 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriptycene based organometallic complexes: a new class of acceptor synthons for supramolecular ensembles†
Sourav
Chakraborty
a,
Snehasish
Mondal
a,
Sourav
Bhowmick
a,
Jianqiu
Ma
b,
Hongwei
Tan
b,
Subhadip
Neogi
c and
Neeladri
Das
*a
aDepartment of Chemistry, Indian Institute of Technology Patna, Patna 800 013, Bihar, India. E-mail: neeladri@iitp.ac.in; Fax: +91 612 2277383; Tel: +91 612 2552023
bDepartment of Chemistry, Beijing Normal University, Beijing 100050, People's Republic of China
cDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India
First published on 8th July 2014
Abstract
Preparation and characterization of two new triptycene based polytopic Pt(II) organometallic complexes are being reported. These complexes have three trans-bromobis(trialkylphosphine)platinum(II) units directly attached to the central triptycene unit. These organoplatinum complexes were converted to the corresponding nitrate salts for subsequent use in self-assembly reactions. Characterization of these organometallic triptycene complexes by multinuclear NMR, FTIR, mass spectrometry and elemental analyses is described. The molecular structure of one of the organoplatinum triptycene tripods was determined by single-crystal X-ray crystallography. The potential utility of these organometallic tritopic acceptors as building blocks in the construction of metallasupramolecular cages containing the triptycene motif is explored. Additionally, for the first time, 3,3′-bipyridine has been used as a flexible donor tecton for self-assembly of discrete and finite metallacages using triptycene based tritopic organometallic acceptor units. Triptycene motif containing supramolecules were characterized by multinuclear NMR (including 1H DOSY), mass spectrometry and elemental analyses. Geometry of each supramolecular framework was optimized by employing the PM6 semiempirical molecular orbital method to predict its shape and size.
Introduction
Coordination driven self-assembly has evolved as one of the most popular synthetic protocols in the design of discrete supramolecular abiological structures.1 In this methodology, formation of multiple metal–organic ligand coordination bond is the driving force for the construction of supramolecules with predefined shape and size. This synthetic strategy is also referred to as the “directional bonding” approach because of the extensive use of coordination bonds that have high directional bonding nature.1 Using this coordination driven self-assembly approach, facile and efficient syntheses of a variety of two-dimensional ensembles (metallamacrocycles) as well as three-dimensional nanoscalar discrete moieties (metallacages) have been reported.2Among the various transition metals used in coordination driven self-assembly of supramolecules, square planar Pt(II) based multidentate moieties are among the most popular metal acceptor subunits. Discrete ionic supramolecular architectures are generated when Pt(II) acceptor units are used in conjugation with neutral donor (nitrogen/phosphorus coordinating) linkers.2a,b The coordination driven self-assembly protocol has been efficiently utilized to functionalize the desired supramolecule with various interesting motifs such as ferrocene, carborane, crown ethers, dendrimers and others.3 Additionally, the shape of the supramolecules has been effectively tailored using building blocks of various dimensions.
Triptycene, first synthesized by Bartlett in 1942, has a rigid framework composed of three benzene rings connected via bicycle[2.2.2]octane.4 Supramolecular chemistry of triptycene has been developed by Chen,5 MacLachlan,6 Mastalerz7 and others,8 wherein a variety of supramolecular hosts have been designed and applied in molecular host–guest chemistry.5a,b,6a In this context, discrete metallasupramolecular frameworks containing triptycene motifs are rare.9 A thorough literature search has revealed that there are a handful of organometallic complexes derived from triptycene.10 Consequently, the use of organometallic triptycene derivatives as building blocks in supramolecular chemistry has not been reported to date.
In continuation of our research interest to develop triptycene based linkers11 and their use as tectons in supramolecular chemistry, herein we report the synthesis of two new tritopic organoplatinum compounds having a triptycene core. Complexes 1 and 2 were synthesized by multiple oxidative-addition of Pt(PR3)3 (R = Me/Et) with tribromo triptycene. Subsequent bromide abstraction with AgNO3 results in the formation of the corresponding trinitrate salts 3 and 4. Complexes 1–4 represent unique examples of triptycene based organoplatinum molecules that are structurally rigid with predefined bite angles and thus they qualify to act as metal-containing acceptor subunits for use in self-assembly via a directional bonding approach. All the organoplatinum complexes (1–4) have been fully characterized by FT-IR and multinuclear NMR spectroscopy, mass spectrometry, and elemental analyses. In addition, the organometallic complex 1 has been structurally characterized by single crystal X-ray diffraction analysis.
These multitopic triptycene based organometallic linkers (3 and 4) are potential building blocks for the construction of discrete metallasupramolecular architectures. To illustrate this point and using the “ligand-directed” approach,12a facile self-assembly of two new triptycene based [3 + 2] cationic metallacages (5 and 6) has been achieved by reacting 3,3′-bipyridine with organometallic tritopic acceptor linkers 3 and 4 in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometric ratio. Supramolecules 5 and 6 are unique examples of charged molecular cages that have been self-assembled from triptycene based organometallic tectons. Also for the first time, flexible 3,3′-bipy has been employed as a donor tecton for the design of a supramolecular cage. These newly synthesized metallacages were completely characterized by multinuclear NMR spectroscopy, 1H DOSY, mass spectrometry (ESI-TOF-MS), and elemental analysis techniques. Geometry optimizations (by employing a PM6 semiempirical molecular orbital method) suggest that these triptycenes containing supramolecules are of nanoscalar dimensions.
:2 stoichiometric ratio. Supramolecules 5 and 6 are unique examples of charged molecular cages that have been self-assembled from triptycene based organometallic tectons. Also for the first time, flexible 3,3′-bipy has been employed as a donor tecton for the design of a supramolecular cage. These newly synthesized metallacages were completely characterized by multinuclear NMR spectroscopy, 1H DOSY, mass spectrometry (ESI-TOF-MS), and elemental analysis techniques. Geometry optimizations (by employing a PM6 semiempirical molecular orbital method) suggest that these triptycenes containing supramolecules are of nanoscalar dimensions.
