 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[NiFe] hydrogenases: how close do structural and functional mimics approach the active site?
Sandeep
Kaur-Ghumaan
*ab and
Matthias
Stein
*b
aDepartment of Chemistry, University of Delhi, Delhi-110007, India. E-mail: skaur@chemistry.du.ac.in; Tel: +91-9811137848
bMax-Planck-Institute for Dynamics of Complex Technical Systems, Molecular Simulations and Design Group, Sandtorstrasse 1, 39106 Magdeburg, Germany. E-mail: matthias.stein@mpi-magdeburg.mpg.de; Fax: +49-391-6110403
First published on 20th May 2014
Abstract
Hydrogen is being considered as a versatile alternative fuel with the ever increasing energy demand and oil prices. Hydrogenases (H2ases) found in bacteria, archaea and eukaryotes are very efficient catalysts for biological hydrogen production. An important and unique hydrogenase enzyme is the [NiFe] H2ase, with an unusual heterobimetallic site. Since the determination of its crystal structure, a variety of complexes have been synthesised and studied. Bioinspired and biomimetic complexes have been investigated as potential catalysts. So far, of all the reported complexes only a few of them have been found to be catalytically active. Moreover, most of the reports are on the reverse reaction, e.g. proton reduction rather than dihydrogen oxidation. This perspective article therefore reviews the structural and functional aspects of the very recently reported model complexes that mimic the [NiFe] hydrogenase active site either in structure or function or both.
1. Introduction
With fossil fuels depleting at a very fast rate due to their global consumption, it is evident that the world requires alternative energy sources. One such versatile source being considered is molecular hydrogen (H2), which is also being called a ‘green energy source’: its combustion is emission-free and produces only water. There are various ways of generating hydrogen artificially: steam reforming from hydrocarbons, electrolysis of water releasing H2 from an inert cathode (usually platinum), which is too energy intensive on a large scale use for the present world or by thermolytic water splitting, which is not a cheap and sustainable source either.In nature, certain enzymes termed ‘hydrogenases’ (H2ases) catalyse the reversible oxidation of dihydrogen and play a key role in microbial energy metabolism (in bacteria, algae and archaea).
| H2 ⇄ H+ + H− ⇄ 2H+ + 2e− |
Therefore, the main aim of this perspective is to review the bioinorganic chemistry related to the heterobimetallic [NiFe] H2ase enzyme and the recently reported model complexes that mimic the structure and function of the active site of the enzyme. This is of interest in terms of catalytic mechanism, since the [NiFe] H2ases’ preferred catalytic enzymatic function of heterolytic splitting of dihydrogen rather than proton reduction can be modulated by pH and redox potential.18,19 The elucidation of catalytic mechanisms of model complexes and the fine-tuning of their properties provides an insight into nature's design of the [NiFe] H2ase enzyme vide infra. Non-reactive structural model complexes, on the other hand, do not represent dead ends but point to the sophistication of redox vs. structural design principles in nature.
2. [NiFe] hydrogenase
2.1. Structure of the enzyme
The first X-ray structure of the [NiFe] hydrogenase was reported in 1995 by Volbeda and co-workers.20 The structure from the Desulfovibrio gigas enzyme isolated aerobically was shown to contain Ni as one of the metal ions in the active site. The discovery of a second metal ion in the active site, later identified to be Fe, was a surprise (see Fig. 1). The Fe atom was later shown to remain in its +II oxidation state throughout all accessible redox states (low spin, S = 0, diamagnetic) (Scheme 1). In addition, by FTIR studies the active site was shown to contain carbon monoxide and cyanide ligands coordinating the Fe and the small subunit contains three iron–sulphur centres: one [3Fe–4S] and two [4Fe–4S] clusters, which transfer electrons to and from the active site.21 When isolated aerobically, the [NiFe] enzyme is inactive and requires activation by H2 or other reductants. The ‘as-isolated’ form is a mixture of the ‘unready’ (Ni-A) and the ‘ready’ Ni-B active site states of the hydrogenase. The latter can be activated faster by incubation with molecular hydrogen. The difference in the rate of activation was proposed to be due to the different chemical nature of the ligands in the two states. Ni-B was shown to contain a μ-hydroxo bridging ligand in the active site,22,23 whereas for Ni-A a bridging oxygenic species (O2−, OH− or OOH−) and/or oxidation of a cysteine amino acid was suggested.23–25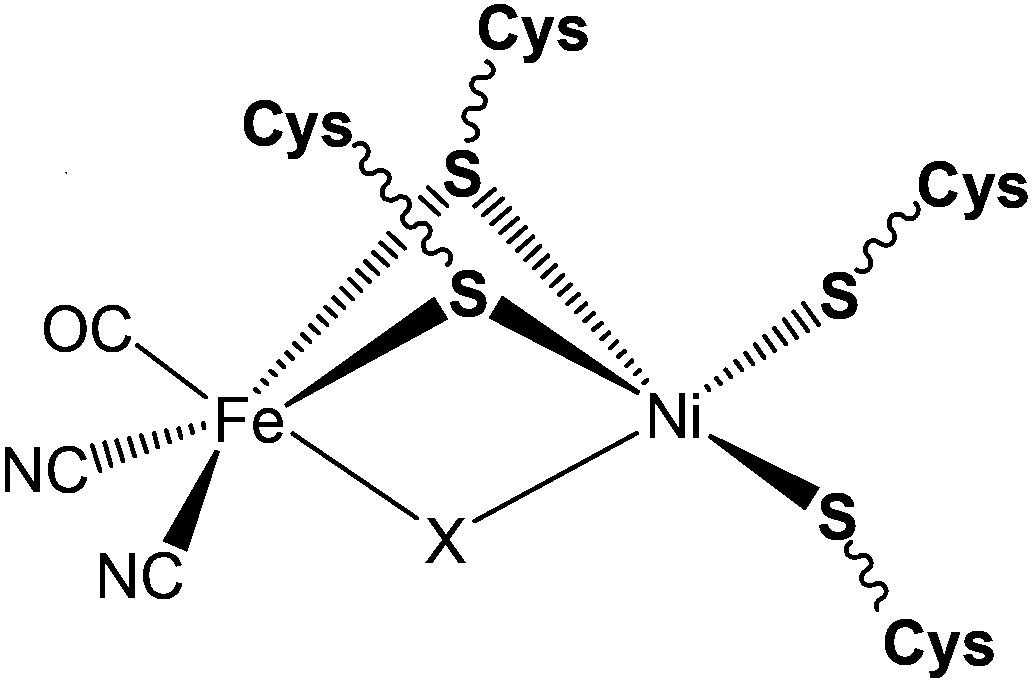 | ||
| Fig. 1 Schematic representation of the ‘as-isolated’ forms of the [NiFe] hydrogenase (X = O2−, OH− or HOO− for Ni-A and OH− for Ni-B). | ||
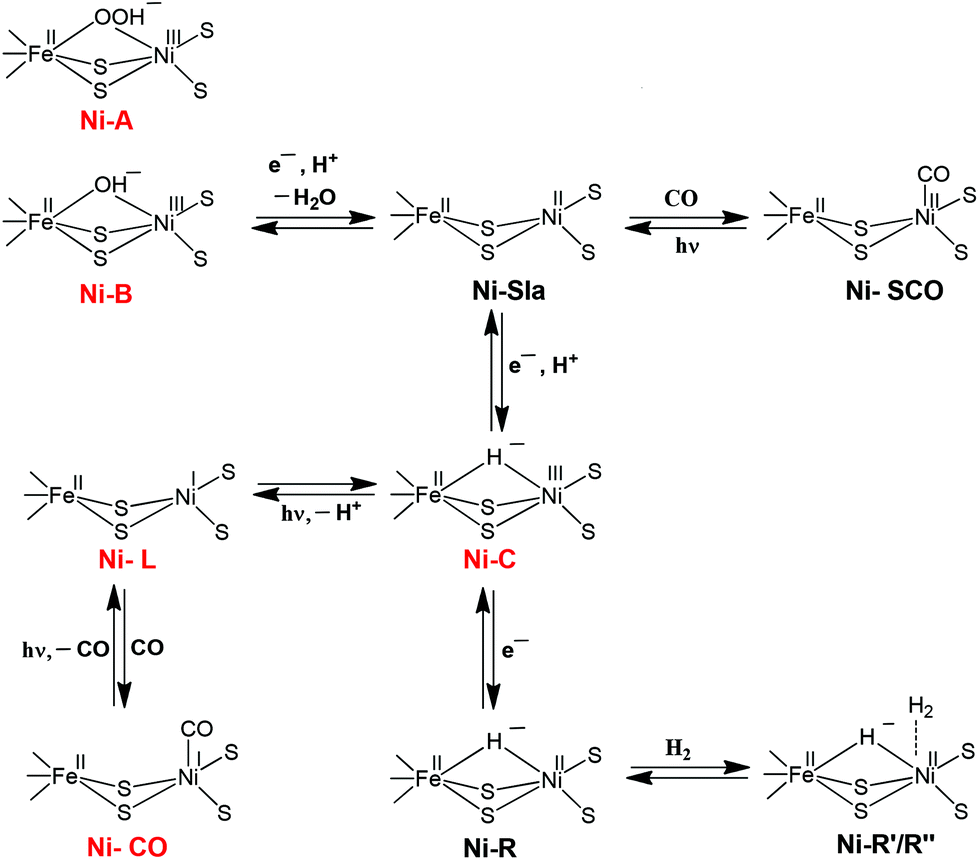 | ||
| Scheme 1 Schematic representation of the different redox states of the active site of the [NiFe] hydrogenase. The EPR-detectable states are shown in red. | ||
In subsequent years, X-ray structures were reported for the Ni–C, Ni–R and Ni–CO states.26–29 In the case of the Ni–CO state (CO inhibited complex), the carbon monoxide complex of the [NiFe] hydrogenase from Desulfovibrio vulgaris Miyazaki F was characterised and studied. CO was shown to bind to the Ni atom only.27 Based on all the reports, the structure of the inactive forms of the heterobimetallic [NiFe] hydrogenase can be depicted as shown in Fig. 1. Besides several reports on the structure of the [NiFe] hydrogenase enzyme, to date various investigations have also been done on the proton and gas channel pathways.5,29 The rates of diffusion within the hydrogenases have been studied using xenon binding crystallography and molecular dynamics simulations.29
2.2. Experimentally tracking the mechanism of dihydrogen splitting by the enzyme
The [NiFe] hydrogenase (H2ase) located in the periplasm30 consists of small and large subunits of 28 and 60 kDa, respectively. The different active site states for the [NiFe] hydrogenase enzyme can be represented as shown in Scheme 1. These different states have either been characterised theoretically by DFT or experimentally by thorough X-ray structure, FTIR, EPR and ENDOR studies. The importance of spectroscopy and electrochemical methods for the characterisation of reactive intermediates have been reviewed elsewhere, from which also the pH-dependence of redox transitions can be rationalised.6,31 In the ‘as-isolated’ states (Ni-A and Ni-B) the active site is in a Ni(III)Fe(II) oxidation state. The oxygenic species, a μ-hydroxo in Ni-B, is removed during the activation process (a pH-dependent one electron reduction). The diamagnetic Ni(II)–SIa is believed to be the catalytically active species.32 Removal of the bridging ligand is a pre-requisite to enable external CO binding to yield the Ni–SCO state. Ni–C is a catalytic intermediate in the reaction cycle and corresponds to a Ni(III)Fe(II) state with a μ-bridging hydride occupying the position between the two metals. Ni-C is light-sensitive and the hydride can be removed by photoreduction to give the Ni–L Ni(I)Fe(II) state.33,34 Exogenous CO can bind to Ni–L to give the paramagnetic Ni(I)–CO state. Ni–R is the fully reduced redox state of the active site.An understanding of the different redox states of the [NiFe] H2ase is very important in order to rationalise the exact reaction mechanism of [NiFe] hydrogenases, which has been a matter of great debate over the past few years. The literature of quantum chemical studies concerning the reaction mechanism of [NiFe] hydrogenases has been reviewed in depth by Siegbahn, Tye and Hall.35 More recent Density Functional Theory (DFT) studies cited here explicitly, however, only mention work after 2007.
Despite different suggestions from various quantum chemical studies, there appears to be a ‘consensus’ mechanism. The actual catalytic cycle of [NiFe] H2ase has been shown to consist of only three states: Ni–SIa, Ni–C and Ni–R. These states can be interconverted to one another by one-electron/one-proton equilibria (Scheme 1).36,37 The first step in the catalytic process involves the attachment of the H2 to the Ni centre. Either oxidative addition or base-assisted heterolytic cleavage of the H2 molecule then leads to a bridging hydride species. It has been proposed that a terminal cysteine at the Ni could be acting as a base and take up the proton. Alternatively, a water molecule bound to the iron has also been proposed.38 The Ni–C (Ni(III)Fe(II)) state carrying the hydride bridge is formed next by concomitant electron transfer to the proximal FeS cluster.39–41 It is then proposed that the Ni–R state, which still carries the hydride, is formed by further reduction of Ni–C by another H2 molecule.42 In the final step, release of another proton and electron occurs to recover the initial Ni–SIa state with an open bridge, ready for the next turnover. This last step could also be preceded by relocation of the proton from the bridge to the terminal cysteine, creating a state that resembles Ni–L.43
In another model (Scheme 2) proposed by Fontecilla-Camps et al.,44 the Ni–R state is formed directly from Ni–SIa with H2. In the catalytic cycle, the hydride remains in the bridge between Ni and Fe and acts as a base for the next incoming H2. In this mechanism, the hydrogenase cycles between Ni–R, Ni–C, and a transient Ni–C state; the latter has a second hydride terminally bound to the Ni. This model proposes that the two protons are released in two subsequent oxidation steps. H2 production has been assumed to occur through the same reverse pathway.
