 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Step-by-step covalent modification of Cr-templated Anderson-type polyoxometalates†
Chang-Gen
Lin
a,
Wei
Chen
a,
De-Liang
Long
b,
Leroy
Cronin
*b and
Yu-Fei
Song
*a
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: songyufei@hotmail.com; songyf@mail.buct.edu.cn; Fax: (+86) 10-64431832
bWestCHEM, Department of Chemistry, University of Glasgow, G12 8QQ, UK. E-mail: Lee.Cronin@glasgow.ac.uk; Web: http://www.croninlab.com Fax: +44 (0)141-330-4888; Tel: +44 (0)141-330-6650
First published on 3rd April 2014
Abstract
A series of tripodal alcohols substituted Anderson-type polyoxometalates (POMs) including mono-substituted (compounds 1 and 2), asymmetrical bi-substituted (compound 3), and symmetrical bi-substituted ones (compounds 4 and 5) have been synthesized under hydrothermal conditions using a pre-designed step-by-step strategy, and compounds 1, 2 and 4 have been fully characterized by single-crystal X-ray diffraction, ESI-MS, and elemental analysis.
Polyoxometalates (POMs) are a large class of discrete metal-oxo clusters of early transition metals of V, Mo, W etc. in their high oxidation states.1 The structural diversity and interesting chemical and physical properties of POMs2 give rise to many potential applications in various fields such as catalysis,3 materials science,4 and chemical biology.5 Previous studies have shown that the development of POM-based organic/inorganic hybrids is one of the most efficient ways to extend the applicability towards multifunctional materials.6 The resulting POM-containing hybrids, which combine not only the advantages of organic moieties and inorganic clusters, but also the close interactions and synergistic effects between them, have attracted tremendous attention.7 Nevertheless, direct covalent functionalization of POMs is not easy, and only a few POM clusters have been covalently modified so far.8–11 Among them, the synthesis of unsymmetrical POMs remains highly challenging.
In this respect the use of hydrothermal reaction processing, with carefully controlled reaction parameters, has proved to be an effective method for the preparation of transition-metal-substituted POMs (TMSPs) and it is widely utilized in POM chemistry.12 However, due to the lack of controllability during the course of hydrothermal reactions, it is hard to predict the desired molecular structures. In recent years, Yang and co-workers successfully tethered (1,1,1-tris(hydroxymethyl)aminomethane (denoted Tris-NH2) and related tripodal alcohol ligands onto a Ni6-substituted POM, leading to the formation of a number of interesting covalently functionalized TMSPs.13
Herein, we present a stepwise method that has been adopted during the preparation of two mono-substituted Anderson-type clusters of compounds 1 and 2 with the molecular formulae of (TBA)3{Cr(OH)3Mo6O18[(OCH2)3CCH2OH]}·12H2O (TBA = tetrabutylammonium cation) and (TBA)3{Cr(OH)3Mo6O18[(OCH2)3CCH3]}·11H2O, one asymmetrical bi-substituted compound 3 with the molecular formula of (TBA)3{CrMo6O18[(OCH2)3CCH2OH][(OCH2)3CCH3]}, and two symmetrical bi-substituted compounds 4 and 5 with the molecular formulae of (TBA)3{CrMo6O18[(OCH2)3CCH2OH]2}·2DMF and (TBA)3{CrMo6O18[(OCH2)3CCH3]2}, under hydrothermal conditions, respectively.