Results and discussion
There are several reports in the literature wherein halogenated arenes have been used as synthons for the design of metal-containing acceptor building blocks for subsequent use in coordination driven self-assembly processes.12 Herein, the triptycene based organoplatinum complexes (1 and 2) were efficiently synthesized by triple oxidative addition13 of Pt(0) to the corresponding 2,7,14-tribromotriptycene14 in good yields (70–73%) as shown in Scheme 1. These triptycene-organoplatinum complexes are air/moisture stable solids with high solubility in common organic solvents. Complexes 1 and 2 were further converted to the corresponding trinitro analogues (3 and 4 respectively) by salt metathesis reaction with AgNO3 in quantitative yields.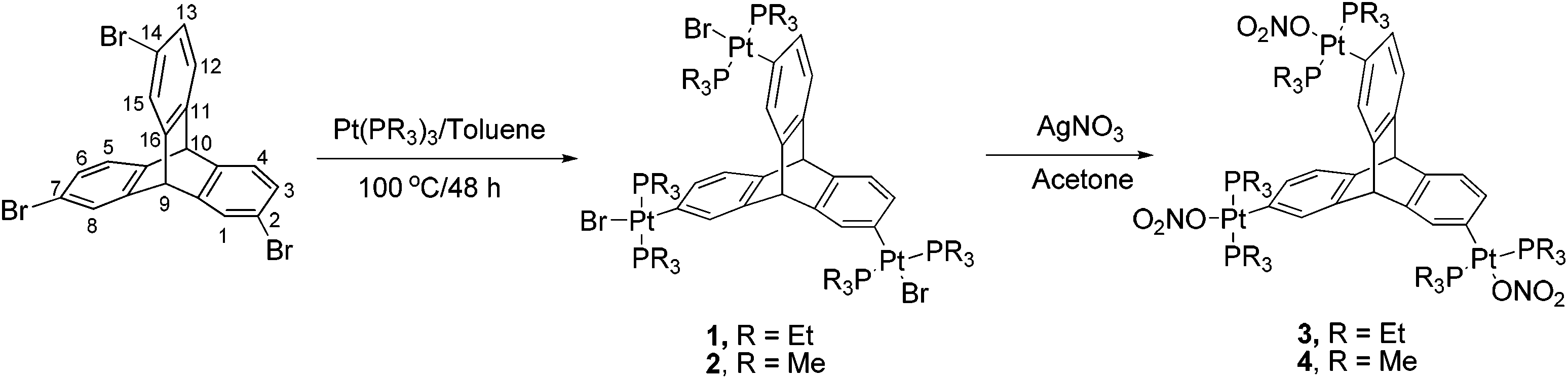 | ||
| Scheme 1 Synthesis of triptycene based organoplatinum complexes (1–4). | ||
In the 1H NMR spectrum of complexes 1–4, two singlet peaks between 4.94 and 5.15 ppm correspond to the bridgehead protons. As expected, the methylene and methyl protons of the PEt3 group of complexes 1 and 3 appear as multiplets in the range 0.85 to 1.62 ppm. In the case of complexes 2 and 4, the methyl protons of the PMe3 group appear as a multiplet centered at 1.06–1.21 ppm due to coupling with phosphorous nuclei.
Organometallic complexes 1–4 were also characterized by 31P{1H} NMR spectroscopy. The 31P NMR spectra of the platinum complexes 1 and 2 show a sharp singlet with concomitant 195Pt satellites. The 31P resonance of the phosphine group appears at 11.92 ppm (1JPPt = 1396 Hz) and −16.09 ppm (1JPPt = 1377 Hz) for complexes 1 and 2, respectively (ESI†). In the case of 31P NMR of complexes 3 and 4, a downfield shift in the peak position compared to the corresponding bromo-derivatives was observed with the signal appearing at 18.97 (1JPPt = 1464 Hz) and −9.76 ppm (1JPPt = 1454 Hz) respectively (ESI†). These 31P NMR shifts are in good agreement with those reported in the literature for the trans-PtP2 system.13 The appearance of a single sharp singlet in the 31P NMR of the complexes 1–4 suggests the chemical equivalence of the phosphines attached to the Pt(II) center and the overall symmetrical structure. Mass spectrometry (ESI-MS) analysis further confirmed the formation of complexes 1–4 (ESI†).
X-ray crystallographic analysis of 1
Attempts were made to grow single crystals of these new organometallic complexes for structural characterization. However, we were successful only in the case of the tripodal PtII3 complex 1. The molecular structure of 1 is shown in Fig. 1. Single crystal X-ray diffraction studies revealed that 1 crystallized together with solvent chloroform molecules in the monoclinic space group Cm (Experimental section). The crystal structure analysis divulges that the platinum center displays slightly distorted square planar geometry with four cis-angles around the platinum centre in the range 89.9–94.1°. No unusual bond lengths and bond angles between atoms were observed in the structural analysis. The average distance between the platinum centres is 9.62 Å.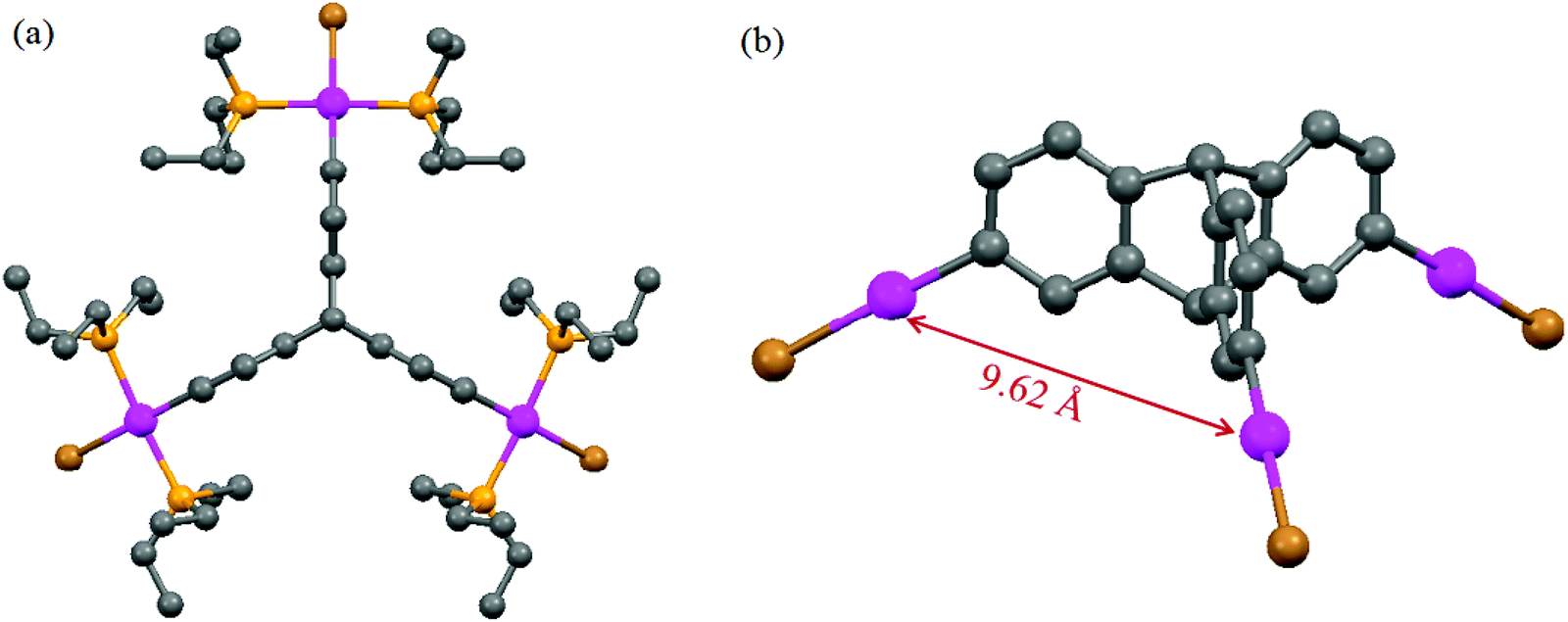 | ||