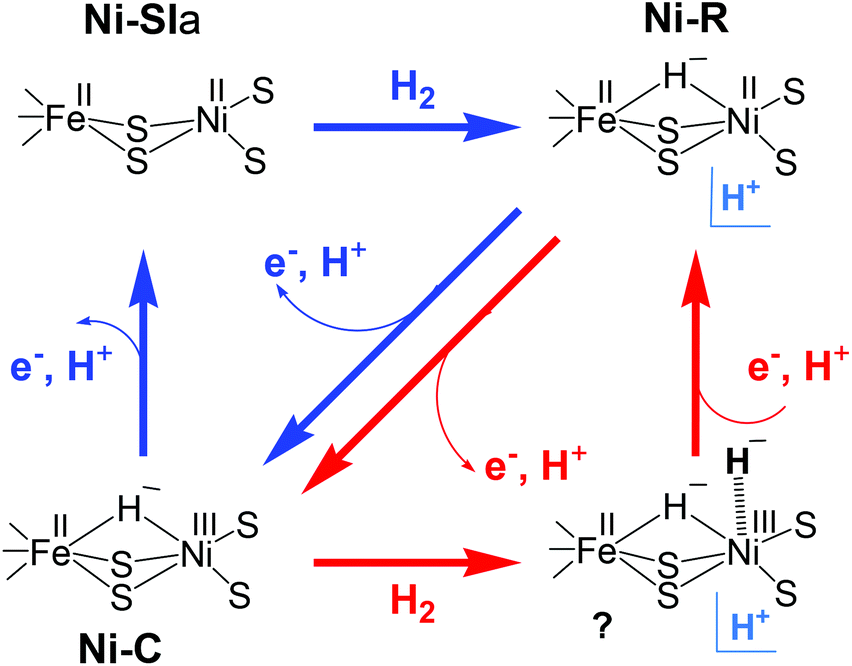 | ||
| Scheme 2 Possible catalytic cycles of [NiFe] hydrogenase adopted from (ref. 44); either Ni–C (red arrows) or Ni–SIa (blue arrows) act as hydrogen acceptors. H+ denotes an unidentified proton acceptor. | ||
The binding of H2 to nickel32 is consistent with the observations that the hydrogen transport channel is directly linked to the nickel centre rather than to iron and that exogenous CO, a competitive inhibitor of H2, binds to nickel. However, different views still exists as to whether the iron atom could also be the site of catalytic turnover.45–47 Though various suggestions for the mechanism of the hydrogen splitting by the [NiFe] hydrogenase enzymes exist, further mechanistic work by means of computation and experiments still needs to be performed in this area to achieve a mechanism that is commonly agreed upon by experimentalists and computational chemists.
2.3. Structural and functional biomimetic/bioinspired models
Reviews by Halcrow and Christou48 and Bouwman and Reedijk49 give an overview of the first period during which mononuclear Ni complexes were developed. This was prior to X-ray crystallography in 1995, before which it was believed that mononuclear Ni represented the active site of the enzyme. During the next period {Fe(CO)x(CN)y}13 and {NiFe} complexes13 were reported. The {NiFe} complexes reported here varied from a range of dinuclear to polymetallic complexes with varied ligand environment (that included S, N, P environments) around the Ni or Fe centres. Dinitrosyl bimetallic {NiFe} complexes have also been synthesised and reported with an aim to utilise the Fe(NO)2 unit as a surrogate for Fe(II)(CO)(CN)2 in the enzyme active site. This was probably due to the intriguing redox and chemical properties of such type of complexes.13,50 It was in 1996 that the first structurally characterised thiolate-bridged {NiFe} carbonyl complex (1) was reported by M. Y. Darensbourg (Fig. 2).51 This heterobimetallic complex was synthesised by S-metallation of N2S2-coordinated Ni(II) complexes with [Fe2(CO)9]. It was 10 years after this that the first syntheses of bridging-dithiolate {NiFe} complexes (2, 3) were reported by Tatsumi et al. (Fig. 2).52 These complexes are considered to be the closest mimics of the enzyme active site so far. Similar other complexes were also reported by the same group, which, however, were found to be thermally unstable in solution and needed to be synthesised and handled at −40 °C.53
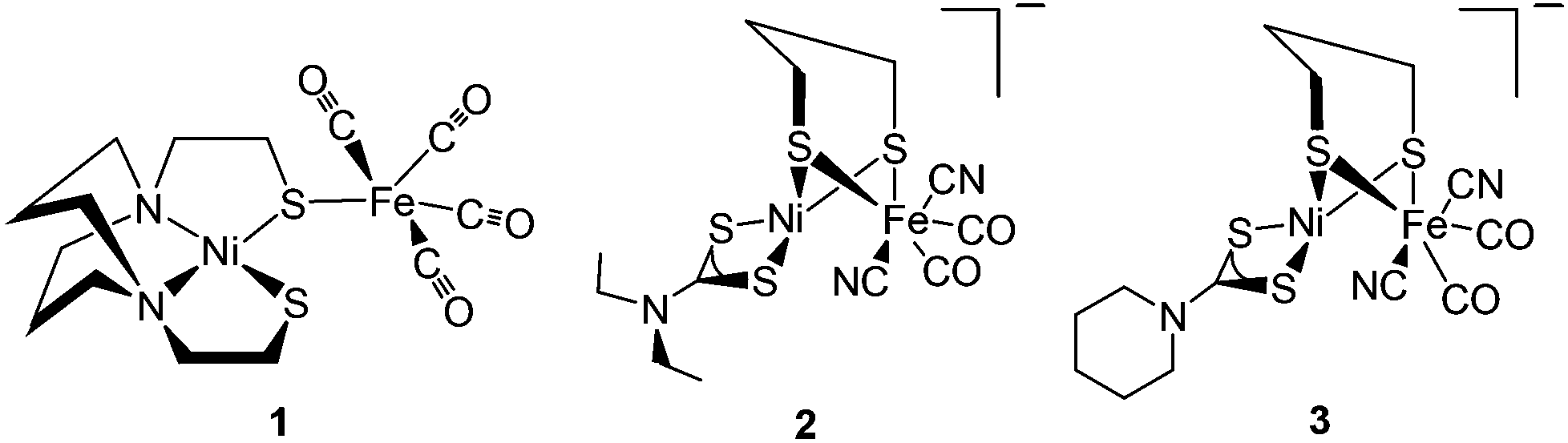 | ||
| Fig. 2 First heterobimetallic thiolate-bridged {NiFe} complexes 1 by Lai (ref. 51) and 2, 3 by Li (ref. 52). | ||
An investigation of the possible catalytic activity of those complexes could not be performed. But whether such complexes can participate in hydrogen uptake or evolution is still a question to be addressed. Moreover, of all the complexes reported up to 2009 only two of them showed catalytic behaviour in the form of proton reduction instead of dihydrogen oxidation.54,55 Complexes of Ni with other metals like Ru,56–63 Mn64,65 and Ge66 have also recently been reported and compared to the [NiFe] H2ase active site, though these bear very little structural resemblance to the enzyme active site. In addition, reports on mononuclear Fe-, Co- and Ni-complexes proposed by DuBois and co-workers67–69 have shown that cleavage of dihydrogen is possible by using such complexes as electrocatalysts. We have also reported mononuclear Fe complexes as mimics of the hydrogenases capable of proton reduction with moderate and strong acids.70
Despite these promising steps in designing biomimetic and bioinspired complexes, some urgent and critical questions still remain. These are, for example, the presence of the second metal atom (Fe) in the active site. The nickel atom undergoes redox chemistry (traced by EPR and electrochemistry) and binds molecular hydrogen.32 The changes of electron density at the Fe metal, however, are only moderate (traced by small differences in CO and CN FTIR frequencies). Why has Nature chosen to accommodate such a complicated heterobimetallic site to confer a reaction that is also feasible by monometallic complexes? From experiments we now have a fair understanding of the fate of only one reaction product from heterolytic hydrogen splitting. It is now commonly agreed upon that Ni–C bears a bridging hydride between the Ni and Fe atoms but where is the second product, the proton? If the heterolytic splitting occurs base-assisted, is one of the (terminal) cysteine ligands or a water molecule acting here? How does Nature enable such a swift and barrierless proton translocation so that it cannot be measured by today's advanced spectroscopic techniques? Last, but not least, Nature has designed a highly active catalyst to enable heterolytic hydrogen splitting at room temperature. Turnover frequencies of more than thousands of molecules H2 per second at 30 °C have been reported.31 Current technological hydrogen activating catalysts rely on precious noble metals and operate at elevated temperatures. Learning from Nature here paves the way for an economic and ecological management of resources and processes.
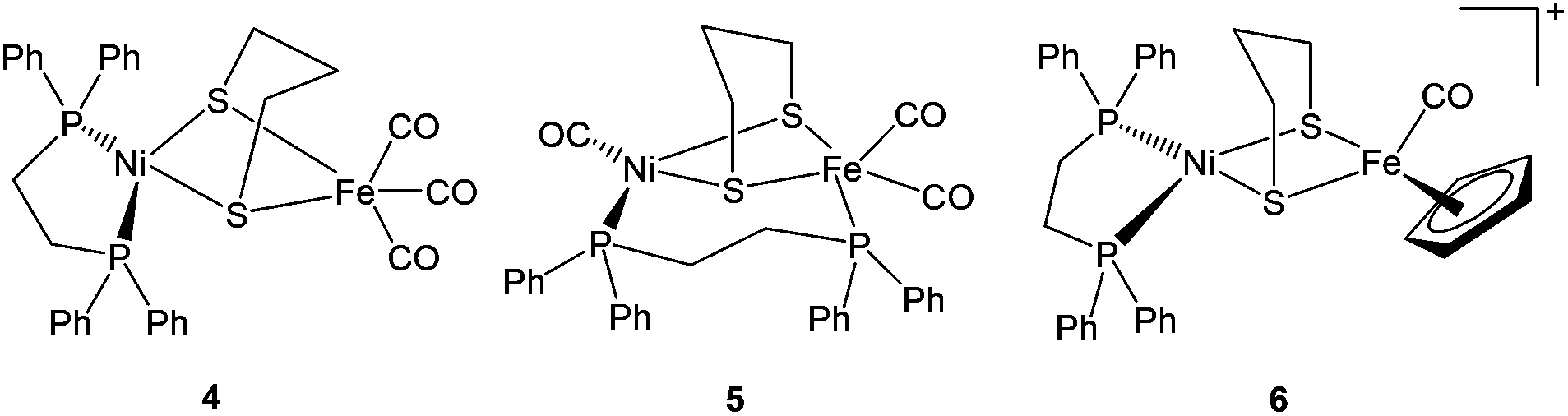 | ||
| Fig. 3 Advanced dithiolate-bridged {NiFe} complexes reported by Zhu et al. (ref. 71). | ||
