohexylphosphane (0.62 g, 2.2 mmol) in hot ethanol (17 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting green solid was collected by filtration, washed with ethanol–dichloromethane–n-hexane (1:1:2, 28 mL) and dried in vacuo. Yield: 0.23 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 45.8, 34.1, 25.6, 16.9, 11.3. Selected IR bands νmax/cm−1: 2923 (m), 2846 (m), 1798 (m, NO), 1756 (m, NO), 1706 (vs, NO), 1442 (m), 1297 (vw), 1264 (w), 1172 (m), 1002 (m), 886 (w), 845 (m), 814 (w), 731 (m), 652 (w). MS-FAB (NBA): m/z calcd for C36H66INOP2Ru = [M − I]+ 819.27, found 820.0
:1, 70 mL), was added to a solution of tricyclopentylphosphane (1.00 g, 4.20 mmol) in hot ethanol (10 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting solid was collected by filtration and dried in vacuo. Yield: 1.20 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 39.2, 20.7, 17.9, 5.0. Selected IR bands νmax/cm−1: 2947 (m), 2863 (m), 1803 (m), 1703 (vs), 1447 (w), 1299 (w), 1230 (w), 1120 (w), 1011 (vw), 906 (w), 724 (vw), 619 (vw). MS-FAB (NBA): m/z calcd for C30H54ClNOP2Ru = [M − Cl]+ 727.39, found 727.8.
:1, 70 mL), was added to a solution of tricyclopentylphosphane (1.00 g, 4.43 mmol) in hot ethanol (10 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting green solid was collected by filtration and dried in vacuo. Yield: 0.965 g, 1.14 mmol, 65%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 36.9, 27.1, 15.4, 15.0. Selected IR bands νmax/cm−1: 2954 (m), 2864 (w), 1828 (NO), 1806 (NO), 1762 (w), 1703 (m), 1447 (vw), 1298 (vw), 1259 (m), 1013 (s), 906 (w), 861 (w), 795 (vs), 703 (w). MS-FAB (NBA): m/z calcd for C30H54BrNOP2Ru = [M − Br]+ 689.19, found 689.7. Elemental analysis (%) calcd for C30H54Br2NOP2Ru: C 46.94, H 7.09, N 1.82. Found: C 46.94, H 6.80, N 1.80.
:1, 30 mL), was added to a solution of tricyclopentylphosphane (1.00 g, 4.20 mmol) in hot ethanol (10 mL). The reaction mixture was kept under refluxing conditions for 45 minutes. The resulting dark green solid was collected by filtration and dried in vacuo. Yield: 1.02 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 49.9, 34.2, 19.2, 16.8, 4.9. Selected IR bands νmax/cm−1: 2943 (w), 2864 (w), 2359 (w) 1750 (s), 1707 (s), 1446 (w), 1299 (w), 1260 (w), 1119 (w), 904 (w), 874 (w), 621 (vw). MS-FAB (NBA): m/z calcd for C30H54INOP2Ru = [M − I]+ 735.18, found 735.4.
:1, 60 mL), was added to a solution of triisopropylphosphane (1.50 g, 9.00 mmol) in hot ethanol (15 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting solid was collected by filtration and dried in vacuo. Yield: 1.17 g, 2.23 mmol, 58.3%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 33.2, 29.6, 27.1. Selected IR bands νmax/cm−1: 2958 (w), 1841 (m), 1804 (s), 1707 (vs), 1455 (m), 1366 (w), 1240 (m), 1061 (m), 882 (m), 655 (vs). MS-FAB (NBA): m/z calcd for C18H42ClNOP2Ru = [M − Cl]+ 522.12, found 522.4. Elemental analysis (%) calcd for C18H42Cl2NOP2Ru: C 41.38, H 8.10, N 2.68. Found: C 41.45, H 8.29, N 2.61.
:1, 70 mL), was added to a solution of tri-isopropylphosphane (2.00 g, 12.5 mmol) in hot ethanol (20 mL). The reaction mixture was kept under refluxing conditions for 30 minutes. The resulting green solid was collected by filtration and dried in vacuo. Yield: 2.30 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 48.1. Selected IR bands νmax/cm−1: 2955 (w), 1756 (m), 1705 (m), 1455 (w), 1365 (w), 1240 (w), 1060 (w), 1028 (w), 930 (w), 883 (m), 655 (vs), 624 (w). MS-FAB (NBA): m/z calcd for C18H42BrNOP2Ru = [M − Br]+ 533.09, found 533.3.