| Fig. 1 (a) Top view and (b) side view (phosphine ligands are omitted) of the crystal structure of complex 1. Solvent molecules and hydrogen atoms are omitted for clarity (grey: C, pink: Pt, orange: P, brown: Br). | ||
Self-assembly of 3D cages
According to the “Molecular Library” model systematized by Stang and co-workers, in order to self-assemble a three-dimensional coordination cage, one of the supramolecular synthons must be tritopic (having three binding sites).15 A trigonal prism is one of the simplest examples of a three-dimensional abiological nanoscalar cage that can be efficiently synthesized using the coordination-driven self-assembly protocol.2b,16 Considering the ligand-directed approach, one strategy to construct a trigonal prism is to self-assemble two units of a planar and rigid tritopic tecton with three units of a complementary tecton having nearly parallel coordination vectors.16 In this regard, Stang and coworkers have shown that if the planar tritopic tecton is replaced by a tetrahedral (109°) tripod, the resulting supramolecular three-dimensional cages assume the shape of a distorted trigonal prism.17 In their design strategy to construct distorted trigonal prisms, Stang and coworkers have utilized rigid anthracene based ditopic tectons (donor/acceptor linkers termed as “molecular clip”) having nearly 0° dihedral angle (within their coordination vectors) as the other building block. The use of flexible ditopic tectons in the molecular self-assembly of a distorted trigonal prism is rather rare, with only a few reports that utilize such modular units.173,3′-Bipyridine is considered to be a flexible ditopic linker due to the two different rotomeric states that it can adopt while acting as a bridging ligand (Scheme 2).
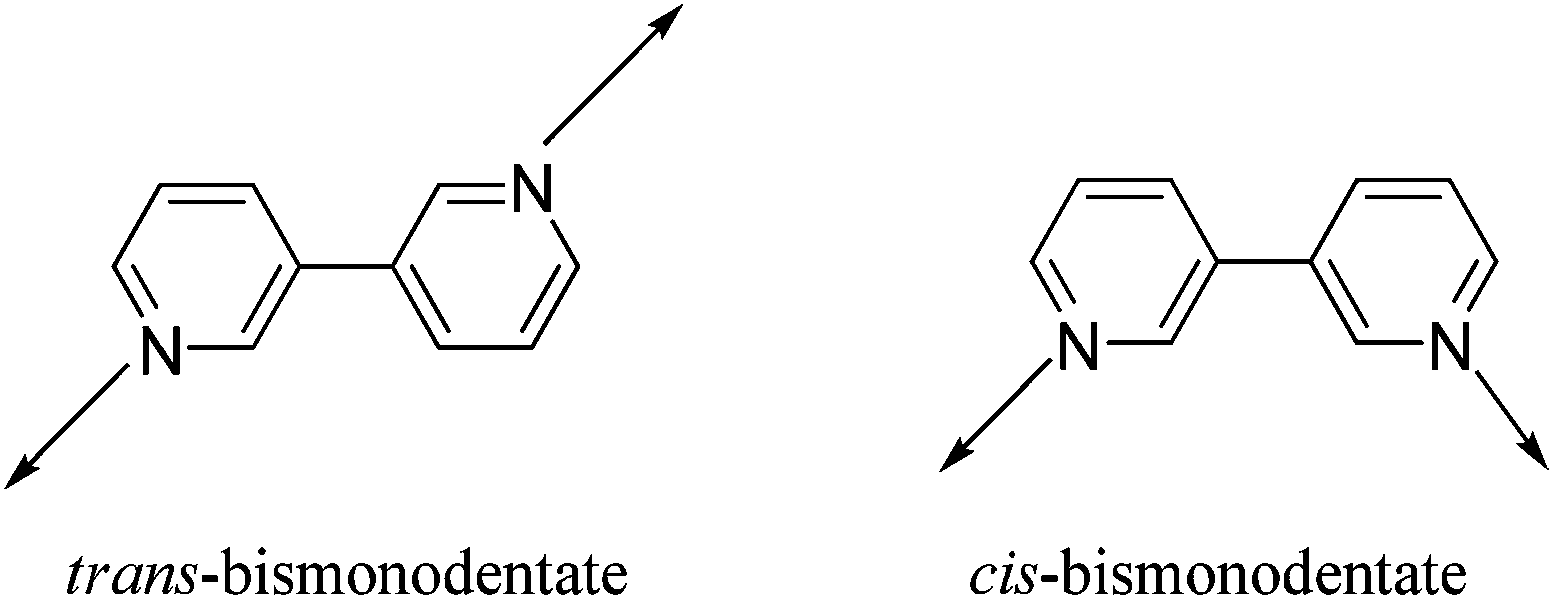 | ||
| Scheme 2 cis- and trans-orientation of 3,3′-bipyridine as a bridging μ-3,3′-bismonodentate ligand. | ||
There are several examples of coordination polymers that contain 3,3′-bipy in both cis- and trans-orientation.18 However, design and self-assembly of discrete supramolecular frameworks using 3,3′-bipy as a donor building block is extremely rare. To the best of our knowledge, there is only one report wherein the flexible 3,3′-bipy was employed for the construction of a discrete metallacycle.18a To date, there is no literature report where 3,3′-bipy has been used for the self-assembly of more complex three-dimensional (3D) metallacages. Herein, for the first time, we utilize 3,3′-bipy as a new flexible ditopic donor tecton in conjugation with a triptycene based non-planar PtII3 acceptor tecton (3 or 4) for the self-assembly of a nanoscalar and triptycene motif containing distorted trigonal prism.
Reaction of organometallic nonplanar acceptor tripods (3 or 4) and 3,3′-bipy in a 2:3 stoichiometric ratio (Scheme 3) resulted in the exclusive formation of the desired discrete 3D metallacages (5 and 6) in high yields. The products obtained in these self-assembly reactions are highly soluble in green solvents such as ethanol and methanol, but sparingly soluble in halogenated solvents such as chloroform or dichloromethane. Products were characterized by multinuclear NMR spectroscopy, mass-spectrometry (ESI-TOF-MS), and elemental analyses.