The linear/macrocyclic Ni/Fe/X (X = S, Se) cluster complexes (7–33) shown in Fig. 4 have been reported as structural mimics of the enzyme active site.73–75 The Fe centre in the complexes is coordinated either to sulphur or selenium ligands. Based on X-ray structure determination, the geometry around the Ni centre was confirmed to be distorted square planar and that around the Fe-centres square pyramidal or octahedral. In the protein X-ray structure analysis, the Ni atom is coordinated by four cysteine residues in a distorted tetrahedral structure or five-coordinate in a square-pyramidal fashion while the Fe atom is tetragonal-pyramidally or octahedrally coordinated by five and six ligands, respectively (see Fig. 1).20
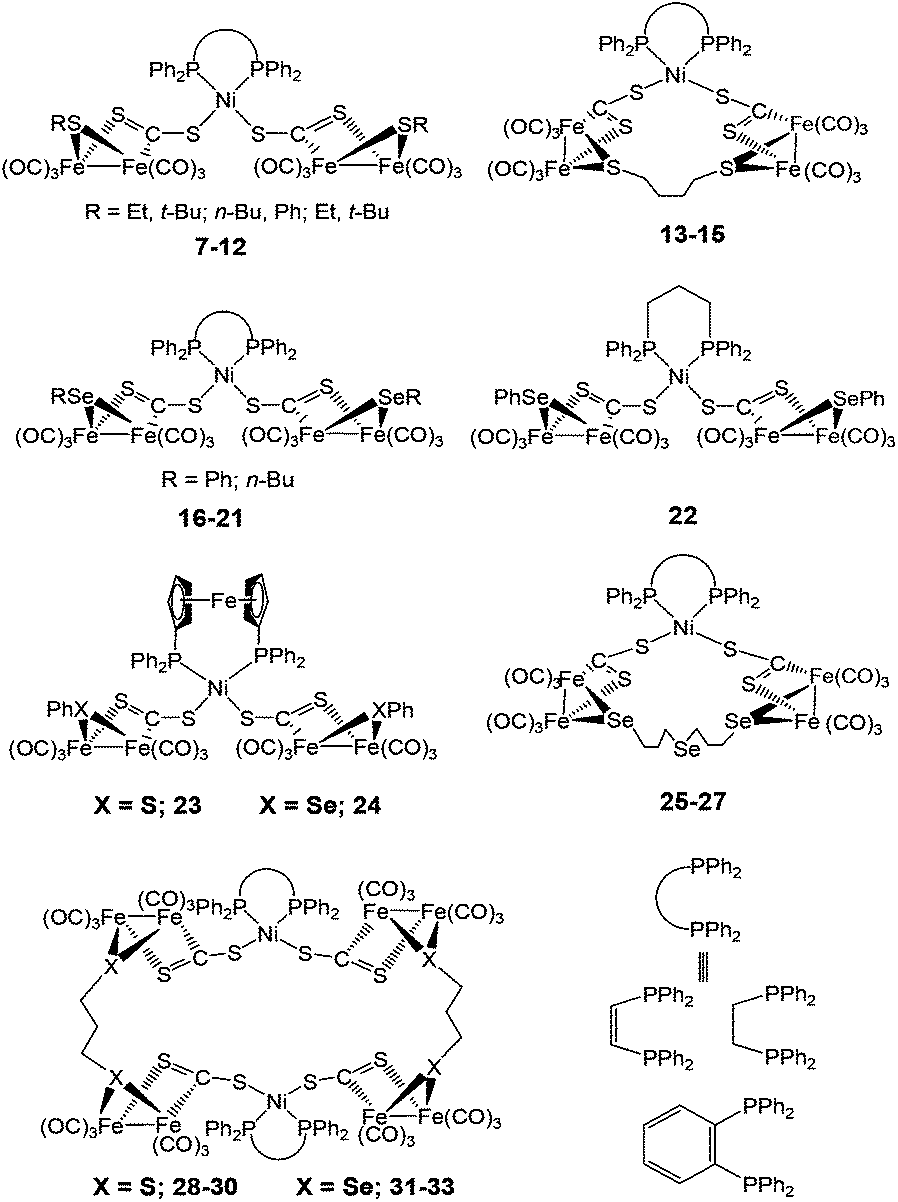 | ||
| Fig. 4 Linear and macrocyclic Ni/Fe/S cluster complexes. | ||
New mixed valence {NiFe} dithiolates, [(diphosphine)Ni(dithiolate)Fe(CO)2L] (34–56) {dithiolate = pdt, edt; diphosphine = dppe, dcpe; L = PPh2(2-pyridyl), PPh3, PCy3, P(OPh)3, P(p-C6H4Cl)3, PPh2(OEt), PPh2(o-C6H4OMe), PPh2(o-C6H4OCH2OMe), P(p-tol)3, P(p-C6H4OMe)3, PMePh2, PHCy2, PPh(NEt2)2, P(i-Pr)3} (dcpe = 1,2-bis(dicyclohexylphosphino)ethane) shown in Fig. 5 have also been reported as structural mimics of the enzyme.76–78
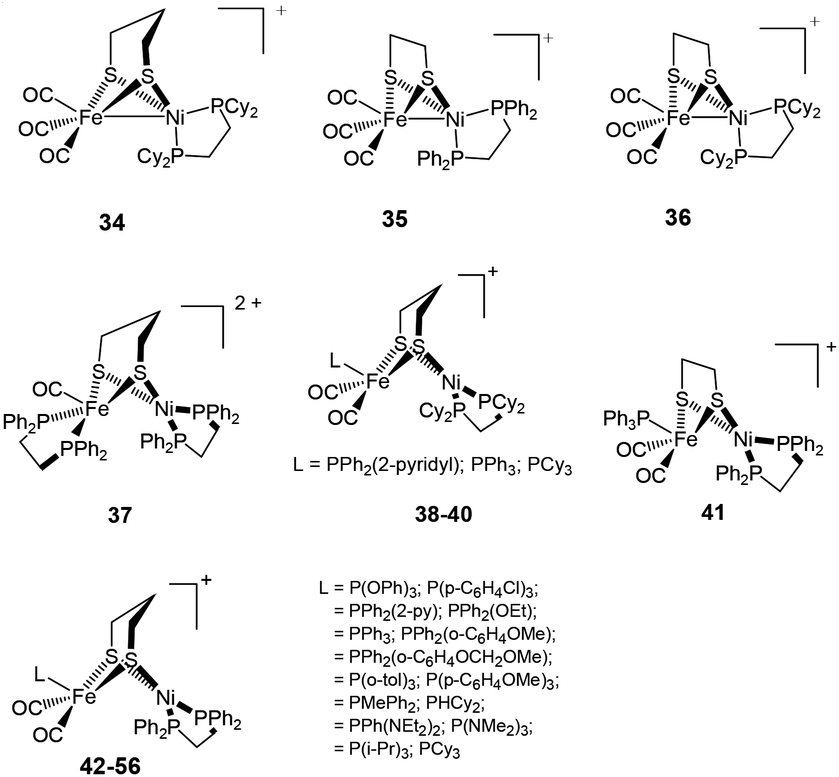 | ||
| Fig. 5 Mixed-valent dithiolate bridged complexes as models for the [NiFe] active site. | ||
The neutral complexes contain Ni(I)Fe(I) centres. On the other hand, the oxidised monocationic complexes [34–56]+ of the general formula [(diphosphine)Ni(dithiolate)Fe(CO)2L]+ have been described as Ni(II)Fe(I) (S = 1/2) species, though this is inverse to the Ni(I)Fe(II) core present in the Ni–L state of the enzyme. It is clear that the assignment of formal oxidation states does not give a picture of electron density distribution in the heterobimetallic complexes. The σ-donating and π-accepting properties of phosphorous and sulphur ligands introduce partial covalency between the metal and its ligands. This makes an assignment of formal oxidation states more complex. The dications (Ni(II)Fe(II)) obtained in some cases (e.g. complex 37), on the other hand, were found to be close mimics of the Ni–SIa state. The remarkable finding was, that the distorted geometry of the Fe(I) centre in [(dppe)Ni(pdt)Fe(CO)2PCy3]+, adopted a “rotated” structure similar to the Hox state of [FeFe] H2ase complexes.79 This was the first demonstration of the introduction of structural features from [FeFe] hydrogenases into {NiFe} model complexes. The complexes were shown to resist protonation in spite of being oxidisable and containing highly basic ligands like phosphine. Moreover, the dicationic complexes (Ni(II)Fe(II)) have a five-coordinate Fe centre that is different from previously reported complexes (Ni(II)Fe(II)) 57–59 (Fig. 6), which bear a coordinatively saturated Fe centre.80,81 More recent examples of mixed-valent complexes include [(dppe)Ni(pdt)XFe(CO)2L]+ (X = Cl−, Br−, I−; L = CO, PPh3, PCy3).82
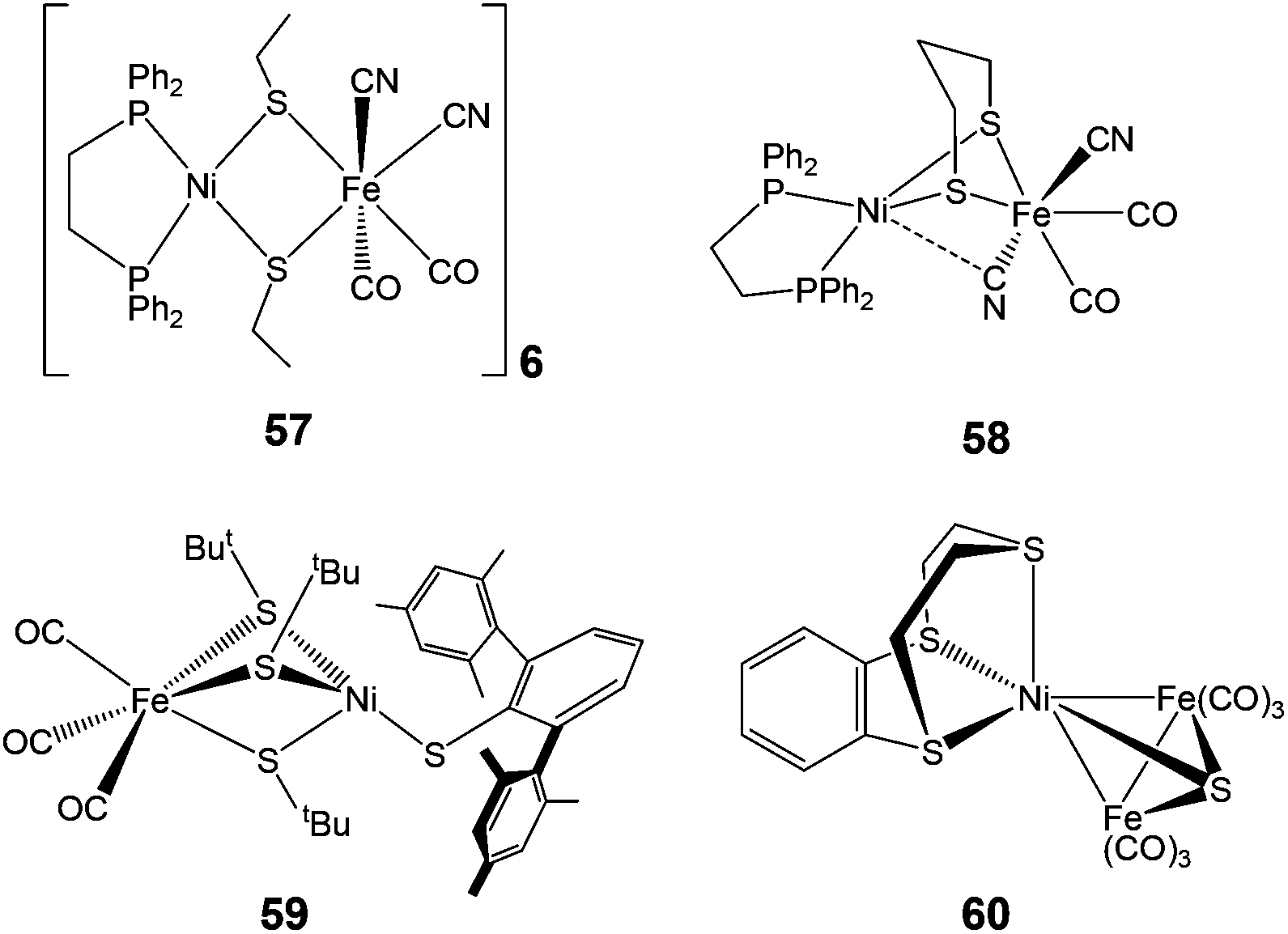 | ||
| Fig. 6 Complexes with coordinatively saturated Fe centre (57–59) and a trinuclear complex (60) bearing structural resemblance to the [NiFe] active site. | ||
Complex 57 features a near-planar diethanethiolate-bridged NiS2Fe rhomb [Ni–S–S–Fe dihedral angle of 170.0°] with cyanide ligands arranged cis to each other. Complex 58 shows a much more folded NiS2Fe rhomb [N–S–S–Fe dihedral angle of 111.69°], two trans CN− ligands, and an unusual semi-bridging CN− between Ni and Fe. This semi-bridging CN− (peak at 2110 cm−1 in the IR spectrum and a short Ni–CN distance of 2.4 Å) was suggested to be similar of the third bridging ligand (X) in oxidised [NiFe] hydrogenases. CO or CNtBu could be added at the Ni site in complex 59. The coordination of CO at the Ni site was found to be reversible. On addition of CO, the additional band observed at 2055 cm−1 in the IR spectrum was assigned as the Ni–CO band, which is comparable to those observed for the CO-inhibited form of the enzyme from Allochromatium vinosum and Desulfovibrio fructosovorans. However, this complex differs from the enzyme in its structural details: three thiolates between the metals in comparison to two in the enzyme and diatomic ligands on Fe i.e. Fe(CO)3 in comparison to CO and CN−i.e. Fe(CO)(CN)2 in the active site.
In contrast to the above heterobimetallic (dinuclear) complexes, Perra et al. have recently reported a trinuclear {NiFe} complex 60 (Fig. 6), the structure of which shows a Ni(II) bound to three thioether R2–S donors and bridged by a sulfide (S2−) group to two Fe(CO)3 units.83 The Ni–S(sulfide) bond length of 2.165 Å in the complex has been found to be identical to that found in the oxidised inactive form of the [NiFe] hydrogenase from D. vulgaris (2.16 Å). The complex showed an electrochemically reversible redox process (confirmed as a one-electron reduction by coulometry at 233 K) at E = −1.62 V vs. Fc/Fc+ but was found to be unstable in the presence of trifluoroacetic acid. At the beginning of X-ray structural analysis of [NiFe] hydrogenases, a sulfide μ-bridging ligand was also suggested for the oxidised form from D. vulgaris Miyazaki F84 but could not be confirmed by spectroscopic characterisations later. Electrochemically, however, [NiFe] hydrogenases were also shown to react with sulfide.85 Hence, the presence of the bridged sulfide provides an interesting mimic to the enzyme active site.