921 (m), 1048 (vs), 1027 (vs), 880 (m), 673 (m), 648 (w). MS-FAB (NBA): m/z calcd 563.0943 for [RuBr(NO)2(PiPr3)2]+ = [M]+, found 563.0953; m/z calcd 531.0968 for [RuBr(NO)(PiPr3)2]+ = [M − NO]+, found 531.0965.
:1:2 mixture of dichloromethane–ethanol–n-hexane (12 mL) and dried in vacuo. Yield: 1.26 g, 1.59 mmol, 89.3%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 19.6. Selected IR bands νmax/cm−1: 1850 (s, NO), 1599 (vw), 1495 (w), 1482 (w), 1453 (w), 1433 (m), 1408 (w), 1330 (w), 1185 (w), 1143 (w), 1094 (w), 1096 (w), 1030 (w), 1000 (w), 914 (w), 831 (m), 773 (m), 752 (m), 740 (s), 697 (s). MS-FAB (NBA): m/z calcd 754.1 for [RuCl2(NO)(PPh2Bn)2]+ = [M − Cl]+, found 754.0. Elemental analysis (%): calc. C 57.77, H 4.34, Cl 13.46, N 1.77; found: C 58.05, H 4.44, Cl 13.13, N 1.64.
:1, 10 mL) and heated under reflux for 30 minutes. In the course of the reaction a yellow-orange solid precipitated which, after cooling to room temperature, was filtered off and washed with a mixture of ethanol–dichloromethane–n-hexane (1:1:2). The product was freed from all volatile components in vacuo. Yield: 0.610 g, 0.661 mmol, 94.3%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 12.9. Selected IR bands νmax/cm−1: 1847 (s, NO), 1494 (vw), 1453 (vw), 1432 (w), 1408 (vw), 1143 (vw), 1099 (vw), 1069 (vw), 1030 (vw), 830 (m), 772 (m), 750 (m), 739 (s), 697 (vs). MS-FAB (NBA): m/z calcd 844.0 for [RuBr2(NO)(PPh2Bn)2]+ = [M − Br]+, found 844.0. Elemental analysis (%): calc. C 49.43, H 3.71, N 1.52; found: C 50.58, H 3.83, N 1.30.
:1, 60 mL), was added to a solution of tricyclohexylphosphane (2.06 g, 7.35 mmol) in hot ethanol (55 mL). The reaction mixture was kept under refluxing conditions for 4 hours. The resulting solid was collected by filtration and dried in vacuo. Yield: 1.67 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 38.8, 25.2, 17.6. Selected IR bands νmax/cm−1: 2921 (vs), 2846 (vs), 1826 (vs), 1802 (w), 1712 (vs), 1442 (s), 1264 (m), 1195 (m), 1173 (s), 1127 (w), 1002 (s), 899 (m), 847 (s), 734 (m). MS-FAB (NBA): m/z calcd for C36H66ClNOP2Ru = [M]+ 762.3045, found 762.3013; [M − Cl]+ 727.3359, found 727.3315.
:1, 70 mL), was added to a solution of tricyclohexylphosphane (1.15 g, 4.11 mmol) in hot ethanol (50 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting green solid was collected by filtration, washed with ethanol–dichloromethane–n-hexane (1:1:2, 28 mL) and dried in vacuo. Yield: 1.00 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 73.7, 36.5, 24.8, 16.2, 9.3. Selected IR bands νmax/cm−1: 2922 (vs), 2846 (m), 1825 (w), 1802 (m), 1752 (w), 1709 (s), 1442 (m), 1265 (vw), 1172 (m), 1127 (vw), 1001 (m), 886 (w), 846 (m), 732 (m). MS-FAB (NBA): m/z calcd for C36H66BrNOP2Ru = [M]+ 852.2023, found 852.2046; [M − Br]+ 773.2888, found 773.2847.