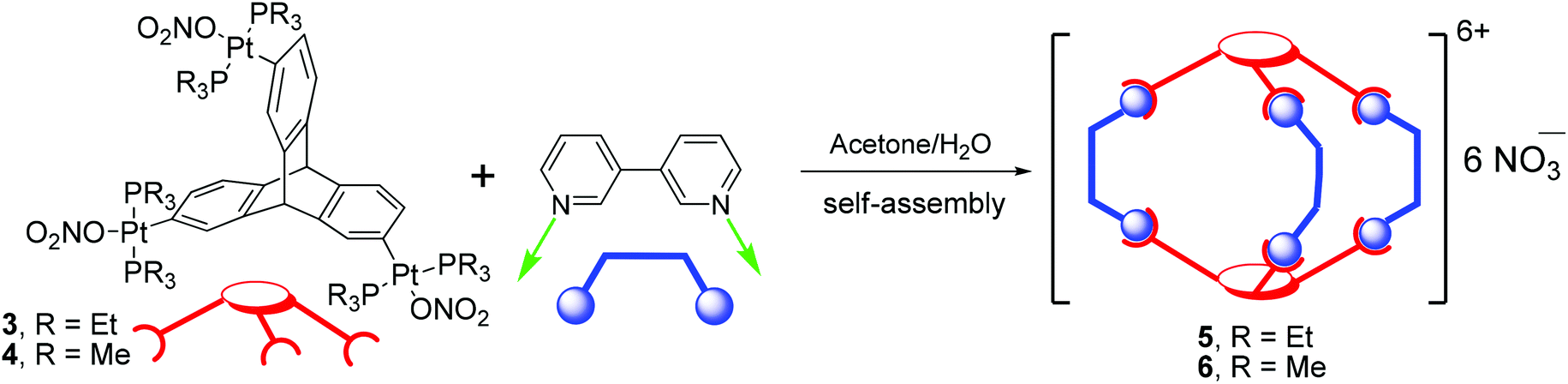 | ||
| Scheme 3 Synthesis of metallasupramolecular cages 5 and 6. | ||
The formation of discrete supramolecular species was evident from NMR spectroscopic studies in the case of these self-assembly reactions. The appearance of a sharp singlet peak (δ 13.59 ppm for 5 and −14.48 ppm for 6) in the 31P{1H} NMR spectrum of each assembly with concomitant Pt satellites (5, 1JPPt = 1345 Hz and 6, 1JPPt = 1357 Hz) clearly indicated the formation of a single highly symmetrical molecular cage in which all the phosphorus nuclei are equivalent (Fig. 2a and ESI†). Additionally, a significant decrease in the coupling of accompanying 195Pt satellites of the products (5 or 6) in comparison with that of the respective organometallic precursors (3 and 4) suggests ligand–metal coordination.
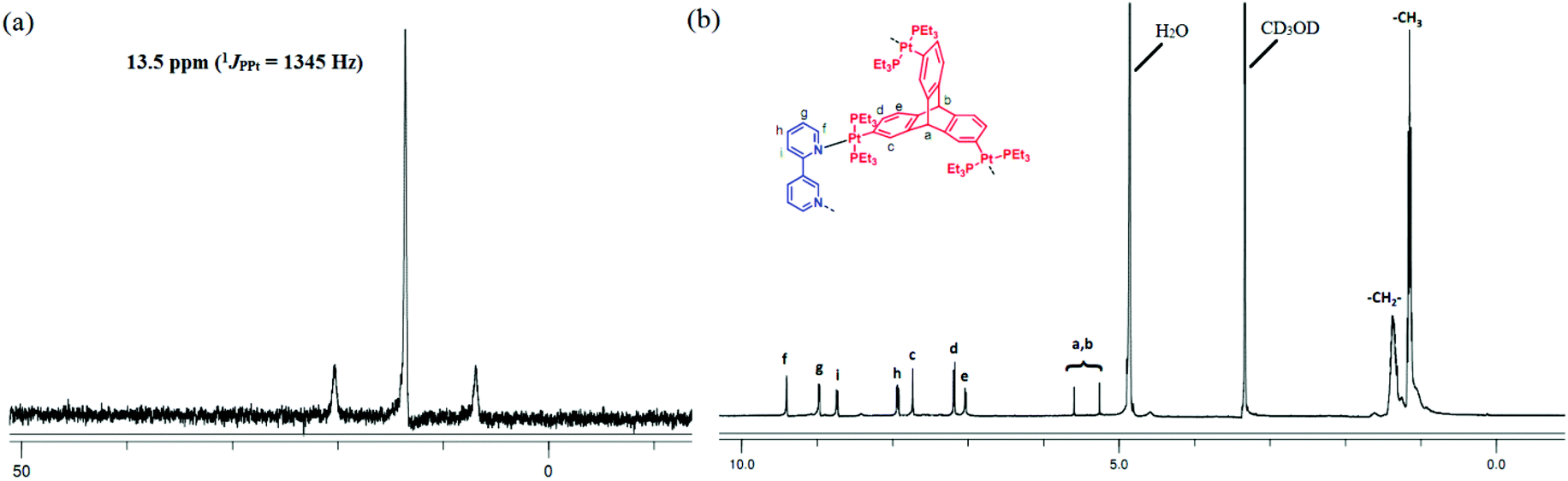 | ||
| Fig. 2 (a) 31P{1H} NMR and (b) 1H NMR spectra of cage 5 in CD3OD. | ||
Similarly, the simplicity of the 1H NMR spectrum of both 5 and 6 and observance of sharp signals hint at the formation of finite and discrete supramolecular frameworks. A representative proton NMR spectrum of 5 is shown in Fig. 2b. All signals observed in the 1H NMR spectrum were assigned. Peaks at δ 7.91, 8.73, 8.97 and 9.40 ppm correspond to 3,3′-bipy moieties. Peaks at 5.25 and 5.59 ppm correspond to bridgehead protons of the triptycene motifs. Peaks in the range 1.45–1.30 ppm are due to the ethyl groups of the PEt3 ligand coordinated to Pt(II) centers. The integration ratio of signals due to 3,3′-bipy and that due to the triptycene core suggests incorporation of the 3,3′-bipy units as well as triptycene organometallic motifs in the product 5 in 3:2 stoichiometric ratio. Similarly, in the case of 6, all proton resonances were assigned precisely (ESI†) and the integration ratio of peaks due to 3,3′-bipy and the PtII3 acceptor 4 indicated their self-assembly in 3:2 stoichiometric ratio. Furthermore, these organometallic cages advocate a single trace in the 1H DOSY NMR (ESI†), indicating the formation of single product and rule out the possibility of larger cages or oligomers in solution.