2.3.3.1. Proton reduction. A number of dinuclear complexes of Ni with Fe or Ru, Mn, etc have been reported very recently. An important example is the Ni(I)Fe(I) complex [NiFe(pdt)(dppe)(CO)3] 4 (Fig. 3) which was first synthesised and characterised by Zhu et al.71 Rauchfuss and coworkers reported the protonated form of the complex [4H]+ as the first example of a Ni–Fe hydride relevant to the [NiFeS2(μ-H)] core in the active site.86,87 This has also been related to the Ni–R state of the hydrogenase. In [4H]+ the Ni centre was found to be square pyramidally coordinated whereas the Fe centre was quasi-octahedral; this was taking into account the bridging hydride ligand. Complex [4H]+ exhibited a reduction at −1.20 V vs. Fc/Fc+ in dichloromethane and showed a catalytic current near −1.37 V in dichloromethane solution of CF3CO2H (pKa = 12.65; E0 ∼ −0.90 V vs. Fc/Fc+).87 This was milder than that seen for the diiron model complexes. Complex 4 and its hydride derivative were recently studied by resonance Raman spectroscopy.88 Based on significant differences between uncoupled metal–hydride vibrational frequencies a slight asymmetry in the metal–hydride bond lengths was suggested. These types of investigations will definitely be helpful in identifying hydride intermediates, be able to characterise the different redox states of the enzyme and hence, facilitate a better understanding of the catalytic mechanism of hydrogen turnover by the enzyme.
Proton reduction studies of complex 4 by replacing one of the CO with monodentate phosphine ligands i.e., [NiFe(pdt)(dppe)(CO)2L] (L = P(OPh)3, PPh2Py, PPh3) to yield 42, 44, 46 have also been reported by Rauchfuss and coworkers (see Fig. 5 and Table 1).86 The X-ray structure of complex [46H]+ shows a highly unsymmetrical bridging hydride in which the Fe–H bond was found to be 0.40 Å shorter than the Ni–H distance (1.89(3) Å). All three complexes undergo reductions near −1.46 V vs. Fc/Fc+. The proton reduction catalysis coincided with the reductive event in the presence of trifluoroacetic acid. The acid independent rate constants for the complexes were in the range of 50–75 s−1. The overpotential for the N-protonated pyridinium complex (260 mV) was lower than that of the complexes [42H]+ and [46H]+ (430 mV). An ECEC (E and C correspond to one-electron reductions and protonations, respectively) reaction sequence was proposed to be followed for the mechanism of hydrogen evolution.
| Catalyst | Acid used |
E
cat![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (V) b (V) |
OPc | Rated (s−1) |
|---|---|---|---|---|
| a The potentials for the reported model complexes have been recorded vs. different reference electrodes. Though the conversion constants in acetonitrile with respect to different reference electrodes are given in literature it is difficult to convert potentials with respect to a particular electrode. This is because the electrochemical experiments have been performed in a range of organic solvents. b GC electrode. c OP = overpotential = |Ecat − EoHA|. d Rate calculated in the acid-independent region cited from ref. 76 and 86. | ||||
| [4H]+ | CF3CO2H | −1.20 | 0.50 | 20 |
| [34H]+ | CH2ClCO2H | −1.46 | 0.59 | 50 |
| [35H]+ | CF3CO2H | −1.23 | 0.49 | 240–310 |
| [36H]+ | CH2ClCO2H | −1.45 | 0.59 | 20 |
| [41H]+ | CH2ClCO2H | −1.45 | 0.54 | 60–120 |
| [42H]+ | CF3CO2H | −1.32 | 0.43 | 50 |
| [44H]+ | CF3CO2H | ∼−1.30 | ∼0.40 | 50 |
| [44H2]2+ | CF3CO2H | −1.15 | 0.26 | 50 |
| [46H]+ | CF3CO2H | −1.30 | 0.41 | 50 |
The hydride derivatives of the complexes [NiFe(pdt)(dcpe)(CO)3] 34, [NiFe(edt)(dppe)(CO)3] 35, [NiFe(edt)(dcpe)(CO)3] 36 and [NiFe(edt)(dppe)(CO)2PPh3] 41 (dcpe = 1,2-bis(dicyclohexylphosphino)ethane) were also reported to be active catalysts for proton reduction (Table 1).76 The edt complexes were found to be more effective in comparison to the pdt complexes, though overpotentials for the proton reduction were similar for all the complexes. The edt–dppe complex 36 was shown to be the most active catalyst. This complex showed an acid independent rate of 300 s−1. Based on the different trends in the basicities and redox potentials of the complexes, the protonation was suggested to occur at the Fe centre whereas the reduction was shared between Ni and Fe. The catalytic cycle was proposed to begin with the reduction of the Ni(II)Fe(II)-hydride, which is not the pathway followed by the biological catalysts.
The neutral complex 61 was synthesised by the reaction of [Ni(xbsms)] (H2xbsms = 1,2-bis(4-mercapto-3,3-dimethyl-2-thiabutyl)benzene) and [Fe(CO)3(BDA)] (BDA = benzylidene) as reported in the literature.89,90 Lubitz and coworkers have shown that this complex can undergo reversible protonation at the terminal thiolate sulfur atom coordinated to Ni to yield {[61H]+} (Fig. 7) by using HBF4·Et2O.91 This was compared with the protonated terminal cysteine for the enzyme that has been proposed theoretically but not yet confirmed experimentally.92–95 Though the protonation is reported to take place at the Ni site, it was reported to also have a significant impact on the electronic structure of the Fe centre as well91 and shows that the two metal sites are coupled to each other. Complex 61 showed a reversible reduction (ΔEp = 160 mV, ipa/ipc ≈ 1) at −1.75 V vs. Fc/Fc+ in dichloromethane at a scan rate of 500 mV s−1 and at −1.81 V vs. Fc/Fc+ (ipa/ipc ≤ 0.1) in acetonitrile at room temperature, respectively.91 The reversibility of the cathodic wave was dependent on the scan rate, solvent and temperature. On the other hand, the cyclic voltammogram of [61H][BF4] displayed only irreversible redox behaviour in both acetonitrile and dichloromethane (Epc = −1.73 V vs. Fc/Fc+) even at higher scan rates. Both complexes were found to be active in electrocatalytic proton reduction in aprotic solvents. Acetonitrile or dichloromethane solutions of 61 showed an increased cathodic current upon addition of increasing amounts of CF3CO2H (up to 100 equiv.). This was indicative of electrocatalytic proton reduction (overpotential of 540 mV). Under the same conditions, a catalytic current was also observed for [61H]+ as the catalyst with an overpotential of 570 mV. However, for [61H]+ the catalytic peak was observed at slightly lower potentials than for 61. Bulk electrolysis experiments (4 h) of CF3CO2H in acetonitrile were carried out on a glassy carbon electrode at −1.60 V vs. Fc/Fc+ in the presence of catalytic amounts of 61 and [61H]+, respectively. An average turnover frequency (TOF) of 5 h−1 and an average turnover number (TON) of 20 were calculated for 61 while for [61H]+, the observed TOF was 8 h−1 and the TON was 32. In addition to hydrogen, traces of CO gas as a product of decomposition were also detected by GC analysis. The behaviour of complex 61i.e. diamagnetic nature and unusually short Ni–Fe bond length (2.426 Å)91 was reported to bear a striking resemblance to complex 4 mentioned in the previous sections.
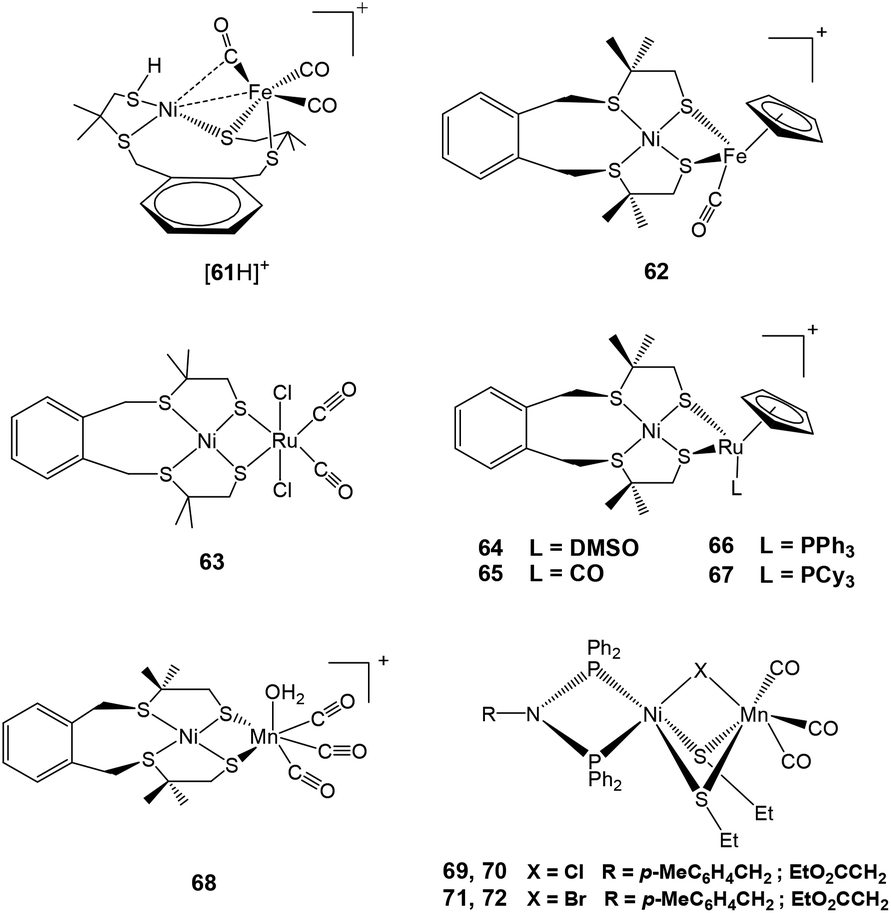 | ||
| Fig. 7 {NiFe}/{NiRu}/{NiMn} complexes reported as catalysts for proton reduction in the presence of acid. | ||
Another complex [Ni(xbsms)FeCp(CO)](BF4) 62 (H2xbsms = 1,2-bis(4-mercapto-3,3-dimethyl-2-thiabutyl)benzene) reported by Artero and coworkers (Fig. 7) has been shown to behave as an electrocatalyst for hydrogen evolution.96 This complex showed similar behaviour as 64 discussed later in this section. Complex 62, with an S4 set of ligands around nickel and a cyclopentadienyl ligand on the iron centre, catalyses hydrogen evolution in DMF in the presence of CF3CO2H at ca. −1.73 V, corresponding to an overpotential of 730 mV. The bulk electrolysis experiment (4 h) carried out in a DMF solution of 62 (7 mmol) and CF3CO2H (0.7 mmol) on a Hg-pool cathode at a controlled potential of −1.83 V showed 20 TON of H2 evolution with a Faradaic yield of 72%. The stability of complex 62 as a catalyst was quite good, this was seen from the rate of catalysis (5 TON h−1) that is sustained over the 4 h electrolysis experiment.
The first member of the series of functional bioinspired {NiRu} complexes, [Ni(xbsms)Ru(CO)2Cl2] 63 (Fig. 7) was reported by Oudart and coworkers in 2006.57 Similar dinuclear {NiRu} complexes with arene ligands attached to the Ru centre have also been reported by Reynolds et al.58 Complex 63 was shown to catalyse hydrogen evolution in DMF (−1.66 V vs. Ag/AgCl) on addition of increasing amounts of Et3NHCl as a source of protons.57,59 A complex similar to 63, i.e. [NiCl(xbsms)Ru(CO)3Cl] has also shown proton reduction at −1.52 V vs. Ag/AgCl in DMF.60 The previously proposed mechanism of proton reduction by complex 63 has been recently investigated by DFT modeling studies (Scheme 3).97 DFT calculations have shown the catalytic intermediate to be a semi-bridging {Ni(μ-H)Ru} hydride derivative, protonation of which yields H2 very easily, likely via a {NiRu(H2)} dihydrogen complex.98 The catalytic cycle of 63 was shown to be similar to the proposed enzymatic mechanisms (see above) involving a bridging hydride ligand in the Ni–C state and heterolytic formation of a dihydrogen molecule. It provides a valuable route for the future design of efficient catalysts.
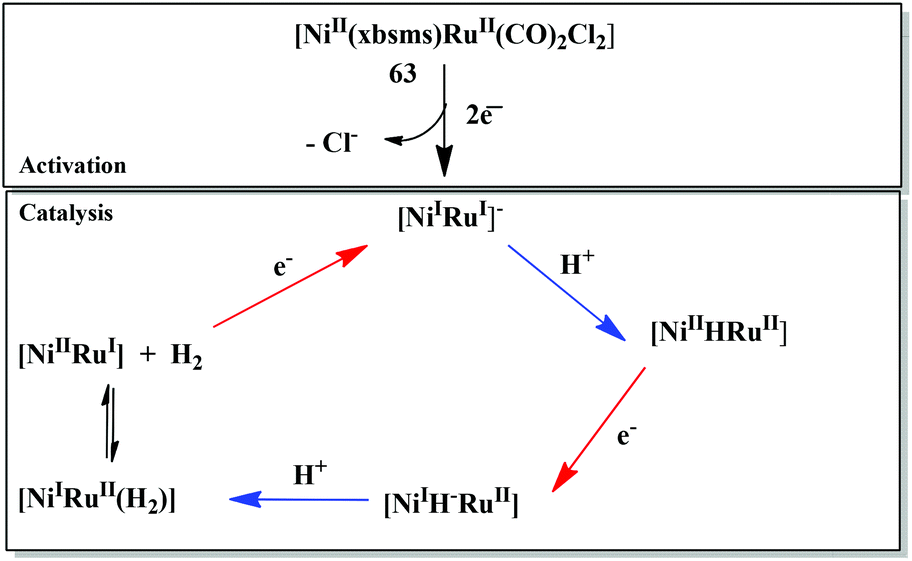 | ||
| Scheme 3 Catalytic mechanism of proton reduction by complex 63 based on DFT calculations (ref. 97). | ||
The catalytic hydrogen production by another {NiRu} mimic of [NiFe] hydrogenase, i.e., [Ni(xbsms)Ru(C6Me6)Cl]+ has been investigated by combined electrochemical and theoretical studies.61 Initially a bridging hydride intermediate is formed in the presence of weak acids. Fast hydrogen evolution from this hydride intermediate then takes place by the formation of a dihydrogen species through a PCET (proton-coupled electron transfer) process. This has been proposed to model the direct conversion between the Ni–C and Ni–R states in the hydrogenase catalytic cycle, which also involves a coupled addition of one electron and one proton.