:1, 24 mL), was added to a solution of tricyclohexylphosphane (0.62 g, 2.2 mmol) in hot ethanol (17 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting green solid was collected by filtration, washed with ethanol–dichloromethane–n-hexane (1:1:2, 28 mL) and dried in vacuo. Yield: 0.23 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 45.8, 34.1, 25.6, 16.9, 11.3. Selected IR bands νmax/cm−1: 2923 (m), 2846 (m), 1798 (m, NO), 1756 (m, NO), 1706 (vs, NO), 1442 (m), 1297 (vw), 1264 (w), 1172 (m), 1002 (m), 886 (w), 845 (m), 814 (w), 731 (m), 652 (w). MS-FAB (NBA): m/z calcd for C36H66INOP2Ru = [M − I]+ 819.27, found 820.0
:1, 70 mL), was added to a solution of tricyclopentylphosphane (1.00 g, 4.20 mmol) in hot ethanol (10 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting solid was collected by filtration and dried in vacuo. Yield: 1.20 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 39.2, 20.7, 17.9, 5.0. Selected IR bands νmax/cm−1: 2947 (m), 2863 (m), 1803 (m), 1703 (vs), 1447 (w), 1299 (w), 1230 (w), 1120 (w), 1011 (vw), 906 (w), 724 (vw), 619 (vw). MS-FAB (NBA): m/z calcd for C30H54ClNOP2Ru = [M − Cl]+ 727.39, found 727.8.
:1, 70 mL), was added to a solution of tricyclopentylphosphane (1.00 g, 4.43 mmol) in hot ethanol (10 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting green solid was collected by filtration and dried in vacuo. Yield: 0.965 g, 1.14 mmol, 65%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 36.9, 27.1, 15.4, 15.0. Selected IR bands νmax/cm−1: 2954 (m), 2864 (w), 1828 (NO), 1806 (NO), 1762 (w), 1703 (m), 1447 (vw), 1298 (vw), 1259 (m), 1013 (s), 906 (w), 861 (w), 795 (vs), 703 (w). MS-FAB (NBA): m/z calcd for C30H54BrNOP2Ru = [M − Br]+ 689.19, found 689.7. Elemental analysis (%) calcd for C30H54Br2NOP2Ru: C 46.94, H 7.09, N 1.82. Found: C 46.94, H 6.80, N 1.80.
:1, 30 mL), was added to a solution of tricyclopentylphosphane (1.00 g, 4.20 mmol) in hot ethanol (10 mL). The reaction mixture was kept under refluxing conditions for 45 minutes. The resulting dark green solid was collected by filtration and dried in vacuo. Yield: 1.02 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 49.9, 34.2, 19.2, 16.8, 4.9. Selected IR bands νmax/cm−1: 2943 (w), 2864 (w), 2359 (w) 1750 (s), 1707 (s), 1446 (w), 1299 (w), 1260 (w), 1119 (w), 904 (w), 874 (w), 621 (vw). MS-FAB (NBA): m/z calcd for C30H54INOP2Ru = [M − I]+ 735.18, found 735.4.
:1, 60 mL), was added to a solution of triisopropylphosphane (1.50 g, 9.00 mmol) in hot ethanol (15 mL). The reaction mixture was kept under refluxing conditions for 1 hour. The resulting solid was collected by filtration and dried in vacuo. Yield: 1.17 g, 2.23 mmol, 58.3%. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 33.2, 29.6, 27.1. Selected IR bands νmax/cm−1: 2958 (w), 1841 (m), 1804 (s), 1707 (vs), 1455 (m), 1366 (w), 1240 (m), 1061 (m), 882 (m), 655 (vs). MS-FAB (NBA): m/z calcd for C18H42ClNOP2Ru = [M − Cl]+ 522.12, found 522.4. Elemental analysis (%) calcd for C18H42Cl2NOP2Ru: C 41.38, H 8.10, N 2.68. Found: C 41.45, H 8.29, N 2.61.
:1, 70 mL), was added to a solution of tri-isopropylphosphane (2.00 g, 12.5 mmol) in hot ethanol (20 mL). The reaction mixture was kept under refluxing conditions for 30 minutes. The resulting green solid was collected by filtration and dried in vacuo. Yield: 2.30 g. 31P NMR (109 MHz, C7H8, 25 °C) δ/ppm: 48.1. Selected IR bands νmax/cm−1: 2955 (w), 1756 (m), 1705 (m), 1455 (w), 1365 (w), 1240 (w), 1060 (w), 1028 (w), 930 (w), 883 (m), 655 (vs), 624 (w). MS-FAB (NBA): m/z calcd for C18H42BrNOP2Ru = [M − Br]+ 533.09, found 533.3.
921 (m), 1048 (vs), 1027 (vs), 880 (m), 673 (m), 648 (w). MS-FAB (NBA): m/z calcd 563.0943 for [RuBr(NO)2(PiPr3)2]+ = [M]+, found 563.0953; m/z calcd 531.0968 for [RuBr(NO)(PiPr3)2]+ = [M − NO]+, found 531.0965.