Mass-spectrometric analysis (ESI-TOF-MS) of 5 and 6 (ESI†) confirmed that the reaction of flexible 3,3′-bipy and the rigid PtII3 tripodal unit in 3:2 stoichiometric ratio results in the self-assembly of the desired [3 + 2] discrete prismatic metallacage over other possible oligomeric/polymeric products. The ESI-TOF-MS spectrum of 5 showed signals attributable to the consecutive loss of nitrate counter anions at m/z = 1903.99 [5-2NO3]2+, 1248.49 [5-3NO3]3+, 920.89 [5-4NO3]4+ and 724.33 [5-5NO3]5+. These peaks were isotopically resolved and are in excellent agreement with the predicted theoretical distribution assuming the formation of the M2L3 [M = PtII3 acceptor 3 or 4, L = 3,3′-bipy] cage (Fig. 3a). Similarly for 6, peaks corresponding to [6-2NO3]2+ at m/z = 1651.04, [6-3NO3]3+ at m/z = 1079.97 and [6-4NO3]4+ at m/z = 794.50 were observed in the ESI-TOF-MS spectrum. The latter two peaks were also isotopically resolved and they matched with the predicted theoretical distribution (Fig. 3b). Thus mass spectroscopy analysis was used to unambiguously confirm the M2L3 composition of the three-dimensional metallacages obtained via self-assembly of two units of triptycene based PtII3 tritopic acceptors (M) and three units of flexible 3,3′-bipy (L). The only possible discrete shape that can be assigned to such a M2L3 cage is that of a distorted trigonal prism.
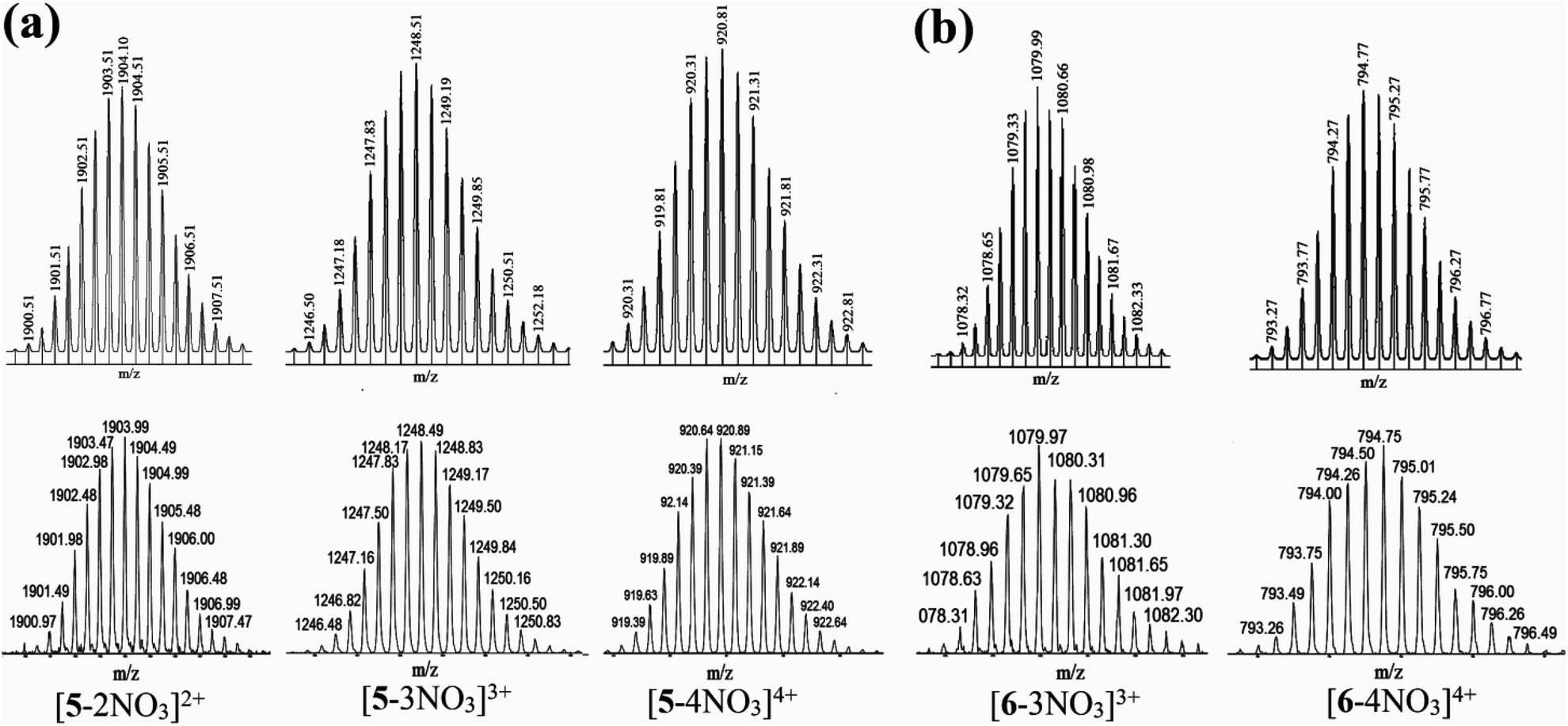 | ||
| Fig. 3 Theoretical (top) and experimental (bottom) ESI-TOF-MS spectra of (a) cage 5 and (b) cage 6. | ||
All attempts to grow diffraction quality single crystals (of 5 and 6) for X-ray structure determination have been unsuccessful to date. However, to obtain further insight into the shape and size of these metallacages (5 and 6), the PM6 semiempirical molecular orbital method19 was employed to optimize their geometry. Both the energy minimized cages have three leaf “clover” like shape and pseudo-D3 symmetry could be observed for these cages. The capped stick model of cages 5 and 6 is shown in Fig. 4. For both cages, each wing of the “clover” extends away from the center by about 10.6 Å. The triangular prismatic cavities inside the two cages have a height of about 6 Å and the longest distance between two platinum atoms is 9.7 Å.
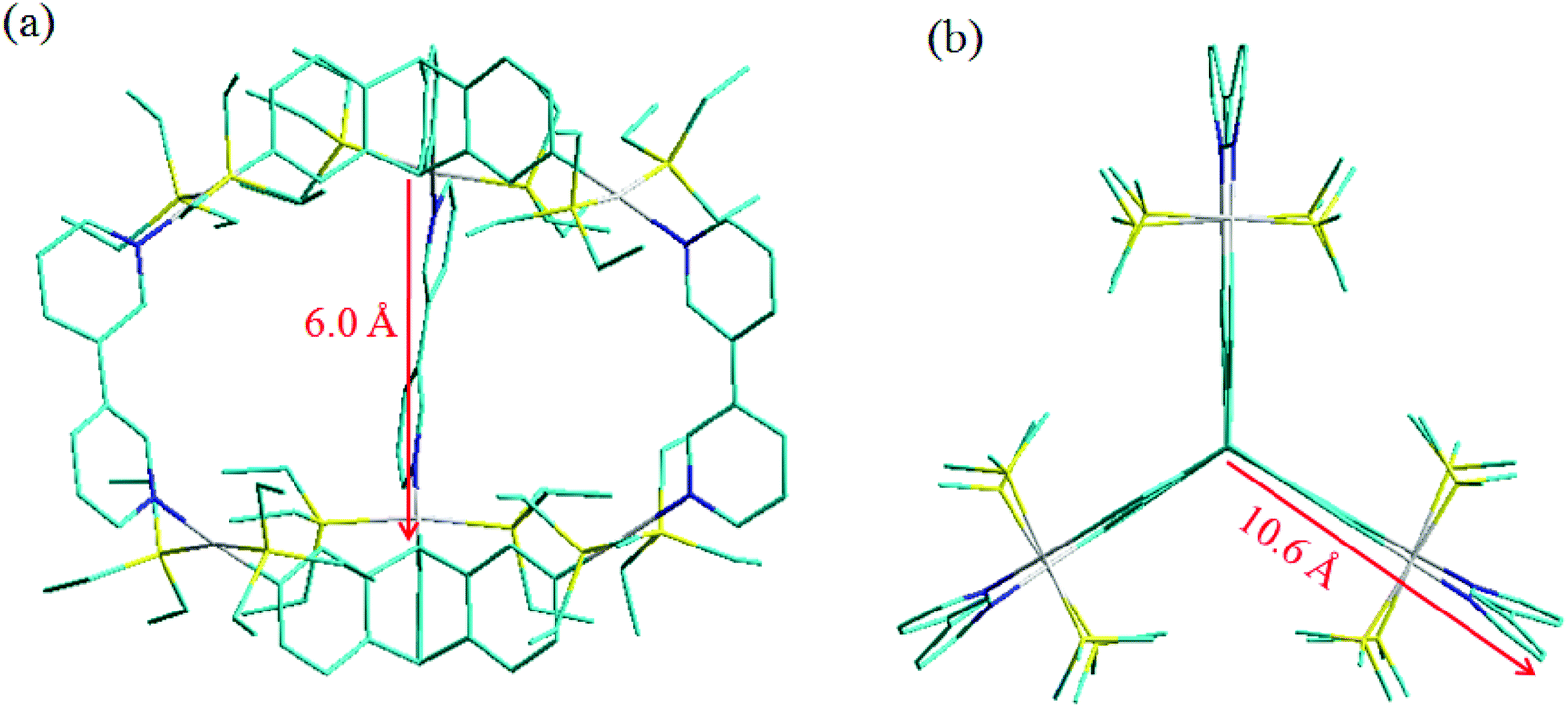 | ||
| Fig. 4 Simulated capped stick molecular model of (a) metallacage 5 and (b) metallacage 6 optimized by PM6 semiempirical molecular orbital methods (cyan: C, yellow: P, white: Pt, blue: N). H atoms are omitted for clarity. | ||
Conclusion
In conclusion, we report the synthesis of two new triptycene based organometallic complexes containing three Pt(II) centers in reasonably high yields. In these complexes, the triptycene core is directly σ-bonded to three trans-bromobis(trialkylphosphine)]platinum(II) units. These triptycene organoplatinum complexes and their corresponding nitrate salts were characterized by FT-IR, multinuclear NMR spectroscopy, mass spectrometry, and elemental analyses. Complex 1 was also structurally characterized by single crystal X-ray diffraction. The presence of triptycene backbone imparts rigidity to these organometallic complexes. Moreover, the divergent spatial orientation of the Pt(II) centers (with respect to each other), renders these linkers potential acceptor building blocks in coordination driven self-assembly for the synthesis of finite supramolecular frameworks. To illustrate this point, two metallacages have been synthesized using these triptycene based organoplatinum complexes. 3,3′-bipy has been employed in conjugation with triptycene based tripodal PtII3 acceptor moieties (3 and 4) for the construction of ionic distorted trigonal nanoprismatic cages (5 and 6). 3,3′-bipy is a flexible donor linker and its use in coordination driven self-assembly reactions might lead to the formation of open chain oligomers.18a,20 However, for the first time, we describe the use of 3,3′-bipy to design three-dimensional discrete supramolecular metallacages in association with triptycene based organometallic tripods. While NMR spectroscopy, including 1H DOSY, of these supramolecules (5 and 6) ruled out the formation of oligomeric species (thereby suggesting the formation of highly symmetrical discrete moieties), mass spectrometric (ESI-TOF-MS) and elemental analyses confirmed their respective composition and purity. The shape and dimension of these nanoscalar frameworks were predicted from 1H DOSY NMR and molecular simulations using PM6 semiempirical molecular orbital methods. To the best of our knowledge, these self-assembled metallacages are the only examples of triptycene based supramolecular distorted and trigonal nanoscalar “platinaprisms” obtained from organometallic triptycene complexes. To summarize, this study provides an efficient and simple synthetic protocol for the facile synthesis of triptycene motif containing supramolecular architectures via coordination driven self-assembly. Additionally the triptycene based organometallic complexes reported herein aim to enrich the molecular library of metal based acceptors used as synthons in directional bonding approach. The use of triptycene based rigid organometallic acceptor linkers in conjunction with various rigid/flexible organic donors has immense potential to incorporate triptycene motifs into self-assembled supramolecular coordination complexes (SCCs) and metal–organic frameworks (MOFs). This in turn, is expected to further develop the range of the directional bonding paradigm in self-assembly. Studies are currently undergoing in our laboratory in these directions.Experimental section
General details
All chemicals and anhydrous solvents used in this work were purchased from commercial sources and used without further purification. 2,7,14-tribromotriptycene,14 3,3′-bipyridine21 and complex tris(triethylphosphine)platinum(0)22 were prepared by following the reported literature procedures. FTIR spectra were recorded in a Shimadzu FTIR spectrophotometer. 1H NMR spectra and 31P NMR spectra were recorded on Bruker 400/500 MHz spectrometers. Elemental analyses were carried out using a CE-440 elemental analyzer (Exeter Analytical Inc.). ESI-MS spectra of the compounds were recorded using a Thermo LCQ Deca XP MAX mass spectrometer. DOSY NMR measurements were performed on a Bruker AV 500 NMR spectrometer using a 5 mm gradient probe at 298 K. DOSY experiments were done with a standard Bruker pulse sequence (ledbpgp2s) with longitudinal eddy current delay.General procedure for the synthesis of complexes 1 and 2
To a 25 ml Schlenk flask containing one equiv. of tri-bromotriptycene and six equiv. of Pt(PR3)3 (R = PEt3 or PMe3), 10 ml anhydrous toluene was added in a glove box. The reaction mixture was stirred for 48 h at 100 °C under nitrogen. The resulting light yellow coloured solution was evaporated to dryness under vacuum in a rotary evaporator. Yellow coloured residue thus obtained was purified by column chromatography over silica gel eluting with dichloromethane–hexane (2.5:1, upto 9:1, v/v) affording pure organometallic complexes 1 and 2 as a white solid.