Other {NiRu} systems reported include [Ni(xbsms)RuCp(L)][PF6] 64–67 (Cp− = cyclopentadienyl; L = DMSO, CO, PPh3, PCy3) (Fig. 7).62 Complex 64 showed one irreversible cathodic monoelectronic wave at −0.93 V with a corresponding flattened anodic wave observed at −0.11 V in the reverse scan. Complex 65 displayed a reversible one-electron reduction at −0.84 V (ΔEp = 90 mV; ipa/ipc ≅ 1). Complexes 66 and 67 displayed irreversible one-electron cathodic waves at −1.16 and −1.22 V followed by a second monoelectronic process at −1.79 and −1.85 V, respectively. The potentials were measured vs. Ag/AgCl, but conversion to the Fc/Fc+ couple for complexes 64–67 can be obtained by subtracting 0.53 V from the measured potentials. All complexes catalysed hydrogen evolution in an organic solvent (DMF) in the presence of a salt. In all cases, the intensity of the catalytic peak increased linearly with increase in concentration of Et3NH+. Complex 64 catalysed hydrogen evolution with the lowest overpotential (660 mV), which is less by about 180 mV than the earlier reported first generation {NiRu} catalysts [Ni(emi)Ru(p-cymene)Cl]− (H2emi = N, N-ethylenebis(2-mercaptoisobutyramide),59 thus representing the most efficient catalyst in this series of compounds. The decrease in overpotential has been attributed to the increased electron density on the metal centres due to the presence of the Cp− ligand. Hydrogen evolution takes place either through a CECE or a CEC mechanism, with an EC pre-catalyst initiation process. The initial reduction leads to elimination of an axial ligand as seen in the case of complex 63.
Recently, Artero and coworkers have reported a {NiMn} complex (Fig. 7), [Ni(xbsms)Mn(CO)3(H2O)]+68 that can catalyse hydrogen evolution from trifluoroacetic acid in DMF at −1.81 V vs. Fc/Fc+ with high overpotential (860 mV).64 The bulk electrolysis experiment (4 h) of the complex in presence of TFA in DMF at constant potential (−1.82 V vs. Fc/Fc+) showed 15.8 TON of H2 evolution with a Faradaic yield of 94%. In the catalytic cycle investigated by electrochemical studies and DFT calculations, the bridging hydride ligand between Ni and Mn was stated to resemble the Ni–C state of the [NiFe] hydrogenase. The proposed electrocatalytic pathways for hydrogen evolution (homolytic or heterolytic) by the complex are shown in Scheme 4.64 The calculated Ni–H and Mn–H bond distances in{NiHMn} are 1.93 Å and 1.63 Å, respectively. Therefore, the Ni–H bond of 1.61 Å in the enzymatic {Ni(III)(μ-H)Fe(II)} centre is considerably shorter than that in {NiHMn}, whereas the Fe–H distance of 1.72 Å is longer than that of Mn–H. However, in the reduced hydride derivative {NiHMn}− the calculated distances were found to be 1.76 Å for Ni–H and 1.66 Å for Mn–H. In addition, in {Ni(H2)Mn}+ and {Ni(H2)Mn} the calculated H–H distances were 0.84 Å and 0.87 Å, respectively. This was found to be in the range for a “true” dihydrogen complex (0.8–1.0 Å).98 The H–H distances were in good agreement with the low enthalpies of H2 binding. In this case the oxidation state changed for both Ni and Mn during the catalytic cycle, whereas the enzyme, the Fe-centre is redox inactive. Moreover, the spin densities on the coordinating sulfur atoms were found to be quite small which is not the case with the enzyme active site.92,99,100
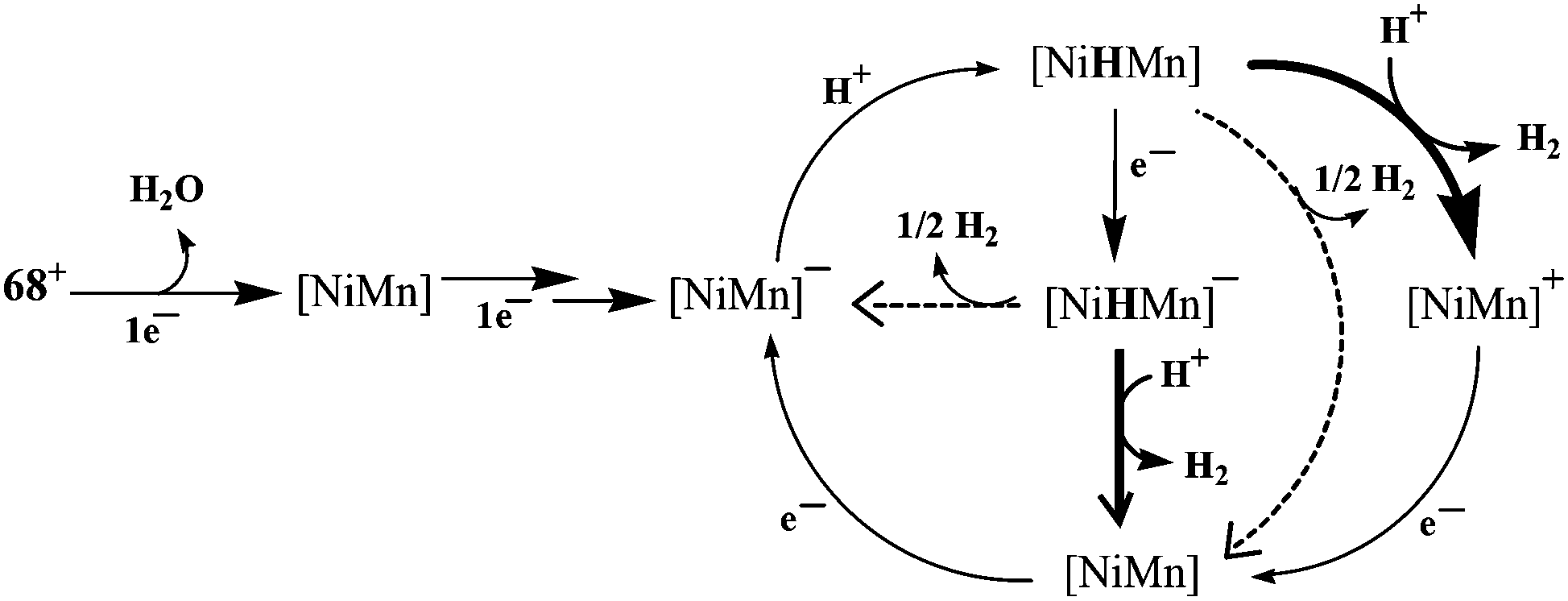 | ||
| Scheme 4 Proposed mechanisms for hydrogen evolution catalysed by [68]+. The homolytic H2-evolution steps are depicted by dotted arrows and the heterolytic steps by solid arrows (adapted from ref. 64). | ||
Dinuclear, neutral {NiMn} complexes [RN(PPh2)Ni(μ-SEt)2(μ-X)Mn(CO)3] 69–72 (X = Cl, Br; R = p-MeC6H4CH2, EtO2CCH2) (Fig. 7) containing a butterfly [NiS2Mn] core have also been reported by Song and coworkers.65 In these complexes, the Ni and Mn centres are bridged by two ethyl thiolates and one chloro or bromo ligand. The Mn(I)(CO)3 fragment in the model complexes is isolobal with the Fe(II)(CO)(CN)2 fragment seen in the active site of the [NiFe] hydrogenase enzyme vide supra. In addition, of the four complexes synthesised and characterised, only 69 (X = Cl, R = p-MeC6H4CH2) and 71 (X = Br, R = p-MeC6H4CH2) were found to reduce protons to dihydrogen in the presence of acetic acid. The overpotential for hydrogen production from acetic acid by complexes 69 (270 mV) and 71 (260 mV) was less than that for model complex [4H]+. For comparison with Artero's complex 68 (860 mV), the overpotentials were calculated by the same first “derivative” technique method as complex 68.64 The values (470 mV for 69 and 480 mV 71) are much lower than that for complex 68, which could probably be due to presence of azadiphosphine-chelated Ni moiety in complexes 69 and 71.
Bouwman and coworkers have shown a unique example of a hexanuclear nickel thiolate metallacrown complex capable of protonation and electrocatalytic dihydrogen evolution in the presence of protic acids such as dichloroacetic acid and chloroacetic acid at −1.5 and −1.6 V vs. Ag/AgCl in DMF, respectively.101 In this case, the protonation has been proposed to take place on the thioether sulfurs available in the metallacrown. Moreover, the complex is capable of proton reduction when immobilised on the surface of a pyrolytic graphite electrode (reduction on the surface of the modified electrode occurs at 220 mV more positive potential in comparison to the unmodified electrode).
The electrochemical catalytic hydrogen evolution by all the [NiFe] H2ase mimics discussed in this perspective is summarised in Table 2. From the table one can conclude that: (i) electrochemistry has been performed in a range of organic solvents, which is related to the stability of the model complexes in the presence or absence of acids; (ii) almost all of the complexes reduce protons at a high overpotential; and (iii) as compared to the enzyme active site the turnover numbers for the catalytic cycles are still very low.