Previous studies have demonstrated that tripodal alcohols such as Tris-NH2 with their unique molecular structures can be capped onto Anderson clusters.9 Encouraged by the successful preparation of asymmetrical Mn-Anderson clusters,9c,d we tried to apply our approach to graft different tripodal alcohols onto Anderson clusters (Na3[Cr(OH)6Mo6O18], abbreviated as CrMo6). As the first trial, pentaerythritol and CrMo6, in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, were mixed in aqueous solution for 24 hours under hydrothermal conditions. Subsequently, tetrabutylammonium bromide (TBA-Br) was added to the solution yielding the product as pink crystals in a yield of 78% from the mother liquor in a few days. Compound 2 was prepared in a similar way to compound 1, except that 1,1,1-tris(hydroxylmethyl)ethane (Tris-CH3) was used instead of pentaerythritol. The molecular structures of compounds 1 and 2 have been determined by single-crystal X-ray diffraction (single-crystal XRD, Table S1†). In the case of compounds 1 and 2, both compounds crystallised in an orthorhombic system adopting the chiral space group P212121.‡ The chirality might be caused by the asymmetric mono-substitution of the achiral tripodal alcohols on the surface of the Anderson plate. Interestingly the compounds exist only as monomers without the internal μ2-oxo ligands being protonated, and this is quite different from what has been reported in the literature.11,14
:1, were mixed in aqueous solution for 24 hours under hydrothermal conditions. Subsequently, tetrabutylammonium bromide (TBA-Br) was added to the solution yielding the product as pink crystals in a yield of 78% from the mother liquor in a few days. Compound 2 was prepared in a similar way to compound 1, except that 1,1,1-tris(hydroxylmethyl)ethane (Tris-CH3) was used instead of pentaerythritol. The molecular structures of compounds 1 and 2 have been determined by single-crystal X-ray diffraction (single-crystal XRD, Table S1†). In the case of compounds 1 and 2, both compounds crystallised in an orthorhombic system adopting the chiral space group P212121.‡ The chirality might be caused by the asymmetric mono-substitution of the achiral tripodal alcohols on the surface of the Anderson plate. Interestingly the compounds exist only as monomers without the internal μ2-oxo ligands being protonated, and this is quite different from what has been reported in the literature.11,14
The asymmetric unit of compound 1 consists of three TBA cations, one {Cr(OH)3Mo6O18[(OCH2)3CCH2OH]}3− anion, and twelve crystallized water molecules. Compound 1 shows a typical Anderson structure, in which six edge-sharing {MoO6} octahedra form a hexagon around the central {CrO6} unit (Fig. 1a). Moreover, the μ3-hydroxo moiety of one side of the B-type Anderson plate is replaced by three μ3-alkoxo ligands, while the other side of Anderson is left unaffected. The crystal structure of compound 2 is similar to that of compound 1, and the only difference is the appended organic moiety (Fig. 1b).
 | ||
| Fig. 1 Single-crystal X-ray crystallographic structure of (a) {Cr(OH)3Mo6O18[(OCH2)3CCH2OH]}3− in compound 1; (b) {Cr(OH)3Mo6O18-[(OCH2)3CCH3]}3− in compound 2. Ball and stick representation with Cr pink, Mo dark teal, C gray, O red. H atoms have been omitted for clarity. | ||
Electrospray ionisation mass spectrometry (ESI-MS) has been proved to be a successful technique for the characterisation and elucidation of cluster transformation mechanisms in POM chemistry. The molecular structures and compositions of compounds 1 and 2 were also confirmed by ESI-MS. Fig. 2 shows the ESI-MS spectrum of compound 1, and the peaks observed at m/z = 1585.67 (calcd 1585.42) could be assigned to {(TBA)4{Cr(OH)3Mo6O18[(OCH2)3CCH2OH]}2}2− (Fig. S1†). As for compound 2, the peak observed at m/z = 1568.99 (cald 1568.92) corresponds to {(TBA)4{Cr(OH)3Mo6O18[(OCH2)3CCH3]}2}2− (Fig. S2†).
 | ||
| Fig. 2 ESI-MS spectrum of 1 with peaks at m/z 1585.67 attributed to {(TBA)4[Cr(OH)3Mo6O18{(OCH2)3CCH2OH}2]}2−. | ||
The above results indicate that CrMo6 tends to form mono-substituted POM species in the presence of an equivalent amount of tripodal alcohols under hydrothermal reactions. In order to control the functionalization, and to synthesize asymmetrical bi-substituted POMs, pure crystals of mono-substituted compound 1 were used subsequently for further modification. Compound 1 and Tris-CH3 in a molar ratio of 1:1 were dissolved in a water–acetonitrile mixed solvent and reacted under hydrothermal conditions. Accordingly, the asymmetric bi-substituted compound 3 was obtained as a TBA salt and characterized by ESI-MS spectroscopy (Fig. 3a). The example peaks of compound 3 observed at m/z = 1653.08 (cald 1652.98) were assigned to {(TBA)2{CrMo6O18[(OCH2)3CCH3][(OCH2)3CCH2OH]}}− (Fig. S3†).
