 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insertion of alkynes into Pt–X bonds of square planar [PtX2(N^N)] (X = Cl, Br, I) complexes†
Michele
Benedetti
*a,
Vincenza
Lamacchia
b,
Daniela
Antonucci
a,
Paride
Papadia
a,
Concetta
Pacifico
b,
Giovanni
Natile
b and
Francesco P.
Fanizzi
*a
aDipartimento di Scienze e Tecnologie Biologiche ed Ambientali, Università del Salento, Via Monteroni, I-73100 Lecce, Italy. E-mail: fp.fanizzi@unisalento.it; michele.benedetti@unisalento.it; Fax: +39 0832 298626; Tel: +39 0832 298867
bDipartimento di Chimica, Università degli Studi di Bari, Via E. Orabona 4, I-70125 Bari, Italy
First published on 3rd April 2014
Abstract
The reactivity with acetylene of [PtX2(Me2phen)] (X = Cl, Br, I) complexes has been investigated. Whereas the chlorido species [PtCl2(Me2phen)] exhibits negligible reactivity at short reaction times, the bromido and iodido species [PtBr2(Me2phen)] and [PtI2(Me2phen)] lead initially to formation of Pt(II) five-coordinate complexes, [PtX2(η2-CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH)(Me2phen)], that evolve to four-coordinate alkenyl complexes of the type [PtX(η1-E-CH
CH)(Me2phen)], that evolve to four-coordinate alkenyl complexes of the type [PtX(η1-E-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHX)(Me2phen)]. The alkenyl complexes, in the presence of excess acetylene, establish an equilibrium with the five-coordinate alkyne–alkenyl species [PtX(η1-E-CHCHX)(η2-CHCH)(Me2phen)] (X = Br, I). The π-bonded acetylene can be exchanged with free olefins or CO, affording the new alkene–alkenyl or carbonyl–alkenyl complexes [PtX(η1-E-CHCHX)(η2-olefin)(Me2phen)] and [PtX(η1-E-CHCHX)(CO)(Me2phen)]. The five-coordinate geometry of the alkyne–alkenyl and alkene–alkenyl complexes was assessed from NMR data and is fully consistent with that of a previously determined X-ray structure of [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)].
CHX)(Me2phen)]. The alkenyl complexes, in the presence of excess acetylene, establish an equilibrium with the five-coordinate alkyne–alkenyl species [PtX(η1-E-CHCHX)(η2-CHCH)(Me2phen)] (X = Br, I). The π-bonded acetylene can be exchanged with free olefins or CO, affording the new alkene–alkenyl or carbonyl–alkenyl complexes [PtX(η1-E-CHCHX)(η2-olefin)(Me2phen)] and [PtX(η1-E-CHCHX)(CO)(Me2phen)]. The five-coordinate geometry of the alkyne–alkenyl and alkene–alkenyl complexes was assessed from NMR data and is fully consistent with that of a previously determined X-ray structure of [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)].
Introduction
The synthesis of Zeise's anion, [PtCl3(η2-CH2CH2)]−, the first organometallic complex containing a metal-bonded alkene, was first reported in 1825.1 Zeise's anion analogues, with metal bonded alkynes, have been reported only since 1955 (Bukhovet's salt).2 Studying the chemistry of such complexes, Theophanides and Kong were able to isolate cationic [PtCl(η2-alkyne)(N^N)]+ and neutral [PtCl2(η2-alkyne)(N^N)] alkyne derivatives.3 In the following years it was demonstrated, for the latter neutral complexes, to have five-coordinate, trigonal bipyramidal, structures.4,5 Nearly at the same time other trigonal bipyramidal complexes, with a metal bonded alkene, of the formula [PtRX(η2-CF2CF2)L2] (R = carbanion; X = halide; L = tertiary phosphine or arsine) were reported by Clark and Puddephatt.6 The structure of the early alkyne and alkene complexes of Pt(II) was generally assigned on the basis of IR data, and only after quite a few years Puddephatt reported the first single crystal X-ray structure of a trigonal bipyramidal Pt(II) complex with an η2-coordinated alkyne, trans-[PtCl(Me)(η2-CF3CCCF3)(AsMe3)2].7 Several pentacoordinate complexes, similar to those reported by Clark and Puddephatt, and having two halogens in apical positions and one chelate dinitrogen ligand and one ethylene in the trigonal plane, have been reported since 1973.8 Their structures, initially assigned on the basis of NMR and IR data, were subsequently confirmed by single crystal X-ray diffraction analysis.9 A key feature for the stabilization of the trigonal bipyramidal five-coordinate structure was found to be the presence of an alkene and a bidentate ligand in the trigonal plane.10 Moreover, it was observed that the stability of the five-coordinate species is greater with olefins bearing electron withdrawing substituents (therefore characterized by enhanced π acidity) and with bidentate ligands able to introduce high sterical crowding in the trigonal plane. Such a result can be rationalized by considering that (i) an enhanced metal to olefin back donation will reduce the electron charge in the trigonal plane where, apart from the lone pairs of the three donors, there are also two pairs of platinum d-electrons and (ii) a five-coordinate species can accommodate a bulky chelate ligand in the trigonal plane much better than a four-coordinate species can do in the square-plane. The role of the metal-to-olefin back donation in the stabilization of the metal–olefin bonding interaction was also supported by the enhanced stabilization of anionic square planar Pt(II) complexes bearing η2-olefins, with respect to neutral or cationic complexes, which instead exhibit reduced stability and increased reactivity.11–20
In previous work we synthesized and characterized complexes with the Me2phen ligand, of the type [PtX2(Me2phen)] (X = halogen),21 showing considerable distortions from the regular square planar arrangement and an unusual chemical and electrochemical behavior, with respect to analogous complexes in which the phenanthroline has no substituents in the 2,9 positions.22 Because of steric interactions between ortho substituents of the phenanthroline and halogen ligands in cis positions, the square planar [PtX2(Me2phen)] complexes have a great tendency to add an external L ligand (L = CO, PPh3, DMSO, DMS, py, n-PrNH2, alkene, alkyne) to give the corresponding addition product.23,24 The reaction pathway is likely to contemplate the dissociation of one end of the N-donor chelate ligand, with formation of a tricoordinate intermediate, able to add an additional ligand (Scheme 1). Then, depending upon the electron withdrawing properties of the entered ligand, the Me2phen can remain monodentate, giving a tetra-coordinate complex21,24 or can coordinate the second end, to give a pentacoordinate, trigonal bipyramidal, species.25 Although it was not expected, intermediate cases are also possible.21,23,24
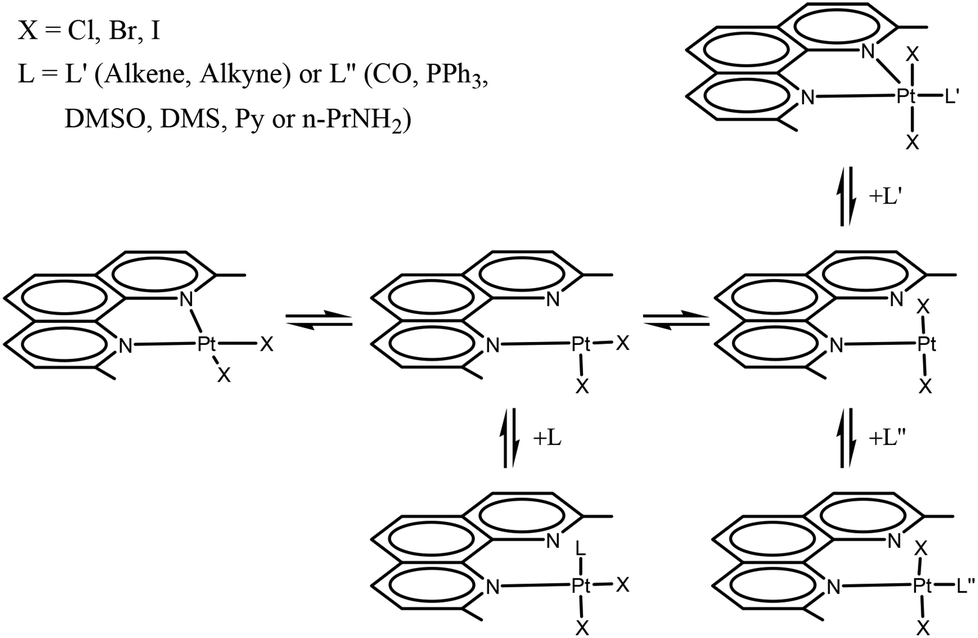 | ||
| Scheme 1 Intramolecular steric interactions in [PtX2(Me2phen)], (X = Cl, 1; Br, 2; I, 3), favor the dissociation of one end of the N-donor chelate ligand, with formation of tricoordinate intermediates, able to add an extra ligand L, to give a pentacoordinate [PtX2(L′)(Me2phen)] species or a square planar cis- or trans-[PtX2(L′′)(η1-Me2phen)], depending upon the electron withdrawing properties of the additional ligand. | ||
Therefore, when L is a very strong π acceptor ligand (such as an alkene or an alkyne; this class of ligands is indicated as L′ in Scheme 1), the electron density on the metal is strongly reduced by the π back-donation from the metal to the ligand. In this case both Me2phen nitrogen donors are coordinated to the metal core, in a five-coordinated trigonal bipyramidal structure, having the halogens in the apical positions and the Me2phen and L′ ligands in the equatorial plane. The observed Pt–N bonding distances are generally longer than the corresponding distances in [PtX2(Me2phen)] square planar species.
In contrast, in the case of ligands with weak or no π-acceptor capacity (such as py or n-PrNH2; this class of ligands is indicated as L′′ in Scheme 1), the second N-donor of the Me2phen remains uncoordinated and the [PtX2L′′(Me2phen)] complex is four-coordinate, square planar. However, also in the latter case the two halves of Me2phen are magnetically equivalent because of rapid exchange, in the NMR time scale, of the two nitrogens of Me2phen on the single platinum coordination site. Because of fast phenanthroline flipping, it was possible to distinguish between five-coordinate and four-coordinate [PtX2L(η1-Me2phen)] species only by X-ray diffraction studies in the solid state (where the flipping of the ligand is frozen) or by low-temperature NMR experiments. A somewhat intermediate case between five- and four-coordinate species was found when the L ligand had the π-acceptor capacity lower than ethylene.21,23,24
The distortion of the square planar complexes [PtX2(Me2phen)] depends chiefly on the steric interaction of the two ortho methyl substituents of Me2phen with the cis ligands, therefore the size of the halogen ligands plays a role. In the case of addition of an L′ ligand to give the pentacoordinate species [PtX2L′(Me2phen)], the iodide species is more reactive than the bromo species and this latter is more reactive than the chloro analogues. Both kinetic and thermodynamic data for formation of the [PtX2L′(Me2phen)] five coordinate species have been reported for the three halides and the barrier to rotation of the π ligand around the metal ligand bond was evaluated.25,26
Interestingly, in the case of acetylene addition to the square planar [PtBr2(Me2phen)] complex, the equilibrium constant for [PtBr2(η2-HCCH)(Me2phen)] formation could not be measured because of further reaction with the excess acetylene. In the present work we report further investigation, arising from that original observation, leading to the discovery of an interesting alkyne insertion reaction into a Pt–X bond, giving σ bonded alkenyl complexes.
Results and discussion
At short reaction times in chlorinated solvents, the chlorido species [PtCl2(Me2phen)] (1) exhibits negligible reactivity with acetylene, whereas the iodido species (3) undergoes fast alkyne uptake with formation of the previously described five coordinate complex [PtI2(η2-CHCH)(Me2phen)] (4) (Scheme 2, Fig. 1S†).26 Interestingly, performing the reaction on the bromido species [PtBr2(Me2phen)] (2), and following the reaction course by 1H-NMR, the instantaneous formation of a mixture was observed, characterized by the presence of two new Me2phen systems. One system was characterized by two Me resonances, one at 3.25 ppm coupled with 195Pt (4JPt–H = 7 Hz) and the other at 2.81 ppm. The second system had just one single Me signal at 3.46 ppm. It is evident that the first system contains an asymmetric Me2phen moiety (5) while the second system contains a symmetric chelate ligand (6) (Scheme 2, Table 1, Fig. 1 and 2S,† complex 6 can be isolated as shown in the Experimental section). This spectrum also contained four doublets ascribable to two, 195Pt coupled, vinylic systems. On the basis of the integral consistency with other signals, one vinylic system (5.89 and 4.63 ppm) could be ascribed to the asymmetric species (5), while the second vinylic system (7.55 and 5.08 ppm) could be ascribed to the symmetric species (6) (Table 1). Finally, a singlet, strongly coupled with 195Pt (3.73 ppm, 2JH–Pt = 68 Hz), could be ascribed to a π coordinated acetylene of the symmetric species (6), Table 1.
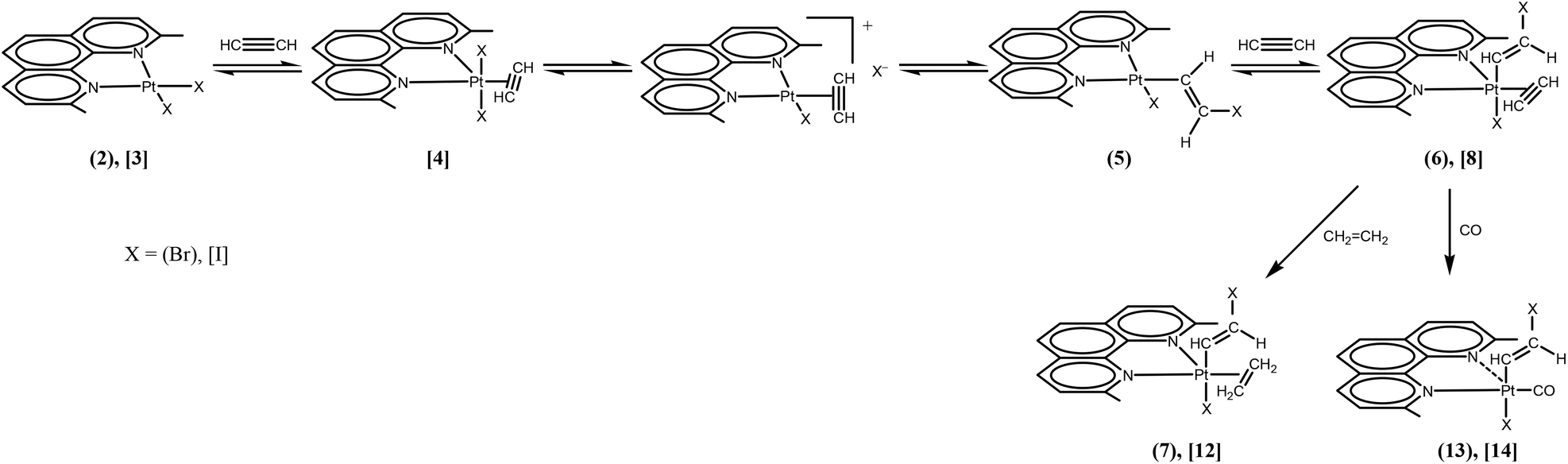 | ||
| Scheme 2 A possible mechanism for the formation of acetylene–alkenyl complexes. The coordinated alkyne can be easily substituted by other π-acceptor ligands (CH2CH2 and CO are shown). | ||
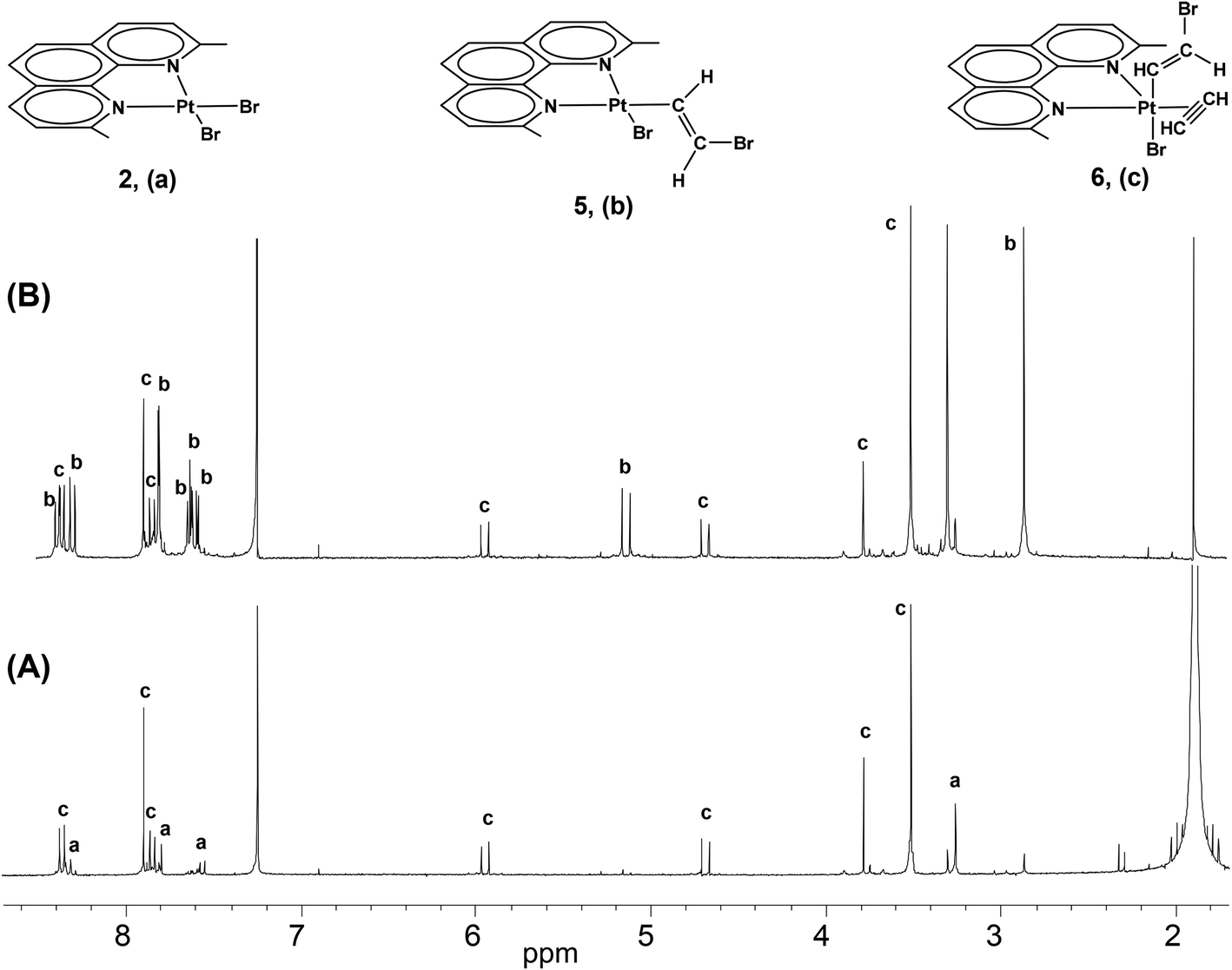 | ||
| Fig. 1 (A) 1H NMR spectrum of [PtBr2(Me2phen)] (2), (CDCl3 solution) treated with a large excess of acetylene (signal at 1.95 ppm) at room temperature. Signals (a) refer to the starting complex 2, while signals (c) refer to the reaction product [PtBr(η1-E-CHCHBr)(η2-CHCH)(Me2phen)] (6). In (B) the same reaction is carried on with a reduced amount of acetylene so to shift the equilibrium toward the intermediate species [PtBr(η1-E-CHCHBr)(Me2phen)] (5), signals (b). | ||
CHBr)(Me2phen)] (5) and [PtX(η1-E-CHCHX)(η2-CHCH)(Me2phen)] (X = Br, 6; I, 8)
| Complex | Me2phena | |||||
|---|---|---|---|---|---|---|
| Me(2,9) | CH(3,8) | CH(4,7) | CH(5,6) | η2-CHCH |
η1-E-CHCHX |
|
| a The (3JH–H), [JH–Pt] and [nJPt–C] values are given, where assigned. c = cis, g = geminal. | ||||||
| 5 | 3.25 [7] | 7.57 d(8) | 8.33 d(8) | 7.75 | — | 7.55 d(14) [62] CHα,g |
| 2.81 | 7.55 d(8) | 8.25 d(8) | 5.08 d(14) [27] CHβ,c | |||
| {28.50} | {126.85} | {136.31} | {125.50} | {118.39 Cα} | ||
| {29.52} | {127.10} | {136.71} | {93.64 Cβ} | |||
| 6 | 3.46 | 7.79 d(8) | 8.31 d(8) | 7.84 | 3.73[68] CH | 5.89 d(13) [42] CHα,g |
| 4.63 d(13) [35] CHβ,c | ||||||
| {28.94} | {125.95} | {137.86} | {125.86} | {32.87} | {119.01 [856] Cα} | |
| {93.88 [62] Cβ} | ||||||
| 8 | 3.50 | 7.84 d(8) | 8.36 d(8) | 7.90 | 3.73 [68] CH | 6.26 d(14) [37] CHα,g |
| 4.55 d(14) [38] CHβ,c | ||||||
It was also observed that in the presence of a strong excess of acetylene the equilibrium could be shifted toward the symmetric compound (6). On the basis of previous observations, the first species (5), containing an asymmetric Me2phen, is consistent with a square planar complex, containing a chelated Me2phen, a bromide, and a σ-bonded β-bromo-alkenyl ligand ([PtBr(η1-E-CHCHBr)(Me2phen)] (5)). Besides the asymmetry of the chelated Me2phen, also consistent with this structure is the observation of 195Pt coupling (7 Hz) only for one Me (that cis to the σ-bonded alkenyl and trans to the bromido ligand) while the second Me (that trans to the σ bonded alkenyl) is expected to have a negligible coupling with 195Pt due to the lengthening of the Pt–N bond. The second species 6, containing a symmetric Me2phen, is consistent with a trigonal bipyramidal complex containing a Br− and a β-bromo-alkenyl in apical positions, and a Me2phen and an acetylene in the trigonal plane ([PtBr(η1-E-CHCHBr)(η2-CHCH)(Me2phen)] (6)). The different position of the alkenyl ligand in compound 5 (in the square-planar coordination plane) and in compound 6 (in the apical position of a pentacoordinate system in which the Me2phen is in the trigonal plane), results in a considerable deshielding of the alkenylic protons in complex 5 (7.55α,geminal and 5.08β,cis ppm) with respect to complex 6 (5.89α,geminal and 4.63β,cis ppm, Table 1). This effect is more pronounced for protons in the α position, geminal to the metal, with respect to protons in the β position. An analogous shift of alkenylic protons, to lower frequency, was observed on passing from the square planar Pt(II) complex trans-[PtCl(η1-Z-CHCHCl)(PPh2Me)2] to the octahedral Pt(IV) species trans-[PtCl3(η1-Z-CHCHCl)(PPh2Me)2].27
In both complexes 5 and 6, the 1H-NMR data (Table 1) also show a 14 Hz coupling constant between the two alkenyl protons. This is a clear indication that the two hydrogen atoms are in trans positions with respect to the double bond.28,29 Moreover, the coupling constants between β-proton and 195Pt (42 and 62 Hz for complexes 5 and 6, respectively) are smaller than corresponding coupling constants in complexes in which the β-proton and 195Pt are trans to one another (≈ 100–200 Hz30), thus confirming the relative cis position of Pt and β-proton.
As expected for five-coordinate complexes, the π-acetylene protons undergo a high frequency shift of about 1 ppm with respect to free alkyne and are coupled with 195Pt (2JPt–H in the range of 50–65 Hz). A similar effect, but to a smaller extent, is observed for protons on carbons in the α position to the triple bond.2613C-NMR data of complexes 5 and 6 are in agreement with the suggested structures (Table 1).
The four-coordinate alkenyl species [PtBr(η1-E-CHCHBr)(Me2phen)] (5), formed by alkyne insertion in the Pt–Br bond of [PtBr2(Me2phen)] (2), reacts further with acetylene to form the alkyne–alkenyl five-coordinate product [PtBr(η1-E-CHCHBr)(η2-CHCH)(Me2phen)] (6). The equilibrium between 5 and 6 can be shifted toward the five-coordinate complex by using a strong excess of free alkyne (Scheme 2, Fig. 1).
By exchanging the acetylene with ethylene in compound 6, the compound [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7) can be obtained. The NMR spectra (1H, 13C, [1H–1H]-NOESY and [1H–13C]-HSQC) show the pattern typical of a symmetrically coordinated Me2phen (Fig. 2–4, 3S†). A 1H singlet, resonating at 3.31 ppm, accounting for six protons, and giving a cross peak with a carbon at 28.76 ppm (3JPt–C = 124 Hz), can be attributed to the two Me2phen methyls. A 1H singlet at 7.87 ppm, accounting for two protons, giving a cross peak with a carbon at 126.22 ppm, can be assigned to Me2phen CH's in positions 5 and 6. Two 1H doublets, at 7.78 and 8.34 ppm, accounting for two protons each and giving cross peaks with two carbons at 126.22 and 137.87 ppm can be attributed to Me2phen CH's in positions 3 and 8 and in positions 4 and 7, respectively. Two second order multiplets at 3.32 and 2.64 ppm (belonging to an AA′XX′ system), are coupled with 195Pt (2JPt–H = 68 and 65 Hz, respectively) and give HSQC cross peaks with a carbon at 32.25 ppm (1JPt–C = 343 Hz). These signals are assigned to the η2-coordinated ethylene. Finally, two proton doublets at 5.98 (2JPt–H = 41 Hz) and 4.66 ppm (3JPt–H = 48 Hz) and giving cross peaks with two carbons at 119.01 (1JPt–C = 882 Hz) and 94.32 (2JPt–C = 78 Hz) ppm, respectively, are assigned to the α and β protons of the η1-coordinated alkenyl.
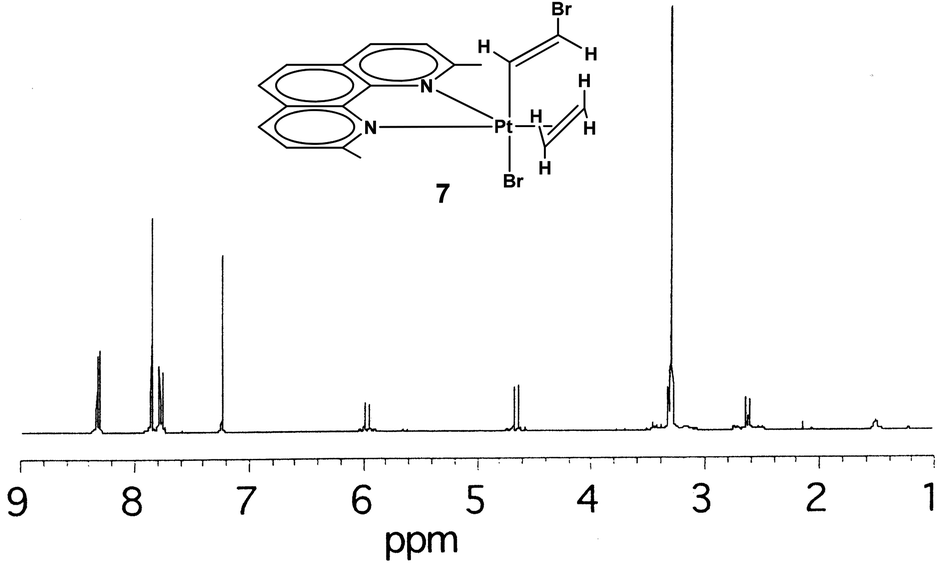 | ||
| Fig. 2
1H NMR spectrum of complex [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7). | ||
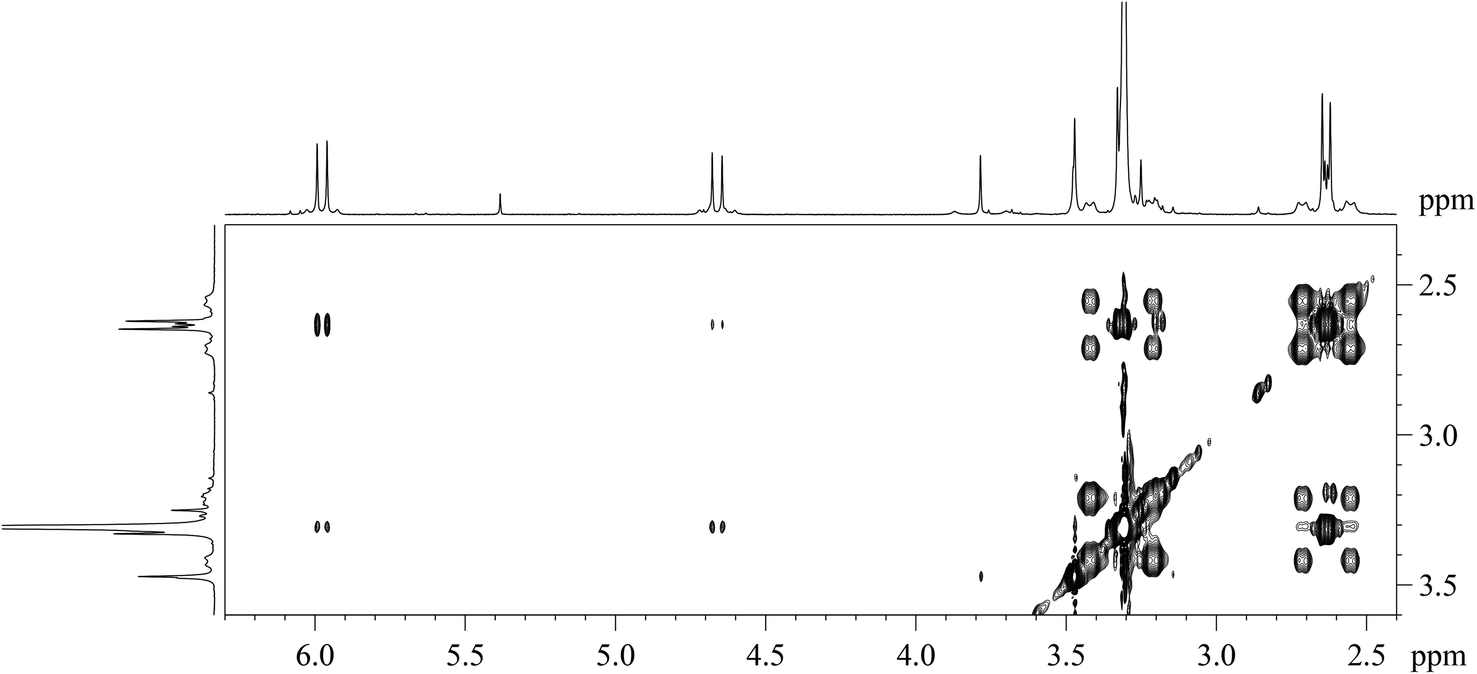 | ||
| Fig. 3 Expansion of the 2D [1H,1H]-NOESY spectrum (CDCl3, alkene–vinylic region) of [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7). | ||
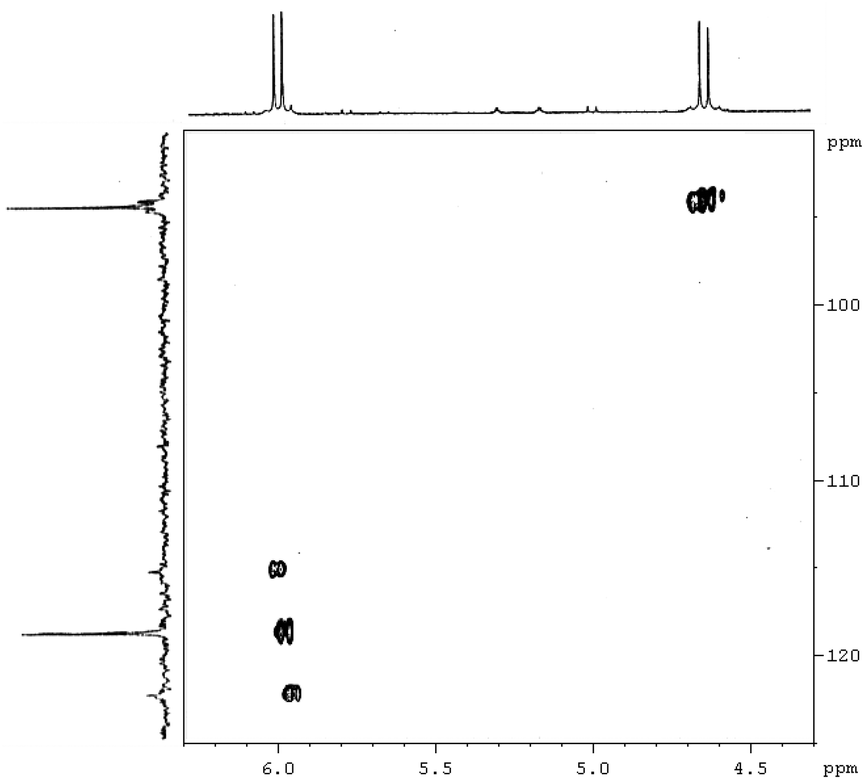 | ||
| Fig. 4 Expansion of the 2D [1H,13C]-HSQC spectrum (vinylic region) of complex [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7). | ||
The Me2phen and alkenyl 1H NMR signals of complex 7 are only slightly shifted with respect to the corresponding signals in complex 6. Moreover, the 2,9 methyl signal of Me2phen (δ = 3.31 ppm) has [1H,1H]-NOESY cross-peaks with both alkenyl protons (δ = 5.98 and 4.66 ppm). A 2D [1H,1H]-NOESY spectrum (Fig. 3) allowed assigning the two sets of alkene protons for [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7). The non-equivalence of the alkene protons is generated by the two different apical ligands. Only the most shielded proton signal of the coordinated alkene (δ = 2.64 ppm) shows cross-peaks with the protons of the bromo-alkenyl moiety, (the more intense peak is with the CHα proton). Therefore, the signal at 2.64 ppm is assigned to the pair of olefin protons facing the alkenyl ligand, while the more deshielded signal at 3.32 ppm is assigned to the protons facing the bromido ligand.
Altogether the NMR data are consistent with a complex of the type [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7), with bromido and β-bromo-vinylic ligands in apical positions, and the Me2phen and the π-ethylene in the trigonal plane (Table 2 and Fig. 2–4, 3S†), similar to previously reported Pt(II) complexes with trigonal bipyramidal geometries.31–39
CHX)(η2-CH2CH2)(Me2phen)], X = Br, (7) and I, (12); syn and anti-[PtBr(η1-E-CHCHBr)(η2-propene)(Me2phen)] (10); syn and anti-[PtBr(η1-E-CHCHBr)(η2-cis-2-butene)(Me2phen)] (11); [PtBr(η1-E-CHCHBr)(CO)(Me2phen)] (13); [PtI(η1-E-CHCHI)(CO)(Me2phen)] (14)
| Complex | Me2phena | |||||
|---|---|---|---|---|---|---|
| Me(2,9) | H(3,8) | H(4,7) | H(5,6) | η2-CH2CHR |
η1-E-CHCHX |
|
| a The (3JH–H), [JH–Pt] and [nJPt–C] values are given, where assigned. b syn and anti refer to the structures with the methyl substituent(s) of the π bonded alkene pointing to the alkenyl or bromine, bounded to platinum in axial positions, respectively. c The signals of phenanthroline protons were not assigned to the respective syn and anti isomers. | ||||||
| 7 | 3.31 | 7.78 d(8) | 8.34 d(8) | 7.87 | 3.32 dd (4) [68] 2CH | 5.98 d(13)[41] CHα,g |
| 2.64 dd (4) [65] 2CH | 4.66 d(13)[48] CHβ,c | |||||
| {28.76 [124]} | {126.22} | {137.87} | {126.22} | {32.25 [343]} | {119.01 [882] Cα} | |
| {94.32 [78] Cβ} | ||||||
| 12 | 3.30 | 7.78 d(8) | 8.33 d(8) | 7.87 | 3.49 dd (4) [82] 2CH | 6.28 d(13)[24] CHα,g |
| 2.63 dd (4) [66] 2CH | 4.53 d(13)[38] CHβ,c | |||||
| {30.07 [106]} | {127.12} | {138.77} | {127.12} | {31.85 [342]} | {131.45 [850] Cα} | |
| {65.00 [56] Cβ} | ||||||
| syn-10b,c | 3.42 | 7.81 d(8) | 8.33 d(8) | 7.85 | 2.67 d (11) [34] CHc | 6.27 d(13)[35] CHα,g |
| 3.36 | 7.79 d(8) | 8.33 d(8) | 7.84 | 4.02 m [91] CHg | 4.73 d(13)[32] CHβ,c | |
| 3.33 | 7.75 d(8) | 8.30 d(8) | 3.53 d(8) [88] CHt | |||
| 3.30 | 8.29 d(8) | 1.26 d(6) [69] CH3 | ||||
| anti-10b,c | 3.36 d(11) CHc | 5.97 d(13)[28] CHα,g | ||||
| 3.07 m[63] CHg | 4.67 d(13)[32] CHβ,c | |||||
| 2.74 d(8) [30] CHt | ||||||
| 1.65 d(6) [57] CH3 | ||||||
| syn-11b,c | 3.42 | 7.77 d(8) | 8.29 d(8) | 7.83 | 4.14 qd(5,2) [96] CH | 6.36 d(13)[39] CHα,g |
| 3.36 | 7.76 d(8) | 8.29 d(8) | 7.82 | 1.19 dd(5,2) [61] CH3 | 4.62 d(13)[28] CHβ,c | |
| anti-11b,c | 3.14 qd(5,2) [83] CH | 5.96 d(13)[26] CHα,g | ||||
| 1.59 dd(5,2)[53] CH3 | 4.68 d(13)[33] CHβ,c | |||||
| 13 | 3.38 | 7.74 d(8) | 8.32 d(8) | 7.83 | — | 6.41 d(14)[54] CHα,g |
| 5.77 d(14)[48] CHβ,c | ||||||
| 14 | 3.30 | 7.78 d(8) | 8.33 d(8) | 7.87 | — | 6.28 d(13)[24] CHα,g |
| 4.53 d(13)[36] CHβ,c | ||||||
The single crystal X ray structure of complex 7 was also preliminarily reported (see Fig. 6S and Tables 1S, 2S†).40
The reaction with acetylene, performed in the case of the iodo species [PtI2(Me2phen)] (3), shows the immediate formation of a five-coordinate species with the alkyne π-bonded to platinum, [PtI2(η2-CHCH)(Me2phen)]; which, subsequently, converts completely, in the presence of a large excess of tetra(n-Bu)ammonium iodide, Bu = butyl, into another species characterized by vinylic protons at 6.26 and 4.55 ppm. Most likely the latter species is the alkyne–alkenyl complex [PtI(η1-E-CHCHI)(η2-CHCH)(Me2phen)] (8), (Fig. 1S†). In this case the excess iodide, in solution, seems necessary to favor a faster formation of the reactive unobserved intermediate [PtI(η1-E-CHCHI)(Me2phen)]). In this case the formation of the alkyne–alkenyl complex is not quantitative even operating in the presence of excess acetylene. For long reaction times, it is possible to observe complete consumption of [PtI2(η2-CHCH)(Me2phen)] (2 days) and formation of a brown precipitate. The 1H-NMR spectrum of the CDCl3 soluble fraction showed several 1H-NMR signals in the vinylic region, as is generally observed in the case of formation of polymeric species. In contrast, if the reaction between [PtI2(Me2phen)] (3) and acetylene is performed in the presence of a large excess of N(n-Bu)4I (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the pentacoordinate alkyne–alkenylic product [PtI(η1-E-CHCHI)(η2-CHCH)(Me2phen)] (8) is obtained in quantitative yield and the reaction is faster (complete consumption of the starting species 3 in ≈2.5 h). Unlike the bromo species 2, the iodo species [PtI2(Me2phen)] (3) reacts with acetylene without detectable formation of the intermediate square planar alkenylic species [PtI(η1-E-CHCHI)(Me2phen)] (9). This is probably due to the higher reactivity of complex 9, with respect to the analogous bromo derivative, for addition of an η2-alkyne and direct formation of the five-coordinate species, a consequence of the greater steric hindrance of iodine with respect to bromine.
:1), the pentacoordinate alkyne–alkenylic product [PtI(η1-E-CHCHI)(η2-CHCH)(Me2phen)] (8) is obtained in quantitative yield and the reaction is faster (complete consumption of the starting species 3 in ≈2.5 h). Unlike the bromo species 2, the iodo species [PtI2(Me2phen)] (3) reacts with acetylene without detectable formation of the intermediate square planar alkenylic species [PtI(η1-E-CHCHI)(Me2phen)] (9). This is probably due to the higher reactivity of complex 9, with respect to the analogous bromo derivative, for addition of an η2-alkyne and direct formation of the five-coordinate species, a consequence of the greater steric hindrance of iodine with respect to bromine.
Interestingly, the pentacoordinate akyne–alkenyl complexes can be transformed in the more stable alkene–alkenyl complexes [PtX(η1-E-CHCHX)(η2-alkene)(Me2phen)], X = Br (alkene = ethylene, 7; propene, syn and anti-10; cis-2-butene, syn and anti-11, Table 2) or I (alkene = ethylene, 12), and carbonyl–alkenyl complexes [PtX(η1-E-CHCHX)(CO)(Me2phen)], X = Br (13) or I (14), by simply saturating the solution of the alkyne–alkenyl complex with the alkene or CO, respectively (Scheme 2, Table 2).
In the case of propene and cis-2-butene, due to the hindered rotation of the η2-coordinated alkene, a 1:1 mixture of the two possible isomers was obtained. NMR data referring to the alkene–alkenyl complexes are reported in Table 2. The 1H NMR spectra of the alkene–alkenyl complexes of 1-propene (10) and of cis-2-butene (11) have been interpreted on the basis of bidimensional COSY and NOESY spectra (Fig. 4S and 5S†). For five-coordinated alkenyl complexes, on the basis of 1H NMR data (integrated signals), the equilibrium constant for the ethylene–acetylene exchange reaction (T = 25 °C; Keq ≈ 6.3), Scheme 2, was also calculated.
A possible mechanism for the formation reaction of the alkyne–alkenyl complexes is depicted in Scheme 2. First, a pentacoordinate [PtX(η1-E-CHCHX)(η2-CHCH)(Me2phen)] complex can be formed from the square planar [PtX2(Me2phen)] complex, via dissociation of one end of Me2phen and formation of a T-shaped intermediate, as previously demonstrated in similar complexes, followed by addition of the alkyne.16,17,41 The formed five-coordinate complex can then be in equilibrium with a square planar cationic species formed by spontaneous dissociation of a halide (X−).42 The X− nucleophile can therefore give an exo nucleophilic attack on the π-bonded alkyne, giving a square planar complex with a platinum σ-bonded β-halogeno-vinylic group having the metal and the halogen in trans positions with respect to the double bond. Further alkyne addition gives finally the alkyne–alkenyl complex. The known higher reactivity, as a nucleophile, of the bromide with respect to iodide in organic solvents, accounts for the slower reaction of the iodo species 3, with respect to the bromo species 2, for formation of the β-halogen-alkenyl derivative. The proposed mechanism for alkyne insertion into a Pt–X (X = Br, I) bond is different from that proposed for the insertion in the Pt–O bond of five-coordinated alkene derivatives.43 Interestingly, the nucleophilic attack of X− on the π-bonded alkyne extends to halides the possibility of acting as nucleophiles toward unsaturated molecules π bonded to cationic complexes of platinum(II).8–11,13,15,23,25,28,29,41,42
Conclusions
In this work we have investigated the reactivity of [PtX2(Me2phen)] (X = Cl, 1; Br, 2; I, 3) complexes with acetylene. The chloro species, [PtCl2(Me2phen)], exhibits negligible reactivity while both the bromo, [PtBr2(Me2phen)], and iodo, [PtI2(Me2phen)], species give formation of five-coordinate species of the type [PtX2(η2-CHCH)(Me2phen)] (X = Br, I) which can evolve to square planar alkenyl complexes of the type [PtX(η1-E-CHCHX)(Me2phen)] (X = Br, I). In the presence of excess acetylene, the square planar alkenyl complexes are in equilibrium with the pentacoordinate alkyne–alkenyl species [PtX(η1-E-CHCHX)(η2-CHCH)(Me2phen)] (X = Br, I). The latter derivatives can be converted to alkene–alkenyl or carbonyl–alkenyl complexes, [PtBr(η1-E-CHCHBr)(η2-olefin)(Me2phen)] and [PtX(η1-E-CHCHX)(CO)(Me2phen)], by exchanging acetylene with free olefins or CO, respectively. The five coordinate geometry of the alkyne–alkenyl and alkene–alkenyl complexes assessed by NMR data, was consistent with that preliminarily obtained by single crystal X ray diffraction for [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)]. A possible mechanism for formation of the alkyne–alkenyl complexes is based on an exo nucleophilic attack of a free halide anion upon the π-bonded alkyne in a cationic platinum species, followed by further alkyne addition to the resulting square planar alkenyl compound.
Experimental
Reagents and methods
Reagents and solvents were commercially available and used as received, without further purification. Elemental analyses were performed with a CHN Eurovector EA 3011. NMR spectra were recorded with DRX500, DPX400 or DPX300 Avance Bruker instruments, equipped with probes for inverse detection and with a z gradient for gradient-accelerated spectroscopy. 1H and 13C NMR spectra were referenced to TMS; the residual proton signal of the solvent was used as an internal standard. Complexes [PtCl2(Me2phen)] (1), [PtBr2(Me2phen)] (2), and [PtI2(Me2phen)] (3), were synthesized with previously reported methods.25Synthesis of compounds
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHBr)(η2-CH
CHBr)(η2-CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CH)(Me2phen)], 6.
A solution of [PtBr2(Me2phen)] (2) (56 mg, 0.1 mmol) in chloroform (10 cm3) was saturated with acetylene, under magnetic stirring, for 2.5 h, at room temperature. The yellow color of the solution gradually turned orange. After 1 h the [PtBr(η1-E-CHCHBr)(η2-CHCH)(Me2phen)] (6) alkyne–alkenyl complex was isolated by addition of n-hexane, causing precipitation of a pink solid which was collected by filtration, washed with diethyl ether, and dried in dry air (yield 38.7 mg, 60%). Anal. Calc. for C18H16Br2N2Pt (6): C, 35.1; H, 2.6; N, 4.6; Br, 26.0. Found: C, 35.5; H, 2.7; N, 4.5; Br 26.1%.
CHI)(η2-CHCH)(Me2phen)], 8.
The iodo species [PtI(η1-E-CHCHI)(η2-CHCH)(Me2phen)] (8) was characterized by 1H NMR in CDCl3. 1–3 mg of [PtI2(Me2phen)] (3) were solubilized in 1 cm3 of CDCl3 containing a large excess (thirty equivalents) of tetra(n-Bu)ammonium iodide and the solution saturated with acetylene gas. The quantitative formation of the complex (total disappearance of the starting product) was observed after 2.5 h.
CHBr)(η2-CH2CH2)(Me2phen)], 7.
A solution of [PtBr2(Me2phen)] (2) (56 mg, 0.1 mmol) in chloroform (10 cm3) was saturated with acetylene gas and left standing for 2.5 h at room temperature under magnetic stirring. The yellow color of the solution turned orange. Later the solution was saturated with ethylene gas and the alkene–alkenyl complex 7 was precipitated by addition of n-hexane, the solid was removed by filtration, washed with diethyl ether, and dried by dry air (yield 38.8 mg, 60%). Anal. Calc. for C18H18Br2N2Pt (7): C, 35.0; H, 2.9; N, 4.5; Br, 25.9. Found: C, 34.8; H, 2.7; N, 4.3; Br, 26.2%.
CHBr)(η2-olefin)(Me2phen)] (olefin = propene, 10; cis-2-butene, 11).
The bromo species 10 and 11 were directly formed in CDCl3 and characterized by NMR. In a typical experiment 1–3 mg of [PtBr2(Me2phen)] (2) were dissolved in 1 mL of CDCl3 and the solution was saturated with acetylene gas and left standing for 2.5 h at room temperature under magnetic stirring. The yellow color of the solution turned orange. Then the solution was saturated with propene or cis-2-butene. The quantitative formation of complex 10 or 11, respectively, was observed, by NMR, after about 2.5 h (see Table 2).
CHI)(η2-CH2CH2)(Me2phen)] (12).
A solution of [PtI2(Me2phen)] (3) (50 mg, 0.075 mmol) in chloroform (10 cm3) containing a large excess (thirty times) of tetra(n-Bu)ammonium iodide was saturated with acetylene gas and left standing at room temperature for 2.5 h under magnetic stirring. Later the solution was saturated with ethylene gas and the alkene–alkenyl complex (12) was precipitated by addition of n-hexane, filtered, washed with diethyl ether and dried in dry air (yield 30.5 mg, 55%). Anal. Calc. for C18H18I2N2Pt (12): C, 30.4; H, 2.6; N, 3.9; I, 35.7. Found: C, 29.9; H, 2.5; N, 3.6; I, 35.6%.
CHX)(CO)(Me2phen)] (X = Br, 13; I, 14).
A solution of [PtBr2(Me2phen)] (56 mg, 0.1 mmol) or [PtI2(Me2phen)] (66 mg, 0.1 mmol) and a large excess (thirty equivalents) of tetra(n-Bu)ammonium iodide, in chloroform (10 cm3) was saturated with acetylene gas and left under magnetic stirring for 2.5 h at room temperature, meanwhile the yellow color of the solution gradually turned orange. The solution was then saturated with carbon monoxide and left under stirring for 3 h. The carbonyl–alkenyl complex (13 or 14) was precipitated by addition of n-hexane, filtered, washed with diethyl ether, and dried in dry air (yield 38 mg, 60%, 13 or 44 mg, 60%, 14). Anal. Calc. for C17H14Br2N2OPt (13): C, 33.1; H, 2.3; N, 4.5; Br, 26.0. Found: C, 32.9; H, 2.3; N, 4.6; Br, 25.7%. Anal. Calc. for C17H14I2N2OPt (14): C, 28.7; H, 2.0; N, 3.9; I, 35.7. Found: C, 29.1; H, 2.1; N, 3.8; I, 35.5%.
CH)(Me2phen)], 6.
A solution of [PtBr2(Me2phen)] (2) (56 mg, 0.1 mmol) in chloroform (10 cm3) was saturated with acetylene, under magnetic stirring, for 2.5 h, at room temperature. The yellow color of the solution gradually turned orange. After 1 h the [PtBr(η1-E-CHCHBr)(η2-CHCH)(Me2phen)] (6) alkyne–alkenyl complex was isolated by addition of n-hexane, causing precipitation of a pink solid which was collected by filtration, washed with diethyl ether, and dried in dry air (yield 38.7 mg, 60%). Anal. Calc. for C18H16Br2N2Pt (6): C, 35.1; H, 2.6; N, 4.6; Br, 26.0. Found: C, 35.5; H, 2.7; N, 4.5; Br 26.1%.
CHI)(η2-CHCH)(Me2phen)], 8.
The iodo species [PtI(η1-E-CHCHI)(η2-CHCH)(Me2phen)] (8) was characterized by 1H NMR in CDCl3. 1–3 mg of [PtI2(Me2phen)] (3) were solubilized in 1 cm3 of CDCl3 containing a large excess (thirty equivalents) of tetra(n-Bu)ammonium iodide and the solution saturated with acetylene gas. The quantitative formation of the complex (total disappearance of the starting product) was observed after 2.5 h.
CHBr)(η2-CH2CH2)(Me2phen)], 7.
A solution of [PtBr2(Me2phen)] (2) (56 mg, 0.1 mmol) in chloroform (10 cm3) was saturated with acetylene gas and left standing for 2.5 h at room temperature under magnetic stirring. The yellow color of the solution turned orange. Later the solution was saturated with ethylene gas and the alkene–alkenyl complex 7 was precipitated by addition of n-hexane, the solid was removed by filtration, washed with diethyl ether, and dried by dry air (yield 38.8 mg, 60%). Anal. Calc. for C18H18Br2N2Pt (7): C, 35.0; H, 2.9; N, 4.5; Br, 25.9. Found: C, 34.8; H, 2.7; N, 4.3; Br, 26.2%.
CHBr)(η2-olefin)(Me2phen)] (olefin = propene, 10; cis-2-butene, 11).
The bromo species 10 and 11 were directly formed in CDCl3 and characterized by NMR. In a typical experiment 1–3 mg of [PtBr2(Me2phen)] (2) were dissolved in 1 mL of CDCl3 and the solution was saturated with acetylene gas and left standing for 2.5 h at room temperature under magnetic stirring. The yellow color of the solution turned orange. Then the solution was saturated with propene or cis-2-butene. The quantitative formation of complex 10 or 11, respectively, was observed, by NMR, after about 2.5 h (see Table 2).
CHI)(η2-CH2CH2)(Me2phen)] (12).
A solution of [PtI2(Me2phen)] (3) (50 mg, 0.075 mmol) in chloroform (10 cm3) containing a large excess (thirty times) of tetra(n-Bu)ammonium iodide was saturated with acetylene gas and left standing at room temperature for 2.5 h under magnetic stirring. Later the solution was saturated with ethylene gas and the alkene–alkenyl complex (12) was precipitated by addition of n-hexane, filtered, washed with diethyl ether and dried in dry air (yield 30.5 mg, 55%). Anal. Calc. for C18H18I2N2Pt (12): C, 30.4; H, 2.6; N, 3.9; I, 35.7. Found: C, 29.9; H, 2.5; N, 3.6; I, 35.6%.
CHX)(CO)(Me2phen)] (X = Br, 13; I, 14).
A solution of [PtBr2(Me2phen)] (56 mg, 0.1 mmol) or [PtI2(Me2phen)] (66 mg, 0.1 mmol) and a large excess (thirty equivalents) of tetra(n-Bu)ammonium iodide, in chloroform (10 cm3) was saturated with acetylene gas and left under magnetic stirring for 2.5 h at room temperature, meanwhile the yellow color of the solution gradually turned orange. The solution was then saturated with carbon monoxide and left under stirring for 3 h. The carbonyl–alkenyl complex (13 or 14) was precipitated by addition of n-hexane, filtered, washed with diethyl ether, and dried in dry air (yield 38 mg, 60%, 13 or 44 mg, 60%, 14). Anal. Calc. for C17H14Br2N2OPt (13): C, 33.1; H, 2.3; N, 4.5; Br, 26.0. Found: C, 32.9; H, 2.3; N, 4.6; Br, 25.7%. Anal. Calc. for C17H14I2N2OPt (14): C, 28.7; H, 2.0; N, 3.9; I, 35.7. Found: C, 29.1; H, 2.1; N, 3.8; I, 35.5%.
Acknowledgements
The University of Salento (Italy) and the PON 254/Ric. Potenziamento del “CENTRO RICERCHE PER LA SALUTE DELL'UOMO E DELL'AMBIENTE” Cod. PONa3_00334 and the Consorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici (CIRCMSB), Bari (Italy), are acknowledged for financial support.References
- W. C. Zeise, Pogg. Ann. Phys., 1827, 9, 632 Search PubMed.
- S. V. Bukhovets, Izv. Sekt. Platiny i Drug. Blagorod. Metal. Inst. Obshch. Neorg. Khim. Akad. Nauk. USSR, 1955, 29, 55 Search PubMed.
- T. Theophanides and P. C. Kong, Can. J. Chem., 1970, 48, 1084–1092 CrossRef CAS PubMed.
- J. Chatt, Chimica Inorganica, Accad. Nazl. Licei, Roma, 1961, p. 113 Search PubMed.
- E. O. Greaves, C. J. L. Lock and P. M. Maitlis, Can. J. Chem., 1968, 46, 3879–3891 CrossRef CAS.
- H. C. Clark and R. J. Puddephatt, Inorg. Chem., 1970, 9, 2670–2675 CrossRef CAS.
- B. W. Davies, R. J. Puddephatt and N. C. Payne, Can. J. Chem., 1972, 50, 2276–2284 CrossRef CAS.
- L. Cattalinis, F. Gasparrini, L. Maresca and G. Natile, J. Chem. Soc., Chem. Commun., 1973, 369–370 RSC.
- L. Maresca, G. Natile, M. Calligaris, P. Delise and L. Randaccio, J. Chem. Soc., Dalton Trans., 1976, 2386–2390 RSC.
- L. Maresca and G. Natile, Comments Inorg. Chem., 1993, 14(6), 349–366 CrossRef CAS.
- M. Benedetti, F. P. Fanizzi, L. Maresca and G. Natile, Chem. Commun., 2006, 1118–1120 RSC.
- V. M. Vecchio, M. Benedetti, D. Migoni, S. A. De Pascali, A. Ciccarese, S. Marsigliante, F. Capitelli and F. P. Fanizzi, Dalton Trans., 2007, 5720–5725 RSC.
- C. R. Barone, M. Benedetti, V. M. Vecchio, F. P. Fanizzi, L. Maresca and G. Natile, Dalton Trans., 2008, 5313–5322 RSC.
- M. Benedetti, D. Antonucci, D. Migoni, V. M. Vecchio, C. Ducani and F. P. Fanizzi, ChemMedChem, 2010, 5, 46–51 CrossRef CAS PubMed.
- M. Benedetti, C. R. Barone, D. Antonucci, V. M. Vecchio, A. Ienco, L. Maresca, G. Natile and F. P. Fanizzi, Dalton Trans., 2012, 41, 3014–3021 RSC.
- M. Benedetti, D. Antonucci, S. A. De Pascali, C. R. Girelli and F. P. Fanizzi, J. Organomet. Chem., 2012, 714, 60–66 CrossRef CAS PubMed.
- M. Benedetti, D. Antonucci, S. A. De Pascali, G. Ciccarella and F. P. Fanizzi, J. Organomet. Chem., 2012, 714, 104–108 CrossRef CAS PubMed.
- M. Benedetti, D. Antonucci, C. R. Girelli, F. Capitelli and F. P. Fanizzi, Inorg. Chim. Acta, 2014, 409, 427–432 CrossRef CAS PubMed.
- M. Benedetti, C. R. Girelli, D. Antonucci, S. A. De Pascali and F. P. Fanizzi, Inorg. Chim. Acta, 2014, 413, 109–114 CrossRef CAS PubMed.
- P. Papadia, A. Ciccarese, J. A. Miguel-Garcia, P. M. Maitlis and F. P. Fanizzi, J. Inorg. Biochem., 2005, 690, 2097–2105 CAS.
- R. J. H. Clark, F. P. Fanizzi, G. Natile, C. Pacifico, G. Van Rooyen and D. A. Tocher, Inorg. Chim. Acta, 1995, 235, 205–213 CrossRef CAS.
- F. P. Fanizzi, G. Natile, M. Lanfranchi, A. Tiripicchio, F. Laschi and P. Zanello, Inorg. Chem., 1996, 35, 3173–3182 CrossRef CAS PubMed.
- F. P. Fanizzi, L. Maresca, G. Natile, M. Lanfranchi, A. Tiripicchio and G. Pacchioni, J. Chem. Soc., Chem. Commun., 1992, 4, 333–335 RSC.
- F. P. Fanizzi, M. Lanfranchi, G. Natile and A. Tiripicchio, Inorg. Chem., 1994, 33, 3331–3339 CrossRef CAS.
- F. P. Fanizzi, F. P. Intini, L. Maresca, G. Natile, M. Lanfranchi and A. Tiripicchio, J. Chem. Soc., Dalton Trans., 1991, 1007–1015 RSC.
- F. P. Fanizzi, G. Natile, M. Lanfranchi, A. Tiripicchio and G. Pacchioni, Inorg. Chim. Acta, 1998, 275–276, 500–509 CrossRef CAS.
- B. F. G. Johnson, J. Lewis, J. D. Jones and K. A. Taylor, J. Chem. Soc., Dalton Trans., 1974, 34 RSC.
- G. Lorusso, G. Boccaletti, N. G. Di Masi, F. P. Fanizzi, L. Maresca and G. Natile, Eur. J. Inorg. Chem., 2004, 24, 4751–4754 CrossRef.
- C. R. Barone, C. Coletti, R. J. McQuitty, N. J. Farrer, G. Lorusso, L. Maresca, A. Marrone, G. Natile, C. Pacifico, S. Parsons, N. Re, P. J. Sadler and F. J. White, Dalton Trans., 2013, 42, 6840–6851 RSC.
- (a) A. R. Siedle, W. B. Gleason and R. A. Newmark, Organometallics, 1986, 5, 1969 CrossRef CAS; (b) H. C. Clark, G. Ferguson, A. B. Goel, E. G. Janzen, H. Ruegger, P. Y. Siew and C. S. Wong, J. Am. Chem. Soc., 1986, 108, 6961–6972 CrossRef CAS; (c) P. J. Stang, M. H. Kowalski, M. D. Schiavelli and D. Longford, J. Am. Chem. Soc., 1989, 111, 3347–3356 CrossRef CAS.
- V. G. Albano, D. Braga, V. De Felice, A. Panunzi and A. Vitagliano, Organometallics, 1987, 6, 517–525 CrossRef CAS.
- M. E. Cucciolito, V. De Felice, A. Panunzi and A. Vitagliano, Organometallics, 1989, 8, 1180–1187 CrossRef CAS.
- V. De Felice, M. Funicello, A. Panunzi and F. Ruffo, J. Organomet. Chem., 1991, 403, 243–252 CrossRef CAS.
- F. Giordano, F. Ruffo, A. Saporito and A. Panunzi, Inorg. Chim. Acta, 1997, 264, 231–237 CrossRef CAS.
- V. De Felice, B. Giovannitti, A. Panunzi, F. Ruffo and D. Tesauro, Gazz. Chim. Ital., 1993, 123, 65–69 CAS.
- V. G. Albano, G. Natile and A. Panunzi, Coord. Chem. Rev., 1994, 133, 67–114 CrossRef CAS.
- F. Giordano, B. Panunzi, A. Roviello and F. Ruffo, Inorg. Chim. Acta, 1995, 239, 61–66 CrossRef CAS.
- V. G. Albano, C. Castellari, M. E. Cucciolito, A. Panunzi and A. Vitigliano, Organometallics, 1990, 9, 1269–1276 CrossRef CAS.
- V. G. Albano, M. Monari, I. Orabona, F. Ruffo and A. Vitagliano, Inorg. Chim. Acta, 1997, 265, 35–46 CrossRef CAS.
- X-ray structure of [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7). An X-ray single crystal structure of the complex [PtBr(η1-E-CHCHBr)(η2-CH2CH2)(Me2phen)] (7) has been deposited at the Cambridge Crystallographic Data Centre by Francesco P. Fanizzi, Luciana Maresca,
Giovanni Natile, and Concetta Pacifico in 1999 (CCDC-122701). We give here a brief description of complex 7 since its X-ray structure was never described. The asymmetric unit includes two crystallographically independent molecules, their structures (A and B) are shown in Fig. 6S† while Crystal Data and selected bonding distances and angles are reported in Tables 1S and 2S,† respectively. None of the two molecules has (crystallographically imposed) symmetry elements, but molecule A has approximate Cs symmetry. In both molecules the Pt atom is characterized by a trigonal bipyramidal geometry, similar to that found in analogous Pt(II) alkene complexes.15,21,23,24,29 The apical positions are occupied by one bromide and the α carbon atom of a β-bromo-vinyl group, while the equatorial positions are kept by the nitrogen atoms of the Me2phen ligand and by the ethylene carbons. In both molecules the Me2phen ligand is approximately planar and slightly inclined with respect to the trigonal plane, probably because of steric interaction between the Me2phen methyl and the alkene ligand. In the case of molecule A the inclination is larger (7.9(3)°) with the Me2phen methyl directed towards the alkenyl ligand. In the case of molecule B the inclination is smaller (5.1(3)°) with the Me2phen methyl directed towards the bromido ligand; probably a steric interaction between one of the methyl groups of Me2phen (C19) and the β carbon of the β-bromo-vinyl group (C36) is responsible for such an inclination. The length of the Pt–C and CC bonds of ethylene are comparable to those found in other five-coordinate Pt(II) complexes.36.
- M. Benedetti, C. R. Barone, C. R. Girelli, F. P. Fanizzi, G. Natile and L. Maresca, J. Chem. Soc., Dalton Trans., 2014, 3669–3675 RSC.
- L. Maresca and G. Natile, Comments Inorg. Chem., 1994, 16, 95–112 CrossRef CAS.
- H. E. Bryndza, Organometallics, 1985, 4, 406–408 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt00679h |
| This journal is © The Royal Society of Chemistry 2014 |