| Catalystb | Acid used, solvent |
E
1/2 or Epcb (V) |
E
pcc (V) |
OPd | TOF (h−1) | TON after 4 h | Ref. |
|---|---|---|---|---|---|---|---|
| On addition of acid | |||||||
| a The potentials for the reported model complexes have been recorded vs. different reference electrodes. Though the conversion constants in acetonitrile with respect to different reference electrodes are given in literature, it is difficult to convert potentials with respect to a particular electrode. This is because the electrochemical experiments have been performed in a range of organic solvents. b Electrochemical data mentioned only for proton reduction. c GC electrode. d OP = overpotential = |Ecat − EoHA|. e Reference electrode: Ag/AgCl/KClaq. f Catalytic peak potential with 1.5 equiv. acid; the potential shifts towards more negative values upon increasing the addition of the acid. g After 3 h. h After 0.5 h. | |||||||
| 1–3, 5–33, 37–40, 43, 45, 47–59, 70, 72 | — | — | — | — | — | — | — |
| 60 | Unstable in presence of trifluoroacetic acid, dichloromethane | −1.62 | — | — | — | — | 83 |
| 61 | CF3CO2H, acetonitrile | −1.81 | Same | 0.540 | 5.0 | 20 | 91 |
| [61H]+ | CF3CO2H, acetonitrile | −1.73 | Slightly lower | 0.570 | 8.0 | 32 | 91 |
| 62 | CF3CO2H, DMF | −1.43; −2.01 | −1.83; −1.73 | 0.730 | 5.0 | 20 | 96 |
| 63 | Et3NHCl, DMFe | −0.90 | −1.50 | — | 5.0 | 15g | 57 |
| 64 | Et3NHCl, DMFe | −0.93 | −1.39f | 0.660 | 4.3 | 13g | 62 |
| 65 | Et3NHCl, DMFe | −0.84 | −1.60f | 0.870 | 5.3 | 15.8g | 62 |
| 66 | Et3NHCl, DMFe | −1.16; −1.79 | −1.65f | 0.910 | 1.2 | 3.6g | 62 |
| 67 | Et3NHCl, DMFe | −1.22; −1.85 | −1.66f | 0.840 | 1.0 | 3g | 62 |
| 68 | CF3CO2H, DMF | −1.53; −1.83 | −1.81 | 0.860 | 4.0 | 15.8 | 64 |
| 69 | Acetic acid, acetonitrile | −1.26; −1.58 | −1.80 | 0.270 | — | 10.5h | 65 |
| 71 | Acetic acid, acetonitrile | −1.25; −1.56 | −1.79 | 0.260 | — | 9.1h | 65 |
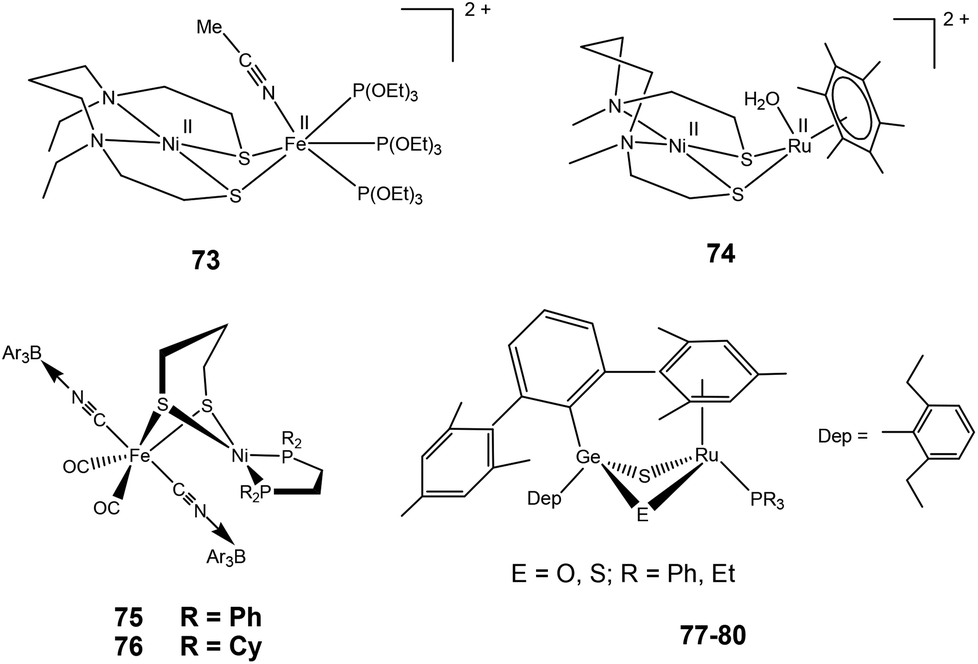
2.3.3.2. Dihydrogen activation. Very recently, Ogo and coworkers have reported a functional model complex ([Ni(II)(X′)Fe(II)(MeCN){P(OEt)3}3](BPh4)2 {[73](BPh4)2, where X′ = N,N′-diethyl-3,7-diazanonane-1,9-dithiolato, Et = ethyl, Ph = phenyl} (Fig. 8) based on a [NiFe] core.102 In previous reports, a similar {NiRu} model complex ([Ni(II)(X)(H2O)(μ-H)Ru(II)(C6Me6)](NO3) 74 {X = N,N′- dimethyl-3,7-diazanonane-1,9-dithiolato and Me indicates a methyl group} was shown to activate H2 in water at room temperature.63 In 73, Ru was replaced with Fe, the aryl ligand with three triethylphosphite {P(OEt)3} ligands, and, crucially, sodium methoxide (MeONa) was used as a base instead of water. Complex [73]2+ was found to heterolytically activate H2 in MeCN/MeOH at room temperature and atmospheric pressure (Scheme 5). The hydride complex ([Ni(II)(X′)(μ-H)Fe(II){P(OEt)3}3](BPh4)), [73]+ was formed on abstraction of a proton from bound H2 by a strong base (MeONa). The Ni atom in the hydride complex [73]+ adopted a square-planar structure and the Ni⋯Fe distance was 2.79 Å as determined by different spectroscopic techniques. This distance is longer than the Ni⋯Fe separation in the reduced state of [NiFe] H2ase28 and that in the catalytically active {NiFe} complex [4H]+ reported by Rauchfuss and coworkers (discussed earlier). Complex [73]+ is bifunctional in the sense that it can perform both hydride and electron transfers. This is similar to the bidirectional activity of the [NiFe] H2ases. On the other hand, the earlier {NiRu} complex [Ni(II)(L)(H2O)(μ-H)Ru(II)(C6H6)](NO3) 74 can only perform electron transfer, whereas complex ([Ni(II)(dppe)(μ-H)Fe(II)(pdt)(CO)3](BF4) can catalyse the proton reduction reaction. The water soluble {NiRu} complex [74]2+ has been extensively characterised by Ogo et al.,56,103,104 see also ref. 105 for an overview. A bridging hydride was observed by neutron diffraction for this complex upon reaction with hydrogen in water. The Ni–Ru distance was 2.74 Å in the hydride complex whereas it was 3.16 Å without the hydride ligand. This change in distance is similar to that observed in the crystal structures of the native [NiFe] hydrogenases (Ni–Fe distances of the oxidised and reduced state were 2.69–2.80 Å and 2.55 Å, respectively).26,28 For complex [74]2+, a dihydride species has been proposed during the catalytic cycle (studied by pH-dependent hydrogen isotope exchange reactions), suggesting that in the enzyme the hydrogen could be bound to the active site in several ways.
 | ||
| Fig. 8 Reported complexes capable of hydrogen oxidation. | ||
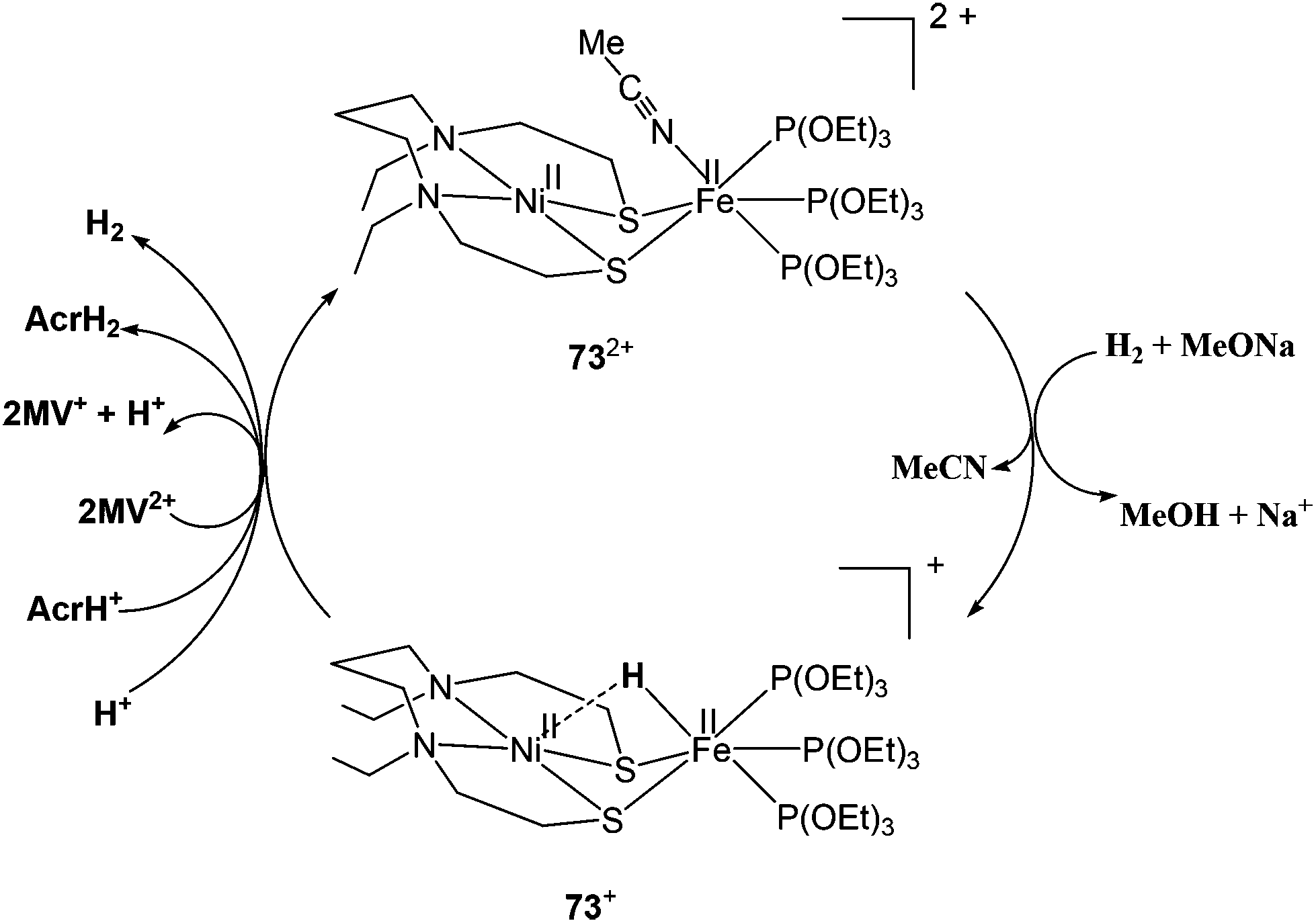 | ||
| Scheme 5 Heterolytic activation of hydrogen by complex [73]2+ (see ref. 102). | ||
Rauchfuss and coworkers have recently reported {NiFe} model complexes [(CO)2(CNBArF3)2Fe(pdt)Ni(dxpe)] (dxpe = dppe and dcpe) 75–76 (Fig. 8)106 formed from the reaction of [(CO)2(CN)2Fe(pdt)Ni(dxpe)] with Lewis acid B(C6H5)3(BArF3). Hydrido derivatives [75H]− and [76H]− are generated upon decarbonylation using amine oxides followed by reacting with hydrogen. Oxidation of hydrogen is catalysed by complex [75H]− in the presence of a base as seen by electrochemical investigations. The anionic hydrides have hydridic character107 and oxidise at mild potentials (vs. Fc/Fc+) in comparison to the cationic Ni–Fe hydrides.76,86,102 Hence, these anionic hydrides participate in dihydrogen bonding. Similar dihydrogen bonding has also been seen for the terminal hydrides of the model complexes reported for the [FeFe] hydrogenase enzyme.108
Tatsumi and coworkers have reported hetero dinuclear {RuGe} complexes 77–80 (Fig. 8) for which they have shown the conversion of protonated {μ-S/μ-SH}+ complex to a hydride {μ-S/μ-H}+ complex upon treatment with H2.109 The reaction was found to be slower than the {μ-S/μ-OH}+ complex. The {μ-S/μ-SH}+ complex equilibrated with the {μ-S/μ-OH}+ complex in the presence of water and then reacted with H2 more easily to form the {μ-S/μ-H}+ complex though this was still slower than the case of the {μ-S/μ-OH}+ complex (Scheme 6). It was also shown that the {μ-S/μ-OH}+ complexes could be quickly converted to {μ-S/μ-SH}+ complexes upon reacting with H2S. The complexes were shown to activate H2 heterolytically, the reaction of H2 and {μ-S/μ-OH}+ complex was reversible, and the {μ-S/μ-OH}+ complex could further convert H2 into two protons and electrons. Moreover, the reactivity pattern of the {μ-S/μ-SH}+ and {μ-S/μ-OH}+ complexes towards H2 was compared to the ‘unready’ and ‘ready’ states of [NiFe] hydrogenase. It was proposed that the active site may be converted by H2S (produced by sulphate-reducing bacteria in their metabolism) into the Ni-‘B’ or Ni-‘S’ states having protonated bridging cysteines μ-SH, similar to the results reported by Tatsumi and co-workers for complexes 77–80.109
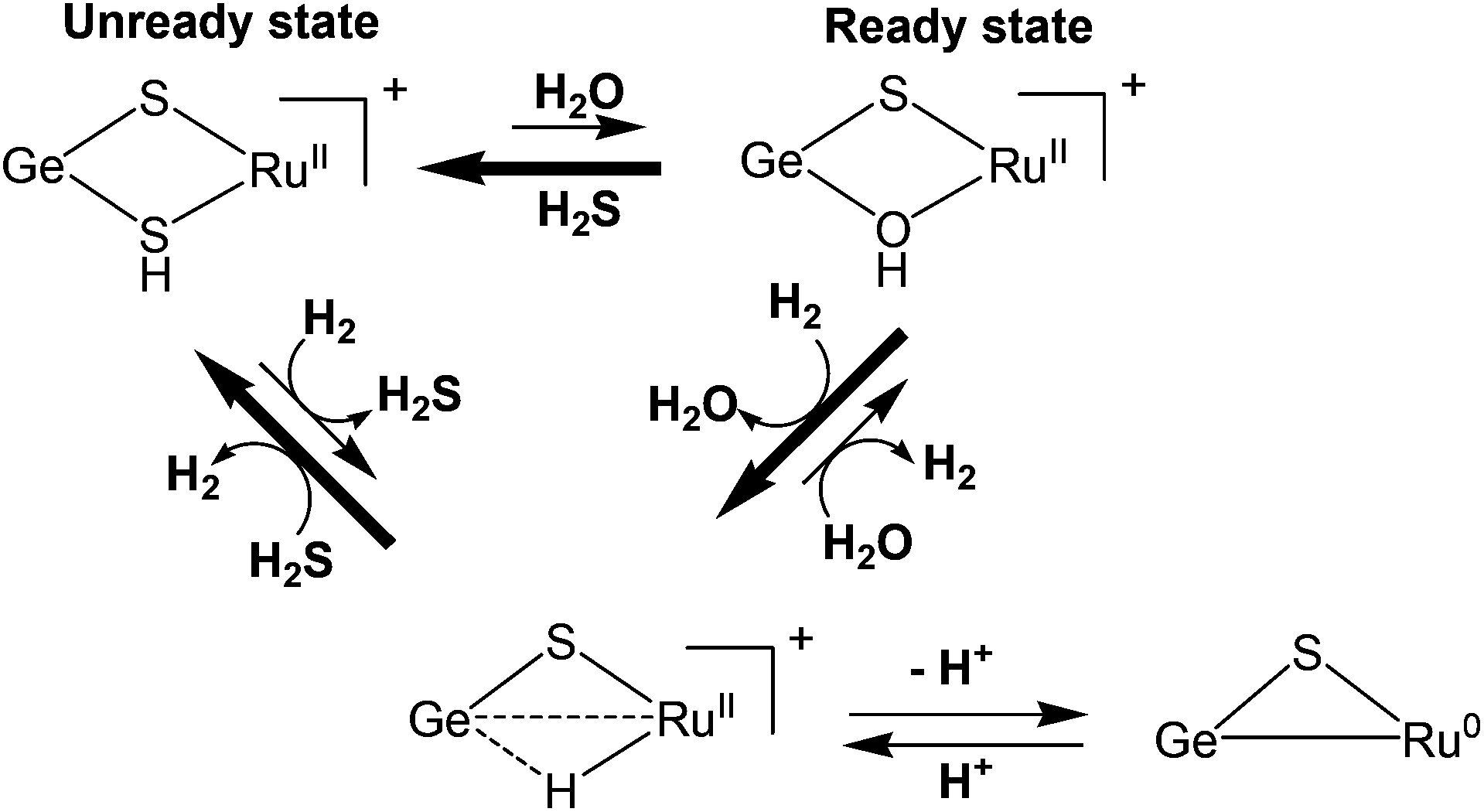 | ||
| Scheme 6 Dihydrogen activation by {RuGe} complexes [77–80] (see ref. 109). | ||
In all of the above reported dinuclear complexes ({NiFe}, {NiRu}, {NiMn}) functioning as electrocatalysts for proton reduction it can be seen that bridging hydride species are involved in the catalytic cycle of hydrogen production. On the other hand, terminal hydride ligands bound to the Fe centre have been reported for complex 73 by Ogo et al.102 Another such example is a mononuclear Fe complex reported by Bullock et al.69 A third possibility for binding is protonation of the sulphur atom of the thiol ligand as seen in the dinuclear complex [61H]+ reported by Lubitz and co-workers91 and also seen by us in a mononuclear Fe-complex [Fe(bdt)(CO)2(PMe3)2] (bdt = 1,2-benzenedithiolate).70 Recently, a dinuclear {FeFe} complex has been reported by Liu et al. as the first diprotonated [FeFe] H2ase model bearing the S-proton and Fe-hydride.110 As seen from all the examples discussed the synthesis of heterodinuclear complexes as new catalysts for hydrogen production or activation is both promising and challenging at the same time. Controlling the nuclearity in nickel thiolate complexes is complicated. This requires appropriate design of the precursor for the second metal site. Also most of the {NiFe} complexes have an iron centre that is coordinatively saturated with low reactivity. This could also be the reason for the instability of the {NiFe} complexes. However, the {NiRu} complexes with the heavier ruthenium metal as the second metal site seem to be more stable and possess higher turnover numbers than the {NiFe} complexes mimicking the active site and are also known to be thermally more stable in solution than the dinuclear {NiFe} complexes. The presence of monoatomic non-bulky ligands at the Ru centre and its non-rigidity in solution could be the reasons that enable some of the {NiRu} complexes to explore diverse, possibly less endothermic, and more efficient catalytic pathways. The {NiRu} complexes could, therefore, be valuable alternatives for the design of new electrocatalysts for hydrogen production and for investigating the mechanism of hydrogen production and uptake catalysed by the active sites of the [NiFe] hydrogenases.