 | ||
| Fig. 3 ESI-MS spectra of compounds 3 (a) and 4 (b). | ||
To further confirm the asymmetrical structure of compound 3, bi-substituted symmetrical compounds 4 and 5 were prepared using the same method. Although these three compounds are distinguished only by one –CH2– unit from their organic linkers, ESI-MS spectra of compounds 4 and 5 are totally different from that of compound 3 (Fig. 3b and S5†). For compound 4, peaks at m/z = 1667.05 (cald 1666.96) could be assigned as {(TBA)2{CrMo6O18[(OCH2)3CCH2OH]2}}− (Fig. S4†), while peaks of compound 5 observed at m/z = 1635.03 (cald 1634.97) correspond to {(TBA)2{CrMo6O18[(OCH2)3CCH3]2}}− (Fig. S5†). It should be noted that compounds 4 and 5 could also be obtained with a relatively higher yield in a molar ratio of pentaerythritol:CrMo6 and Tris-CH3:CrMo6 = 2:1, respectively. Block pink crystals of compounds 4 and 5 could be obtained by ether diffusion into the DMF or acetonitrile solutions of the corresponding compounds. The molecular structure of compound 4 has been determined by single-crystal XRD and belongs to the well-known analogue of the MnIII-Anderson structure ({MnMo6O18[(OCH2)3CCH2OH]2}3−),9a with two tripodal alcohols capped onto both sides of the Anderson clusters (Fig. 4). The coordination geometry of the central CrIII ion of compound 4 is regular, with a mean Cr–O distance of 1.95(8) Å, and O–Cr–O bond angles between 86° and 94°.
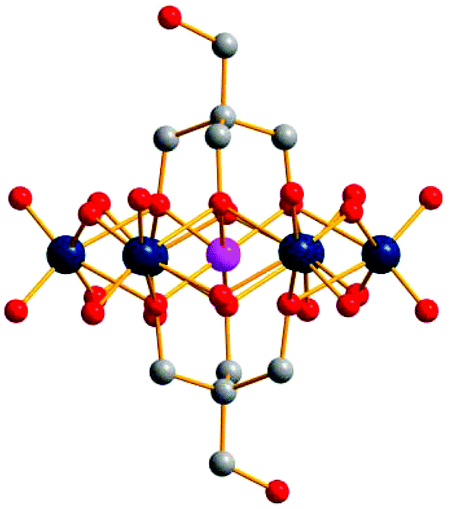 | ||
| Fig. 4 Single-crystal X-ray crystallographic structure of compound 4 ({CrMo6O18[(OCH2)3CCH2OH]2}3−). Ball and stick representation with Cr pink, Mo dark teal, C gray, O red. H atoms have been omitted for clarity. | ||
FT-IR spectra of compounds 1–5 were very similar, with typical Anderson-type POM vibrational bands9a such as ca. 940, 920, and 665 cm−1, which correspond to the vibrations of Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Mo–O–Mo groups. The bands between 1020 and 1130 cm−1 could be assigned to C–O stretching vibrations indicating that tripodal alcohol had been successfully tethered onto the Anderson clusters (Fig. S6†). Usually, B-type Anderson anions such as [Cr(OH)6–Mo6O18]3− give rise to peaks with very weak intensity at 576 and 527 cm−1, which are assigned to asymmetrical stretching of Cr–O and bending vibrations of Mo–O–Mo and O–Cr–O bonds.15 However, the FT-IR spectra of mono-substituted and bi-substituted chromium Anderson clusters are different in the region of 400–600 cm−1 meaning that FT-IR may be useful for distinguishing between compounds before subsequent reactions. Taking compounds 1 and 3 as examples, compound 1 exhibits weak vibrations at 565 and 488 cm−1, while compound 3 shows distinct medium vibrations at 565, 513, and 460 cm−1 (Fig. S7†). The different splitting peak numbers and significant shifts of compounds 1–5 in the range of 400–600 cm−1 clearly demonstrated that the tripodal alcohols had been successfully attached to the Anderson clusters.
O and Mo–O–Mo groups. The bands between 1020 and 1130 cm−1 could be assigned to C–O stretching vibrations indicating that tripodal alcohol had been successfully tethered onto the Anderson clusters (Fig. S6†). Usually, B-type Anderson anions such as [Cr(OH)6–Mo6O18]3− give rise to peaks with very weak intensity at 576 and 527 cm−1, which are assigned to asymmetrical stretching of Cr–O and bending vibrations of Mo–O–Mo and O–Cr–O bonds.15 However, the FT-IR spectra of mono-substituted and bi-substituted chromium Anderson clusters are different in the region of 400–600 cm−1 meaning that FT-IR may be useful for distinguishing between compounds before subsequent reactions. Taking compounds 1 and 3 as examples, compound 1 exhibits weak vibrations at 565 and 488 cm−1, while compound 3 shows distinct medium vibrations at 565, 513, and 460 cm−1 (Fig. S7†). The different splitting peak numbers and significant shifts of compounds 1–5 in the range of 400–600 cm−1 clearly demonstrated that the tripodal alcohols had been successfully attached to the Anderson clusters.
Conclusions
In summary, we have demonstrated that it is feasible to covalently functionalize POM clusters through a pre-designed synthetic strategy using a controllable hydrothermal approach. This led us to develop a series of organic–inorganic POMs including the mono-substituted, symmetrical bi-substituted and asymmetrical bi-substituted. All the compounds have been synthesized in good yields using a step-by-step method (Scheme 1) and extensively characterized. Further work will explore the post-modification of the functionalized POMs and their self-assembly behavior in solution. | ||
| Scheme 1 The synthetic procedure for the preparation of compounds 1–5 under hydrothermal conditions. Colour code: CrO6 pink polyhedron, MoO6 green polyhedron, C gray, O red. H atoms have been omitted for clarity. | ||
This research was supported by the National Science Foundation of China (21222104), EPSRC (grants EP/H024107/1; EP/I033459/1; EP/J015156/1) and the Fundamental Research Funds for the Central Universities (RC1302). LC thanks the Royal-Society Wolfson Foundation for a Merit Award, and the University of Glasgow.
Notes and references
- (a) L. Cronin and A. Müller, Chem. Soc. Rev., 2012, 41, 7333 RSC; (b) C. L. Hill, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed; (c) D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC; (d) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef CAS PubMed; (e) D.-L. Long and L. Cronin, Dalton Trans., 2012, 41, 9815 RSC.
- (a) M. T. Pope, in Comprehensive Coordination Chemistry II, ed. A. G. Wedd, Elsevier, Oxford, 2004, vol. 4, pp. 635–678 Search PubMed; (b) C. L. Hill, in Comprehensive Coordination Chemistry II, ed. A. G. Wedd, Elsevier, Oxford, 2004, vol. 4, pp. 679–759 Search PubMed; (c) A. Müller, P. Kögerler and A. W. M. Dress, Coord. Chem. Rev., 2001, 222, 193 CrossRef.
- (a) M. V. Vasylyev and R. Neumann, J. Am. Chem. Soc., 2004, 126, 884 CrossRef CAS PubMed; (b) N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944 CrossRef CAS PubMed; (c) S. Zhao, L. Liu and Y.-F. Song, Dalton Trans., 2012, 41, 9855 RSC.
- (a) Y.-F. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384 RSC; (b) C. Streb, C. Ritchie, D. L. Long, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2007, 46, 7579 CrossRef CAS PubMed; (c) H. D. Zeng, G. R. Newkome and C. L. Hill, Angew. Chem., Int. Ed., 2000, 39, 1771 CrossRef.
- B. Hasenknopf, Front. Biosci., 2005, 10, 275 CrossRef CAS PubMed.
- (a) A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837 RSC; (b) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- (a) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed; (b) M. Santoni, A. K. Pal, G. S. Hanan, A. Proust and B. Hasenknopf, Inorg. Chem., 2011, 50, 6737 CrossRef CAS PubMed.
- (a) Z. Peng, Angew. Chem., Int. Ed., 2004, 43, 930 CrossRef CAS PubMed; (b) D. Li, J. Song, P. C. Yin, S. Simotwo, A. J. Bassler, Y. Y. Aung, J. E. Roberts, K. I. Hardcastle, C. L. Hill and T. Liu, J. Am. Chem. Soc., 2011, 133, 14010 CrossRef CAS PubMed.
- (a) B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Eur. J. Inorg. Chem., 2002, 1081 CrossRef CAS; (b) Y. F. Song, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2007, 46, 3900 CrossRef CAS PubMed; (c) Y. F. Song, D. L. Long, S. E. Kelly and L. Cronin, Inorg. Chem., 2008, 47, 9137 CrossRef CAS PubMed; (d) C. Yvon, A. Macdonell, S. Buchwald, A. J. Surman, N. Follet, J. Alex, D.-L. Long and L. Cronin, Chem. Sci., 2012, 4, 3810 RSC; (e) M. H. Rosnes, C. Musumeci, C. P. Pradeep, J. S. Mathieson, D. L. Long, Y. F. Song, B. Pignataro, R. Cogdell and L. Cronin, J. Am. Chem. Soc., 2010, 132, 15490 CrossRef CAS PubMed; (f) J. Thiel, D. M. Yang, M. H. Rosnes, X. L. Liu, C. Yvon, S. E. Kelly, Y. F. Song, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2011, 50, 8871 CrossRef CAS PubMed.
- (a) J. Li, I. Huth, L. Chamoreau, B. Hasenknopf, E. Lacôte, S. Thorimbert and M. Malacria, Angew. Chem., Int. Ed., 2009, 48, 2035 CrossRef CAS PubMed; (b) C. Boglio, K. Micoine, É. Derat, R. Thouvenot, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, J. Am. Chem. Soc., 2008, 130, 4553 CrossRef CAS PubMed; (c) C. P. Pradeep, D. L. Long, G. N. Newton, Y. F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388 CrossRef CAS PubMed; (d) C. P. Pradeep, M. F. Misdrahi, F. Y. Li, J. Zhang, L. Xu, D. L. Long, T. Liu and L. Cronin, Angew. Chem., Int. Ed., 2009, 48, 8309 CrossRef CAS PubMed.
- P. F. Wu, P. C. Yin, J. Zhang, J. Hao, Z. C. Xiao and Y. G. Wei, Chem.–Eur. J., 2011, 17, 12002 CrossRef CAS PubMed.
- S. T. Zheng and G. Y. Yang, Chem. Soc. Rev., 2012, 41, 7623–7646 RSC.
- (a) S. T. Zheng, J. Zhang, X. X. Li, W. H. Fang and G. Y. Yang, J. Am. Chem. Soc., 2010, 132, 15102 CrossRef CAS PubMed; (b) X. X. Li, S. T. Zheng, W. H. Fang and G. Y. Yang, Inorg. Chem. Commun., 2011, 14, 1541 CrossRef CAS PubMed.
- V. W. Day, J. C. Goloboy and W. G. Kleperer, Eur. J. Inorg. Chem., 2009, 5079 CrossRef CAS PubMed.
- M. H. Alizadeh and A. R. Salimi, J. Mol. Struct. (THEOCHEM), 2007, 809, 1 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures, ESI-MS spectra of compounds 2 and 5 and FT-IR spectra. CCDC 979554–979556. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00033a |
| ‡ Crystal data for 1: C53H144CrMo6N3O37, Mr = 2043.35; orthorhombic; space group P212121; a = 14.924 (3), b = 21.749 (4), c = 25.646 (5) Å; V = 8324 (3) Å3; Z = 4; ρ = 1.630 g cm−3; T = 153 (2) K; R1 = 0.0396; wR2 = 0.0910. Crystal data for 2: C53H142CrMo6N3O35, Mr = 2009.34; orthorhombic; space group P212121; a = 15.164 (3), b = 21.818 (4), c = 25.095 (5) Å; V = 8303 (3) Å3; Z = 4; ρ = 1.607 g cm−3; T = 153 (2) K; R1 = 0.0403; wR2 = 0.0979. Crystal data for 4: C64H140CrMo6N5O28, Mr = 2055.45; monoclinic; space group P21/c; a = 25.2904 (16), b = 13.8087 (9), c = 24.6841 (16) Å; β = 90.966 (4), V = 8556.7 (10) Å3; Z = 4; ρ = 1.596 g cm−3; T = 150 (2) K; R1 = 0.0814; wR2 = 0.2300. Crystallographic details for 1, 2, 3, and 5 may be obtained from CSD quoting CCDC 979554–979556 respectively. |
| This journal is © The Royal Society of Chemistry 2014 |