Notes and references
- J. A. McCleverty, Chem. Rev., 2004, 104, 403–418 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 CrossRef CAS PubMed.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–884 CrossRef CAS PubMed.
- T. E. Bitterwolf, Coord. Chem. Rev., 2006, 250, 1196–1207 CrossRef CAS PubMed.
- D. Schaniel and T. Woike, Phys. Chem. Chem. Phys., 2009, 11, 4391–4395 RSC.
- D. Schaniel, T. Woike, N.-R. Behrnd, J. Hauser, K. W. Krämer, T. Todorova and B. Delley, Inorg. Chem., 2009, 48, 11399–11406 CrossRef CAS PubMed.
- D. Schaniel, T. Woike, B. Delley, D. Biner, K. W. Kramer and H.-U. Güdel, Phys. Chem. Chem. Phys., 2007, 9, 5149–5157 RSC.
- A. Zangl, P. Klüfers, D. Schaniel and T. Woike, Dalton Trans., 2009, 1034–1045 RSC.
- A. Zangl, P. Klüfers, D. Schaniel and T. Woike, Inorg. Chem. Commun., 2009, 12, 1064–1066 CrossRef CAS PubMed.
- H. Giglmeier, T. Kerscher, P. Klüfers, D. Schaniel and T. Woike, Dalton Trans., 2009, 9113–9116 RSC.
- A. F. Vanin, R. R. Borodulin, L. N. Kubrina, V. D. Mikoyan and D. S. Burbaev, Biophysics, 2013, 58, 103–109 CrossRef CAS.
- A. Klein, Y. v. Mering, A. Uthe, K. Butsch, D. Schaniel, N. Mockus and T. Woike, Polyhedron, 2010, 29, 2553–2559 CrossRef CAS PubMed.
- J. Mason, D. M. P. Mingos, D. Sherman and R. W. M. Wardle, J. Chem. Soc., Chem. Commun., 1984, 1223–1225 RSC.
- D. Michael, P. Mingos, D. Sherman and S. Bott, Transition Met. Chem., 1987, 12, 471–475 CrossRef.
- C. G. Pierpont, D. G. Van Derveer, W. Durland and R. Eisenberg, J. Am. Chem. Soc., 1970, 92, 4760–4762 CrossRef CAS.
- C. G. Pierpont and R. Eisenberg, Inorg. Chem., 1972, 11, 1088–1094 CrossRef CAS.
- L. K. Bell, J. Mason, D. M. P. Mingos and D. G. Tew, Inorg. Chem., 1983, 22, 3497–3502 CrossRef CAS.
- D. M. Mingos, D. Sherman and I. Williams, Transition Met. Chem., 1987, 12, 493–496 CrossRef CAS.
- L. A. Watson, M. Pink and K. G. Caulton, J. Mol. Catal. A: Chem., 2004, 224, 51–59 CrossRef CAS PubMed.
- M. R. Crimmin, R. G. Bergman and F. D. Toste, Angew. Chem., Int. Ed., 2011, 50, 4484–4487 CrossRef CAS PubMed.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS PubMed.
- N. G. Connelly, T. Damhus, R. M. Hartshorn and A. T. Hutton, Nomenclature of inorganic chemistry: IUPAC recommendations 2005, The Royal Society of Chemistry, Cambridge, 2005 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. R. Rossi and R. Hoffmann, Inorg. Chem., 1975, 14, 365–374 CrossRef CAS.
- M. H. B. Stiddard and R. E. Townsend, J. Chem. Soc. D, 1969, 1372–1372 RSC.
- D. J. Hodgson, N. C. Payne, J. A. McGinnety, R. G. Pearson and J. A. Ibers, J. Am. Chem. Soc., 1968, 90, 4486–4488 CrossRef CAS.
- J. R. Durig, W. A. McAllister, J. N. Willis and E. E. Mercer, Spectrochim. Acta, 1966, 22, 1091–1100 CrossRef CAS.
- M. J. Cleare and W. P. Griffith, J. Chem. Soc. A, 1967, 1144–1147 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Computational Molecular Science, 2012, 2, 73–78 CrossRef CAS.
- J. Cirera, E. Ruiz and S. Alvarez, Chem. – Eur. J., 2006, 12, 3162–3167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of crystallographic studies. CCDC 985278–985281, 985283–985289, 986283–986284. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01506a |
| This journal is © The Royal Society of Chemistry 2014 |