3. Conclusions
The [NiFe] hydrogenase is a well-characterised enzymatic hydrogen converting enzyme. To date, a significant amount of information has been accumulated for the enzyme redox states and reaction mechanism for the reversible heterolytic splitting of dihydrogen at the [NiFe] H2ase active site. This information has been obtained by using a wide range of spectroscopic techniques (X-ray crystallography, FTIR, EPR, ENDOR, HYSCORE, electrochemical redox titrations, UV-Vis) and by performing DFT calculations. Though this has led to huge progress in the design, synthesis and characterisation of structural and functional models of the active site of [NiFe] hydrogenases, there are hardly any complexes that mimic the enzyme active site both in structure and function simultaneously. So far, it has also been difficult to obtain model complexes that mimic the enzyme as far as the structural coordination of the metal centres is concerned. The developments in this direction have been reviewed recently by Simmons and Artero.111 The variety of complexes reported so far include NiS4 complexes, mononuclear Fe/Co/Ni complexes with phosphine ligands, thiolate-bridged {NiFe} carbonyl complexes, and very recently the {NiRu}/{NiMn} heterobimetallic complexes. Looking at the synthetic routes of the model complexes reported so far, some explanation can be given regarding the difficulty to reproduce the enzyme active site in a test tube. Recent success, however, has been made in a modular assembly of features of the [NiFe] hydrogenase active site: model complexes have been synthesised by reacting Ni–thiolate/dppe and Fe-inorganic ligand/thiolate precursors. The combination of Fe with various inorganic ligands is feasible and bridging dithiolates are now accessible. Recently, the biological mixture of two cyanides and one CO was obtained.106 In some {NiFe} complexes, Cp− has been used in place of CO ligands on the Fe centre as well, which led to lower overpotential for proton reduction but this needs further investigation. Also the formation of bridging hydrides as observed in Ni–C is feasible. Ogo102 and Manor106 were the first to present Ni–C/Ni–R-like {NiFe} heterodinuclear complexes with a bridging thiolate and a bridging hydride each. Nevertheless, in both complexes the hydride was asymmetrically bound towards the iron atom. The differences in metal–hydride distances were minor in Manor's106 and more pronounced in Ogo's102 complexes. Hence, the nature and electronic properties of the terminal ligands to the Ni centre must also be critical. Phosphine ligands like dppe have replaced the N-donors employed in the early generation of models due to their soft donor nature considering them to be better replacements for the cysteine sulfurs of the enzyme. However, as seen from detailed investigations of the electronic structure of model complexes, phosphines apparently do not seem to be perfect substitutes for cysteine amino acids. Furthermore, phosphine ligands are also employed as substitutes for the small inorganic ligands CO and CN− at the iron site (see for example 37). This is certainly an oversimplification and the phosphine ligands are too soft and polarisable here. On the other hand, at the Ni site (e.g. in compounds 75 and 76) the dppe ligands are not soft and polarisable enough to replace cysteinate coordination (large and soft S). Some very recent examples have generated structural models for the mixed-valence Ni–L state Ni(I)Fe(II) of the enzyme, however, with an inverted Ni(II)Fe(I) core redox activity and spin density distribution.78 In most of the reported heterobimetallic complexes Ni is coordinatively saturated, redox inactive and the second metal (Fe or Ru) acts as the catalytic site. This is contrary to the biological system. From our knowledge of the enzyme active site, detailed spectroscopic and computational insight into the reaction mechanism, we can come up with the following requirements for a functional {NiFe} mimic. The electronic structure of the [NiFe] active site is polarisable with an uneven distribution of electron density and ‘softness’. A ‘push-and-pull’ polar site model can be suggested: the iron site has to have strong, non-polarisable ligands which on the one hand ensure a low-spin state, on the other hand make the iron rich in electron density but hardly polarisable. In contrast, the Ni atom has to be surrounded by large and polarisable ligands which allow a significant distribution of electron density onto the ligands (nitrogen and phosphorous are not suitable). Furthermore, in model complexes, sometimes protonation of a sulfur ligand species can be observed, see for example [61H]+. Such a situation is very difficult to observe in the biological system (see above). This indicates that if cysteine ligand protonation occurs transiently, proton translocation to further acceptors must be immediate and barrierless. In model complexes, sometimes strong bases are required to take up the proton from the heterolytic hydrogen splitting such as MeONa and DBU (1,8-diazabicycloundec-7-ene). This, again, is not required in the enzyme and supports the idea of an immediate proton translocation path to avoid any recombination reactions between the hydride and the proton. Also none of the models has been able to reproduce the Ni(III)Fe(II) oxidation states of the oxidised enzyme. Strategies have to be found to stabilise the higher nickel oxidation states.Very efficient reversible hydrogen oxidising mononuclear Ni and Fe complexes from the DuBois group68,69 contain bidentate diposphine ligands and proton-accepting amine nitrogens in close vicinity. Apparently, systematic modifications of the steric and electronic properties of the ligands allow a control of the catalytic bias towards either hydrogen oxidation or production. Simultaneous control of the hydride binding ability of the metal centre and the proton acceptor ability of the pendant base are critical features. Such a chemical design was inspired by [FeFe] hydrogenases, which possess an azadithiolate bridging ligand.
The stability of a hydrogen converting complex can be improved by grafting the synthetic complex on a carbon nanotube112 or incorporating it into a metal–organic framework (MOF).113
Direct release of molecular hydrogen from a particulate photocatalyst using visible light has been obtained from nanoparticles of a mixed oxide of rhodium and chromium on a solid solution of gallium and zinc nitrogen oxide (Ga1−xZnx)(N1−xOx).114 Assembly of a molecular hydrogen producing system using Ni(II)acetate and 2-mercaptoethanol in aqueous solution containing triethanolamine (as sacrificial agent), followed by addition of Erythrosin B as the photosensitiser (PS) could contribute to the development of economically viable solar hydrogen production systems.115 A bio-nanoengineered combination between the chemical design of advanced molecular complexes with heterogeneous116 or metal-free polymeric117 photocatalysts for water splitting from visible light is an attractive possibility for a sustainable hydrogen economy.
Hence, a significant amount of research is still required as far as fulfilling the characteristics of the enzyme active site by the {NiFe} model complexes is concerned. Due to the difficulties involved with the {NiFe} complexes and in their efforts to understand the chemistry behind the active site several researchers have also synthesised and characterised stable {NiRu} complexes that have been found to be catalytically active. Two important complexes have been reported recently which have been found to participate in dihydrogen oxidation.
However, many aspects still remain to be explored; intermediates in the catalytic cycle need to be modelled to obtain insight into the geometric and electronic structure contribution towards the reactivity of the [NiFe] H2ase enzyme. All these efforts will enhance our understanding of the proton reduction and dihydrogen oxidation process, though it may not result in a cheap catalyst to generate H2 from H2O. An affordable process may be based on less structured, amorphous solids with some incorporated, less expensive metal ions.
Note added in proof
After submission of this article, mixed-valence {NiRu} complexes [(cymene)Ru(II)(μ-pdt)Ni(I)(dcpe)]+ and [(cymene)Ru(II)(μ-pdt)Ni(I)(dppe)]+ were reported recently. They are structurally related to the Ni–L state of [NiFe] hydrogenases and also show the correct distribution of unpaired spin, namely on the Ni atom (G. M. Chambers, J. Mitra, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2014, 53, 4243).Acknowledgements
S.K.-G. is grateful to the Department of Science & Technology (DST), Indo-German Science & Technology Centre (IGSTC), India, and the Max Planck Society, Germany for a Max-Planck-India Visiting Fellowship. This work was supported by the Max-Planck-Society for Advancement of Science and the “Research Centre Dynamic Systems: Biosystems Engineering (CDS)” funded by the Federal State of Saxony-Anhalt. Financial support from DST (SR/S1/IC-28/2011) and University of Delhi is also gratefully acknowledged. Part of this work was supported by the COST Action CM1305 ECOSTBio (Explicit Control Over Spin-states in Technology and Biochemistry).Notes and references
- T. Hiromoto, K. Ataka, O. Pilak, S. Vogt, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer, S. Shima and U. Ermler, FEBS Lett., 2009, 583, 585 CrossRef CAS PubMed.
- S. Dey, P. K. Das and A. Dey, Coord. Chem. Rev., 2012, 257, 42 CrossRef PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853 CrossRef CAS.
- A. L. De Lacey, V. Fernández, M. Rousset and R. Cammack, Chem. Rev., 2007, 107, 4304 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273 CrossRef CAS PubMed.
- W. Lubitz, E. Reijerse and M. Van Gastel, Chem. Rev., 2007, 107, 4331 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592 RSC.
- A. F. Wait, C. Brandmayr, S. T. Stripp, C. Cavazza, J. C. Fontecilla-Camps, T. Happe and F. A. Armstrong, J. Am. Chem. Soc., 2011, 133, 1282 CrossRef CAS PubMed.
- R. A. Kinney, C. T. Saouma, J. C. Peters and B. M. Hoffman, J. Am. Chem. Soc., 2012, 134, 12637 CrossRef CAS PubMed.
- A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J.-M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607 CrossRef CAS PubMed.
- C. Tard and C. J. Picket, Chem. Rev., 2009, 109, 2245 CrossRef CAS PubMed.
- F. Gloaguen and T. B. Rauchfuss, Chem. Soc. Rev., 2009, 38, 100 RSC.
- Y. Ohki and K. Tatsumi, Eur. J. Inorg. Chem., 2011, 973 CrossRef CAS.
- S. Canaguier, V. Artero and M. Fontecave, Dalton Trans., 2008, 315 RSC.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127 CrossRef CAS PubMed.
- S. E. Lamle, S. P. J. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2004, 126, 14899 CrossRef CAS PubMed.
- A. Parkin, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2006, 128, 16808 CrossRef CAS PubMed.
- A. Volbeda, M.-H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580 CrossRef CAS PubMed.
- K. A. Bagley, E. C. Duin, W. Roseboom, S. P. J. Albracht and W. H. Woodruff, Biochemistry, 1995, 34, 5527 CrossRef CAS.
- M. van Gastel, M. Stein, M. Brecht, O. Schröder, F. Lendzian, R. Bittl, H. Ogata, Y. Higuchi and W. Lubitz, J. Biol. Inorg. Chem., 2006, 11, 41 CrossRef PubMed.
- A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. J. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg. Chem., 2005, 10, 239 CrossRef CAS PubMed.
- A. Volbeda, E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989 CrossRef CAS.
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557 CrossRef CAS.