X-ray crystallography
Slow evaporation of chloroform solution of 1 at ambient temperature provides the X-ray quality crystals. The crystals are very sensitive to air and loose crystallinity immediately when taken out of the mother liquor. Data collection could only be done at low temperature by wrapping in oil prior to mounting. Single crystal X-ray data on 1 were collected at 100 K on a Bruker SMART APEX CCD diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The linear absorption coefficients, scattering factors for the atoms, and the anomalous dispersion corrections were taken from International Tables for X-ray Crystallography. Data integration and reduction were processed with SAINT23a software. An empirical absorption correction was applied to the collected reflections with SADABS23b using XPREP.23c The structure was solved by the direct method using SHELXTL23d and was refined on F2 by the full-matrix least-squares technique using the SHELXL-9723e program package. Apart from two and half CHCl3 solvent molecules, the unit cell contains several other disordered solvent molecules, which could not be modeled as discrete atomic sites. Therefore, we employed PLATON/SQUEEZE24 and the structure was then refined again using the data generated.Crystal data
1·2.5CHCl3: C61H106Br3Cl15P6Pt3, M = 2382.03, monoclinic, space group Cm, a = 16.482(5), b = 29.135(5), c = 10.874(5) Å, β = 112.156(5)°, V = 4836.0(3) Å3, T = 100 K, Z = 2, 12376 reflections measured, 5790 unique (Rint = 0.0556) which were used in all calculations. The final wR(F2) was 0.1272 (all data). CCDC 953632.
Authors’ contribution
S.C. synthesized all new triptycene based organometallic complexes and the ionic metallacages reported in this manuscript. S.B. and S.M. assisted in the synthesis of some of the literature reported triptycene precursors. S.C. grew single crystals of organometallic complex 1 and S.N. carried out its single crystal X-ray crystallographic analysis. J.M. and H.T. optimized the energy minimized geometry of the metallacages 5 and 6 using the PM6 semiempirical molecular modelling method. All authors have contributed to compiling the manuscript and have approved the final manuscript.Acknowledgements
N.D. thanks the Department of Science and Technology, Govt. of India, New Delhi (DST no.: SR/FT/CS-028/2009) and the Indian Institute of Technology Patna, for financial support. The authors acknowledge Dr P.R. Rajamohanan, Central NMR Facility, CSIR-NCL, Pune for recording DOSY NMR spectra. The authors also acknowledge Dr Hai-Bo Yang (East China Normal University) for helpful discussions. S.C. and S.B thank IIT Patna for an Institute Research Fellowship. S.M. thanks UGC, New Delhi for a Junior Research Fellowship. S.N. thanks IIT Kanpur for a Post Doctoral Fellowship. The authors also acknowledge SAIF-Panjab University and SID, IISc Bangalore for providing analytical facilities.References
- (a) J. W. Steed, D. R. Turner and K. J. Wallace, Core Concepts in Supramolecular Chemistry and Nano chemistry, Wiley, West Sussex, U.K., 2007 Search PubMed; (b) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763 CrossRef CAS PubMed; (c) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd, United Kingdom, 2009 Search PubMed; (d) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS; (e) B. Olenyuk, J. A. Whiteford, A. Fechtenkotter and P. J. Stang, Nature, 1999, 398, 796 CrossRef CAS PubMed; (f) K. N. Raymond, Nature, 2009, 460, 585 CrossRef CAS.
- (a) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (c) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (d) M. Yoshizawa and M. Fujita, Bull. Chem. Soc. Jpn., 2010, 83, 609 CrossRef CAS; (e) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (f) C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618 CrossRef CAS PubMed; (g) D. Samanta and P. S. Mukherjee, Chem. Commun., 2014, 50, 1595 RSC; (h) N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354 RSC; (i) P. Thanasekaran, C.-C. Lee and K.-L. Lu, Acc. Chem. Res., 2012, 45, 1403 CrossRef CAS PubMed; (j) B. Therrien, Chem. – Eur. J., 2013, 19, 8378 CrossRef CAS PubMed; (k) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464 CrossRef CAS PubMed; (l) J. K. Clegga, F. Li and L. F. Lindoy, Coord. Chem. Rev., 2013, 257, 2536 CrossRef PubMed; (m) M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815 RSC.
- (a) N. Das, A. M. Arif, P. J. Stang, M. Sieger, B. Sarkar, W. Kaim and J. Fiedler, Inorg. Chem., 2005, 44, 57984 Search PubMed; (b) N. Das, P. J. Stang, A. M. Arif and C. F. Campana, J. Org. Chem., 2005, 70, 10440 CrossRef CAS PubMed; (c) F. Huang, H.-B. Yang, N. Das, U. Maran, A. M. Arif, H. W. Gibson and P. J. Stang, J. Org. Chem., 2006, 71, 6623 CrossRef CAS PubMed; (d) H.-B. Yang, B. H. Northrop, Y.-R. Zheng, K. Ghosh and P. J. Stang, J. Org. Chem., 2009, 74, 7067 CrossRef CAS PubMed; (e) Q.-J. Li, G.-Z. Zhao, L.-J. Chen, H. Tan, C.-H. Wang, D.-X. Wang, D. A. Lehman, D. C. Muddiman and H.-B. Yang, Organometallics, 2012, 31, 7241 CrossRef CAS; (f) N. Das, A. Ghosh, O. M. Singh and P. J. Stang, Org. Lett., 2006, 8, 1701 CrossRef CAS PubMed; (g) N. Das, A. Ghosh, A. M. Arif and P. J. Stang, Inorg. Chem., 2005, 44, 7130 CrossRef CAS PubMed; (h) B. H. Northrop, H.-B. Yang and P. J. Stang, Chem. Commun., 2008, 5896 RSC.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649 CrossRef CAS.
- (a) C.-F. Chen, Chem. Commun., 2011, 47, 1674 RSC and references therein; (b) Y. Jiang and C.-F. Chen, Eur. J. Org. Chem., 2011, 6377 CrossRef CAS and references therein; (c) C.-F. Chen and Y.-X. Ma, Iptycene Chemistry: from Synthesis to Applications, Springer-Verlag, Berlin, 2013 Search PubMed.
- (a) J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301 RSC and references therein; (b) A. K. Crane, B. O. Patrick and M. J. MacLachlan, Dalton Trans., 2013, 42, 8026 RSC; (c) A. K. Crane, E. Y. L. Wong and M. J. MacLachlan, CrystEngComm, 2013, 15, 9811 RSC.
- (a) M. Mastalerz, Synlett, 2013, 781 CrossRef CAS PubMed and references therein; (b) G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 1 CrossRef.
- L. Zhao, Z. Li and T. Wirth, Chem. Lett., 2010, 39, 658 CrossRef CAS.
- S. Chakraborty, S. Mondal, Q. Li and N. Das, Tetrahedron Lett., 2013, 54, 1681 CrossRef CAS PubMed.
- (a) C. Azerraf and D. Gelman, Organometallics, 2009, 28, 6578 CrossRef CAS; (b) C. Azerraf and D. Gelman, Chem. – Eur. J., 2008, 14, 10364 CrossRef CAS PubMed; (c) A. Ishii, N. Nakata, R. Uchiumi and K. Murakami, Angew. Chem., Int. Ed., 2008, 47, 2661 CrossRef CAS PubMed; (d) A. Ishii, H. Kamon, K. Murakami and N. Nakata, Eur. J. Org. Chem., 2010, 1653 CrossRef CAS; (e) N. Nakata, R. Uchiumi, T. Yoshino, T. Ikeda, H. Kamon and A. Ishii, Organometallics, 2009, 28, 1981 CrossRef CAS; (f) Y. Yamaguchi, N. Nakata and A. Ishii, Eur. J. Inorg. Chem., 2013, 30, 5233 CrossRef; (g) S. Toyota, H. Okuhara and M. Ōki, Organometallics, 1997, 16, 4012 CrossRef CAS; (h) H.-B. Yang, K. Ghosh, N. Das and P. J. Stang, Org. Lett., 2006, 8, 3991 CrossRef CAS PubMed.
- (a) S. Mondal, S. Chakraborty, S. Bhowmick and N. Das, Macromolecules, 2013, 46, 6824 CrossRef CAS; (b) S. Chakraborty, S. Mondal and N. Das, Inorg. Chim. Acta, 2014, 413, 214 CrossRef CAS PubMed.
- (a) S. Leininger, M. Schmitz and P. J. Stang, Org. Lett., 1999, 1, 1921 CrossRef CAS; (b) C. J. Kuehl, C. L. Mayne, A. M. Arif and P. J. Stang, Org. Lett., 2000, 2, 3727 CrossRef CAS PubMed; (c) Y. K. Kryschenko, S. R. Seidel, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2003, 125, 5193 CrossRef CAS PubMed; (d) H.-B. Yang, A. M. Hawkridge, S. D. Huang, N. Das, S. D. Bunge, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2007, 129, 2120 CrossRef CAS PubMed.
- J. Manna, C. J. Kuehl, J. A. Whiteford and P. J. Stang, Organometallics, 1997, 16, 1897 CrossRef CAS.
- C. Zhang and C.-F. Chen, J. Org. Chem., 2006, 71, 6626 CrossRef CAS PubMed.
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS PubMed.
- (a) S. Neogi, Y. Lorenz, M. Engeser, D. Samanta and M. Schmittel, Inorg. Chem., 2013, 52, 6975 CrossRef CAS PubMed; (b) J. Freudenreich, J. Furrer, G. Suss-Fink and B. Therrien, Organometallics, 2011, 30, 942 CrossRef CAS; (c) S. Ghosh, B. Gole, A. K. Bar and P. S. Mukherjee, Organometallics, 2009, 28, 4288 CrossRef CAS; (d) S. Mirtschin, A. Slabon-Turski, R. Scopelliti, A. H. Velders and K. Severin, J. Am. Chem. Soc., 2010, 132, 14004 CrossRef CAS PubMed; (e) K.-T. Youm, S. T. Nguyen and J. T. Hupp, Chem. Commun., 2008, 3375 RSC; (f) M. Wang, V. Vajpayee, S. Shanmugaraju, Y.-R. Zheng, Z. Zhao, H. Kim, P. S. Mukherjee, K.-W. Chi and P. J. Stang, Inorg. Chem., 2011, 50, 1506 CrossRef CAS PubMed; (g) K. Kumazawa, K. Biradha, T. Kusukawa, T. Okano and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 3909 CrossRef CAS PubMed.
- (a) Y. K. Kryschenko, S. R. Seidel, D. C. Muddiman, A. I. Nepomuceno and P. J. Stang, J. Am. Chem. Soc., 2003, 125, 9647 CrossRef CAS PubMed; (b) K.-W. Chi, C. Addicott, Y. K. Kryschenko and P. J. Stang, J. Org. Chem., 2004, 69, 964 CrossRef CAS PubMed; (c) Y.-R. Zheng, H.-B. Yang, K. Ghosh, L. Zhao and P. J. Stang, Chem. – Eur. J., 2009, 15, 7203 CrossRef CAS PubMed; (d) H.-B. Yang, K. Ghosh, N. Das and P. J. Stang, Org. Lett., 2006, 8, 3991 CrossRef CAS PubMed.
- (a) A. Khutia, P. J. Sanz Miguel and B. Lippert, Bioinorg. Chem. Appl., 2010 DOI:10.1155/2010/169054; (b) C. J. Adams, M. F. Haddow, D. J. Harding, T. J. Podesta and R. E. Waddington, CrystEngComm, 2011, 13, 4909 RSC; (c) R. L. LaDuca Jr., M. Desciak, M. Laskoski, R. S. Rarig Jr. and J. Zubieta, Dalton Trans., 2000, 2255 RSC; (d) G. F. Cavichioli, D. P. Martin, A. J. Ursini and R. L. LaDuca, J. Mol. Struct., 2008, 881, 107 CrossRef CAS PubMed; (e) L. L. Johnston, A. J. Ursini, N. P. Oien, R. M. Supkowski and R. L. LaDuca, Inorg. Chim. Acta, 2007, 360, 3619 CrossRef CAS PubMed.
- J. J. P. Stewart, J. Mol. Model, 2007, 13, 1173 CrossRef CAS PubMed.
- (a) S. Hiraoka and M. Fujita, J. Am. Chem. Soc., 1999, 121, 10239 CrossRef CAS; (b) P. K. Bowyer, V. C. Cook, N. G. Naseri, P. A. Gugger, D. A. Rae, G. F. Swiegers, A. C. Willis, J. Zank and S. B. Wild, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4877 CrossRef CAS PubMed.
- M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli and M. Montanucci, Synthesis, 1984, 736 CrossRef CAS.
- T. Yoshida, T. Matsuda, S. Otsuka, G. W. Parshall and W. G. Peet, Inorg. Synth., 1990, 28, 122 CrossRef CAS.
- (a) SAINT+, version 6.02, Bruker AXS, Madison, WI, 1999 Search PubMed; (b) G. M. Sheldrick, SADABS, Empirical Absorption Correction Program, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (c) XPREP, 5.1 ed., Siemens Industrial Automation Inc., Madison, WI, 1995 Search PubMed; (d) G. M. Sheldrick, SHELXTL, Reference Manual, version 5.1, Bruker AXS, Madison, WI, 1997 Search PubMed; (e) G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 31P{1H} NMR spectra for complexes 1–4 and metallacages 5, 6. ESI-MS spectra of complexes 2–4. ESI-TOF-MS spectra and DOSY NMR spectra of metallacages 5 and 6. X-ray crystallographic file (CIF) for 1. CCDC 953632 for 1. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt01298d |
| This journal is © The Royal Society of Chemistry 2014 |