 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pincer versus pseudopincer: isomerism in palladium(II) complexes bearing κ3C,S,C ligands†
Jan Christopher
Bernhammer
ab,
Gilles
Frison
*b and
Han Vinh
Huynh
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmhhv@nus.edu.sg; Fax: +65 6779 1691; Tel: +65 6516 2670
bLaboratoire de Chimie Moleculaire, Department of Chemistry, Ecole Polytechnique and CNRS, 91128 Palaiseau Cedex, France. E-mail: gilles.frison@polytechnique.org; Fax: +331 6933 4803; Tel: +33 1 6933 4834
First published on 14th April 2014
Abstract
In NHC pincer complexes incorporating a hemilabile donor site, there exists an equilibrium between the true pincer form and a pseudopincer coordination isomer. The influence of the NHC moieties on this isomerism has been studied by DFT calculations.
Tridentate, pincer and pincer-type ligands have gained increasing popularity due to the robustness and thermal stability the rigid, κ3-mer or κ3-(pseudo)-mer coordination geometry of these ligands confers on their complexes. Transition metal complexes incorporating pincer ligands have been shown to be versatile and useful catalysts in organic synthesis.1 Beyond catalysis, pincer complexes have also been utilized successfully for the development of new materials.2 The fine-tuning of the steric and electronic properties of pincer ligands – be it by modifications in the backbone or changes to the nature of the donor sites – has attracted considerable research interest, in order to provide coordination spheres for each and every application.3 While catalysis often involves the coordination sites on the metal centre not occupied by the pincer ligand, there are also cases in which partial decoordination of a hemilabile donor takes place. The advantage is the rapid creation of a coordinatively and electronically unsaturated metal centre, open for incoming substrates, while retaining the tethered donor functionality in the vicinity in order to allow for rapid recoordination and stabilisation of the active species. As a result, highly active yet stable catalysts are obtained.4 Frequently, the donor functionalities in the side chains of the pincer ligands are hemilabile sites,5 but cases in which the central donor moiety exhibits hemilabile behaviour have also been reported.6 However, to control hemilabile behaviour, the governing geometric and electronic factors still need to be understood.7
N-heterocyclic carbenes (NHCs) have become ubiquitously employed ligands for transition metal catalysts due to their strong σ-basicity, the robustness of their complexes, and the ease with which they can be synthesized.8 Their steric bulk and electronic properties can be fine-tuned rapidly by modifying the backbone and side chains,9 and additional donor functionalities can be introduced in the side chains, which give rise to chelating and pincer-type ligands. Donor moieties based on heteroatoms such as nitrogen, oxygen, phosphorus, and sulfur have been reported.10,11 Especially the soft thioether functionalities are known to exhibit hemilabile behavior in their complexes.12
It has been shown that in the presence of coordinating counteranions, complexes of CSC pincer ligands are isolated either as κ2-trans-C,C pseudopincer complexes or in their κ3-mer-C,S,C pincer form.13 The electron-donating abilities of the carbene moieties were suggested to have a pivotal influence on this isomerism. To study this electronic effect, we examined the electronic properties of NHCs with markedly different donor strengths and the preference for either the pincer or the pseudopincer form by means of computational methods.
The relative Gibbs free energies ΔG of a neutral pseudopincer species A and the corresponding cationic pincer C and a bromide ion are difficult to obtain due to the significant impact of solvation on the ion pair. Instead, the Gibbs free energy ΔGR of the reaction of a pincer and a pseudopincer complex was used as an indication of the preference for the pincer form (Scheme 1). During this fictitious reaction, the number of ionic species remains constant throughout the reaction, and solvation effects as well as the Gibbs free energy of the bromide counteranion can be neglected as they are almost identical for the starting materials and products.
 | ||
| Scheme 1 Homodesmotic pincer/pseudopincer exchange reaction. | ||
The resulting scale of pincer preference energy is arbitrary in the sense that the absolute values will depend on the choice of the reference system. We decided to use complexes B and D featuring dichloroimidazolin-2-ylidene moieties as the probes.
This system was the most weakly donating ligand under scrutiny, and the respective complexes are known to prefer the pseudopincer form. A negative value for ΔGR will thus signify a higher preference for the pincer form in comparison to the reference system. Both the pincer and pseudopincer forms of all examined complexes were optimised at the B3LYP/cc-pVDZ level of theory,14–16 and their respective Gibbs free energies were calculated at the same level of theory (see ESI† for computational details).
To avoid the influence of variations in steric bulk associated with changing the substituents at nitrogen and to limit the computational cost, NHC side chains were simplified to sterically unassuming methyl groups, and the effect of five vastly different NHC backbones (cf. Fig. 2) was examined, two of which have already been studied experimentally.13
The optimised structures are comparable to the reported molecular structures.13 All complexes have a distorted square planar coordination geometry of the palladium center (Fig. 1). In the pincer complexes, the metallacycles adopt a distorted boat conformation and the planes defined by the NHC rings are almost coplanar, while they showed a twist of 26–43° in the corresponding pseudopincer forms. From these optimized structures, ΔGR can be obtained.
 | ||
| Fig. 1 Optimised pseudopincer and pincer geometries based on the dichloromidazolin-2-ylidene moiety. | ||
No experimental donor strength values are available for the ligands in the calculated complexes. However, it has been shown that the Kohn–Sham eigenvalue of the σ-lone pair orbital ε(σ-HOMO) correlates well with the donor strength.17 To limit the computational cost, orbital energies were calculated for truncated ligands at the B3LYP/auc-cc-pVTZ//B3LYP/cc-pVDZ level (Fig. 2).14,16,18 In all cases, the highest orbital with σ-symmetry was found to be the lone pair at Ccarbene (Fig. 3).
 | ||
| Fig. 2 Truncated ligands with different NHC moieties. | ||
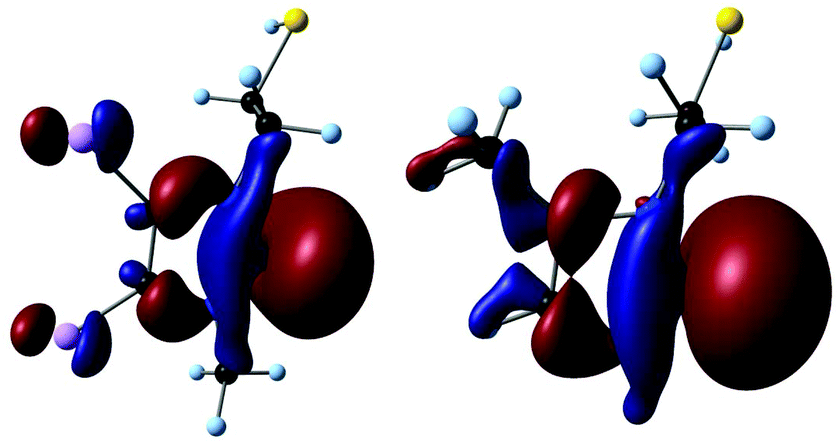 | ||
| Fig. 3 σ-HOMO of NHCs 1 (HOMO−1) and 5 (HOMO). | ||
Orbital energies for the σ-lone pair orbital were found to range from −6.56 to −5.52 eV, and this confirmed the expected increase in donor strength from 1 to 5 (Table 1). The preference for the pincer form, represented by ΔGR of the test reaction, increased in the same order, and Fig. 4 shows the strong correlation between donor strength and ΔGR (R2 = 0.9931).
 | ||
| Fig. 4 Correlation of ε(σ-HOMO) [eV] and ΔGR [kJ mol−1]. | ||
| NHC moiety | ε(σ-HOMO) [eV] | ΔGR [kJ mol−1] |
|---|---|---|
| Cl2-imidazole | −6.56 | 0.0 |
| 1,2,4-Triazole | −6.52 | −5.4 |
| Imidazole | −6.12 | −26.5 |
| Imidazoline | −5.94 | −32.8 |
| Pyrazole | −5.52 | −57.0 |
It is apparent that more strongly donating carbene moieties in the CSC ligand favour the pincer form. The cationic pincer complexes are better stabilised by stronger NHCs than the neutral pseudopincer form, and this difference in stabilization increases with the donor strength. Additionally, entropy and solvation also favour the formation of the ionic pincer over the pseudopincer form. On the other hand, the neutral pseudopincer complexes are preferred if the NHC moieties transfer less electron density towards the metal atom due to the fact that bromide is a better ligand than the thioether.
In a more formalistic approach, the [PdBr(NHC)2]+ fragment can be understood as a Lewis acid interacting with either the thioether moiety or a bromido ligand as a Lewis base. The internal charge redistribution in the Lewis acid upon charge transfer from the Lewis base is energetically more favourable with more weakly donating NHC moieties, which allows the coordination of the more strongly donating bromido ligand.19
In summary, the relationship between donor strength and pincer/pseudopincer isomerism has been unambiguously demonstrated to be in good agreement with experimental observations. Calculated or experimentally determined ε(σ-HOMO) values can thus serve as an indication of the hemilability of the central donor site in similar complexes, allowing for a more rational ligand design and the synthesis of tailor-made pincer complexes.
We thank the Vice-president for external relations in École Polytechnique for support through the international internship program and the National University of Singapore and the Singapore Ministry of Education for financial support (WBS R-143-000-483-112) and a SINGA scholarship for J. C. B. This work was performed using HPC resources from GENCI-CINES (grant 2013-x086894).
Notes and references
- The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. Jensen, Elsevier, Amsterdam CrossRef CAS PubMed; G. Rivera and R. H. Crabtree, N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, ch. 9, p. 223 CrossRef CAS PubMed; D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610 CrossRef CAS PubMed; N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048 CrossRef PubMed; G. van Koten, J. Organomet. Chem., 2013, 730, 156 CrossRef PubMed; K. J. Szabó, Top. Organomet. Chem., 2013, 40, 203 CrossRef.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750 CrossRef CAS; J. L. Hawk and S. L. Craig, Top. Organomet. Chem., 2013, 40, 319 CrossRef.
- G. van Koten, Pure Appl. Chem., 1989, 61, 1681 CrossRef CAS; I. Moreno, R. SanMartin, B. Inés, F. Churruca and E. Domínguez, Inorg. Chim. Acta, 2010, 363, 1903 CrossRef PubMed; D. Gelman and S. Musa, ACS Catal., 2012, 2, 2456 CrossRef; D. M. Roddick, Top. Organomet. Chem., 2013, 40, 49 CrossRef.
- J. C. Jeffrey and T. B. Rauchfuss, Inorg. Chem., 1979, 18, 2658 CrossRef CAS; C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233 CrossRef; P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680 CrossRef; H. V. Huynh, C. H. Yeo and G. K. Tan, Chem. Commun., 2006, 3833 RSC.
- L. Canovese, F. Visentin, G. Chessa, P. Uguagliati, C. Santo, G. Bandoli and L. Maini, Organometallics, 2003, 22, 3230 CrossRef CAS; M. Bassetti, A. Capone and M. Salamone, Organometallics, 2004, 23, 247 CrossRef; D. Vuzman, E. Poverenov, L. J. W. Shimon, Y. Diskin-Posner and D. Milstein, Organometallics, 2008, 27, 2627 CrossRef; G. R. Fulmer, W. Kaminsky, R. A. Kemp and K. I. Goldberg, Organometallics, 2011, 30, 1627 CrossRef; A. J. Ruddy, S. J. Mitton, R. McDonald and L. Turculet, Chem. Commun., 2012, 48, 1159 RSC.
- G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, R. L. Woodward, A. S. Weller and M. C. Willis, Angew. Chem., Int. Ed., 2006, 45, 7618 CrossRef CAS PubMed; C. Zhao, M. C. Jennings and R. J. Puddephatt, Dalton Trans., 2008, 1243 RSC; G. L. Moxham, H. Randell-Sly, S. K. Brayshaw, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2008, 14, 8383 CrossRef PubMed.
- E. Poverenov, M. Gandelman, L. J. W. Shimon, H. Rozenberg, Y. Ben-David and D. Milstein, Organometallics, 2005, 24, 1082 CrossRef CAS; R. Lindner, B. van den Bosch, M. Lutz, J. N. H. Reek and J. I. van der Vlugt, Organometallics, 2011, 30, 499 CrossRef.
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS; D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef PubMed; W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef; C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef PubMed; F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef PubMed; L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705 CrossRef PubMed.
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef PubMed; T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef PubMed.
- D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Organometallics, 2006, 25, 4850 CrossRef CAS; F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2006, 25, 5927 CrossRef; S. Warsink, P. Hauwert, M. A. Siegler, A. L. Spek and C. J. Elsevier, Appl. Organomet. Chem., 2009, 23, 225 CrossRef; J. DePasquale, M. Kumar, M. Zeller and E. T. Papish, Organometallics, 2013, 32, 966 CrossRef; H. V. Huynh and C.-S. Lee, Dalton Trans., 2013, 42, 6803 RSC; X. Liu and P. Braunstein, Inorg. Chem., 2013, 52, 7367 CrossRef PubMed.
- M. Bierenstiel and E. D. Cross, Coord. Chem. Rev., 2011, 255, 574 CrossRef CAS PubMed; D. Yuan and H. V. Huynh, Molecules, 2012, 17, 2491 CrossRef PubMed.
- S. J. Roseblade, A. Ros, D. Monge, M. Alcarazo, E. Álvarez, J. M. Lassaletta and R. Fernández, Organometallics, 2007, 26, 2570 CrossRef CAS; C. Fliedel, G. Schnee and P. Braunstein, Dalton Trans., 2009, 2474 RSC; H. V. Huynh, C. H. Yeo and Y. X. Chew, Organometallics, 2010, 29, 1479 CrossRef; C. Fliedel and P. Braunstein, Organometallics, 2010, 29, 5614 CrossRef; L. Canovese, F. Visentin, C. Levi, C. Santo and V. Bertolasi, J. Organomet. Chem., 2013, 732, 27 CrossRef PubMed; D. Krishnan, S. A. Pullarkat, M. Wu, Y. Li and P.-H. Leung, Chem. – Eur. J., 2013, 19, 5468 CrossRef PubMed.
- H. V. Huynh, D. Yuan and Y. Han, Dalton Trans., 2009, 7262 RSC; D. Yuan, H. Tang, L. Xiao and H. V. Huynh, Dalton Trans., 2011, 40, 8788 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113 CrossRef CAS PubMed; K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283 CrossRef; K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef CAS PubMed; D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358 CrossRef PubMed.
- R. Tonner, G. Heydenrych and G. Frenking, Chem. – Asian J., 2007, 2, 1555 CrossRef CAS PubMed; R. Tonner, G. Heydenrych and G. Frenking, ChemPhysChem, 2008, 9, 1474 CrossRef PubMed; J. C. Bernhammer, G. Frison and H. V. Huynh, Chem. – Eur. J., 2013, 19, 12892 CrossRef PubMed.
- R. A. Kendall and T. H. Dunning Jr., J. Chem. Phys., 1992, 96, 6796 CrossRef CAS PubMed.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt01047g |
| This journal is © The Royal Society of Chemistry 2014 |