- H. Ogata, S. Hirota, A. Nakahara, H. Komori, N. Shibata, T. Kato, K. Kano and Y. Higuchi, Structure, 2005, 13, 1635 CrossRef CAS PubMed.
- H. Ogata, Y. Mizoguchi, N. Mizuno, K. Miki, S. Adachi, N. Yasuoka, T. Yagi, O. Yamauchi, S. Hirota and Y. Higuchi, J. Am. Chem. Soc., 2002, 124, 11628 CrossRef CAS PubMed.
- Y. Higuchi, H. Ogata, K. Miki, N. Yasuoka and T. Yagi, Structure, 1999, 7, 549 CrossRef CAS.
- F. Leroux, S. Dementin, B. Burlatt, L. Cournac, A. Volbeda, S. Champ, L. Martin, B. Guigliarelli, P. Bertrand, J. C. Fontecilla-Camps, M. Rousset and C. Leger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11188 CrossRef CAS PubMed.
- E. C. Hatchikian, M. Bruschi and J. Le Gall, Biochem. Biophys. Res. Commun., 1978, 82, 451 CrossRef CAS.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366 CrossRef CAS PubMed.
- M. Stein and W. Lubitz, Curr. Opin. Chem. Biol., 2002, 6, 243 CrossRef CAS.
- C. Bagyinka, J. P. Whitehead and M. J. Maroney, J. Am. Chem. Soc., 1993, 115, 3576 CrossRef CAS.
- M. Stein, E. van Lenthe, E. J. Baerends and W. Lubitz, J. Am. Chem. Soc., 2001, 123, 5839 CrossRef CAS.
- P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414 CrossRef CAS PubMed.
- S. J. George, S. Kurkin, R. N. F. Thorneley and S. P. J. Albracht, Biochemistry, 2004, 43, 6808 CrossRef CAS PubMed.
- S. Kurkin, S. J. George, R. N. F. Thorneley and S. P. J. Albracht, Biochemistry, 2004, 43, 6820 CrossRef CAS PubMed.
- M. Stein and W. Lubitz, J. Inorg. Biochem., 2004, 98, 862 CrossRef CAS PubMed.
- M. Brecht, M. van Gastel, T. Buhrke, B. Friedrich and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 13075 CrossRef CAS PubMed.
- S. Foerster, M. van Gastel, M. Brecht and W. Lubitz, J. Biol. Inorg. Chem., 2005, 10, 51 CrossRef CAS PubMed.
- S. Forester, M. Stein, M. Brecht, H. Ogata, Y. Higuchi and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 83 CrossRef PubMed.
- A. Volbeda and J. C. Fontecilla-Camps, Top Organomet. Chem., 2006, 17, 57 CrossRef CAS.
- A. L. de Lacey, V. M. Fernández and M. Rousset, Coord. Chem. Rev., 2005, 249, 1596 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volveda, Nature, 2009, 460, 814 CrossRef CAS PubMed.
- J. M. Keith and M. B. Hall, Inorg. Chem., 2010, 49, 6378 CrossRef CAS PubMed.
- H. Wu and M. B. Hall, C. R. Chim., 2008, 11, 790 CrossRef CAS PubMed.
- A. Pardo, A. L. De Lacey, V. M. Fernández, H.-J. Fan, Y. Fan and M. B. Hall, J. Biol. Inorg. Chem., 2006, 11, 286 CrossRef CAS PubMed.
- M. A. Halcrow and G. Christou, Chem. Rev., 1994, 94, 2421 CrossRef CAS.
- E. Bouwman and J. Reedijk, Coord. Chem. Rev., 2005, 249, 1555 CrossRef CAS PubMed.
- C.-H. Hsieh, R. B. Chupik, S. M. Brothers, M. B. Hall and M. Y. Darensbourg, Dalton Trans., 2011, 40, 6047 RSC.
- C.-H. Lai, J. H. Reibenspies and M. Y. Darensbourg, Angew. Chem., Int. Ed. Engl., 1996, 35, 2390 CrossRef CAS.
- Z. L. Li, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2005, 127, 8950 CrossRef CAS PubMed.
- Y. Ohki, K. Yasumura, K. Kuge, S. Tanino, M. Ando, Z. Li and K. Tatsumi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7652 CrossRef CAS PubMed.
- Q. Wang, J. E. Barclay, A. J. Blake, E. S. Davies, D. J. Evans, A. C. Marr, E. J. L. McInnes, J. McMaster, C. Wilson and M. Schröder, Chem. – Eur. J., 2004, 10, 3384 CrossRef CAS PubMed.
- D. Sellmann, F. Lauderbach, F. Geipel, F. W. Heinemann and M. Moll, Angew. Chem., Int. Ed., 2004, 43, 3141 CrossRef CAS PubMed.
- S. Ogo, R. Kabe, K. Uehara, B. Kure, T. Nishimura, S. C. Menon, R. Harada, S. Fukuzumi, Y. Higuchi, T. Ohhara, T. Tamada and R. Kuroki, Science, 2007, 316, 585 CrossRef CAS PubMed.
- Y. Oudart, V. Artero, J. Pécaut and M. Fontecave, Inorg. Chem., 2006, 45, 4334 CrossRef CAS PubMed.
- M. A. Reynolds, T. B. Rauchfuss and S. R. Wilson, Organometallics, 2003, 22, 1619 CrossRef CAS.
- Y. Oudart, V. Artero, J. Pécaut, C. Lebrun and M. Fontecave, Eur. J. Inorg. Chem., 2007, 2613 CrossRef CAS.
- Y. Oudart, V. Artero, L. Norel, C. Train, J. Pécaut and M. Fontecave, J. Organomet. Chem., 2009, 694, 2866 CrossRef CAS PubMed.
- S. Canaguier, V. Fourmond, C. U. Perotto, J. Fize, J. Pécaut, M. Fontecave, M. J. Field and V. Artero, Chem. Commun., 2013, 49, 5004 RSC.
- S. Canaguier, L. Vaccaro, V. Artero, R. Ostermann, J. Pécaut, M. J. Field and M. Fontecave, Chem. – Eur. J., 2009, 15, 9350 CrossRef CAS PubMed.
- S. Ogo, Chem. Commun., 2009, 3317 RSC.
- V. Fourmond, S. Canaguier, B. Golly, M. J. Field, M. Fontecave and V. Artero, Energy Environ. Sci., 2011, 4, 2417 CAS.
- L.-C. Song, J.-P. Li, Z.-J. Xie and H.-B. Song, Inorg. Chem., 2013, 52, 11618 CrossRef CAS PubMed.
- T. Matsumoto, Y. Nakaya, N. Itakura and K. Tatsumi, J. Am. Chem. Soc., 2008, 130, 2458 CrossRef CAS PubMed.
- M. R. DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62 RSC.
- E. S. Wiedner, J. Y. Yang, W. G. Dougherty, W. S. Kassel, R. M. Bullock, M. R. DuBois and D. L. DuBois, Organometallics, 2010, 29, 5390 CrossRef CAS.
- T. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228 CrossRef CAS PubMed.
- S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, M. Stein and S. Ott, Angew. Chem., Int. Ed., 2010, 49, 8033 CrossRef CAS PubMed.
- W. Zhu, A. C. Marr, Q. Wang, F. Neese, D. J. E. Spencer, A. J. Blake, P. A. Cooke, C. Wilson and M. Schröder, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18280 CrossRef CAS PubMed.
- G. Davidson, S. B. Choudhury, Z. Gu, K. Bose, W. Roseboom, S. P. J. Albracht and M. J. Maroney, Biochemistry, 2000, 39, 7468 CrossRef CAS PubMed.
- L.-C. Song, Y.-L. Li, L. Li, Z.-C. Gu and Q.-M. Hu, Inorg. Chem., 2010, 49, 10174 CrossRef CAS PubMed.
- L. Li, L.-C. Song, M.-M. Wang, Q.-L. Li and H.-B. Song, Organometallics, 2011, 30, 4899 CrossRef CAS.
- L.-C. Song, L. Li, Y.-Y. Hu and H.-B. Song, J. Organomet. Chem., 2013, 743, 123 CrossRef CAS PubMed.
- M. E. Carroll, B. E. Barton, D. L. Gray, A. E. Mack and T. B. Rauchfuss, Inorg. Chem., 2011, 50, 9554 CrossRef CAS PubMed.
- D. Schilter, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2012, 51, 8931 CrossRef CAS PubMed.
- D. Schilter, M. J. Nilges, M. Chakrabarti, P. A. Lindahl, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2012, 51, 2338 CrossRef CAS PubMed.
- M. T. Stiebritz and M. Reiher, Inorg. Chem., 2010, 49, 5818 CrossRef CAS PubMed.
- J. Jiang, M. Maruani, J. Solaimanzadeh, W. Lo, S. A. Koch and M. Millar, Inorg. Chem., 2009, 48, 6359 CrossRef CAS PubMed.
- Y. Ohki, K. Yasumura, M. Ando, S. Shimokata and K. Tatsumi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3994 CrossRef CAS PubMed.
- D. Schilter and T. B. Rauchfuss, Dalton Trans., 2012, 41, 13324 RSC.
- A. Perra, Q. Wang, A. J. Blake, E. S. Davies, J. McMaster, C. Wilson and M. Schröder, Dalton Trans., 2009, 925 RSC.
- Y. Higuchi, T. Yagi and N. Yasuoka, Structure, 1997, 5, 1671 CrossRef CAS.
- K. A. Vincent, N. A. Belsey, W. Lubitz and F. A. Armstrong, J. Am. Chem. Soc., 2006, 128, 7448 CrossRef CAS PubMed.
- B. E. Barton and T. B. Rauchfuss, J. Am. Chem. Soc., 2010, 132, 14877 CrossRef CAS PubMed.
- B. E. Barton, M. Whaley, T. B. Rauchfuss and D. L. Gray, J. Am. Chem. Soc., 2009, 131, 6942 CrossRef CAS PubMed.
- H. S. Shafaat, K. Weber, T. Petrenko, F. Neese and W. Lubitz, Inorg. Chem., 2012, 51, 11787 CrossRef CAS PubMed.
- J. A. W. Verhagen, D. D. Ellis, M. Lutz, A. L. Spek and E. Bouwman, J. Chem. Soc., Dalton Trans., 2002, 1275 RSC.
- J. A. S. Howell, B. F. G. Johnson, P. L. Josty and J. J. Lewis, J. Organomet. Chem., 1972, 39, 329 CrossRef CAS.
- K. Weber, T. Krämer, H. S. Shafaat, T. Weyhermüller, E. Bill, M. van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 20745 CrossRef CAS PubMed.
- L. De Gioia, P. Fantucci, B. Guigliarelli and P. Bertrand, Inorg. Chem., 1999, 38, 2658 CrossRef CAS.
- S. Niu, L. M. Thomson and M. B. Hall, J. Am. Chem. Soc., 1999, 121, 4000 CrossRef CAS.
- S. Li and M. B. Hall, Inorg. Chem., 2001, 40, 18 CrossRef CAS.
- F. Dole, A. Fournel, V. Magro, E. C. Hatchikian, P. Bertrand and B. Guigliarelli, Biochemistry, 1997, 36, 7847 CrossRef CAS PubMed.
- S. Canaguier, M. Field, Y. Oudart, J. Pécaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876 RSC.
- L. Vaccaro, V. Artero, S. Canaguier, M. Fontecave and M. J. Field, Dalton Trans., 2010, 39, 3043 RSC.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152 CrossRef CAS PubMed.
- P. Amara, A. Volbeda, J. C. Fontecilla-Camps and M. J. Field, J. Am. Chem. Soc., 1999, 121, 4468 CrossRef CAS.
- L. De Gioia, P. Fantucci, B. Guigliarelli and P. Bertrand, Int. J. Quantum Chem., 1999, 73, 187 CrossRef CAS.
- R. Angamuthu and E. Bouwman, Phys. Chem. Chem. Phys., 2009, 11, 5578 RSC.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682 CrossRef CAS PubMed.
- B. Kure, T. Matsumoto, K. Ichikawa, S. Fukuzumi, Y. Higuchi, T. Yagi and S. Ogo, Dalton Trans., 2008, 4747 RSC.
- T. Matsumoto, B. Kure and S. Ogo, Chem. Lett., 2008, 37, 970 CrossRef CAS.
- H. Ogata, W. Lubitz and Y. Higuchi, Dalton Trans., 2009, 7577 RSC.
- B. C. Manor and T. B. Rauchfuss, J. Am. Chem. Soc., 2013, 135, 11895 CrossRef CAS PubMed.
- J. W. Raebiger, A. Miedaner, C. J. Curtis, S. M. Miller, O. P. Anderson and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 5502 CrossRef CAS PubMed.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843 CrossRef CAS PubMed.
- T. Matsumoto, N. Itakura, Y. Nakaya and K. Tatsumi, Chem. Commun., 2011, 47, 1030 RSC.
- Y.-C. Liu, K.-T. Chu, R.-L. Jhang, G.-H. Lee and M.-H. Chiang, Chem. Commun., 2013, 49, 4743 RSC.
- T. R. Simmons and V. Artero, Angew. Chem., Int. Ed., 2013, 52, 6143 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métaye, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997 CrossRef CAS PubMed.
- D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef PubMed.
- W. Zhang, J. Hong, J. Zheng, Z. Huang, J. Zhou and R. Xu, J. Am. Chem. Soc., 2011, 133, 20680 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |